

 This work is licensed under a
This work is licensed under a Abstract
Pheochromocytomatosis, a very rare form of pheochromocytoma recurrence, refers to new, multiple, and often small pheochromocytomas growing in and around the surgical resection bed of a previous adrenalectomy for a solitary pheochromocytoma. We here report a case of pheochromocytomatosis in a 70-year-old female. At age 64 years, she was diagnosed with a 6-cm right pheochromocytoma. She underwent laparoscopic right adrenalectomy, during which the tumor capsule was ruptured. At age 67 years, CT of abdomen did not detect recurrence. At age 69 years, she began experiencing episodes of headache and diaphoresis. At age 70 years, biochemical markers of pheochromocytoma became elevated with normal calcitonin level. CT revealed multiple nodules of various sizes in the right adrenal fossa, some of which were positive on metaiodobenzylguanidine (MIBG) scan. She underwent open resection of pheochromocytomatosis. Histological examination confirmed numerous pheochromocytomas ranging 0.1–1.2 cm in size. Next-generation sequencing of a panel of genes found a novel heterozygous germline c.570delC mutation in TMEM127, one of the genes that, if mutated, confers susceptibility to syndromic pheochromocytoma. Molecular analysis showed that the c.570delC mutation is likely pathogenic. Our case highlights the typical presentation of pheochromocytomatosis, a rare complication of adrenalectomy for pheochromocytoma. Previous cases and ours collectively demonstrate that tumor capsule rupture during adrenalectomy is a risk factor for pheochromocytomatosis. We also report a novel TMEM127 mutation in this case.
Learning points:
Pheochromocytomatosis is a very rare form of pheochromocytoma recurrence.
Pheochromocytomatosis refers to new, multiple and often small pheochromocytomas growing in and around the surgical resection bed of a previous adrenalectomy for a solitary pheochromocytoma.
Tumor capsule rupture during adrenalectomy predisposes a patient to develop pheochromocytomatosis.
Surgical resection of the multiple tumors of pheochromocytomatosis is recommended.
Pheochromocytoma recurrence should prompt genetic testing for syndromic pheochromocytoma.
Background
Recurrence of pheochromocytoma after complete surgical resection is an important concern. Pheochromocytoma recurrence can be in the form of new tumor formation (in a quarter of patients), local recurrence (also in a quarter of patients) or metastasis (in the remaining half of patients), and that the 5-year cumulative recurrence risk is ~5% and the life-time risk ~20–25% (1). Syndromic and extra-adrenal pheochromocytomas have higher risks of recurrence. A fourth form of recurrence, pheochromocytomatosis, is very rare and has only been reported in slightly over 10 cases (2, 3, 4, 5). Pheochromocytomatosis refers to new, multiple and often small pheochromocytomas growing in and around the surgical resection bed of a previous adrenalectomy for a solitary pheochromocytoma. In most cases, pheochromocytomatosis is likely caused by tumor cell seeding as a result of capsule rupture or friability of the original pheochromocytoma. In a few cases, pheochromocytomatosis occurs in a background of a familial syndrome, such as multiple endocrine neoplasia type 2 (MEN2) or neurofibromatosis type 1 (NF1) (4, 5). We here report a case of pheochromocytomatosis in a patient with a novel germline pathogenic mutation of TMEM127, one of the genes that, if mutated, confers susceptibility to syndromic pheochromocytoma.
Case presentation
A 70-year-old female presented for diagnosis and management of recurrent symptoms of pheochromocytoma. Six years before presentation, at age 64 years, she had begun experiencing episodic sweating and severe headache. Biochemical testing showed markedly elevated levels of plasma metanephrine at 16.2 nmol/L (normal <0.5), plasma normetanephrine 14.2 nmol/L (<0.9), and 24-hour urine total metanephrines at 11,810 µg (224–832); imaging showed a 6-cm right adrenal mass. She underwent laparoscopic right adrenalectomy at an outside hospital. Intraoperatively, the tumor appeared well encapsulated. During manipulation of the lower pole of the tumor, the capsule was ruptured. The tumor was otherwise completely removed. Grossly, the tumor was 6-cm, encapsulated and did not display lymphovascular invasion. There was no detailed description of the tumor capsule. On cut surfaces, the tumor appeared soft, friable and hemorrhagic. Microscopically, the tumors cells exhibited morphology typical for pheochromocytoma and were immunoreactive with chromogranin A. In some focal areas, mitotic index was higher than usual (highest mitotic index 4/10 high-power fields), and spindle tumor cell morphology or marked nuclear polymorphism was found. The Ki-67 labeling index was 5%. She did not experience any complications after that surgical operation; her preoperative symptoms resolved. Two months postoperatively, 24-hour urine total metanephrines returned to normal level (383 µg). She had no biochemical follow-up for pheochromocytoma recurrence. Three years later, at age 67 years, CT of abdomen did not detect recurrence. One year before presentation (nearly 5 years after the adrenalectomy), at age 69 years, the patient had begun experiencing recurrent episodes of headache and diaphoresis, especially after exercise. Other significant medical history included right breast cancer and lumpectomy at age 56 years, hypothyroidism, type 2 diabetes and hypertension. There was no family history of adrenal or other endocrine tumors. Her mother had recurrent skin cancer, a sister had breast cancer at age 74 years, a paternal aunt had colon cancer in her 60s and a paternal uncle had stomach cancer in his 70s.
Investigation
Physical examination findings were unremarkable except for hypertension (147/101 mmHg on carvedilol). Her plasma metanephrine level was 3.1 nmol/L (<0.5), plasma normetanephrine 2.8 nmol/L (<0.9) and calcitonin <5.0 pg/mL (<7.6). As she only had one remaining adrenal gland, subclinical adrenal insufficiency was a concern. Her morning cortisol level was 1.2 µg/dL (5.0–25.0) and ACTH 9.1 pg/mL (10–60); ACTH stimulation test confirmed adrenal insufficiency. MRI showed normal pituitary, and other pituitary hormones were normal. She started corticosteroid replacement. CT of abdomen and pelvis revealed multiple nodules of various sizes in and near the resection bed, and the left adrenal was unremarkable (Fig. 1). MRI demonstrated that the nodules exhibited slightly high signal on T2 imaging, slight enhancement upon gadolinium administration, and no drop of signal on out-of-phase imaging, consistent with features of pheochromocytoma. Metaiodobenzylguanidine (MIBG) scan identified several nodules near the right liver and previous resection bed (Fig. 2). FDG-PET/CT was also performed, but these nodules were not FDG-avid (Fig. 3). A diagnosis of pheochromocytomatosis was made.
Figure 1.

Axial images of CT of abdomen and pelvis with intravenous contrast. A to F: superior to inferior sections. Note the multiple small masses in the right adrenalectomy bed and along inferior liver border (arrows).
Figure 2.

Axial images of MIBG scan with single-photon emission computed tomography (SPECT). Upper panels, axial section corresponding to panel D in Figure 1. Lower panels, axial section corresponding to panel F in Fig. 1. Left, CT images; middle, MIBG scan with SPECT; and right, overlap of left and middle images. Note the masses are labeled with MIBG.
Figure 3.

Axial images of FDG-PET/CT. The section corresponds to panel D in Figure 1. Left, CT image; middle, FDG-PET image; and right, overlap of CT and FDG-PET image. Note that masses are not FGD-avid. Normal gut FDG avidity exists.
Treatment
After preparation with alpha blockade, she underwent open resection of pheochromocytomatosis. Hydrocortisone was given intravenously upon anesthesia induction. Intraoperatively, multiple tumor deposits were found adhering to the inferior margin of the liver and the superior pole of the right kidney, which were removed en block in a piece of fibroadipose tissue containing the tumors (Fig. 4); a tumor deposit abutting the inferior vena cava was also found, which was removed separately. At the end of operation, no other tumor deposits were visible or palpable. The patient tolerated the procedure well.
Figure 4.
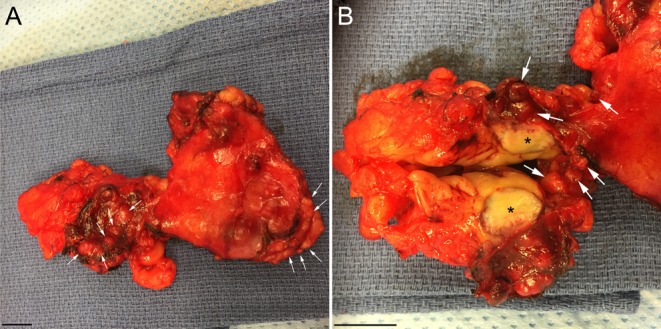
Gross images of a fibroadipose tissue block containing the tumors. Left, the whole tissue block; right, enlarged view of the left part of the tissue block cut in half. Arrows, individual tumors. Asterisk, the largest tumor (1.2-cm). Bar, 1 cm.
Gross examination of the resected surgical specimens identified many yellow or tan, well-circumscribed masses, 0.5–1.2-cm in greatest dimension. Histological examination confirmed that all these masses were pheochromocytoma and further revealed a 0.1-cm pheochromocytoma and a few even smaller ones (Fig. 5). Approximately 70% of tumor cells displayed a spindle morphology. Central necrosis was also identified. Mitotic figures were infrequent (<1 per 20 high-power fields). Capsular or vascular invasion was not found, nor was lymph node metastasis. The tumor cells were immunoreactive with chromogranin A and synaptophysin. Ki67 labeling index was 2%.
Figure 5.

Histopathologic images. (A) Low-power view showing multiple tumor deposits in adipose tissue. Hematoxylin & eosin, 2×. (B) Each tumor deposit is composed of nests of cells ranging from oval to spindle, without overt cytologic atypia or significant mitotic activity. Hematoxylin & eosin, 10×. (C) There is focal necrosis in a few tumor deposits. Hematoxylin & eosin, 10×. (D) The tumor cells are strongly and diffusely positive for synaptophysin. Immunoperoxidase, 2×. Bar, 1 mm.
Outcome and follow-up
The patient recovered uneventfully. Her preoperative symptoms resolved. Two months after the operation, her blood pressure improved (123/74 mmHg on carvedilol) and her plasma metanephrine level was 0.34 nmol/L and plasma normetanephrine 1.5 nmol/L. Tiny tumor implants likely remained after resection. As she unlikely had gross disease and she had well-controlled hypertension, she did not undergo MIBG scan and was not treated with alpha blockade or calcium channel blockers postoperatively. The patient will undergo biochemical surveillance every 3 months and abdominal imaging every 6 months. If there is clear evidence of gross tumor recurrence, additional therapies such as alpha blockade, MIBG radiotherapy and external beam radiation will be considered.
Because of the family history of cancers and personal history of breast cancer and recurrent pheochromocytoma, the patient underwent a CancerNext-Expanded genetic test (Ambry Genetics, Aliso Viejo, CA, USA), which uses next-generation sequencing to detect mutations in 49 genes: APC, ATM, BAP1, BARD1, BRCA1, BRCA2, BRIP1, BMPR1A, CDH1, CDK4, CDKN2A, CHEK2, EPCAM, FH, FLCN, GREM1, MAX, MEN1, MET, MITF, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, NF1, PALB2, PMS2, POLD1, POLE, PTEN, RAD50, RAD51C, RAD51D, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, SMAD4, SMARCA4, STK11, TMEM127, TP53, TSC1, TSC2 and VHL (the underlined genes are the 10 most commonly found, in mutant forms, in patients with pheochromocytoma or paraganglioma). Most genes (including the 10 underlined ones) were sequenced in full. A novel heterozygous c.570delC mutation in TMEM127 was found. Molecular analysis (with the EMBOSS software) showed that this mutation causes frameshift in TMEM127 mRNA translation and use of an alternative stop codon, resulting in replacement of the C-terminus of the TMEM127 protein with a different and longer peptide (p.T191RFS*116). A known pathogenic mutation in TMEM127, c.572delC, is associated with bilateral pheochromocytoma in 2 monozygotic twin sisters (6, 7). The c.570delC and c.572delC mutations have identical predicted deleterious effects on TMEM127 mRNA translation; the c.570delC mutation is thus likely pathogenic. No deletions or duplications of the tested pheochromocytoma susceptibility genes were found. The patient’s children were recommended to undergo genetic testing for the same mutation.
Discussion
Long-term follow-up of patients after complete surgical resection of pheochromocytoma is important to detect recurrence and is recommended by consensus guidelines (1, 8). Although the specific biochemical tests, imaging modalities and duration of follow-up are controversial, yearly biochemical testing, periodic imaging and continuous clinical monitoring are generally considered prudent; it should be noted, however, that no prospective studies have been done to study the effectiveness of a given surveillance protocol. Patients at higher risks of recurrence such as those with syndromic, multiple or extra-adrenal tumors, at a young age or with family history, or with aggressive histological features should be more stringently followed up. Our case highlights an often-overlooked risk factor of pheochromocytoma recurrence, tumor capsule rupture during adrenalectomy. The patient described here apparently does not have any traditional risk factors for recurrence except questionable and mild aggressive histological features. She nonetheless developed recurrence due to pheochromocytomatosis as a result of tumor capsule rupture during the original adrenalectomy.
We compare our case of pheochromocytomatosis with previous ones in this paragraph. Pheochromocytoma tumor seeding and growth at previous surgical resection sites are very rare events. In 2001, the term pheochromocytomatosis was coined to describe this phenomenon (3). Pheochromocytomatosis occasionally happens after open adrenalectomy (only 2 cases so far reported) but mostly occurs after laparoscopic adrenalectomy (2, 3, 4, 5). Tumor capsule rupture or probable spillage of tumor cells during manipulation of friable, hemorrhagic tumors is a universal feature in all cases of pheochromocytomatosis. Small pheochromocytomas may not have a clear capsule. The median age at the diagnosis of the original pheochromocytoma is 46 years (range: 27–63), and the median tumor size is 6 cm (range: 2.5–11). The median duration from the original adrenalectomy to the diagnosis of pheochromocytomatosis is 60 months (range: 24–156). Our patient is the oldest so far reported at the diagnosis of the original pheochromocytoma but her original tumor size and time to pheochromocytomatosis are both typical. In retrospect, since her tumor capsule was ruptured, she should have been followed more closely for recurrence. Up to 40% of all pheochromocytomas and paragangliomas are hereditary (9). Our patient is the third case of pheochromocytomatosis with a genetic mutation (the other two with MEN2 and NF1, respectively). The patient’s initial presentation is typical for pheochromocytoma associated with TMEM127 mutation, namely unilateral pheochromocytoma (10). The lack of family history is not surprising as penetrance of pheochromocytoma in TMEM127 mutation carriers is typically low (32% at age 51–65 years). The etiology of her secondary adrenal insufficiency is not clear.
The clinical course of pheochromocytomatosis is generally benign. After surgical removal of gross tumors, most patients enjoy years of remission. Although tiny tumor implants likely remain after resection of pheochromocytomatosis, adjuvant treatments such as high-dose iodine-131 MIBG radionuclide therapy or external beam radiation are not proven to reduce the likelihood of recurrence. In our view, cure of pheochromocytomatosis is unlikely and another gross recurrence is probable; there are no data, however, on the likelihood and timing of a second gross recurrence after debulking of the gross tumors. The long-term surveillance of patients with pheochromocytomatosis or with germline mutations should be individualized (8). As TMEM127 mutations have only been known to cause adrenal pheochromocytoma, we recommend biochemical testing and abdominal imaging for following our patient; some experts recommend whole-body MRI surveillance every 2–3 years for patients with TMEM127 mutations (9).
In summary, our case underscores the typical presentation of pheochromocytomatosis, a rare complication of adrenalectomy for pheochromocytoma. Previous cases and ours collectively demonstrate that tumor capsule rupture during adrenalectomy is a risk factor for pheochromocytomatosis. We also report a novel TMEM127 mutation in this case.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Written informed consent has been obtained from the patient for publication of the submitted article and accompanying images.
Author contribution statement
All authors contributed to the writing of this paper. R Yu, D Sharaga, C Donner, M J Livhit and M W Yeh provided clinical care, and C F Palma Diaz provided histological service to this patient.
References
- 1.Amar L, Lussey-Lepoutre C, Lenders JW, Djadi-Prat J, Plouin PF, Steichen O. 2016. Recurrence or new tumors after complete resection of pheochromocytomas and paragangliomas: a systematic review and meta-analysis. European Journal of Endocrinology 175 R135–R145. ( 10.1530/EJE-16-0189) [DOI] [PubMed] [Google Scholar]
- 2.Brennan MF, Keiser HR. 1982. Persistent and recurrent pheochromocytoma: the role of surgery. World Journal of Surgery 6 397–402. ( 10.1007/BF01657665) [DOI] [PubMed] [Google Scholar]
- 3.Li ML, Fitzgerald PA, Price DC, Norton JA. 2001. Iatrogenic pheochromocytomatosis: a previously unreported result of laparoscopic adrenalectomy. Surgery 130 1072–1077. ( 10.1067/msy.2001.118373) [DOI] [PubMed] [Google Scholar]
- 4.Boscà Robledo A, Ponce Marco JL, Belda Ibáñez T, Meseguer Anastasio MF, Gómez Gavara C. 2010. Pheochromocytomatosis: a risk after pheochromocytoma surgery. American Surgeon 76 E122–E124. [PubMed] [Google Scholar]
- 5.Rafat C, Zinzindohoue F, Hernigou A, Hignette C, Favier J, Tenenbaum F, Gimenez-Roqueplo AP, Plouin PF, Amar L. 2014. Peritoneal implantation of pheochromocytoma following tumor capsule rupture during surgery. Journal of Clinical Endocrinology and Metabolism 99 E2681–E2685. ( 10.1210/jc.2014-1975) [DOI] [PubMed] [Google Scholar]
- 6.Patócs A, Lendvai NK, Butz H, Liko I, Sapi Z, Szucs N, Toth G, Grolmusz VK, Igaz P, Toth M, et al. 2016. Novel SDHB and TMEM127 mutations in patients with pheochromocytoma paraganglioma syndrome. Pathology and Oncology Research 22 673–679. ( 10.1007/s12253-016-0050-0) [DOI] [PubMed] [Google Scholar]
- 7.Tóth G, Patócs A2, Tóth M. 2016. Hereditary phaeochromocytoma in twins. Orvosi Hetilap 157 1326–1330. ( 10.1556/650.2016.30513) [DOI] [PubMed] [Google Scholar]
- 8.Plouin PF, Amar L, Dekkers OM, Fassnacht M, Gimenez-Roqueplo AP, Lenders JW, Lussey-Lepoutre C, Steichen O. 2016. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. European Journal of Endocrinology 174 G1–G10. ( 10.1530/EJE-16-0033) [DOI] [PubMed] [Google Scholar]
- 9.Favier J, Amar L, Gimenez-Roqueplo AP. 2015. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nature Reviews Endocrinology 11 101–111. ( 10.1038/nrendo.2014.188) [DOI] [PubMed] [Google Scholar]
- 10.Toledo SP, Lourenço DM, Lucon AM, Baena ME, Castro CC, Bortolotto LA, Zerbini MC, Siqueira SA, Toledo RA, et al. 2015. Penetrance and clinical features of pheochromocytoma in a six-generation family carrying a germline TMEM127 mutation. Journal of Clinical Endocrinology and Metabolism 100 E308–E318. ( 10.1210/jc.2014-2473) [DOI] [PubMed] [Google Scholar]