

Abstract
Nicotinamide (NAM) is the amide of nicotinic acid and a predominant precursor for NAD+ biosynthesis via the salvage pathway. Sirt1 is a NAD+-dependent deacetylase, playing an important role in regulating cellular functions. Although hepatoprotective effect of NAM has been reported, the underlying mechanism remains elusive. ER stress, induced by saturated fatty acids, in specific palmitate, plays a pathological role in the development of nonalcoholic fatty liver disease. This study aims to determine the effect of NAM on palmitate-induced ER stress in hepatocytes and to elucidate molecular mechanisms behind. Both HepG2 cells and primary mouse hepatocytes were exposed to palmitate (conjugated to BSA at a 2:1 M ratio), NAM, or their combination for different durations. Cellular NAD+ level, Sirt1 expression/activity, ER stress, as well as cAMP/PKA/CREB pathway activation were determined. NAM increased Sirt1 expression and enzymatic activity, which contributes to the ameliorative effect of NAM on palmitate-triggered ER stress. NAM increased intracellular NAD+ level in hepatocytes, however, blocking the salvage pathway, a pathway for NAD+ synthesis from NAM, only partially prevented NAM-induced Sirt1 upregulation while completely prevented NAD+ increase in response to NAM. Further mechanistic investigations revealed that NAM elevated intracellular cAMP level via suppressing PDE activity, leading to downstream PKA and CREB activation. Importantly, cAMP/PKA/CREB pathway blockade abolished not only NAM-induced Sirt1 upregulation, but also its protective effect against ER stress. Our results demonstrate that NAM protects hepatocytes against palmitate-induced ER stress in hepatocytes via upregulating Sirt1. Activation of the cAMP/PKA/CREB pathway plays a key role in NAM-induced Sirt1 upregulation.
Keywords: nicotinamide, Sirt1, NAD+, deacetylation, cAMP, PKA
1. Introduction
Lipotoxicity, the cellular dysfunction or cell death induced by lipid accumulation in cell types other than adipocytes, plays a pathological role in the development of obesity-related non-alcoholic fatty liver disease (NAFLD) [1]. Palmitate, a 16-C saturated fatty acid, is one of the most abundant free fatty acids (FFAs) in the circulation and a major inducer of lipotoxicity in hepatocytes [2]. ER stress plays a mechanistic role in palmitate-induced cellular dysfunction in hepatocytes, including cell death and insulin resistance (IR) [3]. Sirt1 is a NAD+-dependent deacetylase catalyzing the deacetylation of protein targets at a specific lysine residue by hydrolyzing NAD+ to produce a de-acetylated protein, acetyl-ADP-ribose, and nicotinamide (NAM) [4, 5]. It plays an important role in regulating cellular functions via modifying acetylation status of a wide range of molecules, including enzymes, histones, and transcriptional regulators [6]. Impaired hepatic Sirt1 signaling contributes to the pathogenesis of both alcoholic liver disease and NAFLD [7-11]. Sirt1 activation has been reported to protect hepatocytes against palmitate-triggered ER stress [12].
Intracellular NAD+ content plays a key role in controlling Sirt1 expression and activity. The biosynthesis of NAD+ in mammalian cells occurs through two major pathways: a de novo pathway and a salvage pathway [13, 14]. The de novo pathway (the Preiss-Handler pathway) utilizes tryptophan to generate NAD+ through kynurenine pathway, while the salvage pathway regenerates NAD+ from nicotinic acid, NAM, or NAM riboside present in the metabolites of NAD+ or dietary sources. In mammalian cells, most NAD+ is synthesized by the salvage pathway from NAM [4, 5]. The fluctuation in NAD+ availability in response to either energy status change or changes in NAD+ biosynthetic or degradation process leads to altered Sirt1 expression and activity. The increase of cellular NAD+ level derived from either low energy conditions, such as fasting, caloric restriction, and exercise, or enzymatic activation of NAM phophoribosyltransferase (Nampt), the rate limiting enzyme for NAD+ biosynthesis from NAM via the salvage pathway, upregulates Sirt1 activity [15-21]. By contrast, high-energy diet consumption reduces cellular NAD+ concentration, leading to suppressed Sirt1 activation [22-24].
Other than well-documented canonical mechanism of modulating cellular NAD+ level to regulate Sirt1, accumulating evidence supports that Sirt1 activity is also regulated at expression level, which is NAD+-independent. Among a growing list of transcription factors reported to control the expression of Sirt1, cyclic AMP response-element-binding protein (CREB), a transcription factor whose activation is mediated by PKA in response to increased intracellular cAMP level, has recently emerged to play a critical role in the regulation of Sirt1 expression [25, 26].
NAM is the amide of nicotinic acid (vitamin B3 / niacin). It is the general product of Sirt1-catalyzed deacetylation reactions and was reported to inhibit Sirt1 enzyme activity with either competitive or noncompetitive mechanism [27-29]. Paradoxically, as NAM also serves as the predominant endogenous precursor for NAD+ biosynthesis via the salvage pathway, several recent studies have demonstrated that NAM supplementation in cell culture increased intracellular NAD+ level in a variety of cell types, leading to Sirt1 activation [30, 31]. Nevertheless, the effect of NAM on Sirt1 expression and activity in hepatocytes, as well as its role in protecting hepatocytes against harmful cellular stresses, have not been well-established. In this study, we conducted in vitro cell culture studies to determine these events. We demonstrated that NAM supplementation upregulated Sirt1 expression and enzymatic activity in hepatocytes and ameliorated ER stress induced by palmitate exposure. Intriguingly, our mechanistic investigations revealed for the first time that NAM-induced Sirt1 upregulation was independent of its NAD+-incrementing action, but involving cAMP/PKA/CREB pathway activation.
2. Methods and materials
2.1. Cells culture
HepG2 cells was obtained from the American Type Culture Collection (Manassas, VA). Primary mouse hepatocytes were obtained from Celsis In Vitro Technologies (M91684). Both HepG2 cells and primary hepatocytes were cultured in DMEM (Sigma-Aldrich, D5648) containing 10% (v/v) fetal bovine serum, 2 mmol/l glutamine (Sigma-Aldrich, G3126), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified O2/CO2 (95:5) atmosphere. All experiments were repeated at least 3 times.
2.2. Gene silencing by siRNA
Transient gene silencing was attained by transfection either scrambled siRNA (Control, sc-37007, Santa Cruz Biotechnology, Santa Cruz, CA, USA) or siRNAs for target genes (CREB: sc-29281, Sirt1: sc-40986, Santa Cruz Biotechnology, Santa Cruz, CA, USA) into HepG2 cells using siPORT lipid transfection reagent according to the manufacturer's instructions. Gene silencing was verified by detecting protein with immunoblotting analysis after transient transfection with siRNA.
2.3. Cell lysates and Western blotting detections
Total proteins from hepatocyte were obtained using Western lysis buffer consisting of the following: 20 mM TrisHCl, pH 7.4, 150 mM NaCl, 10% glycerol, 2% Nonidet P-40, 1 mM EDTA, pH 8.0, 20 mM sodium fluoride, 30 mM sodium pyrophosphate, 0.2% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, 1 mM sodium vanadate, 50 μM leupeptin, and 5 μM aprotinin. Samples were incubated on ice with frequent vortexing for 15 minutes and centrifuged for 20 minutes at 18,000 g. The protein content of each supernatant was quantified via a protein assay reagent from Bio-Rad Laboratories (Hercules, CA) in accordance with the manufacturer's instructions. Proteins were separated by SDS-PAGE and transferred to 0.45 μm Immobilin-P polyvinylidene difluoride membrane (PerkinElmer Life Sciences). After transfer, membranes were blocked in 5% (wt. /vol.) nonfat dry milk in PBS-0.1% Tween 20 and probed with anti-phospho-PKA (5566), phospho-CREB (9191s), phospho-Perk (3179) from Cell Signaling, anti-beta-actin (sc-1615), anti-Sirt1 (sc-15404) from Santa Cruz Biotechnology, anti-alpha-tubulin (600-401-880) from Rockland Inc. Horseradish peroxidase conjugated secondary antibodies and enhanced chemiluminescence substrate kit (Thermo Scientific, IL) were used in detection of specific proteins.
2.4. Quantitative real-time RT-PCR
Total RNA from hepatocytes was isolated with a phenol-chloroform extraction. For each sample, 1.0 μg total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). The cDNA was amplified in MicroAmp Optical 96-well reaction plates with a SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) on an Applied Biosystems Prism 7000 sequence detection system. Relative gene expression was calculated after normalization by a house-keeping gene (mouse or human 18S rRNA).
2.5. Intracellular NAD+ Measurement
NAD+ level in cell lysate were measured using NAD/NADH quantification kit in accordance with the manufacturer's instructions (Biovision Inc, Milpitas, CA). All data are representative of at least three independent experiments.
2.6. Intracellular cAMP measurement
Intracellular cAMP level in cell lysate were measured using a cAMP enzyme immunoassay kit (Cayman Chemical, Ann Arbor, Michigan) in accordance with the manufacturer's instructions.
2.7. Intracellular Phosphodiesterase (PDE) activity assay
Hepatocytes were treated with NAM for 2 hours and PDE activity was measured using a commercially available assay kit according to the manufacturer's instruction (ab139460, Abcam, Cambridge, MA).
2.8. Immunoprecipitation (IP)
Cells lysates were obtained in IP buffer (150 mM NaCl, 50 mM Tris-HCl, 1% NP-40, PH7.8, and mammalian cell specific protease inhibitor cocktail) and adjusted to the same concentration of 1 mg/ml, then cell lysates (200 μg protein) were incubated with primary antibody (1 mg/ml) overnight at 4°C with constant rotation, followed by adding Protein A/G plus Agarose (Santa Cruz Biotechnology, Santa Cruz, CA) 50 μl to incubate for 1-2 hour at 4°C. The lysates were then centrifuged at 4,000 g for 1 minute, the pelleted beads were then washed 4 times with IP buffer, and immunoprecipitated proteins were eluted from beads by preparing 1×sample for Western Blotting (12.5 μl 4×loading buffer, DTT 5 μl, lysis buffer 37.5 μl). Beads with immunoprecipitated proteins were incubated at 95°C for 5 minutes. After a brief centrifugation, the supernatants (immunoprecipitation samples) were subject to Western Blotting.
2.9. Statistical analysis
All data were expressed as Mean ± SD. Statistical analysis was performed using a one-way ANOVA and was analyzed further by Post-hoc test with Fisher's least significant difference (LSD). Differences between treatments were considered to be statistically significant at p<0.05.
3. Results
3.1. NAM upregulates Sirt1 expression and activity in hepatocytes
The effect of NAM on Sirt1 expression was determined primarily in HepG2 cells and key findings were validated in mouse primary hepatocytes. In HepG2 cells, an eight-hour exposure of NAM increased Sirt1 expression, at both mRNA and protein level, in a dose-dependent manner (Fig. 1A & B). Similar effects were observed when mouse primary hepatocytes were used (Fig. 1C). Time-course changes of Sirt1 protein abundance in response to NAM exposure in HepG2 cells were shown in Fig. 1D. NAM markedly increased Sirt1 expression in 2 hours, and this increase was maintained throughout the 16 hours of the investigation. The effect of NAM on Sirt1 enzymatic activity was determined via detecting the effect of NAM on acetylation status of FOXO1, a well-established substrate of Sirt1-catalyzed deacetylation reaction. Immunoprecipitation result showed that, in comparison to control cells, HepG2 cells exposed to NAM for 4 hours manifested an evident decrease in lysine acetylation level of FoxO1 (Fig. 1E).
Figure 1.
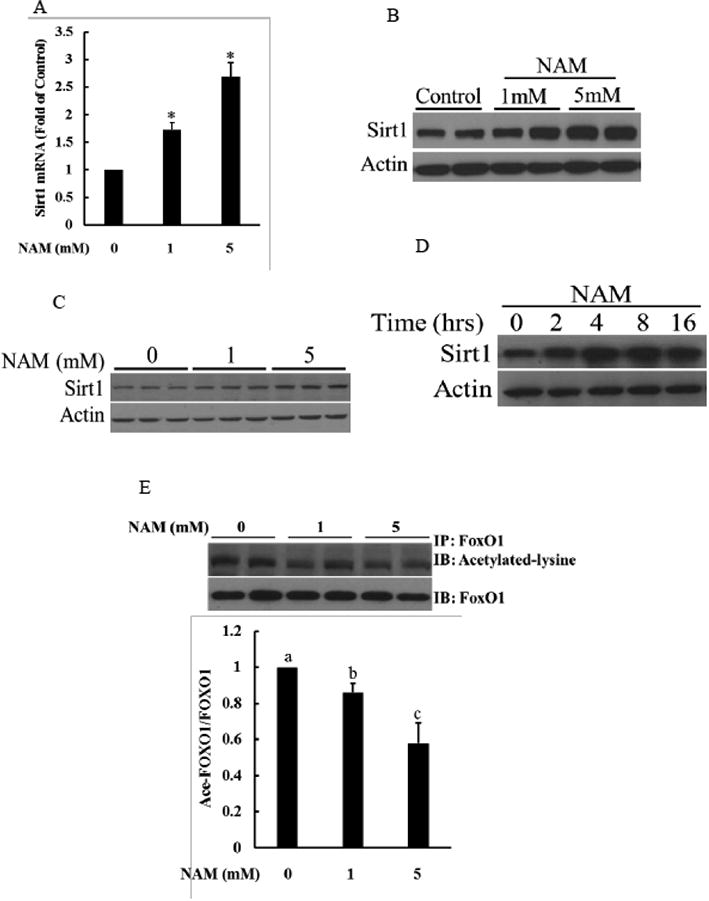
NAM supplementation increases Sirt1 expression and activity in hepatocytes. A & B. NAM increases Sirt1 gene expression in a dose-dependent manner in HepG2 cells. HepG2 cells were treated with NAM with indicated doses for 8 hours. Sirt1 expressions at both mRNA and protein level were determined. All values are denoted as means ± SD from three or more independent studies. * p < 0.05 vs. control. C. NAM upregulates Sirt1 expression in a dose-dependent fashion in primary mouse hepatocytes. Mouse primary hepatocytes were treated with NAM for indicated doses for 8 hours. Sirt1 protein abundance were determined by Western blot. D. Time-course changes of Sirt1 expression in response to NAM supplementation. HepG2 cells were treated with 5 mM NAM for indicated time periods. Sirt1 protein abundance were determined by Western blot. E. NAM increases Sirt1 activity. Sirt1 activity was determined by FoxO1 lysine-acetylated status. Lysine acetylation of immunoprecipated FoxO1 protein were measured in HepG2 cells treated with or without NAM for 4 hours. The pull-down proteins were subjected to Western blot and probed with anti-acetyl lysine to determine FoxO1 acetylation status. All values are denoted as means ± SD from three or more independent studies. Bars with different letters differ significantly (p < 0.05).
3.2. NAM increases intracellular NAD+ levels in hepatocytes
Sirt1 is a NAD+-dependent deacetylase. In mammalian cells NAM is readily converted to NAD+ through the salvage pathway. To determine the effect of NAM supplementation on intracellular NAD contents in hepatocytes, HepG2 cells were treated with NAM (1 and 5 mM) for 2 and 8 hours, respectively, and intracellular NAD+ and NADH concentrations were measured as previously described by us [32]. As shown in Fig. 2, only the higher dose of NAM (5 mM) led to the significant elevations of intracellular NAD+ and NAD+/NADH ratio after a 2-hour NAM exposure, however, they were significantly increased by both concentrations of NAM at the 8-hour time point. At 5 mM, intracellular NAD+ concentration was increased by close to 60% in comparison to control cells after an 8-hour exposure.
Figure 2.
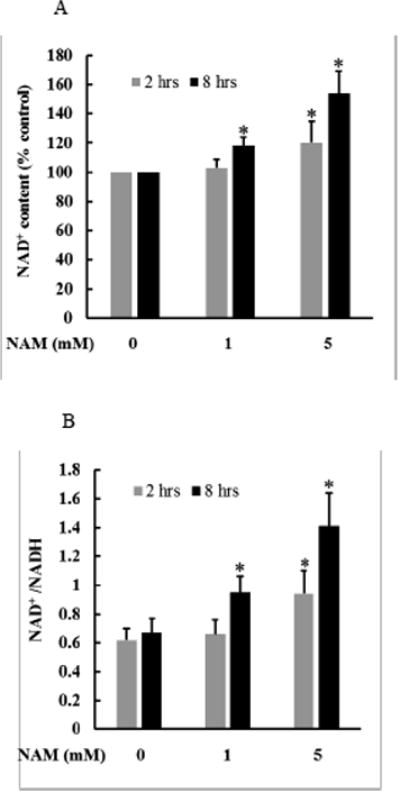
The effect of NAM supplementation on intracellular NAD content. HepG2 cells were treated with NAM (1 and 5 mM) for 2 and 8 hours, respectively. Intracellular NAD+ and NADH levels were determined. A. NAM increases cellular NAD+ concentration. B. NAM increases cellular NAD+/NADH ratio. * p < 0.05 vs. control. All values are denoted as means ± SD from three or more independent studies.
3.3. Cellular NAD+ increase only partially contributes to NAM-induced Sirt1 upregulation
NAM phosphoribosyltransferase (Nampt) is the rate-limiting enzyme in the salvage pathway for NAD+ biosynthesis from NAM, which converts NAM to NAM mononucleotide (NMN) [33], followed by NAD+ generation (Fig. 3A). To determine the potential mechanistic involvement of NAD+ increment in NAM-induced Sirt1 upregulation, HepG2 cells were pretreated with FK866, a potent inhibitor of Nampt, for 2 hours before NAM supplementation. The inclusion of FK866 in culture media not only lowered basal intracellular NAD+ levels, but also abolished NAD+ increase in NAM-treated cells, both being prevented by the addition of NMN (Fig. 3B), the direct precursor for NAD biosynthesis (Fig. 3A). Interestingly, FK866 only partially reduced NAM-induced Sirt1 upregulation (Fig. 3C). Although NMN supplementation at 0.5 mM resulted in a similar extent of intracellular NAD+ increase as NAM at 5 mM did (Fig. 3B), a lesser extent of increase in Sirt1 expression was induced and FK866 had no effect on NMN-induced Sirt1 upregulation (Fig. 3C).
Figure 3.
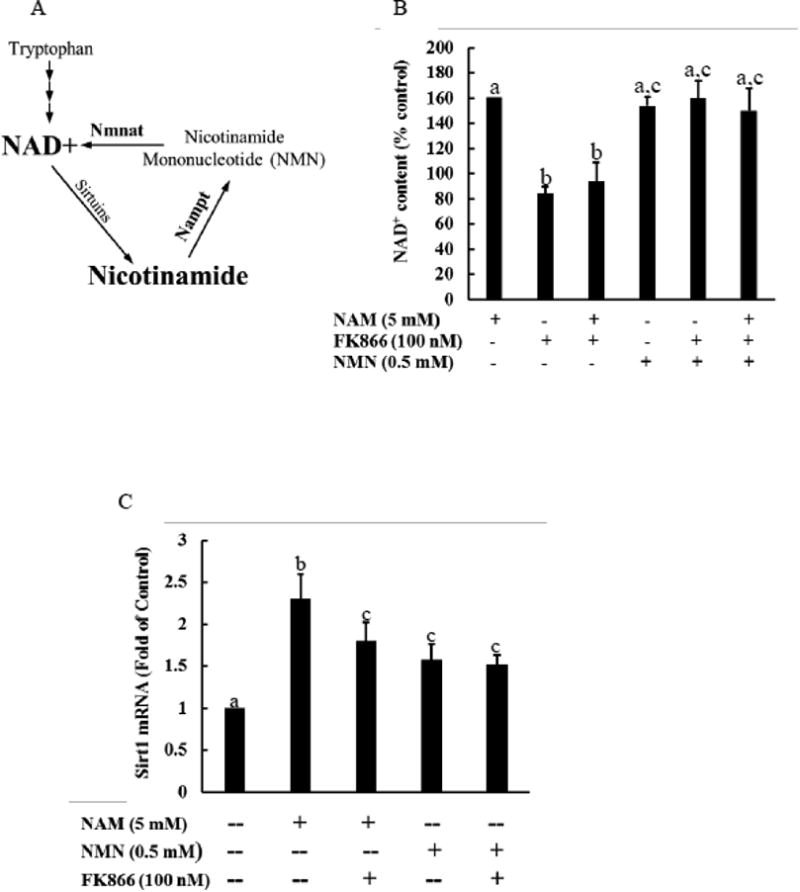
Increase in intracellular NAD+ level partially contributes to NAM-induced Sirt1 upregulation. HepG2 cells were treated with NAM at 5 mM for 8 hours with/without 2-hour pretreatment with either FK866, a specific inhibitor of Nampt, or NMN, a direct product of Nampt-catalyzed reaction from NAM in the salvage pathway. A. the de novo pathway, which uses tryptophan as the precursor, and the salvage pathway, which utilizes NAM as the precursor, for NAD+ biosynthesis. Nampt: nicotinamide phophoribosyltransferase, the rate-limiting enzyme of salvage pathway. B. The inhibition of Nampt via FK866 prevents NAM-induced NAD+ increase, which is recovered by NMN supplementation. C: FK866 partially prevents NAM-induced Sirt1 upregulation. All values are denoted as means ± SD from three or more independent studies. Bars with different letters differ significantly (p < 0.05).
3.4. NAM increases intracellular cAMP levels in hepatocytes via suppressing PDE activity
Intracellular cAMP has been recently emerging as an important factor mediating Sirt1 activity through a NAD+-independent mechanism [25, 26]. To determine if this can be a mechanism attributing to NAM-mediated Sirt1 upregulation, we first examined the effect of NAM supplementation on intracellular cAMP level. In comparison to control cells, both HepG2 cells (Fig. 4A) and primary mouse hepatocytes (Fig. 4B) exposed to NAM manifested a dose-dependent increase of intracellular cAMP levels. A significant elevation of intracellular cAMP levels was observed at as early as 1 hour after NAM supplementation (Fig. 4A & B). Intracellular cAMP concentration is controlled by its production catalyzed by adenylyl cyclase (AC) and degradation by phosphodiesterase (PDE). Both AC activation and PDE inhibition lead to intracellular cAMP elevation [30, 31]. To delineate the mechanism involved in NAM-induced cAMP increase, HepG2 cells were initially pretreated with either SQ22536, a selective AC inhibitor, or forskolin, an AC activator, for 2 hours before NAM supplementation. While AC inhibition failed to abolish NAM-induced intracellular cAMP elevation, AC activation by forskolin further increased intracellular cAMP level in NAM-treated cells (Fig. 4C). These results suggested that AC activity is not involved in NAM's impact on intracellular cAMP changes. We thus subsequently measured the effect of NAM on PDE enzymatic activity. In comparison to control cells, a two-hour NAM exposure significantly lowered enzymatic activity of PDE (Fig. 4D).
Figure 4.
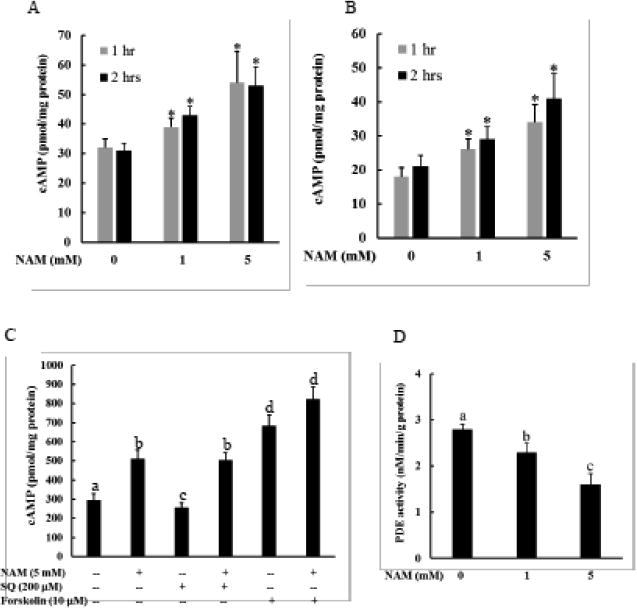
NAM supplementation increases intracellular cAMP level. Both HepG2 cells (A) and primary mouse hepatocytes (B) were treated with NAM (1 and 5 mM) for indicated time periods. Intracellular cAMP levels were determined. NAM elevates intracellular cAMP concentration in a dose-dependent manner. * p < 0.05 vs. control. All values are denoted as means ± SD from three or more independent studies. C. SQ22536 fails to attenuate NAM-induced cAMP elevation, whereas forskolin aggravates cAMP increase induced by NAM supplementation. HepG2 cells were treated with NAM with/without a 2-hour pretreatment of either SQ22536, a specific adenylyl cyclase (AC) inhibitor, or forskolin, a potent stimulator of AC, for 2 hours and intracellular cAMP concentration was determined. All values are denoted as means ± SD from three or more independent studies. Bars with different letters differ significantly (p < 0.05). D. NAM suppresses PDE activity. HepG2 cells were treated with NAM for 2 hours and intracellular PDE activity was determined. All values are denoted as means ± SD from three or more independent studies. Bars with different letters differ significantly (p < 0.05).
3.5. Activation of cAMP/PKA/CREB pathway contributes to NAM-induced Sirt1 upregulation
The main downstream events followed by an increase of cellular cAMP concentration include PKA phosphorylation (activation), which subsequently phosphorylates/activates transcription factor CREB, leading to the upregulation of target genes/proteins. Interestingly, a recent study showed that CREB controls Sirt1 expression [25, 26]. To determine whether cAMP/PKA/CREB pathway activation is mechanistically involved in NAM-induced Sirt1 expression, we first examined the effect of NAM on PKA and CREB phosphorylation status in HepG2 cells. As shown in Fig, 5A, both PKA and CREB was activated by NAM exposure, manifested by a substantial increase of corresponding phosphorylated protein abundance. Enhanced CREB phosphorylation was associated with increased nuclear CREB protein abundance (Fig. 5B). Importantly, NAM's effects were all dampened by the pretreatment with H89, a specific inhibitor of PKA (Fig. 5A & B). Furthermore, we blocked cAMP/PKA/CREB pathway with either H89 pretreatment or CREB siRNA transfection, before NAM supplementation, followed by the measurement of Sirt1 expression. Both PKA inhibition by H89 (Fig. 5C) and CREB knockdown by siRNA transfection (Fig. 5D) abolished NAM-induced Sirt1 upregulation in HepG2 cells (Fig. 5E). Similar result was observed with H89 pretreatment in primary mouse hepatocytes (Fig. 5F).
Figure 5.
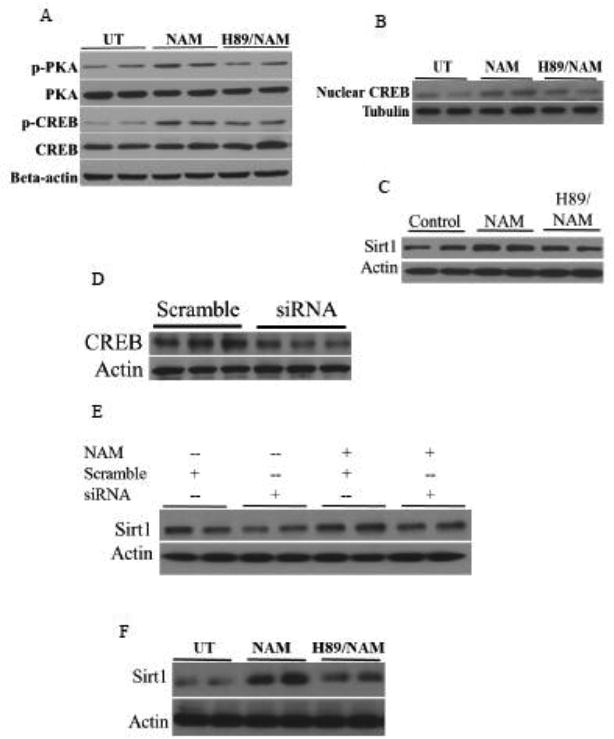
Activation of cAMP/PKA/CREB pathway contributes to NAM-induced Sirt1 upregulation. HepG2 cells were treated with 5 mM NAM, with or without 2-hour H89 pretreatment, for 2 hours and PKA and CREB activations were determined. A. PKA and CREB phosphorylations. B. Nuclear CREB protein abundance. C-E. Both H89 pretreatment (C) and CREB siRNA transfection (D & E) abolishes NAM-induced Sirt1 upregulation. HepG2 cells were treated with 5 mM NAM with or without a 2-hour pretreatment of H89, a specific PKA inhibitor, or overnight transfection of siRNA for CREB. CREB and Sirt1 protein abundance after an 8-hour NAM treatment were determined by Western blot. F. Sirt1 protein abundance in primary mouse hepatocytes. Mouse primary hepatocytes were treated with NAM, with or without a 2-hour H89 pretreatment, for 8 hours. Sirt1 protein abundance were determined by Western blot.
3.6. NAM ameliorates palmitate-induced ER stress via a Sirt1-dependent mechanism
Previous report demonstrated that Sirt1 activation protected hepatocytes against palmitate-triggered UPR response and ER stress [12]; therefore, the Sirt1-enhancing property raised the possibility that NAM may confer protection against palmitate-induced UPR response and ER stress in hepatocytes. To test this possibility, we first exposed HepG2 cells to palmitate (conjugated to BSA at a 2:1 M ratio), with/without a 2 hour pretreatment with NAM, for 16 hours. UPR and ER stress induction was characterized by measuring CHOP and GRP78 expressions at both mRNA and protein levels, as well as protein phosphorylation of PERK and JNK, a downstream target of Ire 1a activation. Palmitate exposure dose-dependently increased CHOP and GRP78 gene expressions (Fig. 6A) and protein abundance (Fig. 6B). Similarly, both PERK and JNK phosphorylation was markedly enhanced by palmitate exposure (Fig. 6C). NAM pretreatment alleviated palmitate-triggered ER stress induction (Fig. 6A-C). To determine the mechanistic involvement of Sirt1 upregulation in NAM's beneficial effect against ER stress, HepG2 cells were either pretreated with Sirtinol, a selective chemical inhibitor of Sirt1, or transfected with siRNA for Sirt1 before NAM and palmitate exposure. The Sirt1 inhibition abrogated the ameliorative effect of NAM on palmitate-triggered ER stress, evidenced by the recovery of CHOP and GRP78 expressions, as well as PERK and JNK phosphorylation (Fig. 6A-C).
Figure 6.
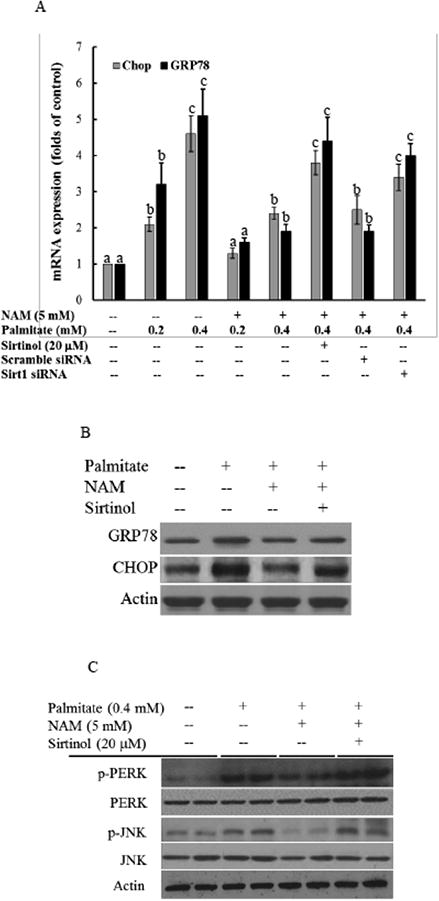
NAM ameliorates palmitate-triggered ER stress in hepatocytes in a Sirt1-dependent mechanism. A & B. Gene expressions and protein abundance of CHOP and GRP78, two ER stress markers. B. PERK and JNK activation. HepG2 cells were treated with palmitate with or without NAM in the presence or absence of Sirtinol, a selective Sirt1 inhibitor, or of overnight siRNA transfection for 16 hours. CHOP and GRP78 expressions were determined by qRT-PCR and Western blot, respectively. P-PERK, p-JNK, PERK, and JNK proteins were detected by Western blot. All values are denoted as means ± SD from three or more independent studies. Bars with different letters differ significantly within same gene (p < 0.05).
3. Discussion
In the present study, we examined the effect of exogenous NAM supplementation on Sirt1 expression and activity in hepatocytes and investigated underlying mechanisms behind, as well as its contribution to NAM-conferred protection against palmitate-triggered ER stress. We demonstrated for the first time to our knowledge that NAM supplementation protected hepatocytes against ER stress in response to palmitate exposure via Sirt1 upregulation. NAM supplementation led to enhanced Sirt1 expression and enzymatic activity, concomitant with a significant increase of intracellular NAD+ level. Nevertheless, although Nampt inhibition abrogated NAM-induced NAD+ increase, it failed to abolish Sirt1 upregulation, suggesting that intracellular NAD+ increment only partially contributes to NAM-mediated Sirt1 upregulation. Further mechanistic investigations unraveled that NAM supplementation led to a significant elevation of intracellular cAMP level, concomitant with enhanced PKA and CREB phosphorylation/activation, the canonical signal transduction pathway in response to cellular cAMP increase. Importantly, we found that the blockade of this pathway abolished not only NAM-induced Sirt1 upregulation, but also NAM's protection against palmitate-triggered ER stress, suggesting that the cAMP/PKA/CREB pathway activation plays a mechanistic role in this event.
NAM is the amide of nicotinic acid (vitamin B3 / niacin). As the predominant precursor for cellular NAD+ biosynthesis via the salvage pathway [4, 5], NAM supplementation has been reported to increase Sirt1 activity in a variety of cell types [30, 31] and protected immune cells against stress-triggered cell death [34]. Paradoxically, as NAM is the common product of Sirt1-mediated deacetylation reactions, it is also a widely employed as a chemical inhibitor of Sirt1 [27-29]. The factors leading to these conflicted observations may derive from cell type, dosage, and exposure duration, and remain to be clarified. In this study, we clearly demonstrated that NAM at less than 5 mM upregulated Sirt1 expression and activity in hepatocytes, which plays a mechanistic role in the protective effect of NAM against palmitate-triggered ER stress.
Although it is currently not clear as to how the catalytic activity of Sirt1 is precisely regulated, several previous studies have demonstrated that changes in intracellular NAD+ levels are sufficient to alter Sirt1 activity under a variety of conditions [15-24]. In the current study, the robust increases of intracellular NAD+ levels were observed when hepatocytes were treated with NAM. Moreover, the NAM-induced NAD+ elevation was abolished by Nampt inhibition, indicating that exogenous NAM supplementation enhances NAD+ biosynthesis via the salvage pathway. It was in fact our initial hypothesis that NAM induces Sirt1 upregulation via this canonical mechanism. We indeed showed that NMN, a direct precursor for NAD+ biosynthesis, upregulated Sirt1 expression and protected hepatocytes against palmitate-induced ER stress (Supplementary Fig. S1). However, the hypothesis was not fully supported by our observations in that Sirt1 expression remained to be upregulated in response to NAM addition under the circumstance of the salvage pathway blockage by Nampt inhibitor, although which completely abrogated NAM-induced intracellular NAD+ increase. These results altogether suggest that a NAD+-independent mechanism exits for NAM-induced Sirt1 upregulation.
Emerging evidence supports that the activation of the cAMP/PKA/CREB pathway controls Sirt1 expression and activation independently of changes in intracellular NAD+ levels [24, 25]. In the present study, we examined the potential mechanistic involvement of this pathway in NAM-induced Sirt1 upregulation. Our study revealed for the first time that NAM supplementation elevated intracellular cAMP concentration in hepatocytes and activated cAMP/PKA/CREB signaling pathway. Whereas Nampt inhibition by FK866 failed to abrogate NAM-induced Sirt1 upregulation and protection against palmitate-triggered ER stress, both PKA inhibitor and CREB gene knockdown via siRNA transfection abolished NAM's beneficial effects (Supplementary Fig. S2), indicating that the cAMP/PKA/CREB pathway activation plays a mechanistic role in the regulation of NAM-induced Sirt1 upregulation and resultant protection against ER stress. As mentioned previously, intracellular cAMP concentration is controlled by its production catalyzed by adenylyl cyclase (AC) and degradation, which is catalyzed by phosphodiesterase (PDE), and both AC activation and PDE inhibition lead to intracellular cAMP elevation [35, 36]. In an effort to identify the potential mechanism involved in the enhancing effect of NAM on cellular cAMP concentration, both activator (forskolin) and inhibitor (SQ22536) of AC was utilized. The observations that AC inhibition failed to abolish NAM-induced intracellular cAMP elevation while AC activation aggravates intracellular cAMP accumulation in NAM-treated cells indicated that the enzyme is not the key regulatory point for NAM. The direct measurement of PDE activity confirmed that NAM suppressed PDE activity. This observation is indeed in line with a previous report in which NAM was used as a PDE inhibitor [37].
In summary, our data demonstrate that, in hepatocytes, NAM supplementation protects hepatocytes against palmitate-triggered ER stress via Sirt1 upregulation. Both NAD+-dependent and – independent mechanisms are involved in NAM-induced Sirt1 activation in hepatocytes. Our results suggest that the activation of cAMP/PKA/CREB pathway via suppression PDE activity plays a critical role in NAM's stimulatory effect on Sirt1. These results support that NAM can confer protection against lipotoxicity via acting as either a NAD+ booster or a Sirt1 activator. Further investigations are required to test the potential therapeutic function of NAM in liver diseases with either NAD+ depletion or Sirt1 suppression being a pathological factor.
Supplementary Material
Highlights.
Initial report that nicotinamide protects hepatocytes against palmitate-induced ER stress.
Nicotinamide upregulates Sirt1 expression and activity in hepatocytes
Sirt1 upregulation mechanistically contributes to amelioration of ER stress.
Nicotinamide increases cellular cAMP level via suppressing PDE activity.
The cAMP/PKA/CREB pathway activation contributes to nicotinamide-induced Sirt1 upregulation.
Acknowledgments
This study was supported in part by grants from the National Institutes of Health NIAAA grants R01 AA017442 and NSFC 81470845 (Z Song) and ZJNSF R15H 03005 and NSFC 81473393 (X Dou).
Abbreviation
- NAM
nicotinamide
- PKA
Protein kinase A
- CREB
cyclic AMP response-element-binding protein
- Nampt
nicotinamide phosphoribosyltransferase
- NMN
nicotinamide mononucleotide
- PDE
phosphodiesterase
Footnotes
Conflict of interest: The authors who have taken part in this study declare that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schuppan D, Schattenberg JM. Non-alcoholic steatohepatitis: pathogenesis and novel therapeutic approaches. Journal of Gastroenterology and Hepatology. 2013;1:68–76. doi: 10.1111/jgh.12212. [DOI] [PubMed] [Google Scholar]
- 2.Zámbó V, Simon-Szabó L, Szelényi P, Kereszturi E, Bánhegyi G, Csala M. Lipotoxicity in the liver. World Journal of Hepatology. 2013;5:550–7. doi: 10.4254/wjh.v5.i10.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leamy AK, Egnatchik RA, Shiota M, Ivanova PT, Myers DS, Brown HA, Young JD. Enhanced synthesis of saturated phospholipids is associated with ER stress and lipotoxicity in palmitate treated hepatic cells. J Lipid Res. 2014;55:1478–1488. doi: 10.1194/jlr.M050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imai S, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends in pharmacological sciences. 2010;31:212–220. doi: 10.1016/j.tips.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annual review of pathology. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schug TT, Li X. Sirtuin 1 in lipid metabolism and obesity. Annals of medicine. 2011;43:198–211. doi: 10.3109/07853890.2010.547211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell metabolism. 2009;9:327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, Kellum J, Warnick R, Contos MJ, Sanyal AJ. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell metabolism. 2012;15:665–674. doi: 10.1016/j.cmet.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Timmers S, Konings E, Bilet L, Houtkooper RH, van de Weijer T, Goossens GH, Hoeks J, van der Krieken S, Ryu D, Kersten S, Moonen-Kornips E, Hesselink MK, Kunz I, Schrauwen-Hinderling VB, Blaak EE, Auwerx J, Schrauwen P. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell metabolism. 2011;14:612–622. doi: 10.1016/j.cmet.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.You M, Liang X, Ajmo JM, Ness GC. Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. American journal of physiology Gastrointestinal and liver physiology. 2008;294:G892–898. doi: 10.1152/ajpgi.00575.2007. [DOI] [PubMed] [Google Scholar]
- 11.Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, Odena G, Stevens SM, Jr, You M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology. 2014;146:801–811. doi: 10.1053/j.gastro.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung TW, Lee KT, Lee MW, Ka KH. SIRT1 attenuates palmitate-induced endoplasmic reticulum stress and insulin resistance in HepG2 cells via induction of oxygen-regulated protein 150. Biochem Biophys Res Commun. 2012;422:229–32. doi: 10.1016/j.bbrc.2012.04.129. 422. [DOI] [PubMed] [Google Scholar]
- 13.Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome--a key determinant of cancer cell biology. Nature reviews Cancer. 2012;12:741–752. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- 14.Abdellatif M. Sirtuins and pyridine nucleotides. Circulation research. 2012;111:642–656. doi: 10.1161/CIRCRESAHA.111.246546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 16.Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes & development. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashida S, Arimoto A, Kuramoto Y, Kozako T, Honda S, Shimeno H, Soeda S. Fasting promotes the expression of SIRT1, an NAD+ -dependent protein deacetylase, via activation of PPARalpha in mice. Molecular and cellular biochemistry. 2010;339:285–292. doi: 10.1007/s11010-010-0391-z. [DOI] [PubMed] [Google Scholar]
- 18.Graham TE, Saltin B. Estimation of the mitochondrial redox state in human skeletal muscle during exercise. Journal of applied physiology. 1989;66:561–566. doi: 10.1152/jappl.1989.66.2.561. [DOI] [PubMed] [Google Scholar]
- 19.Chabi B, Adhihetty PJ, O'Leary MF, Menzies KJ, Hood DA. Relationship between Sirt1 expression and mitochondrial proteins during conditions of chronic muscle use and disuse. Journal of applied physiology. 2009;107:1730–1735. doi: 10.1152/japplphysiol.91451.2008. [DOI] [PubMed] [Google Scholar]
- 20.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell metabolism. 2010;11:213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell metabolism. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim HJ, Kim JH, Noh S, Hur HJ, Sung MJ, Hwang JT, Park JH, Yang HJ, Kim MS, Kwon DY, Yoon SH. Metabolomic analysis of livers and serum from high-fat diet induced obese mice. Journal of proteome research. 2011;10:722–731. doi: 10.1021/pr100892r. [DOI] [PubMed] [Google Scholar]
- 24.Tao R, Wei D, Gao H, Liu Y, De Pinho RA, Dong XC. Hepatic FoxOs regulate lipid metabolism via modulation of expression of the nicotinamide phosphoribosyltransferase gene. The Journal of biological chemistry. 2011;286:14681–14690. doi: 10.1074/jbc.M110.201061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerhart-Hines Z, Dominy JE, Jr, Blattler SM, Jedrychowski MP, Banks AS, Lim JH, Chim H, Gygi SP, Puigserver P. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+) Molecular cell. 2011;44:851–863. doi: 10.1016/j.molcel.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noriega LG, Feige JN, Canto C, Yamamoto H, Yu J, Herman MA, Mataki C, Kahn BB, Auwerx J. CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability. EMBO reports. 2011;12:1069–1076. doi: 10.1038/embor.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast Sir2 and human SIRT1. Journal of Biological Chemistry. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 28.Hou XY, Xu SQ, Maitland-Toolan KA, Sato K, Jiang BB, Ido YS, Lan F, Walsh K, Wierzbicki M, Verbeuren TJ, Cohen RA, Zang MW. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. Journal of Biological Chemistry. 2008;283:20015–20026. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Enzymology of NAD plus homeostasis in man. Cell Mol Life Sci. 2004;61:19–34. doi: 10.1007/s00018-003-3161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jang SY, Kang HT, Hwang ES. Nicotinamide-induced Mitophagy Event Mediated by High NAD (+)/NADH Ratio and SIRT1 Protein Activation. Journal of Biological Chemistry. 2012;287:19304–19314. doi: 10.1074/jbc.M112.363747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP. Nicotinamide Prevents NAD(+) Depletion and Protects Neurons Against Excitotoxicity and Cerebral Ischemia: NAD(+) Consumption by SIRT1 may Endanger Energetically Compromised Neurons. Neuromol Med. 2009;11:28–42. doi: 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dou XB, Shen C, Wang ZG, Li ST, Zhang XM, Song ZY. Protection of nicotinic acid against oxidative stress-induced cell death in hepatocytes contributes to its beneficial effect on alcohol-induced liver injury in mice. J Nutr Biochem. 2013;24:1520–1528. doi: 10.1016/j.jnutbio.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garten A, Petzold S, Korner A, Imai S, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrin Met. 2009;20:130–138. doi: 10.1016/j.tem.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crowley CL, Payne CM, Bernstein H, Bernstein C, Roe D. The NAD+ precursors, nicotinic acid and nicotinamide protect cells against apoptosis induced by a multiple stress inducer, deoxycholate. Cell Death Differ. 2000;7:314–326. doi: 10.1038/sj.cdd.4400658. [DOI] [PubMed] [Google Scholar]
- 35.Cooper DM. Compartmentalization of adenylate cyclase and cAMP signalling. Biochemical Society transactions. 2005;33:1319–1322. doi: 10.1042/BST0331319. [DOI] [PubMed] [Google Scholar]
- 36.Furman B, Pyne N, Flatt P, O'Harte F. Targeting beta-cell cyclic 3′5′ adenosine monophosphate for the development of novel drugs for treating type 2 diabetes mellitus. A review, The Journal of pharmacy and pharmacology. 2004;56:1477–1492. doi: 10.1211/0022357044805. [DOI] [PubMed] [Google Scholar]
- 37.Yoshimi T, Yasumasu I. Vegetalization of Sea-Urchin Larvae Induced with Camp Phosphodiesterase Inhibitors. Dev Growth Differ. 1978;20:213–218. doi: 10.1111/j.1440-169X.1978.00213.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.