

Abstract
Apixaban is approved for treatment of venous thromboembolism (VTE) and prevention of recurrence. Population pharmacokinetics, pharmacokinetics–pharmacodynamics (anti‐FXa activity), and exposure–response (binary bleeding and thromboembolic endpoints) of apixaban in VTE treatment subjects were characterized using data from phase I–III studies. Apixaban pharmacokinetics were adequately characterized by a two‐compartment model with first‐order absorption and elimination. Age, sex, and Asian race had less than 25% impact on exposure, while subjects with severe renal impairment were predicted to have 56% higher exposure than the reference subject (60‐year‐old non‐Asian male weighing 85 kg with creatinine clearance of 100 mL/min). The relationship between apixaban concentration and anti‐FXa activity was described by a linear model with a slope estimate of 0.0159 IU/ng. The number of subjects with either a bleeding or thromboembolic event was small, and no statistically significant relationship between apixaban exposure and clinical endpoints could be discerned with a logistic regression analysis.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ The efficacy and safety of apixaban for the treatment of VTE and prevention of recurrent VTE have been demonstrated based on results from phase II and phase III studies in which pharmacokinetic and pharmacodynamic data were collected.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The pharmacokinetics and pharmacodynamics of apixaban are described in VTE treatment subjects. In addition, the relationship between apixaban exposure and safety and efficacy outcomes in this population were explored.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ Apixaban exposure in VTE treatment subjects was adequately characterized by a two‐compartment population pharmacokinetic model with first‐order absorption and elimination. This analysis supports the dose recommendation in VTE treatment, as no dose adjustment for apixaban is required based on individual intrinsic factors such as age, sex, race, and renal impairment.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ Reference apixaban exposure and anti‐FXa activity values in this population can help to inform clinical decisions in exceptional situations such as overdose and emergency surgery.
Apixaban is an orally active, selective, and direct reversible inhibitor of the coagulation factor Xa (FXa). It is approved in a number of countries for the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), and for the reduction in the risk of recurrent DVT and PE following initial treatment (hereafter referred to as venous thromboembolism (VTE) treatment).1, 2 Efficacy and safety of apixaban for VTE treatment have been demonstrated in two pivotal phase III studies,1, 2 the AMPLIFY study for acute VTE treatment in subjects with an objectively documented index event of symptomatic, proximal DVT or symptomatic PE, and the AMPLIFY‐EXT study for prevention of recurrent VTE in subjects who had completed ∼6–12 months of anticoagulant therapy for treatment of the index event. These studies demonstrated that the benefit–risk profile of apixaban offers a significant improvement over the current standard of care for subjects requiring treatment of VTE and prevention of recurrence.3
Apixaban exhibits a pharmacokinetic profile characterized by an oral bioavailability of ∼50%, no clinically significant food effect, dose‐proportional increases in exposure over the clinical dose range, and no evident time dependency. It is eliminated by renal and nonrenal pathways including metabolism, biliary excretion, and direct intestinal excretion, with renal clearance accounting for ∼27% of total systemic clearance,4, 5, 6, 7, 8, 9 and a half‐life of ∼12 h. Apixaban is predominantly metabolized by cytochrome P450 3A4 (CYP3A4), with only minor contributions from CYP1A2, 2C8, 2C9, 2C19, and 2J2, with subsequent sulfation by sulfotransferases and is also a substrate for P‐glycoprotein (P‐gp) and breast cancer resistance protein (BCRP).10, 11 Because of the multiple elimination pathways, the potential for comedications to impact the exposure of apixaban is limited. Studies conducted in healthy subjects observed a 2‐fold increase in exposure after coadministration with ketoconazole, a strong inhibitor of both CYP3A4 and P‐gp,12 and a 50% decrease in exposure after coadministration with rifampin, a strong inducer of both CYP3A4 and P‐gp.9 The pharmacodynamic effects of apixaban in clinical studies were consistent with its proposed primary mechanism of action, direct reversible inhibition of FXa. Anti‐FXa activity has been shown to be a more sensitive and precise method for assessing the pharmacodynamic effect of apixaban than other clotting measures.13
The objectives of the present analyses were to describe the pharmacokinetics and pharmacodynamics of apixaban, and to explore the relationship between apixaban exposure and safety and efficacy endpoints in VTE treatment subjects.
METHODS
Study populations and data
All study protocols, their amendments, and informed‐consent documentation for studies included in the analyses were reviewed and approved by Institutional Review Boards, and were conducted in accordance with the codes and guidelines set forth in the Declaration of Helsinki, Good Clinical Practice, and local regulations.
The population pharmacokinetic and pharmacokinetic–pharmacodynamic analyses utilized intensive and sparse data collected in eight phase I studies,14, 15, 16, 17, 18, 19, 20, 21 one phase II DVT study,22 and two phase III VTE treatment clinical trials (Table 1).1, 2 Two blood samples at steady state (Weeks 3 and 12) were collected for measurement of apixaban concentration and anti‐FXa activity in all apixaban‐treated subjects in the phase II study.22 Approximately 10% of the apixaban‐treated subjects in the phase III studies provided apixaban concentrations and anti‐FXa activity data collected ∼2 h prior to dosing (−2), predose (0), and 2‐ and 4‐h postdose at steady state. For eight subjects in the AMPLIFY‐EXT study, serial blood samples were collected at predose, and 1, 2, 3, 4, 6, 10, and 12 h after dosing on one occasion, scheduled at any time between Week 2 and Month 3. Anti‐FXa activity was measured in five of the eight phase I studies. The exposure–response analyses utilized data from subjects with at least one apixaban concentration in the phase II/III studies.
Table 1.
Summary of the studies and data used for population pharmacokinetic and pharmacokinetic–pharmacodynamic analyses
| Study type |
Target population |
Apixaban dose and regimen | Treatment duration | Number of apixaban concentration observations (no. subjects) |
Number of anti‐FXa activity observations (no. subjects) |
|
|---|---|---|---|---|---|---|
| Phase I studiesa | ||||||
| Multiple doses16 | Healthy subjects |
2.5–25 mg b.i.d. 10–25 mg q.d. |
7 days | 1,052 (36) | 0 | |
| Single dose17 | Healthy Caucasian and Japanese subjects | 2.5, 10, 25, and 50 mg | Single dose | 1,440 (24) | 0 | |
| Age and gender15 | Healthy young and elderly male and female subjects | 20 mg single dose | Single dose | 1,121 (79) | 0 | |
| Multiple doses18 | Healthy Japanese subjects | 2.5, 5, and 10 mg b.i.d. | 7 days | 639 (18) | 492 (18) | |
| Multiple doses19 | Healthy Chinese subjects | 10 mg single dose, then 10 mg b.i.d. | Single dose, then 6 days | 356 (12) | 92 (12) | |
| Body weight20 | Healthy subjects of high, normal, and low body weight | 10 mg | Single dose | 693 (55) | 99 (55) | |
| Renal impairment14 | Healthy and renally impaired subjects | 10 mg single dose | Single dose | 523 (32) | 189 (32) | |
| Multiple doses21 | Healthy subjects | 2.5 mg b.i.d. | 4 days | 296 (14) | 272 (14) | |
| Phase II study | ||||||
| Botticelli22 | DVT treatment subjects | 5 and 10 mg b.i.d.‡ | 3 months | 456 (241) | 452 (240) | |
| Phase III studies | ||||||
| AMPLIFY1 | DVT and PE treatment subjects | 10 mg b.i.d. for 7 days followed by 5 mg b.i.d. | 6 months | 1,044 (281) | 931 (253) | |
| AMPLIFY‐EXT2 | Prevention of recurrent DVT and PE subjects | 2.5 and 5 mg b.i.d. | 12 months | 703 (178) | 612 (171) | |
| Total | 8,323 (970) | 3,139 (795) | ||||
b.i.d., twice daily; DVT, deep vein thrombosis; q.d., once daily; VTE, venous thromboembolism.
aThe 20 mg q.d. data (124 subjects) were not included as time since last active dose could not be unambiguously distinguished from time since last placebo dose.
Pharmacokinetic and anti‐FXa activity assays
Apixaban concentration was determined using a validated liquid chromatography tandem mass spectroscopy method (Intertek Pharmaceutical Service, El Dorado Hills, CA). The lower limit of quantification was 1 ng/mL.23 Anti‐FXa activity was measured at Esoterix Coagulation Laboratory (Englewood, CO) using the Diagnostica Stago Rotachrom Heparin assay on a STA‐Compact analyzer (Diagnostica Stago, Parsippany, NJ)24 and is reported in low‐molecular‐weight heparin (LMWH) units (0.1 to 18 IU/mL).
Population pharmacokinetic and pharmacokinetic–pharmacodynamic analyses
Based on prior knowledge,25, 26 a two‐compartment model with first‐order absorption and elimination was fitted to the observed apixaban concentration vs. time data. The effect of renal function (creatinine clearance based on the Cockcroft–Gault (C‐G) formula27) on apparent oral clearance (CL/F) was incorporated in the base model after separation of CL/F into renal (CLR/F) and nonrenal (CLNR/F) components. To avoid the limitations of the C‐G formula for extremes of body weight, a breakpoint value of 150 mL/min was included, as shown below:
where CLi/F is apparent oral clearance for individual i; CLNR/F is the non‐renal component of CLi/F; CLR/F is the renal component of CLi/F; cCrCLi is the creatinine clearance using the C‐G formula for individual i; cCrCLref is the reference value of 100 mL/min; FLAG is a binary indicator which is 1 when cCrCLi is greater than 150 mL/min, otherwise 0.
Based on the less than proportional exposure with doses greater than 10 mg, likely due to dissolution rate‐limited absorption, considering the solubility of apixaban (0.04 mg/mL),7 a reduction in relative bioavailablity (Frel) was also incorporated in the base model using an inhibitory sigmoid maximum drug effect (Emax) function.26
Once the base model was established, the covariates in Table 2 were included simultaneously to form a full model based on prior knowledge.25, 26, 28 In addition to creatinine clearance on CLR/F in the base model, age and sex were further included on CLNR/F, while body weight on CLNR/F was not evaluated, based on previous analyses.25, 26 The impact of Asian race on CL/F of apixaban was evaluated. A potential difference in apixaban pharmacokinetics between VTE treatment subjects and phase I subjects was assessed as a binary subject‐status covariate on CL/F and apparent volume of distribution of the central compartment (Vc/F). Concomitant use of a strong or moderate inhibitor of CYP3A4/P‐gp was included on CL/F. As strong inhibitors of CYP3A4/P‐gp were prohibited from phase II/III studies, this was expected to reflect primarily concomitant use of moderate inhibitors. After evaluating the results of the full model, covariates were removed to define the final model if the impact was found to be neither clinically nor statistically significant, based on assessment of covariate parameter estimates and their 95% confidence intervals (CIs). Continuous covariates were normalized on a typical reference value and included in the model using a power function, and categorical covariate effects were parameterized as a fractional change:
where TVP is the typical value of a model parameter; covmi is the individual continuous covariate; covpi is the individual categorical covariate; θn is an estimated parameter describing the typical pharmacokinetic parameter value for an individual with covariates equal to the reference covariate values (covmi = refm, covpi = 0); θ(m+n) and θ(p+m+n) are estimated parameters describing the magnitude of the covariate–parameter relationships.
Table 2.
Covariate–pharmacokinetic parameter relationships evaluated in the full population pharmacokinetic model (+ indicates the covariate was included on the parameter)
| Covariate | Apparent renal clearance (CLR/F) | Apparent nonrenal clearance (CLNR/F) | Apparent total clearance (CL/F) | Apparent volume of central compartment (Vc/F) |
Absorption rate (Ka) |
|---|---|---|---|---|---|
| Dosing time (diurnal variationa) | NT | NT | NT | NT | + |
| Age | cCrCLb | + | NT | NT | NT |
| Sex | cCrCLb | + | NT | NT | NT |
| Body weight | cCrCLb | NT | NT | + | NT |
| Asian race | NT | NT | + | NT | NT |
| Subject status | NT | NT | + | + | NT |
| Strong or moderate CYP3A4/P‐gp inhibitor | NT | NT | + | NT | NT |
cCrCL, creatinine clearance using Cockcroft‐Gault formula; CYP3A4, cytochrome P450; P‐gp, P‐glycoprotein; NT, not tested.
aMorning administration was defined as a dose taken between 4 am to 11 am (inclusive) while evening administration was defined as a dose taken between 5 pm to 12 am (inclusive).
bBase model included cCrCL on CLR/F, which includes age, sex, and body weight in the calculation.
Based on the relationship estimated from prior analyses,25, 26 a linear model with an intercept of zero was applied to the concentration–anti‐FXa activity data using a simultaneous pharmacokinetic–pharmacodynamic analysis.
Interindividual random effects were described by an exponential error model. The estimate of interindividual variance (IIV) was provided as percent coefficient of variation (CV%). The CV% was estimated as . However, if ω2 was estimated to be greater than 0.16, CV% was calculated as .29 For pharmacokinetic observations, the residual error was described by an additive error model with a log transform‐both‐sides approach.
Simulation of apixaban exposure and anti‐FXa activity in VTE treatment population
To generate predictions of apixaban exposure and anti‐FXa activity, a nonparametric bootstrap using the final population pharmacokinetic model was performed to create 1,000 sets of population pharmacokinetic parameter values from which 1,000 simulated datasets were generated.30 For each of the 1,000 simulated datasets, steady‐state apixaban concentration vs. time profiles were simulated in 700 VTE treatment subjects for each dose regimen of 2.5 mg, 5 mg, and 10 mg b.i.d., using the observed demographic information from 700 subjects in the phase II/III studies to preserve the correlation structure among the covariates. Anti‐FXa activity vs. time profiles were generated by multiplying the simulated apixaban concentrations by the slope parameter estimate from the pharmacokinetic–pharmacodynamic analysis. The median and 5th and 95th percentiles and their 90% CIs were determined for steady‐state daily (0–24 h) area under the apixaban concentration vs. time curve (AUCss), maximum concentration (Cmax), and minimum concentration (Cmin), along with anti‐FXa activity associated with the Cmax and Cmin for each regimen.
Exploratory exposure–response evaluation for bleeding and efficacy endpoints
Subject‐specific predictions of the steady‐state total daily (0–24 h) apixaban exposure (AUCss) to be used in the exposure–response analyses were obtained from the empirical Bayes' predictions of each subject's CL/F value from the final population pharmacokinetic model and total daily dose (AUCss = 2·F·Dose/CL). The following bleeding and efficacy endpoints that occurred during the treatment period, defined as the period from first dose through 2 days after discontinuation of study drug, were analyzed as dichotomous categorical variables representing the occurrence of an event (1 = yes, 0 = no) for any relationship with apixaban exposure (daily AUCss) using a logistic regression model1, 2:
Bleeding endpoint: composite of adjudicated major or adjudicated clinically relevant nonmajor bleeding.
Efficacy endpoint: adjudicated symptomatic VTE or VTE‐related death.
The probability of an event occurring vs. the probability that it does not occur is given by the odds; the log of the odds is known as the logit as shown in the equation below:
where pi denotes the probability of an event occurring for an individual (i); Ii is an indicator variable for subject i taking the value of 1 if the subject has an event and 0 otherwise; β denotes the baseline logit‐probability for the incidence of events (stratified by index event of symptomatic proximal DVT or symptomatic PE); fdrug denotes the function describing the exposure–response relationship.
Considering the difference in population between acute treatment of VTE and prevention of recurrent VTE, one model was developed for the acute treatment studies and another model for the prevention of recurrent VTE study. In addition, the baseline logit‐probability for the incidence of events was stratified by index event of DVT or PE.
Assessment of model adequacy and predictive performance
Assessment of the model goodness‐of‐fit was conducted based on standard goodness‐of‐fit criteria,31 including successful minimization of the objective function, visual inspection of several diagnostic plots of residuals and empirical Bayes predictions of the interindividual random effects, change in the objective function relative to the change in number of parameters, the magnitude and precision of the parameter estimates, as well as changes in both interindividual and residual variability. These diagnostic plots were stratified by dose and study to verify the adequacy of pooling data across studies and doses. The adequacy of the final model and parameter estimates was assessed with a visual predictive check method32 to determine whether the final model could reproduce the observed data from which it was generated. The stability of the models was evaluated throughout the model development process.
Plasma concentration vs. time data were modeled using a population analysis approach with the first‐order conditional estimation with interaction method (NONMEM software system, v. 7.2, Icon Development Solutions, San Antonio, TX,31 and the NM‐TRAN subroutines version III level 1.1, and the PREDPP model library, version IV level 1.1). Postprocessing of NONMEM output to generate goodness‐of‐fit plots and predictive checks and logistic regression analyses were performed using R software (v. 2.15.2; http://www.r-project.org). Visual predictive checks and simulations were conducted using Perl‐Speaks‐NONMEM (PsN v. 3.5.4, http://psn.sourceforge.net/), and Xpose (v. 4.4, http://xpose.sourceforge.net/) was utilized for plotting of simulation results.
RESULTS
Population pharmacokinetic and pharmacokinetic–pharmacodynamic analyses
The population pharmacokinetic analysis used 8,323 apixaban concentrations from 970 subjects. Based on the demographics in VTE treatment subjects (Table 3), reference covariate values for a typical VTE treatment subject were set to: 60‐year‐old non‐Asian male, body weight of 85 kg, and cCrCL of 100 mL/min.
Table 3.
Summary of baseline demographic covariates for population pharmacokinetic analysis dataset
|
Phase I subjects (N = 270) |
VTE treatment subjects (N = 700) |
|
|---|---|---|
| Age (years), n (%) | ||
| Median (min–max) | 33 years (18–85) | 61 years (18–89) |
| < 65 years | 216 (80%) | 440 (63%) |
| 65 to < 75 years | 47 (17%) | 143 (20%) |
| ≥ 75 years | 7 (3%) | 117 (17%) |
| Level of renal impairment, n (%) | ||
| Median (min–max) | 112.8 mL/min (15–318) | 99.2 mL/min (25.3–322) |
| Normal (cCrCL > 80 mL/min) | 216 (80%) | 478 (68%) |
| Mild (50 ≤ cCrCL ≤ 80 mL/min) | 38 (14%) | 161 (23%) |
| Moderate (30 ≤ cCrCL < 50 mL/min) | 8 (3%) | 58 (8%) |
| Severe (15 ≤ cCrCL < 30 mL/min) | 8 (3%) | 3 (0.4%) |
| Body weight (kg), n (%) | ||
| Median (min–max) | 71.2 kg (37.7–175) | 84 kg (46.9–210) |
| ≤ 60 kg | 47 (17%) | 43 (6%) |
| > 60 to < 100 kg | 199 (74%) | 514 (73%) |
| ≥ 100 kg | 24 (9%) | 143 (20%) |
| Sex | 67% Male | 60.3% Male |
| Race, n (%) | ||
| White | 181 (67%) | 636 (91%) |
| Black/African American | 30 (11%) | 16 (2%) |
| Asian | 49 (18%) | 8 (1%) |
| Other | 10 (4%) | 18 (3%) |
| Missing | 0 | 22 (3%) |
cCrCL, creatinine clearance using Cockcroft‐Gault formula; VTE, venous thromboembolism.
Apixaban pharmacokinetics were adequately described with a two‐compartment model with first‐order absorption and elimination. The full model parameters were generally estimated with reasonable precision except for the subject‐status covariate parameters, which were neither statistically significant (95% CIs included zero) nor clinically significant (3.5% increase in CL/F and 7.6% decrease in Vc/F for the VTE treatment subjects compared to healthy subjects). Therefore, these covariates were removed from the final model.
The parameter estimates for the final model are listed in Table 4 . One thousand simulations were performed with the final model to confirm that the observed median (solid red line) was generally contained within the 90% CI around the median of simulated data (pink‐shaded area) (Figure 1). The 5th and 95th percentiles of observed data generally fell within the 90% prediction interval (blue shaded areas) for phase II/III data.
Table 4.
Parameter estimates in the final population pharmacokinetic model
| Fixed effects parameters | Estimate | SE | Description |
|---|---|---|---|
| ka (1/hr) | 0.440 | 0.0209 | Administration of apixaban in the evening resulted in a 46% decrease relative to administration in the morning or afternoon |
| An evening dose | 0.239 | 0.0126 | |
| CLR/Fa (L/hr) | 1.83 | 0.169 | VTE treatment subjects with mild (cCrCL of 65 mL/min), moderate (cCrCL of 40 mL/min), and severe (cCrCL of 15 mL/min) renal impairment would have approximately 35%, 60%, and 85% lower CLR/F, respectively, than a reference VTE treatment subject with normal renal function (cCrCL of 100 mL/min) |
| CLNR/F (L/hr) | 2.52 | 0.162 | |
| Agea | −0.267 | 0.0503 | For example, a 40‐year‐old and 80‐year‐old male VTE treatment subject would have 11% higher and 7% lower CLNR/F relative to a reference VTE treatment male subject who is 60 years old |
| Female | −0.223 | 0.0331 | Female subjects had 22.3% lower CLNR/F relative to male subjects, resulting in 13% lower CL/F, assuming no change in CLR/F |
| CL/F | |||
| Asian subjects | −0.168 | 0.0374 | Asian race resulted in a decrease of 16.8% in CL/F |
| Strong/moderate CYP3A4/P‐gp Inhibitors | −0.203 | 0.051 | Concomitant use of strong or moderate CYP3A4/P‐gp inhibitors resulted in a decrease of 20.3% in CL/F |
| Vc/F (L) | 32.1 | 1.16 | |
| Body weighta | 0.523 | 0.0694 | The effect of baseline body weight on Vc/F was less than directly proportional, with a 24% reduction for a 50‐kg subject and a 20% increase for a 120‐kg subject relative to the reference subject with a body weight of 85 kg |
| Q/F (L/hr) | 1.62 | 0.125 | |
| Vp/F (L) | 19.8 | 1.3 |
CrCL, creatinine clearance using Cockcroft‐Gault formula; CL/F, apparent total clearance; CLNR/F, apparent non‐renal clearance; CLR/F, apparent renal clearance; Ka, absorption rate constant; Q/F, apparent intercompartmental clearance; SE, standard error; Vc/F, apparent volume of central compartment; Vp/F, apparent volume of peripheral compartment; VTE, venous thromboembolism; CYP3A4, Cytochrome P450 3A4; P‐gp, P‐glycoprotein.
aCentered at reference VTE treatment subject values: 60 years, 85 kg with cCrCL = 100 mL/min.
Figure 1.
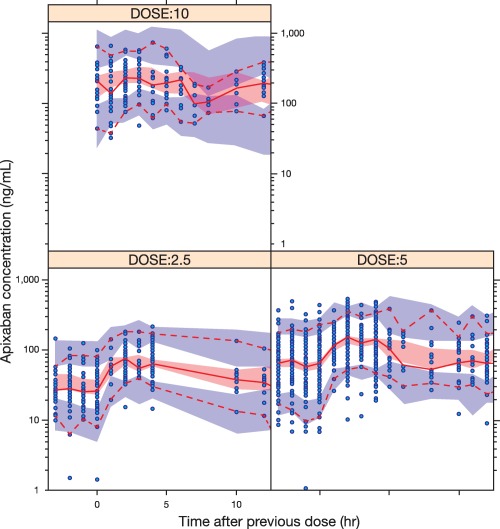
Apixaban concentration–time profile at steady‐state in VTE treatment subjects and visual predictive check. VTE, venous thromboembolism. Open circles represent observed apixaban concentrations; the red solid line represents the median of the observed data and the red dashed lines the 5th and 95th percentiles of the observed data; the shaded areas represent 90% confidence intervals around the 5th (blue, bottom), median (pink), and 95th (blue, top) percentiles of the simulated data.
For a typical VTE treatment subject (60‐year‐old non‐Asian male weighing 85 kg with a cCrCL of 100 mL/min), CLR/F and CLNR/F were estimated to be 1.83 L/hr and 2.52 L/hr, respectively (CL/F = 4.35 L/hr). The estimates of Vc/F, apparent intercompartment clearance (Q/F), and apparent volume of distribution of the peripheral compartment (Vp/F) were 32.1 L, 1.62 L/hr, and 19.8 L, respectively. Apixaban CL/F was affected by sex, age, renal function, Asian race, and inhibitors of CYP3A4/P‐gp with the magnitude of effect generally less than 30%, except for severe renal impairment, which resulted in a 36% decrease in CL/F. The IIV was 50.2%, 23.5%, and 33.1% for the first‐order absorption rate constant (ka), Vc/F, and CL/F, respectively. The shrinkage in CL/F for subjects in the phase I, II, and II studies was calculated to be 31.1%, 48.1%, and 30.0%, respectively.
The pharmacokinetic–pharmacodynamic analysis used 3,139 anti‐FXa activity observations from 795 subjects (Supplementary Figure 1). The slope of the linear relationship between the model‐predicted apixaban plasma concentration and anti‐FXa activity was estimated to be 0.0159 IU/ng. The estimate of IIV for the slope was found to be close to zero and therefore was removed from the final model.
Simulation of apixaban exposure and anti‐FXa activity values in VTE treatment population
A summary of simulated median and 5th and 95th percentiles of steady‐state apixaban exposure and corresponding anti‐FXa activity is presented in Table 5 for the 2.5‐mg, 5‐mg, and 10‐mg b.i.d. doses in VTE treatment subjects. As the final population pharmacokinetic model for apixaban incorporated a separate apixaban absorption rate for morning vs. evening dosing, the Cmax and Cmin represent the daily maximum and minimum values. Apixaban Cmax, Cmin, and daily AUCss increased proportionally following oral administration across the dose range of 2.5–10 mg.
Table 5.
Predicted apixaban steady‐state exposure and anti‐FXa activity in VTE treatment population
|
Steady‐state parameter (units) |
2.5 mg b.i.d. | 5 mg b.i.d. | 10 mg b.i.d. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Median (90% CI) | 5th Percentile (90% CI) | 95th Percentile (90% CI) | Median (90% CI) | 5th Percentile (90% CI) | 95th Percentile (90% CI) | Median (90% CI) | 5th Percentile (90% CI) | 95th Percentile (90% CI) | |
| Daily (0‐24 hours) AUCss (ng*hr/mL) |
1,240 (1,185, 1,301) |
655 (606, 707) |
2,437 (2,240, 2,649) |
2,446 (2,346, 2,554) |
1,293 (1,197, 1,398) |
4,807 (4,433, 5,174) |
4,649 (4,439, 4,875) |
2,456 (2,271, 2,664) |
9,136 (8,445, 9,836) |
|
Cmax
(ng/mL) |
67.0 (63.7, 70.7) |
29.7 (26.9, 33.1) |
153.2 (138.9, 168.4) |
132.3 (125.2, 139.3) |
58.6 (52.9, 64.7) |
302.2 (273.3, 331.9) |
251.2 (237.5, 265.2) |
111.4 (100.2, 123.2) |
572.4 (516.0, 630.1) |
|
Cmin
(ng/mL) |
32.0 (29.9, 34.2) |
11.0 (9.4, 12.6) |
89.5 (79.6, 100.0) |
63.2 (59.0, 67.9) |
21.7 (18.8, 25.3) |
176.5 (156.5, 196.2) |
120.2 (112.1, 129.2) |
41.1 (35.0, 47.6) |
334.5 (295.8, 378.8) |
| Anti‐FXa activity at Cmax (IU/mL) |
1.07 (1.01, 1.12) |
0.47 (0.43, 0.53) |
2.44 (2.21, 2.68) |
2.10 (1.99, 2.21) |
0.93 (0.84, 1.03) |
4.80 (4.35, 5.28) |
3.99 (3.78, 4.22) |
1.77 (1.59, 1.96) |
9.10 (8.20, 10.02) |
| Anti‐FXa activity at Cmin (IU/mL) |
0.51 (0.48, 0.54) |
0.17 (0.15, 0.20) |
1.42 (1.27, 1.59) |
1.00 (0.94, 1.08) |
0.35 (0.30, 0.40) |
2.81 (2.49, 3.12) |
1.91 (1.78, 2.05) |
0.65 (0.56, 0.76) |
5.32 (4.70, 6.02) |
AUCss, area under the concentration–time curve; b.i.d., twice daily; CI, confidence interval; Cmax, peak plasma concentration; Cmin, trough plasma concentration; VTE, venous thromboembolism.
Exploratory exposure–response evaluation for bleeding and efficacy endpoints
The exposure–response analysis dataset included 700 subjects with at least one apixaban concentration in the phase II/III studies. There were five VTE/VTE‐related death events and 25 bleeding events among 522 subjects from the acute VTE treatment studies, and one VTE/VTE‐related death event and eight bleeding events among 178 subjects from the AMPLIFY‐EXT study.
The logistic regression analyses found no statistically significant relationship between apixaban daily AUCss and clinical endpoints (P‐value for any slope estimate >0.1) and thus no further model development was performed. As shown in Figure 2 , the predicted median steady‐state Cmax and Cmin values and their corresponding anti‐FXa activity values were numerically higher for those with bleeding events, and lower for those with efficacy events, than those without events; however, the range of exposure from those subjects with efficacy and bleeding endpoints was entirely contained within the range of exposure from those subjects without events.
Figure 2.
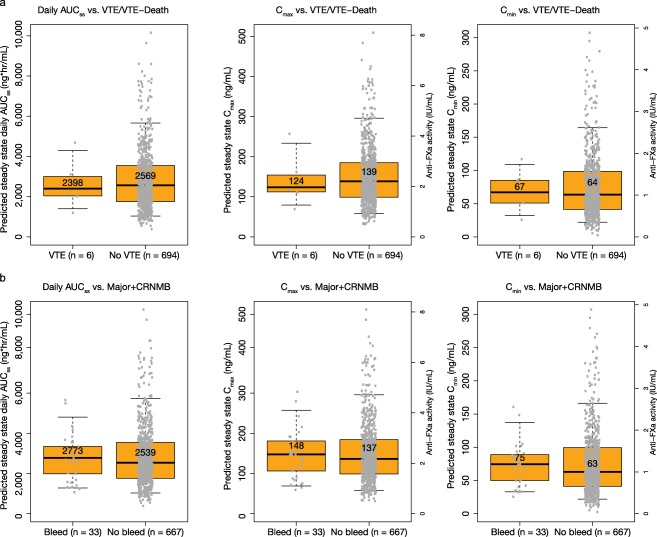
Predicted apixaban steady state daily AUCss, Cmax, and Cmin and anti‐FXa activity values associated with Cmax and Cmin in VTE treatment phase II/III subjects with and without efficacy (a) or bleeding (b) events. AUC, area under the concentration–time curve; CRNMB, clinically relevant nonmajor bleeding; Cmax, peak plasma concentration; Cmin, trough plasma concentration; VTE, venous thromboembolism. Note that there are two y‐axes on each Cmax and Cmin figure: the left axis is for apixaban concentration and the right axis is for anti‐FXa activity. Boxes indicate 25th−75th percentiles, whiskers indicate 5th−95th percentiles, and black horizontal lines represent the median. Numbers inside boxes are median values. Circles are individual predicted values.
DISCUSSION
While the pharmacokinetic profile of apixaban was well characterized in phase I studies, it was important to confirm the findings from the phase I studies and to evaluate potential differences in the target population to inform appropriate dosing in the patient population. Baseline demographics in the VTE treatment subjects included in the pharmacokinetic analysis were similar to those in the overall apixaban‐treated population in the phase III studies. In AMPLIFY and AMPLIFY‐EXT,1, 2 the median age was 58 years and the majority of subjects were non‐Asian males. In addition, ∼60–70% of subjects had normal renal function and ∼20% and ∼5% had mild and moderate renal impairment, respectively. The similarity in demographics between the VTE treatment subjects included in the pharmacokinetic analysis and the overall apixaban‐treated population in the phase III studies indicates that the findings from this analysis are expected to be relevant to the general VTE treatment population.
There was no difference in apixaban pharmacokinetics between phase I subjects and VTE treatment subjects. Apixaban pharmacokinetics were adequately described with a two‐compartment model with first‐order absorption and first‐order elimination. While a 46% slower absorption was identified following evening administration relative to administration earlier in the day, this is not expected to be of clinical relevance for a drug administered twice daily. The estimated Vc/F of ∼32 L for a reference VTE treatment subject was consistent with ∼21 L from phase I studies,7, 8, 9 suggesting limited intracellular distribution.
Covariates that were predictive of apixaban CL/F included renal function, age, sex, Asian race, and concomitant administration of strong or moderate CYP3A4/P‐gp inhibitors, which were generally consistent with observations in the phase I studies and previous analyses, and did not result in differences in dosing recommendations. VTE treatment subjects with mild (cCrCL of 65 mL/min), moderate (cCrCL of 40 mL/min), and severe (cCrCL of 15 mL/min) renal impairment would be predicted to have ∼17%, ∼34%, and ∼56% higher AUC values, respectively, than a reference VTE treatment subject with normal renal function. The results are consistent with those from the phase I study where mild, moderate, and severe renal impairment was associated with an ∼16%, ∼29%, and ∼44% increase in apixaban exposure based on regression analysis, respectively.14 This magnitude of exposure increase was also consistent with the 27% renal contribution to total systemic clearance, assuming renal impairment does not affect other elimination processes. An ∼40% increase in AUC would have been expected in subjects with severe renal impairment, as renal clearance is expected to be close to zero in these subjects.
The impact of age and sex was modest: a 40‐year‐old and 80‐year‐old male VTE treatment subject would be predicted to have ∼6% lower and 5% higher daily AUC values, respectively, compared to a 60‐year‐old reference subject, assuming no change in apparent renal clearance; female subjects are predicted to have an ∼15% higher AUC than male subjects. In phase I studies, a modest impact of sex and age on apixaban exposure was also observed: females had a 15% higher AUC compared to males, and older subjects (≥65 years) had a 32% higher AUC compared to younger subjects (18–40 years), with the difference in renal function between the two age groups partly contributing to the observed difference in exposure.15 A modestly higher AUC (20%) is predicted for Asian subjects compared to non‐Asian subjects. Considering the small magnitude and similar pharmacokinetic results in Japanese and Chinese compared with non‐Asian subjects in the phase I studies,18, 19 the impact of Asian race alone is not considered clinically relevant.
Considering the elimination pathways of apixaban and the results of human drug–drug interaction studies, modulators of CYP3A4 and/or P‐gp activity were expected to influence apixaban clearance. The impact of strong inducers of CYP3A4/P‐gp was not evaluated due to the small number of subjects who took strong inducers with apixaban. The magnitude of the effect for strong or moderate inhibitors of CYP3A4/P‐gp (∼20% reduction on CL/F) was lower than that observed in the phase I clinical studies, where apixaban exposure following coadministration with ketoconazole and diltiazem (a moderate inhibitor of CYP3A4 and an inhibitor of P‐gp) was ∼100% and ∼40% higher, respectively, than that following administration of apixaban alone.12 There were limitations in the population pharmacokinetic dataset that may have prevented an adequate evaluation of the impact of these concomitant medications: few individuals were administered strong inhibitors and there was limited information regarding concomitant medication use, e.g., dose, dosing frequency, timing of administration relative to that of apixaban. Therefore, greater emphasis should be placed on the phase I results regarding the potential impact of CYP3A4/P‐gp inhibitors on apixaban exposure.
Although treatment with apixaban does not require routine monitoring of exposure, information on apixaban exposure or anti‐Factor Xa activity levels may be useful in exceptional situations where this knowledge may help to inform clinical decisions, e.g., overdose and emergency surgery.33 Therefore, the final population pharmacokinetic and pharmacokinetic–pharmacodynamic models were used to generate reference apixaban exposure and anti‐FXa activity values in VTE treatment subjects. The fluctuation between peak and trough concentrations was predicted to be ∼2‐fold. The predicted median daily AUCss for the 5‐mg b.i.d. regimen in the VTE treatment population (2,446 ng*hr/mL) was about 25% lower than that in nonvalvular atrial fibrillation (NVAF) subjects taking apixaban 5 mg b.i.d. (3,280 ng*hr/mL).25 This difference resulted from two factors: 1) NVAF subjects had a 14% lower CL/F compared to phase I subjects; and 2) the NVAF population was slightly older, with a lower cCrCL value compared to the VTE treatment population.
The exposure–response relationships for the bleeding and VTE/VTE‐related death endpoints were explored, but could not be characterized due to the small number of events among VTE treatment subjects with an apixaban concentration measurement. It should be noted that in the AMPLIFY study, apixaban was statistically superior to enoxaparin/warfarin for the primary safety endpoint of adjudicated major bleeding and had a consistently better bleeding profile compared to enoxaparin/warfarin across all bleeding categories: composite major and clinically relevant nonmajor bleeding, minor, and total bleeding.1 In addition, the number of major bleeding events was low and similar across the apixaban and placebo groups in AMPLIFY‐EXT.2 Considering that the range of individual predicted values of daily AUCss, Cmax, and Cmin and corresponding anti‐FXa activity values for subjects with VTE/VTE‐related death or bleeding events was entirely contained within the range of values from subjects without events, no discernable threshold levels could be identified that would predict better or worse safety, or efficacy outcomes for individual subjects.
Without a well‐defined therapeutic exposure range related to clinical endpoints for apixaban in the VTE treatment population, apixaban labels recommend either a reduction in the apixaban dose or avoidance of apixaban in patients receiving strong dual inhibitors of CYP3A4 and P‐gp, based only on the extent of the pharmacokinetic interaction. No dose adjustment is recommended for apixaban when concomitantly administered with moderate inhibitors of CYP3A4 or P‐gp. For intrinsic factors such as age, sex, race, weight, and renal impairment, no dose adjustment or cautionary use of apixaban is recommended, which is supported by subgroup analyses of the pivotal trials that showed maintained benefit–risk profiles of apixaban compared to that of the overall study population.1, 2 However, caution is warranted when multiple factors are present.
In conclusion, apixaban exposure in VTE treatment subjects was adequately characterized by a two‐compartment population pharmacokinetic model with first‐order absorption and elimination. Age, sex, Asian race, and concomitant administration of strong or moderate CYP3A4/P‐gp inhibitors had less than 25% impact on apixaban exposure, while subjects with severe renal impairment were predicted to have 56% higher exposure than the reference subject. The number of subjects with either a bleeding or thromboembolic event was small, and no statistically significant relationship between apixaban exposure and clinical endpoints could be discerned with a logistic regression analysis.
Supporting information
Supplementary Information s01
Supplementary Figure 1. Anti‐FXa activity in LMWH Units versus predicted apixaban plasma concentration
Supplementary Information s03
Acknowledgments
This study was funded by Bristol‐Myers Squibb and Pfizer Inc. Editorial support was provided by Andy Shepherd and Sandi Lusk at Caudex, funded by Bristol‐Myers Squibb and Pfizer Inc.
Disclosures
W.B., K.S., and R.B. are employees of Pfizer Inc., and own stock/stock options. C.F. is an employee of Bristol‐Myers Squibb, and owns stock/stock options. Part of this work was previously presented as a poster at the International Society of Pharmacometrics – 5th American Conference on Pharmacometrics 2014 and is published as an abstract (reference): Byon, W., Sweeney, K., Frost, C. & Boyd, R. Population pharmacokinetic analysis of apixaban in venous thromboembolism treatment patients. Poster T‐060. Presented at ACoP, Las Vegas, Nevada, Oct 12‐15, 2014.
Author Contributions
W.B., K.S., C.F., and R.A.B. wrote the article; W.B. analyzed the data.
Conflict of Interest
The authors declare no conflicts of interest.
References
- 1. Agnelli, G. et al Oral apixaban for the treatment of acute venous thromboembolism. N. Engl. J. Med. 369, 799–808 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Agnelli, G. et al Apixaban for extended treatment of venous thromboembolism. N. Engl. J. Med. 368, 699–708 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Kearon, C. et al Antithrombotic therapy for VTE disease: CHEST guideline. Chest 149, 315–352 (2016). [DOI] [PubMed] [Google Scholar]
- 4. Raghavan, N. et al Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab. Dispos. 37, 74–81 (2009). [DOI] [PubMed] [Google Scholar]
- 5. Wang, L. et al Tissue distribution and elimination of [14C]apixaban in rats. Drug Metab. Dispos. 39, 256–264 (2011). [DOI] [PubMed] [Google Scholar]
- 6. Zhang, D. et al Investigating the enteroenteric recirculation of apixaban, a factor Xa inhibitor: administration of activated charcoal to bile duct‐cannulated rats and dogs receiving an intravenous dose and use of drug transporter knockout rats. Drug Metab. Dispos. 41, 906–915 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Bristol‐Myers Squibb Company PI. Eliquis (apixaban) prescribing information. <http://packageinserts.bms.com/pi/pi_eliquis.pdf> (2014). Accessed 3 March 2016.
- 8. Frost, C. et al Apixaban, a direct factor Xa inhibitor: single‐dose pharmacokinetics and pharmacodynamics of an intravenous formulation [abstract 148]. J. Clin. Pharmacol. 48, 1132 (2008). [Google Scholar]
- 9. Vakkalagadda, B. et al Effect of rifampin on the pharmacokinetics of apixaban, an oral direct inhibitor of factor Xa. Am. J. Cardiovasc. Drugs 16, 119–127 (2016). [DOI] [PubMed] [Google Scholar]
- 10. Wang, L. et al In vitro assessment of metabolic drug‐drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab. Dispos. 38, 448–458 (2010). [DOI] [PubMed] [Google Scholar]
- 11. Zhang, D. et al Characterization of efflux transporters involved in distribution and disposition of apixaban. Drug Metab. Dispos. 41, 827–835 (2013). [DOI] [PubMed] [Google Scholar]
- 12. Frost, C.E. et al Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Br. J. Clin. Pharmacol. 79, 838–846 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barrett, Y.C. , Wang, Z. , Frost, C. & Shenker, A. Clinical laboratory measurement of direct factor Xa inhibitors: anti‐Xa assay is preferable to prothrombin time assay. Thromb. Haemost. 104, 1263–1271 (2010). [DOI] [PubMed] [Google Scholar]
- 14. Chang, M. et al Effect of renal impairment on the pharmacokinetics, pharmacodynamics, and safety of apixaban. J. Clin. Pharmacol. 56, 637–645 (2016). [DOI] [PubMed] [Google Scholar]
- 15. Frost, C.E. et al Effects of age and sex on the single‐dose pharmacokinetics and pharmacodynamics of apixaban. Clin. Pharmacokinet. 54, 651–662 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frost, C. et al Safety, pharmacokinetics and pharmacodynamics of multiple oral doses of apixaban, a factor Xa inhibitor, in healthy subjects. Br. J. Clin. Pharmacol. 76, 776–786 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu, Z. , Nepal, S. , Bragat, A. , Shenker, A. & Frost, C. Single‐dose apixaban pharmacokinetics and pharmacodynamics in healthy male Japanese and Caucasian subjects. Can. J. Clin. Pharmacol. 15, e724 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamahira, N. et al Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple doses of apixaban in healthy Japanese male subjects. Int. J. Clin. Pharmacol. Ther. 52, 564–573 (2014). [DOI] [PubMed] [Google Scholar]
- 19. Cui, Y. et al Single‐ and multiple‐dose pharmacokinetics, pharmacodynamics, and safety of apixaban in healthy Chinese subjects. Clin. Pharmacol. 5, 177–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Upreti, V.V. et al Effect of extremes of body weight on the pharmacokinetics, pharmacodynamics, safety and tolerability of apixaban in healthy subjects. Br. J. Clin. Pharmacol. 6, 908–916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frost, C. et al A randomized direct comparison of the pharmacokinetics and pharmacodynamics of apixaban and rivaroxaban. Clin. Pharmacol. 6, 179–187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buller, H. , Deitchman, D. , Prins, M. & Segers, A. Efficacy and safety of the oral direct factor Xa inhibitor apixaban for symptomatic deep vein thrombosis. The Botticelli DVT dose‐ranging study. J. Thromb. Haemost. 6, 1313–1318 (2008). [DOI] [PubMed] [Google Scholar]
- 23. Pursley, J. et al LC‐MS/MS determination of apixaban (BMS‐562247) and its major metabolite in human plasma: an application of polarity switching and monolithic HPLC column. Bioanalysis 6, 2071–2082 (2014). [DOI] [PubMed] [Google Scholar]
- 24. Barrett, Y.C. et al A randomised assessment of the pharmacokinetic, pharmacodynamic and safety interaction between apixaban and enoxaparin in healthy subjects. Thromb. Haemost. 107, 916–924 (2012). [DOI] [PubMed] [Google Scholar]
- 25. Kowalsk, K. et al Apixaban exposure and anti‐Xa activity in sub‐populations of atrial fibrillation patients: an application of population PK/PD analysis. ACoP5. J. Pharmacokinet. Pharmacodyn. 41, S19 (2014). Abstract M–027. [Google Scholar]
- 26. Leil, T.A. , Frost, C. , Wang, X. , Pfister, M. & LaCreta, F. Model‐based exposure‐response analysis of apixaban to quantify bleeding risk in special populations of subjects undergoing orthopedic surgery. CPT Pharmacometrics Syst. Pharmacol. 3, e136 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cockcroft, D.W. & Gault, M.H. Prediction of creatinine clearance from serum creatinine. Nephron. 16, 31–41 (1976). [DOI] [PubMed] [Google Scholar]
- 28. Gastonguay, M.R. A full model estimation approach for covariate effects: inference based on clinical importance and estimation precision. (Abstract W4354). AAPS J. 6, S1 (2004). [Google Scholar]
- 29. Elassaiss‐Schaap, J , & Heisterkamp, S.H. Variability as constant coefficient of variation: Can we right two decades in error? Abstract 1508. <www.page-meeting.org/?abstract=1508> (2009).
- 30. Keizer, R.J. et al Modeling and simulation workbench for NONMEM: Tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst. Pharmacol. 2, e50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beal, S.L. & Sheiner, L.B. NONMEM User's Guides NONMEM Project Group (University of California, San Francisco, 1992).
- 32. Yano, Y. , Beal, S.L. & Sheiner, L.B. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J. Pharmacokinet. Pharmacodyn. 28, 171–192 (2001). [DOI] [PubMed] [Google Scholar]
- 33.ELIQUIS 5.0 mg Summary of Product Characteristics. Last updated April 2016. <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002148/WC500107728.pdf> Accessed 17 June 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information s01
Supplementary Figure 1. Anti‐FXa activity in LMWH Units versus predicted apixaban plasma concentration
Supplementary Information s03