

Abstract
Single-molecule fluorescence in situ hybridization (smFISH) is a simple and widely used method to measure mRNA transcript abundance and localization in single cells. A comparable single-molecule in situ method to measure mRNA translation would enable a more complete understanding of gene regulation. Here we describe a fluorescence assay to detect ribosome interactions with mRNA (FLARIM). The method adapts smFISH to visualize and characterize translation of single molecules of mRNA in fixed cells. To visualize ribosome–mRNA interactions, we use pairs of oligonucleotide probes that bind separately to ribosomes (via rRNA) and to the mRNA of interest, and that produce strong fluorescence signals via the hybridization chain reaction (HCR) when the probes are in close proximity. FLARIM does not require genetic manipulation, is applicable to practically any endogenous mRNA transcript, and provides both spatial and temporal information. We demonstrate that FLARIM is sensitive to changes in ribosome association with mRNA upon inhibition of global translation with puromycin. We also show that FLARIM detects changes in ribosome association with an mRNA whose translation is upregulated in response to increased concentrations of iron.
Short abstract
We developed a method to image and quantify changes in mRNA translation in fixed cells. The method utilizes RNA in situ hybridization and the hybridization chain reaction to produce bright fluorescence when ribosomes are bound to an mRNA of interest.
Introduction
Gene expression is regulated at both the transcriptional and translational levels. For many genes, changes in mRNA and protein levels are not correlated,1−4 and protein abundance is often dominated by translation rather than transcription.5 Translational regulation is necessary to coordinate the timing, amount, and location of protein synthesis, and is essential to biological processes including cell morphogenesis and migration,6,7 organismal development,8 responses to cell stress,9 and memory formation.10
Ensemble biochemical methods are widely used to measure global changes in mRNA transcription and translation in cells. The most common method for transcriptomic analysis is RNA-seq.11 Ribosome profiling is an extension of RNA-seq in which mRNA fragments bound by ribosomes are isolated and sequenced to assess mRNA translation.3,12,13 The translation efficiency of a particular mRNA can also be determined by fractionating ribosomes using sucrose density gradient centrifugation and measuring the relative abundances of the mRNA in the polysome and nonpolysome fractions.14 These methods measure genome-wide expression levels in populations of cells (although RNA-seq has been modified to measure mRNA expression in single cells as well15). Because these methods require cell disruption, they provide no information about subcellular localization of transcription and translation for particular genes. In order to study local gene regulation, in situ methods that detect mRNA and protein within cells are required.
Various in situ fluorescence imaging techniques have been developed to study transcription and translation in single cells with spatial and temporal resolution.16 Transcription is commonly measured with single-molecule fluorescence in situ hybridization (smFISH), in which mRNAs are probed with fluorophore-labeled DNA oligonucleotides so that their numbers and locations in single cells can be quantified. Several methods have been developed to enhance the brightness and specificity of smFISH.17,18 Recently, the single-molecule hybridization chain reaction (smHCR) has been developed to achieve bright, robust signals for detection of mRNA in cultured cells as well as in thick tissue samples.19 Changes in protein translation can be measured via 35S-methionine labeling,20 bioorthogonal noncanonical amino acid tagging (BONCAT),21,22 or puromycylation.23−25 The latter methods can be combined with the proximity ligation assay (PLA)26 to detect translation of specific proteins in situ,27 but none of these approaches provide information about mRNA abundance or location.
Live-cell fluorescence imaging techniques have utilized dual labeling systems in which an mRNA is genetically tagged with stem-loop recognition elements (MS2 or PP7 stem loops) to which a fluorescently labeled coat protein binds, and the interaction of the mRNA with ribosomes is detected by colocalization of a second label.28−30 Recently, multiple live-cell imaging methods have been developed to simultaneously detect an mRNA and its nascent polypeptide product. Here, a reporter mRNA contains MS2 or PP7 stem loops, and the nascent polypeptide encodes an array of epitope tags to which genetically encoded fluorescent antibodies bind.31−35 Colocalization of fluorescence signals from an mRNA and its nascent polypeptide indicates active translation of that mRNA. These methods are the first to provide single-molecule resolution of mRNA translation events in single living cells. However, they are limited to monitoring reporter mRNAs rather than endogenous transcripts. The cloning required to implement these methods may affect cellular behavior and may not be feasible for all cell types and mRNAs of interest.
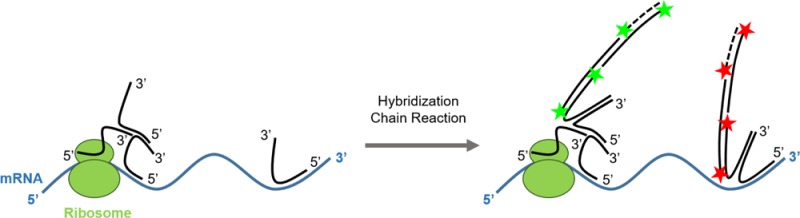
We have developed an imaging method that uses RNA in situ hybridization and the hybridization chain reaction (HCR) to probe translation of unmodified endogenous mRNA transcripts in single fixed cells (Figure 1A). We call this method FLARIM, for fluorescence assay to detect ribosome interactions with mRNA. FLARIM reveals interactions between individual mRNAs and ribosomes to provide a measure of the extent of active translation of the target mRNA species. The method does not require genetic manipulation of cells and can be applied to almost any mRNA of interest. As ribosome profiling extends RNA-seq to quantify mRNAs bound by ribosomes, FLARIM extends smFISH to identify ribosome-bound mRNAs and to monitor the changes in ribosome–mRNA interaction that accompany cellular perturbations. Because FLARIM yields images of fixed cells, the subcellular locations where mRNAs interact with ribosomes can be determined.
Figure 1.

(A) Schematic of method to detect ribosome–mRNA interactions in situ. Multiple DNA oligonucleotide probes are hybridized to ribosomes via rRNA and two different mRNA regions. For illustration purposes, only a single probe per ribosome and mRNA region is shown. Top: When an mRNA is bound by ribosomes, the linker probe can hybridize across the extension sequences of both the ribosome probes and the mRNA interaction probes, and thereby produce a fluorescence signal via HCR. Bottom: When an mRNA is not bound by ribosomes, the linker probe hybridizes weakly to extensions on the ribosome probes and the mRNA interaction probes and can be washed out of cells. (B) NIH 3T3 fibroblasts hybridized with either ribosome probes, mRNA interaction probes to β-actin, or both ribosome and β-actin mRNA interaction probes (top, green, Alexa 546 fluorescence). Cells are simultaneously hybridized with β-actin transcript probes (middle, red, Alexa 488 fluorescence). Nuclei are stained with DAPI (blue). Merge of ribosome–mRNA interaction signals and transcript signals (bottom). Scale bar = 10 μm. (C) Zoom of single mRNA molecules. Red spot: mRNA transcript without ribosome interaction. Yellow spots: mRNA transcripts with ribosome interaction. Scale bar = 1 μm. (D) Fraction of β-actin transcript spots colocalized with ribosome–mRNA interaction spots. Error bars, standard deviation. Data represent two independent experiments, n = 10 or 11 cells per experiment.
Here we introduce and characterize FLARIM in NIH 3T3 mouse fibroblasts. We first demonstrate the method by detecting the interaction of β-actin mRNA with ribosomes and by probing how this interaction changes upon treatment with the translation inhibitor puromycin. Both the fraction of mRNAs bound to ribosomes and the intensities of individual interaction signals decrease. We then examine translational regulation of ferritin heavy chain (FTH1) mRNA in response to added iron. We observe an increase in ribosome–mRNA interaction over a 24-h iron incubation period. We also note a small increase in FTH1 mRNA copy number for cells treated with iron, in contrast to previous reports that FTH1 mRNA levels are unchanged by addition of iron.36,37 FLARIM thus provides both spatial and temporal information about two of the key steps in regulation of gene expression.
Results and Discussion
To visualize ribosome-bound mRNAs, we use pairs of oligonucleotide probes that bind separately to ribosomes (via rRNA) and to the mRNA of interest, and that produce strong fluorescence signals via the hybridization chain reaction (HCR) when in close proximity (Figure 1A). Ribosomes are hybridized with multiple oligonucleotide probes that bind to 18S rRNA. We used the mouse rRNA secondary structure of Holmberg et al.38 to identify regions on the 18S rRNA that are relatively unstructured and that contain bases shown to be accessible for chemical modification. We preferentially targeted our ribosome probes to these regions and designed 24 unique probes in total. We verified that the corresponding sense probes for 18S rRNA do not produce fluorescence via FISH (Figure S1). An mRNA of interest is hybridized with two different sets of oligonucleotide probes: one (designated “mRNA interaction probes”) that pairs with the ribosome probes to form binding sites for a linker probe that carries an HCR initiator, and a second (“mRNA transcript probes”) that separately labels the mRNA transcript with a different HCR initiator. Interaction probes are targeted to the coding sequence (CDS), the region of the mRNA that is translated by ribosomes. Transcript probes are primarily targeted to untranslated regions (5′UTR and 3′UTR) of the mRNA. We targeted each mRNA region with 15–36 probes. Multiple probes are used per target in order to increase the signal-to-noise ratio and to discriminate signal arising from true mRNAs from signal resulting from nonspecific binding of individual probes.39,40 Sequences of all oligonucleotide probes used in this study are listed in Supplemental Table 1.
Each mRNA transcript probe contains an HCR initiator sequence and a 25-nucleotide (nt) region complementary to the mRNA. In HCR, a single-stranded initiator sequence is used to bind and open fluorescently labeled DNA hairpins that then assemble into a fluorescent polymer, resulting in a bright fluorescent signal at the site of amplification. Choi and co-workers have introduced five unique HCR initiator sequences (designated B1–B5), each with a corresponding pair of HCR hairpins for fluorescence amplification.17 We used the B2 HCR initiator and its corresponding hairpins coupled to Alexa Fluor 488 for all mRNA transcript probes (Figure S2, Table S1). Each mRNA interaction probe and each ribosome probe contain a 25-nt region complementary to its target RNA, a 13-nt polyA spacer, and a 22-nt extension sequence. A common extension sequence is used for all mRNA interaction probes, and a different extension sequence is used for all ribosome probes (Figure S2, Table S1).
Extension sequences on the mRNA interaction and ribosome probes are hybridized with an oligonucleotide linker probe bearing a 26-nt binding sequence that spans both extensions when they are in close proximity (15 nt hybridize to the mRNA extensions; 11 nt hybridize to the ribosome extensions) (Figure S2). The binding strength of the linker is tuned with formamide, which lowers the melting temperature of DNA.41 The amount of formamide in solution during the linker hybridization step and subsequent wash steps is adjusted such that the linker remains bound when it spans both extension sequences but not when it hybridizes only one extension (Figure 1A). We determined the appropriate amount of formamide by titrating it in our wash solution and our hybridization buffer during the linker hybridization step. We found that 35% formamide removed the most background signal without comprising hybridization of mRNA transcript probes (Figure S3).
The linker probe also contains an HCR initiator sequence. We used the B3 HCR initiator and its corresponding HCR hairpins coupled to Alexa Fluor 546 to amplify fluorescence signals associated with the linker probe. Signals from the linker probes and from mRNA transcript probes appear as single, diffraction-limited spots when visualized by confocal microscopy (Figure 1C). In the ideal FLARIM scheme, spots that colocalize in the Alexa 488 (shown in red throughout this study) and Alexa 546 (shown in green throughout this study) channels indicate mRNAs bound to ribosomes; spots that appear only in the Alexa 488 channel indicate mRNAs that do not interact with ribosomes. On the basis of the assumption that three nucleotides add ∼1 nm to the length of a DNA probe,42 we estimated that ribosome probes and mRNA interaction probes must be separated by no more than ∼18 nm if they are to produce interaction signals.
We first tested FLARIM in situ in NIH 3T3 fibroblasts, using probes designed for β-actin mRNA. Control experiments in which either the mRNA interaction probes or the ribosome probes were omitted showed little or no labeling from the linker probe, consistent with our expectation that the linker can be effectively washed out of cells when it binds only to an mRNA interaction probe or to a ribosome probe. However, when both probe sets were present, we saw a significant increase in signal from the linker probe (Figure 1B) and substantial colocalization of these signals with those derived from the β-actin transcript probes (Figure 1C). We found on average that 61 ± 4% (n = 21 cells) of β-actin transcripts in the cytoplasm were colocalized with ribosomes.
We examined β-actin transcript probes in control experiments with either ribosome probes or mRNA interaction probes to ensure that signals from the combination of the latter two probe sets showed significantly higher colocalization to β-actin transcripts than to background signals from either probe set alone. Both sets of HCR hairpins were added to all control experiments. Samples with ribosome probes or mRNA interaction probes alone produced punctate Alexa 546 emission to which only 8 ± 1% (n = 10 cells) and 7 ± 2% (n = 11 cells) of Alexa 488 spots colocalized, respectively (Figure 1D, Table S3). We also checked potential background colocalization from the linker probe. Cells treated with the linker probe alone produced spots of Alexa 546 emission that colocalized to fewer than 1% (n = 11 cells) of Alexa 488 spots (Table S3). We conclude that the levels of false positive signals arising from nonspecific binding of the linker, or from the HCR amplification step, are low. As a further check on the method, we analyzed ribosome interaction with β-actin mRNA in cell nuclei, where translation is not expected to occur. We found that only 12 ± 12% (n = 10 cells) of β-actin transcripts in cell nuclei colocalized with ribosome signal, consistent with the results of the control experiments described above (Figure S4). The uncertainty in the measurement of nuclear colocalization arises from the small number (average of 11 ± 4 spots per nucleus for 10 cells) of β-actin mRNA spots in the nucleus. To determine the utility of FLARIM for the study of transcripts characterized by lower copy numbers, we examined actin-related protein 3 (Arp3) mRNA, which is ∼10× less abundant than β-actin mRNA (we measured an average of 190 Arp3 mRNAs per cell, n = 31 cells). As with β-actin, we found that the fraction Arp3 transcripts that colocalized to background signal from ribosome or mRNA interaction probes alone was at least 8× lower than the fraction that colocalized to signal from the combination of both probe sets (Figure S5).
The percentage of cytoplasmic β-actin transcripts observed to bind ribosomes (61 ± 4%) is almost certainly an underestimate. Fluorescence signals generated by smFISH and smHCR invariably yield less than 100% colocalization for probes targeted to the same message.19,39,43 For example, Shah and co-workers found that when using three sets of smHCR probes per transcript, approximately 85% of spots from a single channel colocalized with spots from at least one other channel.19 To set an upper bound on the extent of ribosome–mRNA interaction to be expected in our β-actin experiments, we performed another control experiment in which we replaced the linker probe (which hybridizes to only 15 nt of the mRNA interaction probe) with a linker complementary to 24 nt of the interaction probe (see Figure S6 schematic). This experiment showed 74 ± 3% of Alexa 488 spots to colocalize with spots in the Alexa 546 channel (Figure S6). This result indicates that the measured value of 61 ± 4% is indeed an underestimate of the percent of cytoplasmic β-actin transcripts bound to ribosomes. Our results are consistent with the polysome profiling data of Ventoso and co-workers, who found the fraction of β-actin mRNAs associated with polysomes in NIH 3T3 cells to be 0.72.44
We tested the sensitivity of FLARIM to changes in ribosome–mRNA binding by treating cells with puromycin, a translation inhibitor that causes dissociation of ribosomal subunits from mRNA.45 The effect of puromycin is apparent in a comparison of side-by-side images of treated and untreated cells (Figure 2A). Cells treated with puromycin show less colocalization (yellow) between transcript spots (red) and ribosome-mRNA interaction spots (green). The percentage of β-actin transcripts interacting with ribosomes in the cytoplasm decreased from 61 ± 4% in control cells to 38 ± 6% in puromycin-treated cells (Figure 2B). Furthermore, the intensities of the fluorescence signals associated with single ribosome–mRNA interaction spots shifted to lower values (Figure 2C), indicating a reduction in the number of ribosomes bound per β-actin transcript.
Figure 2.

Translation inhibitor puromycin causes a significant decrease in ribosome–mRNA interaction. (A) Ribosome–mRNA interaction images for β-actin in NIH 3T3 cells that were either untreated (left) or treated (right) with puromycin at 200 μg/mL for 1 h. In the puromycin sample, there is a noticeable decrease in detectable colocalization (yellow) between β-actin mRNA transcript signals (red, Alexa 488 fluorescence) and ribosome–mRNA interaction signals (green, Alexa 546 fluorescence). Top, scale bar = 20 μm. Bottom, scale bar = 2 μm. (B) Fraction of β-actin mRNA transcript spots per cell colocalized with a ribosome–mRNA interaction spot, with and without puromycin treatment. Dots represent single cells. Data represent two independent experiments. n = 7–16 cells per condition per experiment. Error bars, standard deviation. ****P < 0.0001, Student’s t test. (C) Distribution of fluorescence intensities of ribosome–mRNA interaction spots for β-actin, with and without puromycin treatment. Representative data from one experiment. Control, n = 10 cells and 11 314 spots; puromycin, n = 7 cells and 5343 spots.
The observed changes in signal colocalization and signal intensity with puromycin treatment demonstrate the sensitivity of the FLARIM method to perturbations in ribosome association with mRNA. We found no change in the average number of β-actin mRNAs per cell after puromycin treatment (Figure S7). We measured an average of ∼2000 β-actin transcripts per cell, in agreement Schwanhäusser’s estimate of ∼2200 β-actin transcripts per NIH 3T3 cell, as determined by mRNA sequencing.5 We also found no change in the fraction of ribosome–mRNA interaction spots colocalizing with β-actin transcript spots after puromycin treatment (Table S5). To estimate the total change in ribosome interaction with β-actin mRNA after puromycin treatment, we multiplied the fraction of β-actin mRNAs colocalized with ribosomes by the average intensity of the associated Alexa 546 spots. We observed a 2.6-fold decrease in ribosome interaction based on this metric (average of two independent experiments, n = 7–16 cells per condition per experiment). As an additional check on the sensitivity of FLARIM measurements to changes in ribosome association with mRNA, we treated cells with both puromycin and 4E1RCat, which inhibits formation of the translation initiation complex and hence prevents recruitment of the small ribosomal subunit to mRNA.46 The percentage of β-actin transcripts interacting with ribosomes in this experiment dropped to 23 ± 5% (Figure S8), indicating that the signal observed with puromycin treatment alone may have reflected binding of the small ribosomal subunit to the 5′ UTR of β-actin mRNA.
We subjected the FLARIM method to a second test by examining the translational regulation of ferritin synthesis in response to iron treatment.47 Under standard conditions in cell culture, ferritin heavy chain (FTH1) mRNA is translationally repressed by binding of an iron regulatory protein (IRP) to an iron response element (IRE) in the 5′UTR. Upon addition of iron, the IRP is released from the IRE, ribosomes bind to the mRNA, and FTH1 is efficiently translated5,48,49 (Figure 3A). Increases in FTH1 protein levels in mammalian cells in response to elevated iron are attributed to increased translation (not transcription), as the levels of FTH1 mRNA have been shown to remain constant.36,37
Figure 3.

Changes in FTH1 expression in response to added iron. (A) Schematic of translational regulation of FTH1 mRNA by iron. (B) Images illustrating increase in ribosome–mRNA interaction for FTH1 after iron treatment for 4 h (right) compared to a vehicle control (left). In the iron-treated sample, there is a noticeable increase in detectable colocalization (yellow) between FTH1 mRNA transcript signals (red, Alexa 488 fluorescence) and ribosome–mRNA interaction signals (green, Alexa 546 fluorescence). Representative results from three independent experiments are shown. Scale bar = 20 μm. (C) Fraction of FTH1 mRNA transcript spots per cell colocalized with a ribosome–mRNA interaction spot, with and without iron treatment over time. Dots represent single cells. Data represent three independent experiments. n = 10–17 cells per condition per experiment. Error bars, standard deviation. ****P < 0.0001 (one-way ANOVA with Dunnett’s test). (D) Distribution of fluorescence intensities of ribosome–mRNA interaction spots for FTH1 in cells treated with iron for 4 h compared to a vehicle control. Representative results from one experiment. Vehicle, n = 17 cells and 3503 spots; 4 h, n = 15 cells and 9106 spots. (E) Changes in FTH1 mRNA level per cell, with and without iron treatment for 4 h. Error bars, standard deviation. ***P < 0.0002, ****P < 0.0001 (one-way ANOVA with Dunnett’s test).
We used hemin, an iron porphyrin, as the source of iron. When added to cells in culture, hemin rapidly releases iron intracellularly, and has been shown to induce ferritin synthesis.36 We added hemin at a final concentration of 50 μM to cell culture media and fixed cells after different periods of time. Western blotting confirmed that the FTH1 protein level increased upon addition of hemin (Figure S9). In companion FLARIM experiments, we detected a noticeable increase in interaction of the FTH1 mRNA with ribosomes in cells treated with hemin (Figure 3B). After 4 h of treatment, the fraction of FTH1 mRNAs interacting with ribosomes per cell doubled, from 21 ± 4% in vehicle-treated cells to 43 ± 7% in cells incubated with hemin. The extent of increased interaction was essentially constant over 24 h (Figure 3C). The intensities of the fluorescence signals associated with single ribosome–mRNA interaction spots also shifted to higher values (Figure 3D, Figure S10). We found no significant colocalization of FTH1 transcript signals to background signals in control experiments containing only ribosome probes or only mRNA interaction probes (Table S4). We also found no difference in the fraction of ribosome–mRNA interaction spots colocalizing with FTH1 transcript spots between vehicle-treated and iron-treated cells (Table S6).
As discussed previously with respect to β-actin, FLARIM almost certainly provides underestimates of the fractions of ribosome-bound transcripts, owing to imperfect colocalization of the mRNA transcript and interaction probes (Figure S6). Nevertheless, the method reveals distinct increases in ribosome association with FTH1 when translation of the mRNA is upregulated. Our finding that FTH1 transcripts are translated at a lower rate than β-actin transcripts is consistent with the polysome profiling data reported by Ventoso et al., who found that only about 6% of FTH1 mRNAs are associated with polysomes in NIH 3T3 cells in the absence of iron treatment.44
As before, we estimated the change in ribosome interaction with FTH1 mRNA after addition of iron by multiplying the fraction of FTH1 mRNAs colocalized with ribosomes by the average intensity of the associated Alexa 546 spots for each treatment condition. After 4, 12, and 24 h of iron treatment, we observed 2.7-, 2.2-, and 2.3-fold increases, respectively, in ribosome interaction with FTH1 mRNA compared to the vehicle control (average of three independent experiments, n = 10–17 cells per condition per experiment).
We detected an average of ∼1000 FTH1 transcripts per NIH 3T3 cell in our vehicle control condition. In comparison, Schwanhäusser et al. estimated ∼2200 FTH1 transcripts per NIH 3T3 cell in media with no added iron.5 Although several previous studies report that FTH1 mRNA levels in mammalian cells are unchanged upon iron treatment,36,37 we observed a slight but statistically significant (P < 0.0002 at 4 h, P < 0.0001 at 12 and 24 h) increase in the number of FTH1 mRNAs per cell after iron treatment (Figure 3E). On average, cells treated with iron for 4–24 h contained roughly 40% more copies of FTH1 mRNA than untreated cells. This modest increase in mRNA may not have been detectable with previous studies, which used Northern blotting for quantification of FTH1 mRNA abundance.36,37 The increase may also be a unique response in NIH 3T3 cells under our experimental conditions. Studies that suggest unchanged levels of FTH1 mRNA upon treatment with iron have focused on rat liver cells and transgenic mouse fibroblasts.36,37,50 However, an investigation of Friend erythroleukemia cells (FLCs) found that FTH1 mRNA expression increased by up to 10-fold upon treatment with hemin.51 The fact that FLARIM reveals changes in both mRNA interaction with ribosomes and mRNA copy number illustrates the utility of the method in assessing both translational and transcriptional control of gene expression in single cells.
Conclusions
This study shows that changes in ribosome association with endogenous, unmodified mRNAs can be imaged and quantified in situ using standard DNA oligonucleotide probes and HCR. We characterized this method, which we termed FLARIM, in NIH 3T3 mouse fibroblasts. We first measured ribosome–mRNA interactions for β-actin in single cells and detected a decrease in these interactions when cells were treated with the translation inhibitor puromycin. We observed no significant ribosome–mRNA interactions in cell nuclei, where translation is not expected to occur, although a few studies report conflicting evidence.24,52 We also detected increased ribosome binding to FTH1 mRNA when cells were treated with iron, and surprisingly, we noted an increase in FTH1 mRNA levels in concert with the increase in ribosome interaction. Because FLARIM interrogates both transcriptional and translational processes, it has the potential to provide unique insights into the nature of gene regulation in single cells. Although FLARIM was applied only to mouse cells in this study, we designed a nearly identical set of ribosome probes for human 18S rRNA (Table S2) to facilitate FLARIM studies in human cells.
FLARIM is simple and inexpensive, uses commercially available reagents and common laboratory equipment, and requires no genetic manipulation of the cells of interest. Experiments can be completed in 2–3 days from cell fixation to image collection and analysis. Compared to various proximity ligation assays,53,54 which could conceivably be adapted to analyze interactions between ribosomes and mRNA, FLARIM requires fewer steps and is enzyme-free, making it cheaper and easier to modify for different sample types. The method is also amenable to the study of fixed clinical samples, which are inaccessible to techniques that require cloning. We anticipate that FLARIM will be especially useful in studies of local mRNA translation. For example, neurons contain thousands of different mRNA transcripts in their dendrites and or axons,55 and FLARIM is well suited to the monitoring of changes in ribosomal association of these transcripts in response to external stimuli. In similar fashion, studies of local translation during embryonic development56 should prove fruitful.
FLARIM should be applicable to essentially any mRNA of interest; however, it does require that the mRNA be efficiently hybridized with oligonucleotide probes. It is conceivable that short mRNAs may not bind a sufficient number of probes to produce reliable signals. Using a higher number of probes is known to improve the robustness of mRNA detection39 and to increase the ratio of signal to autofluorescence,40 although the number of probes needed for reliable mRNA detection may depend on the target transcript. It is also important to note that FLARIM does not yield a numerically accurate measure of the number of mRNAs being translated in the cell. Rather, it provides an approximate measure of translation, useful for comparisons among samples, along with spatial information and a measure of mRNA copy number. We use ribosome interaction as a proxy for translation, but it is known that mRNAs can be bound by ribosomes without being translated, e.g., in the case of ribosome stalling.57
It should be straightforward to modify the FLARIM method to enable studies of other molecular interactions in single cells. In addition to RNA–RNA interactions, protein–RNA and protein–protein interactions can be revealed by using antibodies conjugated to DNA oligonucleotides or by using aptamer probes.58 Interactions with DNA might be measured by combining the method with DNA FISH techniques. The method is designed in a manner that makes it highly tunable. In adapting the method to detect interactions between different molecular species and in different cell types, the probe sequences and the stringency of the wash buffer can easily be adjusted to lower background and ensure that HCR amplification occurs essentially only from interacting probe pairs. Probe sequences can also be engineered to increase or decrease the maximum distance between probes that allows for signal generation.
Acknowledgments
We thank the National Science Foundation Graduate Research Fellowship Program (NSF GRFP, Grant Number 1144469), the Rose Hills Foundation (Caltech SURF), the German Research Foundation (Grant No. MI 1315/4), and the Programmable Molecular Technology Initiative of the Gordon and Betty Moore Foundation for support of this work. We thank H. M. T. Choi and N. A. Pierce for suggesting the use of a linker probe carrying an HCR initiator as a mechanism for selectively generating signal from cognate probe pairs colocalized by targets in the sample. We thank Florian Mueller for development of FISH-quant and the Broad Institute for development of Cell Profiler for image analysis. We thank Johannes Stegmaier and the Center for Advanced Methods in Biological Image Analysis at the Beckman Institute (CAMBIA) for adapting the XPIWIT software tool for analysis of nuclear transcripts. We thank Andres Collazo for assistance with the LSM 800 confocal microscope in the Biological Imaging Facility of the Beckman Institute at Caltech.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00048.
The authors declare no competing financial interest.
Supplementary Material
References
- Gygi S. P.; Rochon Y.; Franza B. R.; Aebersold R. Correlation between Protein and mRNA Abundance in Yeast. Mol. Cell. Biol. 1999, 19, 1720–1730. 10.1128/MCB.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T.; Thorsson V.; Ranish J. A.; Christmas R.; Buhler J.; Eng J. K.; Bumgarner R.; Goodlett D. R.; Aebersold R.; Hood L. Integrated Genomic and Proteomic Analyses of a Systematically Perturbed Metabolic Network. Science 2001, 292, 929–934. 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- Ingolia N. T.; Ghaemmaghami S.; Newman J. R. S.; Weissman J. S. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science 2009, 324, 218–223. 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y.; Choi P. J.; Li G.; Chen H.; Babu M.; Hearn J.; Emili A.; Xie X. S. Quantifying E. Coli Proteome and Transcriptome with Single-Molecule Sensitivity in Single Cells. Science 2010, 329, 533–538. 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B.; Busse D.; Li N.; Dittmar G.; Schuchhardt J.; Wolf J.; Chen W.; Selbach M. Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473 (7347), 337–342. 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Shestakova E. A.; Singer R. H.; Condeelis J. The Physiological Significance of Beta -Actin mRNA Localization in Determining Cell Polarity and Directional Motility. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (13), 7045–7050. 10.1073/pnas.121146098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttelmaier S.; Zenklusen D.; Lederer M.; Dictenberg J.; Lorenz M.; Meng X.; Bassell G. J.; Condeelis J.; Singer R. H. Spatial Regulation of β-Actin Translation by Src-Dependent Phosphorylation of ZBP1. Nature 2005, 438, 512–515. 10.1038/nature04115. [DOI] [PubMed] [Google Scholar]
- Curtis D.; Lehmann R.; Zamore P. D. Translational Regulation in Development. Cell 1995, 81 (2), 171–178. 10.1016/0092-8674(95)90325-9. [DOI] [PubMed] [Google Scholar]
- Holcik M.; Sonenberg N. Translational Control in Stress and Apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6 (4), 318–327. 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- Costa-mattioli M.; Sossin W. S.; Klann E.; Sonenberg N. Translational Control of Long-Lasting Synaptic Plasticity and Memory. Neuron 2009, 61 (1), 10–26. 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A.; Williams B. A.; McCue K.; Schaeffer L.; Wold B. Mapping and Quantifying Mammalian Transcriptomes by RNA-Seq. Nat. Methods 2008, 5 (7), 621–628. 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Jan C. H.; Williams C. C.; Weissman J. S. Principles of ER Cotranslational Translocation Revealed by Proximity-Specific Ribosome Profiling. Science 2014, 346 (6210), 1257521. 10.1126/science.1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C. C.; Jan C. H.; Weissman J. S. Targeting and Plasticity of Mitochondrial Proteins Revealed by Proximity-Specific Ribosome Profiling. Science 2014, 346 (6210), 748–751. 10.1126/science.1257522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandin V.; Sikström K.; Alain T.; Morita M.; McLaughlan S.; Larsson O.; Topisirovic I. Polysome Fractionation and Analysis of Mammalian Translatomes on a Genome-Wide Scale. J. Visualized Exp. 2014, 87, 1–10. 10.3791/51455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F.; Barbacioru C.; Wang Y.; Nordman E.; Lee C.; Xu N.; Wang X.; Bodeau J.; Tuch B. B.; Siddiqui A.; Lao K.; Surani M. A. mRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6 (5), 377–382. 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- Rodriguez A. J.; Shenoy S. M.; Singer R. H.; Condeelis J. Visualization of mRNA Translation in Living Cells. J. Cell Biol. 2006, 175 (1), 67–76. 10.1083/jcb.200512137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H. M. T.; Beck V. A.; Pierce N. A. Next-Generation in Situ Hybridization Chain Reaction: Higher Gain, Lower Cost, Greater Durability. ACS Nano 2014, 8 (5), 4284–4294. 10.1021/nn405717p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Flanagan J.; Su N.; Wang L. C.; Bui S.; Nielson A.; Wu X.; Vo H. T.; Ma X. J.; Luo Y. RNAscope: A Novel in Situ RNA Analysis Platform for Formalin-Fixed, Paraffin-Embedded Tissues. J. Mol. Diagn. 2012, 14 (1), 22–29. 10.1016/j.jmoldx.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S.; Lubeck E.; Schwarzkopf M.; He T.; Greenbaum A.; Sohn C. H.; Lignell A.; Choi H. M. T.; Gradinaru V.; Pierce N. A.; Cai L. Single-Molecule RNA Detection at Depth via Hybridization Chain Reaction and Tissue Hydrogel Embedding and Clearing. Development 2016, 143 (2016), 2862–2867. 10.1242/dev.138560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S.-E. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteomics 2002, 1 (5), 376–386. 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- Dieterich D. C.; Link A. J.; Graumann J.; Tirrell D. A.; Schuman E. M. Selective Identification of Newly Synthesized Proteins in Mammalian Cells Using Bioorthogonal Noncanonical Amino Acid Tagging (BONCAT). Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (25), 9482–9487. 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich D. C.; Hodas J. J. L.; Gouzer G.; Shadrin I. Y.; Ngo J. T.; Triller A.; Tirrell D. A.; Schuman E. M. In Situ Visualization and Dynamics of Newly Synthesized Proteins in Rat Hippocampal Neurons. Nat. Neurosci. 2010, 13 (7), 897–905. 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starck S. R.; Green H. M.; Alberola-Ila J.; Roberts R. W. A General Approach to Detect Protein Expression In Vivo Using Fluorescent Puromycin Conjugates. Chem. Biol. 2004, 11, 999–1008. 10.1016/j.chembiol.2004.05.011. [DOI] [PubMed] [Google Scholar]
- David A.; Dolan B. P.; Hickman H. D.; Knowlton J. J.; Clavarino G.; Pierre P.; Bennink J. R.; Yewdell J. W. Nuclear Translation Visualized by Ribosome-Bound Nascent Chain Puromycylation. J. Cell Biol. 2012, 197 (1), 45–57. 10.1083/jcb.201112145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt E. K.; Clavarino G.; Ceppi M.; Pierre P. SUnSET, a Nonradioactive Method to Monitor Protein Synthesis. Nat. Methods 2009, 6 (4), 275–277. 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- Söderberg O.; Gullberg M.; Jarvius M.; Ridderstråle K.; Leuchowius K.-J.; Jarvius J.; Wester K.; Hydbring P.; Bahram F.; Larsson L.-G.; Landegren U. Direct Observation of Individual Endogenous Protein Complexes in Situ by Proximity Ligation. Nat. Methods 2006, 3 (12), 995–1000. 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- tom Dieck S.; Kochen L.; Hanus C.; Heumüller M.; Bartnik I.; Nassim-Assir B.; Merk K.; Mosler T.; Garg S.; Bunse S.; Tirrell D. A.; Schuman E. M. Direct Visualization of Newly Synthesized Target Proteins in Situ. Nat. Methods 2015, 12 (5), 411–414. 10.1038/nmeth.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead J. M.; Lionnet T.; Wilbertz J. H.; Wippich F.; Ephrussi A.; Singer R. H.; Chao J. A. An RNA Biosensor for Imaging the First Round of Translation from Single Cells to Living Animals. Science 2015, 347 (6228), 1367–1370. 10.1126/science.aaa3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Buxbaum A. R.; Katz Z. B.; Yoon Y. J.; Singer R. H. Quantifying Protein-mRNA Interactions in Single Live Cells. Cell 2015, 162 (1), 211–220. 10.1016/j.cell.2015.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Z. B.; English B. P.; Lionnet T.; Yoon Y. J.; Monnier N.; Ovryn B.; Bathe M.; Singer R. H. Mapping Translation “Hot-Spots” in Live Cells by Tracking Single Molecules of mRNA and Ribosomes. eLife 2016, 5, e10415. 10.7554/eLife.10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki T.; Lyon K.; Deluca K. F.; Deluca J. G.; English B. P.; Lavis L. D.; Grimm J. B.; Viswanathan S.; Looger L. L.; Lionnet T.; Stasevich T. J.; Zhang Z. Real-Time Quantification of Single RNA Translation Dynamics in Living Cells. Science 2016, 352, 1425–1429. 10.1126/science.aaf0899. [DOI] [PubMed] [Google Scholar]
- Wang C.; Han B.; Zhou R.; Zhuang X. Real-Time Imaging of Translation on Single mRNA Transcripts in Live Cells. Cell 2016, 165 (4), 990–1001. 10.1016/j.cell.2016.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Eliscovich C.; Yoon Y. J.; Singer R. H. Translation Dynamics of Single mRNAs in Live Cells and Neurons. Science 2016, 352, 1430–1435. 10.1126/science.aaf1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X.; Hoek T. A.; Vale R. D.; Tanenbaum M. E. Dynamics of Translation of Single mRNA Molecules In Vivo. Cell 2016, 165 (4), 976–989. 10.1016/j.cell.2016.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichon X.; Bastide A.; Safieddine A.; Chouaib R.; Samacoits A.; Basyuk E.; Peter M.; Mueller F.; Bertrand E. Visualization of Single Endogenous Polysomes Reveals the Dynamics of Translation in Live Human Cells. J. Cell Biol. 2016, 214 (6), 769–781. 10.1083/jcb.201605024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J.; Munro H. Translation of Ferritin Light and Heavy Subunit mRNAs Is Regulated by Intracellular Chelatable Iron Levels in Rat Hepatoma Cells. Proc. Natl. Acad. Sci. U. S. A. 1987, 84 (8), 2277–2281. 10.1073/pnas.84.8.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouault T. A.; Hentze M. W.; Dancis A.; Caughman W.; Harford J. B.; Klausner R. D. Influence of Altered Transcription on the Translational Control of Human Ferritin Expression. Proc. Natl. Acad. Sci. U. S. A. 1987, 84 (18), 6335–6339. 10.1073/pnas.84.18.6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg L.; Melander Y.; Nygård O. Probing the Structure of Mouse Ehrlich Ascites Cell 5.8S, 18S and 28S Ribosomal RNA in Situ. Nucleic Acids Res. 1994, 22 (8), 1374–1382. 10.1093/nar/22.8.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A.; van Den Bogaard P.; Rifkin S. A.; Van Oudenaarden A.; Tyagi S. Imaging Individual mRNA Molecules Using Multiple Singly Labeled Probes. Nat. Methods 2008, 5 (10), 877–879. 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H. M. T.; Chang J. Y.; Trinh L. a; Padilla J. E.; Fraser S. E.; Pierce N. a. Programmable in Situ Amplification for Multiplexed Imaging of mRNA Expression. Nat. Biotechnol. 2010, 28 (11), 1208–1212. 10.1038/nbt.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConaughy B. L.; Laird C. D.; McCarthy B. J. Nucleic Acid Reassociation in Formamide. Biochemistry 1969, 8 (8), 3289–3295. 10.1021/bi00836a024. [DOI] [PubMed] [Google Scholar]
- van Holde K. E.Chromatin, Springer-Verlag: New York, 1989. [Google Scholar]
- Batish M.; van den Bogaard P.; Kramer F. R.; Tyagi S. Neuronal mRNAs Travel Singly into Dendrites. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (12), 4645–4650. 10.1073/pnas.1111226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventoso I.; Kochetov A.; Montaner D.; Dopazo J.; Santoyo J. Extensive Translatome Remodeling during ER Stress Response in Mammalian Cells. PLoS One 2012, 7 (5), e35915. 10.1371/journal.pone.0035915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel G.; Sabatini D. Dissociation of Mammalian Polyribosomes into Subunits by Puromycin. Proc. Natl. Acad. Sci. U. S. A. 1971, 68 (2), 390–394. 10.1073/pnas.68.2.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cencic R.; Hall D. R.; Robert F.; Du Y.; Min J.; Li L.; Qui M.; Lewis L.; Kurtkaya S.; Dingledine R.; Fu H.; Kozakov D.; Vajda S.; Pelletier J. Reversing Chemoresistance by Small Molecule Inhibition of the Translation Initiation Complex eIF4F. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1046–1051. 10.1073/pnas.1011477108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze M. W.; Muckenthaler M. U.; Andrews N. C. Balancing Acts: Molecular Control of Mammalian Iron Metabolism. Cell 2004, 117, 285–297. 10.1016/S0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- Munro H. N. Iron Regulation of Ferritin Gene Expression. J. Cell. Biochem. 1990, 44 (2), 107–115. 10.1002/jcb.240440205. [DOI] [PubMed] [Google Scholar]
- Torti F. M.; Torti S. V. Regulation of Ferritin Genes and Protein. Blood 2002, 99 (10), 3505–3516. 10.1182/blood.V99.10.3505. [DOI] [PubMed] [Google Scholar]
- Zähringer J.; Baliga B. S.; Munro H. N. Novel Mechanism for Translational Control in Regulation of Ferritin Synthesis by Iron. Proc. Natl. Acad. Sci. U. S. A. 1976, 73 (3), 857–861. 10.1073/pnas.73.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coccia E.; Profita V.; Fiorucci G.; Romeo G.; Affabris E.; Testa U.; Hentze M. W.; Battistini A. Modulation of Ferritin H-Chain Expression in Friend Erythroleukemia Cells: Transcriptional and Translational Regulation by Hemin. Mol. Cell. Biol. 1992, 12 (7), 3015–3022. 10.1128/MCB.12.7.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iborra F. J.; Jackson D. A.; Cook P. R. The Case for Nuclear Translation. J. Cell Sci. 2004, 117, 5713–5720. 10.1242/jcs.01538. [DOI] [PubMed] [Google Scholar]
- Jung J.; Lifland A. W.; Zurla C.; Alonas E. J.; Santangelo P. J. Quantifying RNA-Protein Interactions in Situ Using Modified-MTRIPs and Proximity Ligation. Nucleic Acids Res. 2013, 41, e12. 10.1093/nar/gks837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. E. I.; Xie M.; Shu M.; Steitz J. A.; Dimaio D. A Proximity-Dependent Assay for Specific RNA – Protein Interactions in Intact Cells. RNA 2016, 22, 1785–1792. 10.1261/rna.058248.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajigas J.; Tushev G.; Will T. J.; tom Dieck S.; Fuerst N.; Schuman E. M. The Local Transcriptome in the Synaptic Neuropil Revealed by Deep Sequencing and High-Resolution Imaging. Neuron 2012, 74, 453–466. 10.1016/j.neuron.2012.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K. C.; Ephrussi A. mRNA Localization: Gene Expression in the Spatial Dimension. Cell 2009, 136 (4), 719–730. 10.1016/j.cell.2009.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O.; Hegde R. S. Ribosome-Associated Protein Quality Control. Nat. Struct. Mol. Biol. 2016, 23 (1), 7–15. 10.1038/nsmb.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks R. M.; Pierce N. A. Triggered Amplification by Hybridization Chain Reaction. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (43), 15275–15278. 10.1073/pnas.0407024101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.