

Abstract

Bile acids (BAs) are a family of endogenous metabolites synthesized from cholesterol in liver and modified by microbiota in gut. Being amphipathic molecules, the major function of BAs is to help with dietary lipid digestion. In addition, they also act as signaling molecules to regulate lipid and glucose metabolism as well as gut microbiota composition in the host. Remarkably, recent discoveries of the dedicated receptors for BAs such as FXR and TGR5 have uncovered a number of novel actions of BAs as signaling hormones which play significant roles in both physiological and pathological conditions. Disorders in BAs’ metabolism are closely related to metabolic syndrome and intestinal and neurodegenerative diseases. Though BA-based therapies have been clinically implemented for decades, the regulatory mechanism of BA is still poorly understood and a comprehensive characterization of BA-interacting proteins in proteome remains elusive. We herein describe a chemoproteomic strategy that uses a number of structurally diverse, clickable, and photoreactive BA-based probes in combination with quantitative mass spectrometry to globally profile BA-interacting proteins in mammalian cells. Over 600 BA-interacting protein targets were identified, including known endogenous receptors and transporters of BA. Analysis of these novel BA-interacting proteins revealed that they are mainly enriched in functional pathways such as endoplasmic reticulum (ER) stress response and lipid metabolism, and are predicted with strong implications with Alzheimer’s disease, non-alcoholic fatty liver disease, and diarrhea. Our findings will significantly improve the current understanding of BAs’ regulatory roles in human physiology and diseases.
Short abstract
Over 600 bile acid (BA)-interacting proteins including a number of novel targets have been identified through a chemoproteomic approach using clickable and photoreactive BA-based chemical probes in combination with quantitative mass spectrometry.
Introduction
Bile acids (BAs) are a class of important endogenous metabolites that consist of a steroid core and a side chain with a carboxyl group.1 The number and position of hydroxyl groups on the steroid core determine their hydrophobicity. In human, the primary BAs are cholic acid (CA) and chenodeoxycholic acid (CDCA), which are synthesized from cholesterol in liver and are subsequently conjugated with taurine or glycine to increase their water solubility.2 The conjugated BAs are secreted into intestine and undergo deconjugation, dehydroxylation, or isomerization by gut microbiota to form secondary bile acids, including deoxycholic acid (DCA), lithocholic acid (LCA), and ursodeoxycholic acid (UDCA).3 Being typical amphipathic molecules, BAs can facilitate emulsification and absorption of dietary lipid and fat-soluble vitamins. Nearly 95% of the intestinal BAs are reabsorbed in ileum and return back to liver via an active transport system called “enterohepatic circulation”.4 The homeostasis of BAs is maintained by the balance between their synthesis, secretion, and reabsorption.2
Since the discovery of endogenous receptors for BAs such as the nuclear farnesoid X receptor (FXR) and the plasma membrane bound G protein-coupled receptor 5 (TGR5), much effort has been devoted to understand the role of BAs as signaling molecules.5−7 Given that the BA receptors have a broad tissue expression profile and a wide range of downstream target genes, BAs have been shown, via activating these receptors, to regulate numerous physiological processes in the body, such as lipid and lipoprotein metabolism, glucose homeostasis, energy expenditure, carcinogenesis, and intestinal inflammation.8−11 BAs can also regulate protein functions by directly interacting with key metabolic enzymes or ion channels. For example, BAs bind to phospholipase D (PLD) to allosterically stimulate the production of endogenous anandamides,12,13 and target the mitochondrial inner membrane protein ADP/ATP translocase (SLC25A6) to mediate direct mitochondrial destruction of toxic BAs.14 In addition, BAs dynamically interplay with gut microbiota to impact host metabolism and the intricate mechanisms for such crosstalk remain to be unveiled.15,16
Consistent with the important and diverse roles of BAs, disorders in BA signaling are strongly associated with numerous human diseases, such as non-alcoholic fatty liver diseases (NAFLD), type 2 diabetes, diarrhea, and inflammatory bowel disease.17−19 For example, studies show that FXR-deficient mice on chow diet develop steatosis, insulin resistance, and bile acid malabsorption which causes diarrhea.20,21 Conversely, activation of FXR improves diet-induced fatty liver disease and insulin sensitivity, and significantly reduces symptoms in animal models of diarrhea.22−24 BA receptors have therefore become attractive therapeutic targets, and several agonists for FXR and TGR5 have entered clinical trials, aiming to offer new treatment options for these diseases.25,26 Recently, obeticholic acid (OCA), a bile acid derived FXR agonist, has been officially approved for clinical treatment of primary biliary cholangitis (PBC).27 Moreover, BAs can also be de novo synthesized in brain and act as potent antagonists for NMDA and GABAA receptors in the central nervous system.28,29 It has been reported that a secondary BA tauroursodeoxycholic acid (TUDCA) can easily go through the blood–brain barrier and is found to be a strong neuroprotective agent in treatment of Alzheimer’s disease (AD).30,31
Though our knowledge on BA signaling has been greatly improved in the past 20 years, most of the basic research and therapeutic exploration are still focused on FXR and TGR5, the two well-characterized BA receptors. Are there any other novel endogenous receptors for BAs? What are the rest of BA-interacting proteins in cells? These important questions remain to be answered. Current approaches to study BA–protein interactions mainly rely on traditional molecular biotechnologies and genetic mouse models. While they can discover a number of individual BA–protein interactions in a case-by-case manner, the low throughput puts great limitation on gaining deeper and broader understanding of BAs’ mechanism of action.13,14,32−34
Here we aim to perform a comprehensive characterization of BA-interacting proteins in mammalian proteomes using quantitative chemical proteomic technologies. Our approach was inspired by the previous efforts to map cholesterol-binding and lipid-binding proteins in proteomes using activity-based protein profiling (ABPP).35,36 We synthesized multiple clickable, photoreactive BA-based chemical probes and applied them in combination with quantitative proteomics based on stable-isotope labeling by amino acids in cell culture (SILAC) to globally profile BA-interacting proteins directly in living cells. With our ABPP-SILAC strategy, we identified >600 potential BA-interacting proteins with high confidence which include known receptors, transporters, and biosynthetic enzymes of BA. More importantly, our quantitative chemoproteomic profiling uncovered a large number of novel BA-interacting proteins that are functionally enriched in critical metabolic pathways such as endoplasmic reticulum (ER) stress response and fatty acid metabolism, and are strongly linked to vital diseases including Alzheimer’s disease, non-alcoholic fatty liver disease, and diarrhea. Our study provides the first global map of BA-interacting proteins in mammalian cells that will serve a valuable resource for exploring and understanding BA biology.
Results and Discussion
Design and Synthesis of the Clickable and Photoreactive BA Probes
Chemoproteomic probes have emerged as powerful tools to profile the cellular targets of bioactive small molecules in native biological system.37,38 In particular, photoreactive probes have been extensively used in combination with quantitative proteomic strategies to profile noncovalent protein–ligand interactions in cells and tissues.39−42 In order to assess reversible BA-interacting proteins in living cells, a photoreactive group which can establish covalent binding between BA and interacting proteins under UV irradiation needs to be incorporated into a BA scaffold to generate the corresponding probe. Additionally, an alkyne group should be appended, which allows further modification with azide-containing biotin or fluorescein by copper-catalyzed azide–alkyne cycloaddition (CuAAC).43,44 Conceivably, the carboxylic acid moiety at the C-24 position could be modified based on the fact that the endogenous BAs are also conjugated with taurine or glycine at the C-24 position to generate bile salts. A previous report has shown that fluorescein-labeled taurocholate at the C-3 position would retain its original biological activity.45 As ester linkage may be cleaved by endogenous esterases,35 the more stable amide linkage may be adopted in probe design. According to these considerations, ultimately, we designed and prepared a set of structurally diverse BA probes derived from cholic acid structure belonging to primary BAs, each of which contains an alkyne group and a photoreactive diazirine group (Scheme 1). P1 was directly prepared in 85% yield from cholic acid with the minimalist linker L1 developed by Yao and co-workers.46 The synthesis of P2 commenced with the selective oxidation of the 3-hydroxy group of methyl cholate to afford ketone 1 in 92% yield.47 Then the ketone moiety was further transformed to a diazirine moiety using the standard conditions (NH3/HOSA/I2) to furnish compound 2. Hydrolysis of methyl ester followed by amidation with 2-propynylamine smoothly afforded the desired P2 in 95% yield. According to the literature reported protocol,48 we could also transform methyl cholate to 3-amino methyl cholate (4). Then amidation of 4 with the minimalist linker L2 generated P3 in 88% yield. Another two analogues of P1 and P3 containing ester linkage were also synthesized and evaluated. Unfortunately, they failed to label proteins in lysate owning to the unstable ester bond.
Scheme 1. Design and Synthesis of the Clickable and Photoreactive Bile Acid Probes.

Reagents and conditions: (a) L1, BOP, DIPEA, DMF, 24 h, 85%. (b) Ag2CO3 on Celite (50 wt %), toluene, 140 °C, 36 h, 92%. (c) (1) NH3, MeOH, 0 °C, 3 h; (2) HOSA, −15 °C, 2 h, then rt, 14 h; (3) I2, TEA, MeOH, 30 min, 76%. (d) NaOH, MeOH, 36 h, quant. (e) 2-Propynylamine, HBTU, DIPEA, DMF, 20 h, 95%. (f) L2, BOP, DIPEA, DMF, 24 h, 88%.
Gel-Based ABPP Profiling of BA-Interacting Proteins in Living Cells
With these BA probes in hand, we first evaluated their labeling efficiency in living cells by in-gel fluorescence (Figure 1a). HeLa cells were treated with 50 μM of each of the BA probes and shed with 365 nm UV light for 5 min to induce photo-cross-linking. The labeled cell lysates were conjugated with azide-rhodamine via CuAAC and separated by SDS–PAGE. In addition to a negative control that was done without UV cross-linking, two “competition” samples were prepared in parallel where cells were cotreated with 2× and 4× more methyl cholic acids during the probe labeling step. The in-gel fluorescence revealed clear and distinct labeling profiles for each probe (Figure 1b), which suggested that difference in positions of installing the diazirine and alkyne groups indeed affect the probe–protein interactions in different ways. As expected, the labeling events were UV-dependent, confirming that most of the probe–protein interactions are noncovalent and these interactions could be competed off by excessive natural BAs in a dose-dependent manner (100 μM or 200 μM). Among the three BA probes tested, P1 showed the strongest labeling profile overall, however, its labeling was less sensitive to competition of native BA compared to that of P2 and P3. We also separated the probe-labeled lysates into soluble and membrane fractions by ultracentrifugation before conjugation with the fluorescent reporter, and the results showed stronger labeling signals in the membrane fraction (Figure S1), which is consistent with the high lipophilicity of these three BA probes.
Figure 1.

Gel-based profiling of BA-interacting proteins in living cells. (a) Workflow for gel-based ABPP profiling of BA-interacting proteins in living HeLa cells. (b) Evaluation of labeling efficiency of BA probes in living HeLa cells by in-gel fluorescence.
MS-Based ABPP Profiling of BA-Interacting Proteins
We next applied these BA probes in a quantitative chemical proteomic platform to identify BA-interacting proteins in living cells. The technology combines ABPP with SILAC-based quantitative proteomics,49 which allows us to identify and quantify probe-labeling protein targets by liquid chromatography–tandem mass spectrometry (LC–MS/MS). HeLa cells were cultured in “light” and “heavy” SILAC media for several passages, and in order to test whether the metabolic incorporation of heavy amino acids is complete, we combined the light and heavy cells in a 1:1 ratio and digested the mixed proteome for LC–MS/MS analysis. The distribution of the SILAC ratios showed a median value of 1.02 (Figure S2), indicating complete incorporation of the heavy amino acids and readiness of these cells for use in ABPP-SILAC experiments.
We performed two types of ABPP-SILAC experiments with each of the three BA probes in living cells. In a “UV-dependent labeling” experiment (Figure 2a, top panel), we treated light HeLa cells with 50 μM of probe for 1 h and irradiated the cells with UV light for 5 min, while the heavy cells were labeled with the same conditions except that no UV cross-linking was performed. The light and heavy cells were collected and lysed, and their proteomes were mixed with a 1:1 ratio and conjugated to an azide-biotin tag by CuAAC. After enrichment with streptavidin and on-bead digestion by trypsin, we analyzed the digested peptides with LC–MS/MS and quantified SILAC ratios for each protein. We used a light/heavy ratio (Ruv) of 5.0 as the cutoff to exclude any targets due to nonspecific binding to streptavidin and defined the remaining ones as specific “probe-binding” proteins. In parallel, we performed a “BA competition” experiment (Figure 2a, bottom panel) in which cells were cotreated with excessive natural BAs during the probe labeling step. More specifically, we treated light and heavy cells with 50 μM of the BA probe in the presence of either DMSO or 100 μM of methyl cholic acid, respectively. The lysed proteomes were equally combined and then subjected to ABPP-SILAC analysis as described above. We used a competition light/heavy ratio (Rcomp) of 1.5 as the cutoff to define the “BA-sensitive proteins” group, which excluded proteins that could potentially bind to the photoaffinity portion of the probe and therefore fail to be competed by excessive natural BA. In the end, we designated those common targets from both profiling experiments with Ruv ≥ 5.0 and Rcomp ≥ 1.5 as “BA-interacting proteins”.
Figure 2.

MS-based profiling of BA-interacting proteins in living cells by ABPP-SILAC. (a) Scheme for ABPP-SILAC experiments in which UV-dependent labeling and competition by 2× natural BA are quantified, respectively. (b) Venn diagrams showing the number of BA-interacting proteins identified specifically with each BA probe and commonly with all three probes. (c) Representative SILAC ratio plot for all BA-interacting proteins identified with P2. The x-axis is the SILAC ratio quantified in the UV-dependent labeling experiment. The y-axis is the SILAC ratio quantified in the BA-competition experiment. Select examples of known BA-interacting proteins are colored in red, and their extracted ion chromatograms are shown on the right.
For each of the P1, P2, and P3 probes, we performed three biological repeats for both types of profiling experiments, and in order to minimize the variation introduced by the stochastic nature of data-dependent acquisition by LC–MS/MS, only the protein targets that are identified in at least two out of the three replicates are considered for further analysis. The final lists include 564, 681, and 460 proteins for each of the P1, P2, and P3 probes, respectively, and all three probes shared 331 common protein targets (Figure 2b, Table S1). Among these three probes, P2 possessed the greatest number of specific protein targets, likely because the diazirine moiety is installed at the C-3 position on the BA backbone so that P2 can more effectively capture the potential interactions close to the binding pocket (compared to P1) but is less protruding (compared to P3). Consistent with this hypothesis, FXR, one of the best-studied BA receptors, is captured only by P2, but not by the other two probes, suggesting that P2 was a better BA derivative to simulate its interaction with protein targets.
We therefore focused on the “BA-interacting proteins” captured by P2 for further analysis. A quick survey on the list reveals that it indeed includes quite a few proteins that are known to be involved in the pathway of BA metabolism in addition to FXR (Figure 2c, Figure S3, Table S2). For example, the integrin β-1 (ITGB1) is a known TUDCA sensor on the plasma membrane.50 The very long chain acyl-CoA synthetase (SLC27A2)51 is a critical enzyme in the BA biosynthetic pathway, and the epoxide hydrolase 1 (EPHX1)52 is a bifunctional enzyme whose transporter function is responsible for the sodium-dependent uptake of BAs into cells. Furthermore, glutathione S-transferase (MGST3)53 and serum lipoprotein (APOB)54 have been reported to bind BA in order to facilitate their detoxification and transportation. We extracted the SILAC ion chromatographic traces of these proteins, and all of them showed clear UV-dependent and BA-competitive labeling by the probe (Figure 2c). Collectively, these known examples of BA-interacting proteins in our profiling data indicate that the photoaffinity BA probes are capturing physiologically relevant protein–BA interactions in living cells.
Interestingly, we also identified a set of BA–protein interactions which are sensitive to excessive BA competition but show UV-independent probe labeling (group IV in Figure 2c). This suggests that covalent interactions between BA and proteins may exist in cells and will be the subject of further investigation.
Considering the structural similarity between BA and cholesterol, we also compared the BA-interacting proteins identified in our study with those cholesterol-interacting proteins identified in a previous study performed by the Cravatt group35 (Figure S4, Table S3). Both data sets share a reasonable overlap with 146 common targets including a BA biosynthetic enzyme SLC27A2, which is consistent with the fact that cholesterol serves as the precursor for BA biosynthesis. In addition, our study also identified 535 specific BA-interacting targets, among which are well-known BA receptors (FXR, ITGB1), transporters (EPHX1, ABCB1, ABCD3), and binding proteins (PLD, APOE, ALB, etc.), suggesting the high selectivity of BA probe. Further in-depth analysis of these data sets may help delineate functional pathways that are commonly and specifically regulated by the two steroids in cells.
Bioinformatics Analysis of BA-Interacting Proteins
We performed the gene ontology (GO) analysis on these BA interacting proteins regarding their subcellular distributions (Figure 3a).55,56 Major subcellular organelles enriched with BA target proteins are endoplasmic reticulum (23%), mitochondrion (21%), Golgi apparatus (11%), nuclear envelope (5%), cell surface (5%), and peroxisome (4%). The membrane-dominant distribution is consistent with the results from the gel-based profiling that shows stronger labeling signals in the membrane fraction of the proteomes. Furthermore, the diversified subcellular locations of BA-interacting proteins are echoed by the fact that the complete biosynthesis of BA requires 17 individual enzymes and several transporters residing in multiple intracellular compartments.1
Figure 3.

Bioinformatic analysis of BA-interacting proteins. (a). Cellular component analysis of BA-interacting proteins by gene ontology (GO). (b) Functional pathways analysis of BA-interacting proteins by Kyoto Encyclopedia of Genes and Genomes (KEGG). The enriched pathways are ranked based on their P values. (c) Human diseases analysis of BA-interacting proteins by KEGG. The top 5 enriched disease pathways are shown, and their P values are marked with red dots on the contour plot.
We next queried the Kyoto Encyclopedia of Genes and Genomes (KEGG) database to categorize these BA-interacting proteins based on the functional pathways involved (Figure 3b).52,53 The clustering analysis showed that the most functionally enriched pathways include protein processing in ER, lipid metabolisms (including fatty acid, glycerophospholipid, and sphingolipid metabolisms), N-glycan biosynthesis and oxidative phosphorylation, etc., many of which have been reported to be regulated by BA in a receptor-dependent manner.57−60 It is also satisfying to see that the bile secretion pathway is enriched as several known BA transporters are captured by our BA probes in the profiling.
The top functionally enriched pathway, “protein processing in ER”, contains 44 BA-interacting proteins. Gene ontology and functional protein association network analysis revealed that 20 of them are involved in ER stress response caused by accumulation of unfolded or misfolded proteins in ER lumen, and 7 of them are with functions of chaperone binding (Figure S5). It has been reported that a secondary BA, TUDCA, can serve as a chemical chaperone to improve ER folding capacity and reduce ER stress, which resulted in alleviation of hyperglycemia and restoration of glucose homeostasis and insulin sensitivity in livers of diabetic mice.61 However, no exact targets of TUDCA have been reported as participating in the ER stress response. Our profiling data provide a set of potential interacting proteins of TUDCA that could serve as the ER stress responders.
BAs have been known to play important roles in regulating lipid metabolism.62 For example, carnitine palmitoyl transferase 1A (CPT1A), a rate-limiting enzyme of fatty acid metabolism, was reported to be transcriptionally upregulated by BA through the activation of FXR.63 Our profiling data identified a direct interaction between CPT1A and BA, which suggests that the protein function could be also regulated in a post-translational way. Phospholipase D (PLD) is another BA-interacting protein that was identified by our ABPP-SILAC profiling, and the finding is consistent with a previous study showing that BAs can allosterically activate PLD through direct interaction to enhance its dimer assembly.12 ADP-dependent glucokinase (ADPGK) converts glucose to glucose-6-phosphate and plays an essential role in the glycolysis and pentose phosphate pathway.64 Our chemoproteomic profiling study identified ADPGK as a BA-interacting protein and may provide a novel link for BA in direct regulation of glucose metabolism.
Disease Analysis of BA-Interacting Proteins
We analyzed these BA-interacting proteins based on their annotations recorded in the KEGG disease database65 and found that they are strongly implicated with neurodegenerative diseases, non-alcoholic fatty liver diseases, and diarrhea (Figure 3c).
Dysmetabolism of BA has been postulated as a factor for the pathogenesis of neurodegenerative diseases, such as Alzheimer’s disease (AD) and Parkinson disease (PD).66−68 AD is characterized by accumulation of amyloid-β (Aβ) peptides in brain that leads to progressive cognitive deficit.69 TUDCA has been found to be a strong neuroprotective agent for AD, and chronic TUDCA treatment of AD mice for 6 months could significantly reduce amyloid-β deposition and ameliorate memory deficit.31 However, the therapeutic potential of TUDCA in AD pathology has not been fully explored due to the lack of knowledge of exact molecular targets involved. Our chemoproteomic data revealed direct interaction between BA and several proteins in this pathway, including Aβ precursor protein (APP), β-site APP-cleaving enzyme 2 (BACE2), and the lipid-metabolism mediator apolipoprotein E (APOE). It is possible that TUDCA may exert its neuroprotective function by interacting with these proteins to affect APP processing and amyloid-β deposition. In addition, catechol-O-methyltransferase (COMT) is another potential link because this BA-interacting protein plays a prominent role in controlling the metabolism of catecholamine neurotransmitters and the inhibitors of this enzyme can block amyloid-β fibrils.70
Disorders in BA metabolism can lead to non-alcoholic fatty liver diseases (NAFLD), which is strongly associated with obesity, diabetes mellitus, and dislipidemia.71 Our chemoproteomic profiling identified several direct interactions between BA and key proteins that are known to be involved with the development and pathogenesis of NAFLD, including sterol regulatory element binding transcription factor 1 (SREBF1),72 transforming growth factor beta 1 (TGFB1),73 and multiple subunits of NADH:ubiquinone oxidoreductase74 and cytochrome c oxidase.75 Further investigation on how BA binds to and affects functions of these proteins as well as those in lipid metabolism pathways may provide novel links between BA metabolism and NAFLD.
BAs have been known to interplay with microbiota in the intestine to establish a healthy gut environment for the host.76,77 Conversely, pathogenic bacteria such as Vibrio cholerae have evolved sophisticated mechanisms to utilize BAs as nutrients for survival and infection,78,79 causing severe diarrhea.80 Two ER proteins, the ER oxidoreductase ERO1A and the protein transport channel SEC61, are critical host components hijacked by the bacteria to process cholera toxin (CTX) in ER and release it back to cytosol to exert toxicity. Our profiling data reveal that BAs interact with these two proteins and therefore provide a potential link between BA and the Vibrio cholerae infection process.
Validation of BA–Protein Interactions
To confirm these BA–protein interactions identified in our profiling data, we choose six proteins for further validation by Western blotting. Three of these proteins (NTCP, SLC25A6, PLD3) have been previously reported to physically interact with BA, and the other three (CPT1A, ADPGK, COMT) are novel BA binders discovered by our study which have potential functional implications in various diseases as mentioned above. Moreover, each of these six proteins has its distinct subcellular location as annotated according to the knowledge database (Figure 4). Each protein was recombinantly overexpressed with a 6× His epitope sequence in HeLa cells. The cells were treated in situ with the P2 probe, followed by conjugation with an azide-biotin tag and enrichment of the probe-labeled proteins on streptavidin. Immunoblotting of the enriched proteins showed that all of them can be captured by the P2 probe in a UV-dependent manner and these BA–protein interactions can be competed by excess native BA molecules by at least 1.69-fold.
Figure 4.
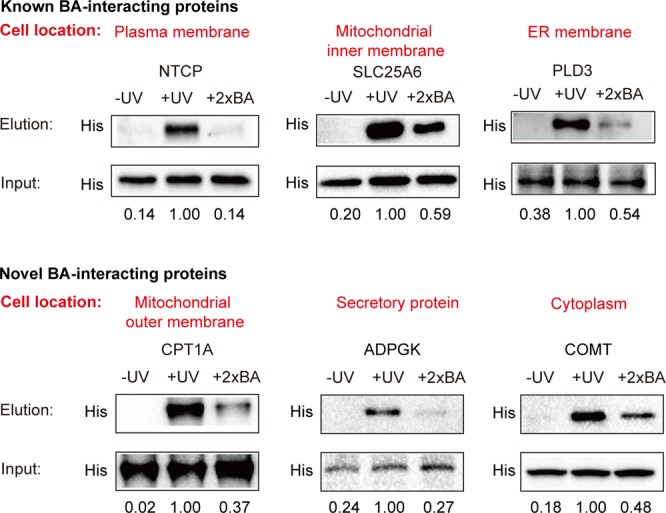
Experimental validation of BA–protein interactions by immunoblotting. Each protein is transiently overexpressed in HeLa cells with a His tag, and the cells are labeled with P2. The labeled proteins are enriched and then eluted for immunoblotting with an anti-His antibody (“Elution”). The samples without UV cross-linking or with 2× BA competition are used as control. Equal loading is validated by immunoblotting of input samples before enrichment (“Input”). Top panel: NTCP (a BA transporter), SLC25A6, and PLD3 are known BA–protein interactions that have been reported before. Bottom panel: CPT1A, ADPGK, and COMT are novel BA–protein interactions discovered in this study. The normalized intensities are shown below each band.
Conclusion
Here we designed and efficiently prepared a number of structurally diverse, clickable, and photoreactive BA probes based on the cholic acid scaffold, and applied them in systematically mapping BA-interacting proteins directly in living cells. These BA probes showed good specificity for known BA receptors, transporters, and biosynthetic enzymes via noncovalent binding. Combined with SILAC-based quantitative proteomic technologies, we successfully identified a large number of novel BA-interacting proteins including chaperone binding proteins involved in ER stress response, key metabolic enzymes in diverse pathways of lipid metabolism, and functional targets strongly associated with metabolic syndrome, neurodegenerative diseases, and diarrhea. These novel BA–protein interactions will provide a rich resource for guiding new functional studies of BA signaling and expand the current understanding of BA-mediated pathways in human metabolism and disease.
Acknowledgments
We thank Prof. Wenhui Li (NIBS) for helpful discussion, Runze Tian for research assistance, and Prof. Jiang Zhou (PKU) for HRMS analysis. We also thank the Computing Platform of the Center for Life Science for supporting the proteomic data analysis. Financial support from NNSFC (21472010, 21521003, 21561142002, and 21625201 to X.L.; and 21472008 and 81490741 to C.W.) and Ministry of Science and Technology of China (2015CB856200 to X.L. and 2016YFA0501500 to C.W.) is gratefully acknowledged. C.W. is supported by a “1000 Talents Plan” Young Investigator Award.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00134.
Author Contributions
§ S.Z. and Q.L. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Monte M. J.; Marin J. J.; Antelo A.; Vazqueztato J. Bile Acids: Chemistry, Physiology, and Pathophysiology. World J. Gastroenterol. 2009, 15, 804–816. 10.3748/wjg.15.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang J. Y. L. Bile Acids: Regulation of Synthesis. J. Lipid Res. 2009, 50, 1955–1966. 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon J. M.; Kang D.; Hylemon P. B. Bile Salt Biotransformations by Human Intestinal Bacteria. J. Lipid Res. 2006, 47, 241–259. 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- Hofmann A. F. The Enterohepatic Circulation of Bile Acids in Mammals: Form and Functions. Front. Biosci., Landmark Ed. 2009, 14, 2584–2598. 10.2741/3399. [DOI] [PubMed] [Google Scholar]
- Makishima M.; Okamoto A. Y.; Repa J. J.; Tu H.; Learned R. M.; Luk A.; Hull M. V.; Lustig K. D.; Mangelsdorf D. J.; Shan B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- Kawamata Y.; Fujii R.; Hosoya M.; Harada M.; Yoshida H.; Miwa M.; Fukusumi S.; Habata Y.; Itoh T.; Shintani Y.; Hinuma S.; Fujisawa Y.; Fujino M. A G Protein-Coupled Receptor Responsive to Bile Acids. J. Biol. Chem. 2003, 278, 9435–9440. 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Hylemon P. B. Bile Acids are Nutrient Signaling Hormones. Steroids 2014, 86, 62–68. 10.1016/j.steroids.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudel T.; Staels B.; Kuipers F. The Farnesoid X Receptor: a Molecular Link between Bile Acid and Lipid and Glucose Metabolism. Arterioscler., Thromb., Vasc. Biol. 2005, 25, 2020–2030. 10.1161/01.ATV.0000178994.21828.a7. [DOI] [PubMed] [Google Scholar]
- Chiang J. Y. L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. 10.1002/cphy.c120023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodoro J. S.; Rolo A. P.; Palmeira C. M. Hepatic FXR: Key Regulator of Whole-Body Energy Metabolism. Trends Endocrinol. Metab. 2011, 22, 458–466. 10.1016/j.tem.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Inagaki T.; Moschetta A.; Lee Y.; Peng L.; Zhao G.; Downes M.; Yu R. T.; Shelton J. M.; Richardson J. A.; Repa J. J.; Mangelsdorf D. J.; Kliewer S. A. Regulation of Antibacterial Defense in the Small Intestine by the Nuclear Bile Acid Receptor. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 3920–3925. 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magotti P.; Bauer I.; Igarashi M.; Babagoli M.; Marotta R.; Piomelli D.; Garau G. Structure of Human N-Acylphosphatidylethanolamine-Hydrolyzing Phospholipase D: Regulation of Fatty Acid Ethanolamide Biosynthesis by Bile Acids. Structure 2015, 23, 598–604. 10.1016/j.str.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margheritis E.; Castellani B.; Magotti P.; Peruzzi S.; Romeo E.; Natali F.; Mostarda S.; Gioiello A.; Piomelli D.; Garau G. Bile Acid Recognition by NAPE-PLD. ACS Chem. Biol. 2016, 11, 2908–2914. 10.1021/acschembio.6b00624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S.; Schmitt S.; Wimmer R.; Aichler M.; Eisenhofer S.; Lichtmannegger J.; Eberhagen C.; Artmann R.; Tookos F.; Walch A. Progressive Stages of Mitochondrial Destruction Caused by Cell Toxic Bile Salts. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 2121–2133. 10.1016/j.bbamem.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Nie Y. C.; Hu J.; Yan X. Cross-Talk between Bile Acids and Intestinal Microbiota in Host Metabolism and Health. J. Zhejiang Univ., Sci., B 2015, 16, 436–446. 10.1631/jzus.B1400327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlstrom A.; Sayin S. I.; Marschall H. U.; Backhed F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- Li T.; Chiang J. Y. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacol. Rev. 2014, 66, 948–983. 10.1124/pr.113.008201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prawitt J.; Caron S.; Staels B. Bile Acid Metabolism and the Pathogenesis of Type 2 Diabetes. Curr. Diabetes Rep. 2011, 11, 160–166. 10.1007/s11892-011-0187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters J. R. F. Bile Acid Diarrhoea and FGF19: New Views on Diagnosis, Pathogenesis and Therapy. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 426–434. 10.1038/nrgastro.2014.32. [DOI] [PubMed] [Google Scholar]
- Wu W.; Liu X.; Peng X.; Xue R.; Ji L.; Shen X.; Chen S.; Gu J.; Zhang S. Bile Acids Override Steatosis in Farnesoid X Receptor Deficient Mice in a Model of Non-Alcoholic Steatohepatitis. Biochem. Biophys. Res. Commun. 2014, 448, 50–55. 10.1016/j.bbrc.2014.04.048. [DOI] [PubMed] [Google Scholar]
- Cariou B.; Van Harmelen K.; Duransandoval D.; Van Dijk T. H.; Grefhorst A.; Abdelkarim M.; Caron S.; Torpier G.; Fruchart J.; Gonzalez F. J. The Farnesoid X Receptor Modulates Adiposity and Peripheral Insulin Sensitivity in Mice. J. Biol. Chem. 2006, 281, 11039–11049. 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- Cipriani S.; Mencarelli A.; Palladino G.; Fiorucci S. FXR Activation Reverses Insulin Resistance and Lipid Abnormalities and Protects Against Liver Steatosis in Zucker (fa/fa) Obese Rats. J. Lipid Res. 2010, 51, 771–784. 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Lee F. Y.; Barrera G. C.; Lee H.; Vales C.; Gonzalez F. J.; Willson T. M.; Edwards P. A. Activation of the Nuclear Receptor FXR Improves Hyperglycemia and Hyperlipidemia in Diabetic Mice. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1006–1011. 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroz M. S.; Keating N.; Ward J. B.; Sarker R.; Amu S.; Aviello G.; Donowitz M.; Fallon P. G.; Keely S. J. Farnesoid X Receptor Agonists Attenuate Colonic Epithelial Secretory Function and Prevent Experimental Diarrhoea in vivo. Gut 2014, 63, 808–817. 10.1136/gutjnl-2013-305088. [DOI] [PubMed] [Google Scholar]
- Schaap F. G.; Trauner M.; Jansen P. L. M. Bile Acid Receptors as Targets for Drug Development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. 10.1038/nrgastro.2013.151. [DOI] [PubMed] [Google Scholar]
- Makri E.; Cholongitas E.; Tziomalos K. Emerging Role of Obeticholic Acid in the Management of Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2016, 22, 9039–9043. 10.3748/wjg.v22.i41.9039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. E. J. Obeticholic Acid for the Treatment of Primary Biliary Cirrhosis. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 1091–1099. 10.1080/17474124.2016.1216784. [DOI] [PubMed] [Google Scholar]
- Mano N.; Goto T.; Uchida M.; Nishimura K.; Ando M.; Kobayashi N.; Goto J. Presence of Protein-Bound Unconjugated Bile acids in the Cytoplasmic Fraction of Rat Brain. J. Lipid Res. 2004, 45, 295–300. 10.1194/jlr.M300369-JLR200. [DOI] [PubMed] [Google Scholar]
- Schubring S. R.; Fleischer W.; Lin J.; Haas H. L.; Sergeeva O. A. The Bile Steroid Chenodeoxycholate is a Potent Antagonist at NMDA and GABA(A) Receptors. Neurosci. Lett. 2012, 506, 322–326. 10.1016/j.neulet.2011.11.036. [DOI] [PubMed] [Google Scholar]
- Ramalho R. M.; Borralho P. M.; Castro R. E.; Sola S.; Steer C. J.; Rodrigues C. M. P. Tauroursodeoxycholic Acid Modulates p53-Mediated Apoptosis in Alzheimer’s Disease Mutant Neuroblastoma Cells. J. Neurochem. 2006, 98, 1610–1618. 10.1111/j.1471-4159.2006.04007.x. [DOI] [PubMed] [Google Scholar]
- Nunes A.; Amaral J. D.; Lo A. C.; Fonseca M. B.; Viana R. J. S.; Callaertsvegh Z.; D'Hooge R.; Rodrigues C. M. P. TUDCA, a Bile Acid, Attenuates Amyloid Precursor Protein Processing and Amyloid-β Deposition in APP/PS1Mice. Mol. Neurobiol. 2012, 45, 440–454. 10.1007/s12035-012-8256-y. [DOI] [PubMed] [Google Scholar]
- Burke C. W.; Lewis B.; Panveliwalla D.; Tabaqchali S. The Binding of Cholic Acid and Its Taurine Conjugate to Serum Proteins. Clin. Chim. Acta 1971, 32, 207–214. 10.1016/0009-8981(71)90334-2. [DOI] [PubMed] [Google Scholar]
- Parks D. J.; Blanchard S. G.; Bledsoe R. K.; Chandra G.; Consler T. G.; Kliewer S. A.; Stimmel J. B.; Willson T. M.; Zavacki A. M.; Moore D. D.; Lehmann J. M. Bile Acids: Natural Ligands for an Orphan Nuclear Receptor. Science 1999, 284, 1365–1368. 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- Fang C.; Filipp F. V.; Smith J. W. Unusual Binding of Ursodeoxycholic Acid to Ileal Bile Acid Binding Protein: Role in Activation of FXRα. J. Lipid Res. 2012, 53, 664–673. 10.1194/jlr.M021733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulce J. J.; Cognetta A. B.; Niphakis M. J.; Tully S. E.; Cravatt B. F. Proteome-Wide Mapping of Cholesterol-Interacting Proteins in Mammalian Cells. Nat. Methods 2013, 10, 259–264. 10.1038/nmeth.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niphakis M. J.; Lum K. M.; Cognetta A. B.; Correia B. E.; Ichu T.; Olucha J.; Brown S. J.; Kundu S.; Piscitelli F.; Rosen H.; Cravatt B. F. A Global Map of Lipid-Binding Proteins and Their Ligandability in Cells. Cell 2015, 161, 1668–1680. 10.1016/j.cell.2015.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niphakis M. J.; Cravatt B. F. Enzyme Inhibitor Discovery by Activity-Based Protein Profiling. Annu. Rev. Biochem. 2014, 83, 341–377. 10.1146/annurev-biochem-060713-035708. [DOI] [PubMed] [Google Scholar]
- Wang C.; Chen N. Activity-Based Protein Profiling. Huaxue Xuebao 2015, 73, 657–668. 10.6023/A15040223. [DOI] [Google Scholar]
- Sumranjit J.; Chung S. J. Recent Advances in Target Characterization and Identification by Photoaffinity Probes. Molecules 2013, 18, 10425–10451. 10.3390/molecules180910425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Koh M.; Koo J. Y.; Lee S.; Park S. B. Investigation of Specific Binding Proteins to Photoaffinity Linkers for Efficient Deconvolution of Target Protein. ACS Chem. Biol. 2016, 11, 44–52. 10.1021/acschembio.5b00671. [DOI] [PubMed] [Google Scholar]
- Pan S.; Zhang H.; Wang C.; Yao S. C. L.; Yao S. Q. Target Identification of Natural Products and Bioactive Compounds Using Affinity-Based Probes. Nat. Prod. Rep. 2016, 33, 612–620. 10.1039/C5NP00101C. [DOI] [PubMed] [Google Scholar]
- Wright M. H.; Sieber S. A. Chemical Proteomics Approaches for Identifying the Cellular Targets of Natural Products. Nat. Prod. Rep. 2016, 33, 681–708. 10.1039/C6NP00001K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective ″Ligation″ of Azides and Terminal Alkynes. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. . [DOI] [PubMed] [Google Scholar]
- Soriano del Amo D.; Wang W.; Jiang H.; Besanceney C.; Yan A.; Levy M.; Liu Y.; Marlow F. L.; Wu P. Biocompatible Copper(I) Catalysts for in vivo Imaging of Glycans. J. Am. Chem. Soc. 2010, 132, 16893–16899. 10.1021/ja106553e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Lecluyse E. L.; Brouwer K. R.; Gan L. S.; Lemasters J. J.; Stieger B.; Meier P. J.; Brouwer K. L. Biliary Excretion in Primary Rat Hepatocytes Cultured in a Collagen-Sandwich Configuration. Am. J. Physiol. 1999, 277, G12–G21. [DOI] [PubMed] [Google Scholar]
- Li Z.; Hao P.; Li L.; Tan C. Y. J.; Cheng X. Z.; Chen G. Y. J.; Sze S. K.; Shen H.; Yao S. Q. Design and Synthesis of Minimalist Terminal Alkyne-Containing Diazirine Photo-Crosslinkers and Their Incorporation into Kinase Inhibitors for Cell- and Tissue-Based Proteome Profiling. Angew. Chem., Int. Ed. 2013, 52, 8551–8556. 10.1002/anie.201300683. [DOI] [PubMed] [Google Scholar]
- Li Q.; Tochtrop G. P. A Stereoselective Synthesis of the Allo-Bile Acids from the 5β-isomers. Tetrahedron Lett. 2011, 52, 4137–4139. 10.1016/j.tetlet.2011.05.140. [DOI] [Google Scholar]
- Zhao Y.; Zhong Z. Oligomeric Cholates: Amphiphilic Foldamers with Nanometer-Sized Hydrophilic Cavities. J. Am. Chem. Soc. 2005, 127, 17894–17901. 10.1021/ja056151p. [DOI] [PubMed] [Google Scholar]
- Ong S.; Mann M. A Practical Recipe for Stable Isotope Labeling by Amino Ccids in Cell Culture (SILAC). Nat. Protoc. 2007, 1, 2650–2660. 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- Gohlke H.; Schmitz B.; Sommerfeld A.; Reinehr R.; Haussinger D. α5 β1-Integrins Are Sensors For Tauroursodeoxycholic Acid in Hepatocytes. Hepatology 2013, 57, 1117–1129. 10.1002/hep.25992. [DOI] [PubMed] [Google Scholar]
- Mihalik S. J.; Steinberg S. J.; Pei Z.; Park J.; Kim D. G.; Heinzer A. K.; Dacremont G.; Wanders R. J. A.; Cuebas D. A.; Smith K. D. Participation of Two Members of the Very Long-Chain acyl-CoA Synthetase Family in Bile Acid Synthesis and Recycling. J. Biol. Chem. 2002, 277, 24771–24779. 10.1074/jbc.M203295200. [DOI] [PubMed] [Google Scholar]
- Von Dippe P.; Zhu Q.; Levy D. Cell Surface Expression and Bile Acid Transport Function of One Topological Form of m-epoxide Hydrolase. Biochem. Biophys. Res. Commun. 2003, 309, 804–809. 10.1016/j.bbrc.2003.08.074. [DOI] [PubMed] [Google Scholar]
- Takikawa H.; Sugiyama Y.; Kaplowitz N. Binding of Bile Acids by Glutathione S-Transferases from Rat Liver. J. Lipid Res. 1986, 27, 955–966. [PubMed] [Google Scholar]
- Ceryak S.; Bouscarel B.; Fromm H. Comparative Binding of Bile Acids to Serum Lipoproteins and Albumin. J. Lipid Res. 1993, 34, 1661–1674. [PubMed] [Google Scholar]
- Huang D. W.; Sherman B. T.; Lempicki R. A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2008, 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang D. W.; Sherman B. T.; Lempicki R. A. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Shen W.; Sun H. Effects of Nuclear Receptor FXR on the Regulation of Liver Lipid Metabolism in Patients with Non-Alcoholic Fatty Liver Disease. Hepatol. Int. 2010, 4, 741–748. 10.1007/s12072-010-9202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne A.; Sharma R.; Duggan G.; Kelleher D.; Long A. Deoxycholic Acid Impairs Glycosylation and Fucosylation Processes in Esophageal Epithelial Cells. Glycobiology 2012, 22, 638–648. 10.1093/glycob/cwr190. [DOI] [PubMed] [Google Scholar]
- Lee M. J.; Whitehouse M. W. Inhibition of Electron Transport and Coupled Phosphorylation in Liver Mitochondria by Cholanic (Bile) Acids and Their Conjugates. Biochim. Biophys. Acta, Gen. Subj. 1965, 100, 317–328. 10.1016/0304-4165(65)90001-2. [DOI] [PubMed] [Google Scholar]
- Barrasa J. I.; Olmo N.; Perezramos P.; Santiagogomez A.; Lecona E.; Turnay J.; Lizarbe M. A. Deoxycholic and Chenodeoxycholic Bile Acids Induce Apoptosis via Oxidative Stress in Human Colon Adenocarcinoma Cells. Apoptosis 2011, 16, 1054–1067. 10.1007/s10495-011-0633-x. [DOI] [PubMed] [Google Scholar]
- Ozcan U.; Yilmaz E.; Ozcan L.; Furuhashi M.; Vaillancourt E.; Smith R. O.; Gorgun C. Z.; Hotamisligil G. S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgharpour A.; Kumar D.; Sanyal A. Hepatol. Int. 2015, 9, 527. 10.1007/s12072-015-9656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krattinger R.; Bostrom A.; Lee S. M. L.; Thasler W. E.; Schioth H. B.; Kullakublick G. A.; Mwinyi J. Chenodeoxycholic Acid Significantly Impacts the Expression of miRNAs and Genes Involved in Lipid, Bile acid and Drug Metabolism in Human Hepatocytes. Life Sci. 2016, 156, 47–56. 10.1016/j.lfs.2016.04.037. [DOI] [PubMed] [Google Scholar]
- Richter S.; Richter J. P.; Mehta S.; Gribble A.; Sutherlandsmith A. J.; Stowell K. M.; Print C. G.; Ronimus R. S.; Wilson W. R. Expression and Role in Glycolysis of Human ADP-Dependent Glucokinase. Mol. Cell. Biochem. 2012, 364, 131–145. 10.1007/s11010-011-1212-8. [DOI] [PubMed] [Google Scholar]
- Kanehisa M.; Goto S.; Furumichi M.; Tanabe M.; Hirakawa M. KEGG for Representation and Analysis of Molecular Networks Involving Diseases and Drugs. Nucleic Acids Res. 2010, 38, D355–D360. 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene C. D.; Rodrigues C. M. P.; Eich T.; Chhabra M.; Steer C. J.; Low W. C. Tauroursodeoxycholic Acid, a Bile Acid, is Neuroprotective in a Transgenic Animal Model of Huntington’s Disease. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 10671–10676. 10.1073/pnas.162362299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalho R. M.; Viana R. J. S.; Low W. C.; Steer C. J.; Rodrigues C. M. P. Bile Acids and Apoptosis Modulation: an Emerging Role in Experimental Alzheimer’s Disease. Trends Mol. Med. 2008, 14, 54–62. 10.1016/j.molmed.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Vang S.; Longley K.; Steer C. J.; Low W. C. The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Glob. Adv. Health Med. 2014, 3, 58–69. 10.7453/gahmj.2014.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman D. M.; Morris J. C.; Goate A. Alzheimer’s Disease: The Challenge of the Second Century. Sci. Transl. Med. 2011, 3, 77sr1. 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serretti A.; Olgiati P. Catechol-o-Methyltransferase and Alzheimer’s Disease: a Review of Biological and Genetic Findings. CNS Neurol. Disord.: Drug Targets 2012, 11, 299–305. 10.2174/187152712800672472. [DOI] [PubMed] [Google Scholar]
- Fuchs M. Non-Alcoholic Fatty Liver Disease: the Bile Acid-Activated Farnesoid x Receptor as an Emerging Treatment Target. J. Lipids 2012, 2012, 934396. 10.1155/2012/934396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan H. Y.; Kim D. Y.; Kim S. J.; Jo H. K.; Kim G. W.; Chung S. H. Betulinic Acid Alleviates Non-Alcoholic Fatty Liver by Inhibiting SREBP1 Activity via the AMPK-mTOR-SREBP Signaling Pathway. Biochem. Pharmacol. 2013, 85, 1330–1340. 10.1016/j.bcp.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Sookoian S.; Gianotti T. F.; Rosselli M. S.; Burgueno A. L.; Castano G. O.; Pirola C. J. Liver Transcriptional Profile of Atherosclerosis-Related Genes in Human Nonalcoholic Fatty Liver Disease. Atherosclerosis 2011, 218, 378–385. 10.1016/j.atherosclerosis.2011.05.014. [DOI] [PubMed] [Google Scholar]
- Petrosillo G.; Portincasa P.; Grattagliano I.; Casanova G.; Matera M.; Ruggiero F. M.; Ferri D.; Paradies G. Mitochondrial Dysfunction in Rat with Nonalcoholic Fatty Liver Involvement of Complex I, Reactive Oxygen Species and Cardiolipin. Biochim. Biophys. Acta, Bioenerg. 2007, 1767, 1260–1267. 10.1016/j.bbabio.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Pessayre D. Role of Mitochondria in Non-Alcoholic Fatty Liver Disease. J. Gastroenterol. Hepatol. 2007, 22, S20–S27. 10.1111/j.1440-1746.2006.04640.x. [DOI] [PubMed] [Google Scholar]
- Jones M. L.; Martoni C.; Ganopolsky J. G.; Labbe A.; Prakash S. The Human Microbiome and Bile Acid Metabolism: Dysbiosis, Dysmetabolism, Disease and Intervention. Expert Opin. Biol. Ther. 2014, 14, 467–482. 10.1517/14712598.2014.880420. [DOI] [PubMed] [Google Scholar]
- Joyce S. A.; Gahan C. G. Bile Acid Modifications at the Microbe-Host Interface: Potential for Nutraceutical and Pharmaceutical Interventions in Host Health. Annu. Rev. Food Sci. Technol. 2016, 7, 313–333. 10.1146/annurev-food-041715-033159. [DOI] [PubMed] [Google Scholar]
- Baumler A. J.; Sperandio V. Interactions between the Microbiota and Pathogenic Bacteria in the Gut. Nature 2016, 535, 85–93. 10.1038/nature18849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plecha S. C.; Withey J. H. Mechanism for Inhibition of Vibrio Cholerae ToxT Activity by the Unsaturated Fatty Acid Components of Bile. J. Bacteriol. 2015, 197, 1716–1725. 10.1128/JB.02409-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Broeck D.; Horvath C. A. J.; De Wolf M. J. S. Vibrio Cholerae: Cholera Toxin. Int. J. Biochem. Cell Biol. 2007, 39, 1771–1775. 10.1016/j.biocel.2007.07.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.