

Abstract
Nicotinamide N-methyltransferase (NNMT) was originally identified as the enzyme responsible for the methylation of nicotinamide (NAM), one of the forms of vitamin B3. Methylated NAM (MNAM) is eventually excreted from the body. Recent evidence has expanded the role of NNMT beyond clearance of excess vitamin B3. NNMT has been implicated in the regulation of multiple metabolic pathways in tissues such as the adipose tissue and liver, as well as cancer cells, through consumption of methyl donors and generation of active metabolites. This review examines recent findings regarding the function of NNMT in physiology and disease and highlights potential new avenues for therapeutic intervention. Finally, key gaps in our knowledge for this enzymatic system and future areas of investigation are discussed.
Keywords: NNMT, NAD+, vitamin B3, methyltransferases, S-Adomet
A brief history of NNMT
NNMT (E.C. 2.1.1.1) catalyzes the methylation of NAM and structurally related compounds using the universal methyl donor S-Adenosyl methionine (SAM), to produce S-adenosyl-L-homocysteine (SAH) and 1-methylnicotinamide (MNAM) [1]. The product of NNMT, MNAM, can be further oxidized by aldehyde oxidase (Aox) into two related compounds, N1-methyl-2-pyridone-5-carboxamide (2py) and N1-methyl-4-pyridone-3-carboxamide (4py), and all three metabolites are eventually excreted in the urine [2] (Figure 1). A secondary NAM clearance pathway starts with the direct oxidation of NAM to NAM N-oxide by cyp2E1, followed by elimination in the urine [3]. Under most conditions, methylation is quantitatively by far the predominant NAM clearance pathway with the exception of an acute pharmacological dose of NAM, which is mainly converted to NAM N-oxide [4,5].
Figure 1. Enzymatic pathway for NAM methylation.
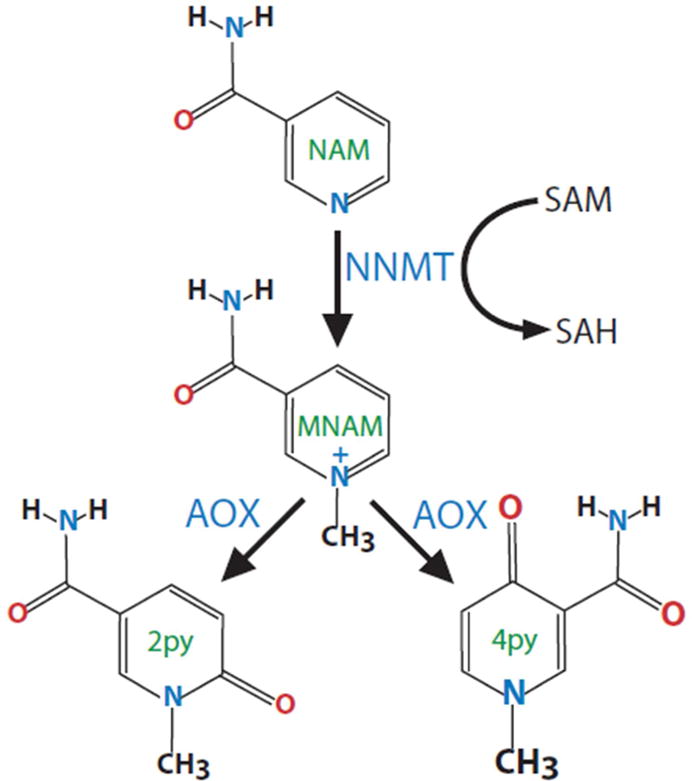
Nicotinamide (NAM) is methylated by nicotinamide N-methyltransferase (NNMT) using the universal methyl donor S-Adenosylmethionine (SAM). The methylated product N1-methylnicotinamide (MNAM) is further oxidized by aldehyde oxidase (Aox) into two related compounds 2py and 4py. All three metabolites are eventually excreted in the urine.
Interest in nicotinamide-methylating activity was prompted by the identification of methylated vitamin B3 metabolites in the urine, in the 1940s [6–8]. In 1951, NNMT was partially purified by Gulio Cantoni, and was the first characterized methyltransferase activity [9]. Although the identity of the methyl donor was not known at that time, Cantoni correctly deduced that ATP and methionine form a high energy intermediate used for the methylation of NAM. He went on and identified SAM as the universal methyl donor a year later [10]. Methylation is considered a minor detoxification pathway and consequently NNMT was classified as a detoxification enzyme. Although NNMT can methylate compounds structurally related to NAM, such as 3-acetylpyridine, quinoline, isoquinoline and others [11], conclusive evidence for the importance of NNMT in detoxification of these xenobiotics is lacking.
The human and mouse NNMT genes were cloned by Weinshilboum and colleagues in 1994 and 1997, respectively [1,12]. The canonical NNMT gene structure (Gene ID: 4837) is relatively simple, with 3 exons and 2 introns on human chromosome 11 and mouse chromosome 9. The human transcribed locus spans ~16.7kbi and produces an mRNA of 1578 base pairs and a protein of 264 amino acids. Alternative NNMT exons and splice variants have been uncovered using RNAseq but they account only for a negligible fraction of the canonical mRNA transcript, and their function, if any, is currently unknowniii. The NNMT protein is relatively well-conserved across mammals, with 85% amino acid identity between humans and miceii and the simplest organism reported to have NAM N-methylation activity is C.elegans [13].
To-date, the best-studied NNMT substrate is NAM (vitamin B3) [11]. NAM is a precursor for nicotinamide adenine dinucleotide (NAD+), a key molecule involved in energy metabolism. The Km of the human NNMT enzyme for NAM is relatively low, approximately 430 μM. Accordingly, NNMT is not saturated under normal conditions, and increasing dietary NAM intake leads to a proportional increase in NAM methylation [14]. Because NAM methylation is irreversible, early studies focused on the methyl donor depletion by high doses of NAM. Indeed, supplementation of diet with 1% NAM (v/v) inhibited the growth of rodent pups, and this inhibition could be prevented by methionine, the principal methyl donor [15]. In adult rodents, depletion of methyl donors by high doses of NAM increased liver steatosis only when diet was limiting in choline. This is because choline can donate its methyl group and prevent depletion of methyl donors which are needed for lipid export from the liver [14–17]. Because of its assignment to the vitamin B3 clearance pathways, NNMT has not been intensely studied in metabolic research. Recent advances have uncovered new important functions for NNMT in metabolism and energy homeostasis, and thus are reshaping our view of this important enzyme.
Box 1. Nomenclature.
Standard naming conventions have been accepted for most of the major intermediates in NAD+ synthesis: nicotinamide (NAM), nicotinamide mononucleotide (NMN), nicotinamide riboside (NR), nicotinamide adenine dinucleotide (NAD) and their corresponding acid analogs: nicotinic acid (NA), nicotinic acid mononucleotide (NAMN), nicotinic acid riboside (NAR) and nicotinic acid dinucleotide (NAAD). However, there is currently no unified nomenclature for several vitamin B3 and structurally related metabolites. For example, N1-methylnicotinamide can be found abbreviated as MNA, meNAM, MNAM or even NMN, creating inconsistency and confusion in the literature [20,53,86,90]. Our abbreviation for the methylated version of nicotinamide, MNAM, is based on nicotinamide (NAM). Accordingly, we call the methylated version of nicotinic acid, also known as trigonelline, MNA. Other compounds in need of name standardization are the oxidized versions of MNAM (usually called 2py and 4py), nicotinamide N-oxide and few other related metabolites. Regardless of our use, the interested community should get together and decide sooner than later on consistent and clear abbreviations for these metabolites as the field is currently expanding.
Genetic variation and NNMT expression
The NNMT gene is strongly expressed in human liveriii,iv [1]. Lower levels of NNMT expression are found in adipose tissue (subcutaneous and visceral), artery (aorta, coronary), muscle, and in various mesenchymal cell types, i.e. fibroblasts. NNMT is expressed at very low levels in the central nervous system and hematopoietic cellsiii. Despite its average high expression in several tissues, NNMT gene expression can vary substantially in the livers and the adipose tissue of inbred recombinant mouse strains and humans, and correlates with metabolic traitsiv, v [18–20]. Limited information exists regarding the regulation of NNMT promoter activity. It has been shown that the transcription factors STAT3 and hepatocyte nuclear factor-1beta (HNF1β) increase NNMT promoter activity in cancer cells [21,22], but beyond these observations the promoter has not been extensively analyzed for other regulatory factors.
The variation in NNMT mRNA expression is in part a consequence of genetic variation. Studies that combine expression profiling and single nucleotide polymorphism (SNP) genotyping provide some clues into genetic variation and its impact on NNMT expression. The Hybrid Mouse Diversity Panel (HMDP)v, a database of ~ 100 recombinant inbred strains, lists several expression quantitative trait loci (eQTLs) with genome wide significance, i.e. SNPs located in the vicinity of the NNMT locus that are associated with NNMT expression (Figure 2A) [23]. The latest release of the GTEx project (v.6p)iii uncovered several eQTLs in the NNMT gene in adipocytes, fibroblasts and lung. The effect size of these eQTLs on NNMT gene expression is between 20–40% in either direction, decreased or increased mRNA expressioniii. Interestingly, the eQTLs in adipose tissue, fibroblasts and the lung do not overlap, suggesting a complex tissue-specific regulation of the NNMT promoter (Figure 2B).
Figure 2. Genetic variation influences NNMT expression in mice and humans.
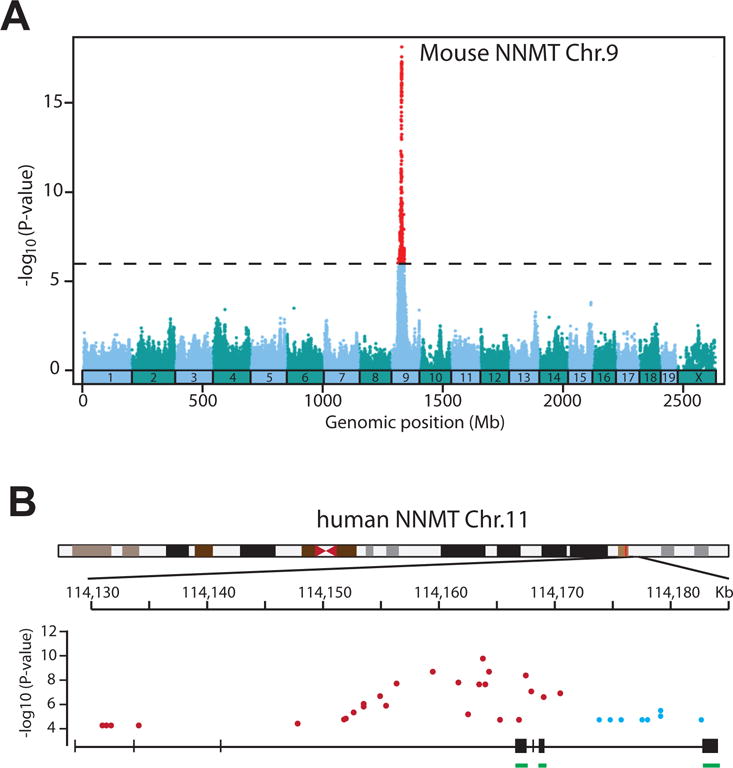
A. eQTLs in the vicinity of the mouse NNMT locus (chr.9) associate strongly with NNMT mRNA levels (adopted from the HMDP database). B. Distribution of eQTLs in the NNMT locus isolated from adipose tissue (blue) and transformed fibroblasts (red). Coding exons are underlined (green) (adopted from GTExiii). −Log10 (P value) genome wide significance.
Genome wide association studies (GWAS) have revolutionized our approach to pinpointing genetic causes of polygenic diseases. A few studies reported associations of SNPs in the NNMT gene with traits as diverse as asthma and brain volume [24,25]. Furthermore, a handful of smaller size genetic association studies reported significant associations between specific polymorphisms in the NNMT gene and metabolic traits such as plasma homocysteine levels and non-alcoholic steatohepatitis (NASH), as well as psychiatric disorders such as schizophrenia and epilepsy (Table 1) [26–30]. In general, the SNPs identified in these studies are located in the regulatory regions surrounding the NNMT gene and are often several kilobases away from the coding sequence. Furthermore, there is no data on the effect of these SNPs on NNMT gene expression. Because of very low NNMT expression in neuronsiii, it is unclear what cell types might be contributing to these central disorders. Clearly more research is needed to clarify the role of genetic variation in the NNMT gene in these disorders.
Table 1.
SNPs in the NNMT locus associated with quantitative traits.
| SNP Id | P-value | Location | Functional class | Reported trait | Ref. |
|---|---|---|---|---|---|
| rs11214966 | 6 ×10-7 | chr11:114,266,397-114,433,396 | downstream | asthma | [24] |
| rs2847476 | 3 ×10-6 | chr11:114,295,244-114,345,343 | downstream | volumetric brain MRI | [25] |
| rs694539 | 0.003 | chr11:114,260,557-114,277,256 | upstream | epilepsy | [28] |
| rs694539 | 0.0059 | chr11:114,260,557-114,277,256 | upstream | schizophrenia | [26,27] |
| rs694539 | 0.009 | chr11:114,260,557-114,277,256 | upstream | NASH | [29] |
| rs694539 | 0.017 | chr11:114,260,557-114,277,256 | upstream | homocysteine | [30] |
Because NNMT metabolizes the NAD+ precursor NAM and the methyl donor SAM, its activity may affect two broad classes of enzymes, the NAD+-dependent enzymes and the SAM-dependent methyltransferases. Below, I break down the metabolic functions of NNMT in two parts based on its enzymatic activity. First, I discuss the metabolism of NAM and the metabolic function of the NNMT product, MNAM. Second, I discuss the consumption of methyl donors by NNMT and its impact on global methylation.
NNMT and NAD+ metabolism
NAD+ synthesis and breakdown
As mentioned above, NAM is a precursor for NAD+, a cofactor best known for donating electrons to the mitochondrial complex I in the electron transport chain, but also a redox factor for multiple oxidoreductases [31]. In addition, enzymes like sirtuins and poly-ADP- ribosyltransferases (PARPs) use NAD+ as a co-substrate to perform deacetylation and ADP-ribosylation reactions, and regulate multiple metabolic processes [32,33]. Both sirtuins and PARPs break NAD+ and release NAM, which inhibits their enzymatic activity [34,35]. Thus, changes in NNMT activity might potentially influence intracellular NAM and NAD+ levels, and the activity of multiple enzymes. This idea however is not supported by theoretical and empirical evidence. The affinity of NNMT for NAM is relatively low and around 430 μM [1]. On the other hand, the affinity of nicotinamide phosphoribosyltransferase (NAMPT), the first and rate-limiting step in NAD+ biosynthesis, for NAM is less than 1 μM [36,37]. For all practical purposes, NAMPT is considered saturated with NAM, and thus it is unlikely that increased NNMT expression would decrease intracellular NAM to levels limiting for NAD+ synthesis by NAMPT. In fact, experimental evidence shows that NNMT does not directly regulate intracellular NAM and NAD+ levels. Adenoviral or antisense NNMT knockdown in vivo did not cause NAM accumulation in the liver or adipocytes, respectively [20,38]. Adenoviral knockdown of NNMT in mouse liver in vivo and in primary hepatocytes in vitro did not change intracellular NAD+ levels [20]. At present, it is not clear why NAM does not accumulate in hepatocytes or adipocytes with loss of NNMT expression. Perhaps, fast equilibration rates of NAM across the plasma membrane minimize changes in steady state intracellular NAM levels. Thus, current evidence does not support the idea that NNMT activity can directly regulate NAD+ and NAM levels, and consequently NAD+-dependent enzymatic activities.
Box 2. NAD+ synthesis, recycling and clearance.
NAD+ is a redox cofactor for multiple enzymatic reactions and also a substrate for sirtuins, mono- and poly-ADP ribosyltransferases (PARPs) and ADP-ribose cyclases (CD38, CD158) [32,33]. Recently, it became apparent that NAD+ levels decline in obesity and aging, and this decline may contribute to the metabolic dysfunction, because restoring tissue NAD+ through supplements ameliorates the metabolic dysregulation caused by high fat diet and aging [81,82,87–89]. Although NAD+ acts on multiple enzymes, Sirt1 and Sirt3 are considered major effectors of its metabolic actions [81,87]. Intracellular NAD+ undergoes cycles of synthesis and breakdown. NAM released from NAD+ breakdown by sirtuins, PARPs and CD38 is recycled through a two-step pathway called the salvage pathway (Box 2 Figure). NAM is converted to nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme for NAD+ synthesis, and subsequently by nicotinamide mononucleotide adenylyl transferases 1–3 (NMNAT1-3) to NAD+. New dietary NAM can also enter into NAD+ synthesis through the salvage pathway. Excess NAM that is not recycled is methylated to MNAM by NNMT. MNAM can be subsequently oxidized by AOX into pyridones (2py, 4py) and excreted through the urine (Box 2 Figure). A minor pathway for NAM clearance involves the direct oxidation of NAM by cyp2E1 to NAM N-oxide, also eliminated in the urine. NAD+ can also be synthesized from other precursors. Nicotinic acid (NA) is incorporated into NAD+ through a three-step reaction called the Preiss-Handler pathway (Box 2 Figure). A minor pathway in tryptophan degradation also leads to NAD+ synthesis (de novo pathway). Finally, a new pathway for incorporation of an intermediate, nicotinamide riboside (NR) into NAD+ has been recently described and involves the phosphorylation of NR by nicotinamide riboside kinases 1 and 2 (NRK1, 2). NR in particular is very effective in raising tissue NAD+ levels and is currently being tested in humans for the treatment of metabolic dysfunction in obesity and aging (ClinicalTrials.gov Identifiers: NCT02303483, NCT02950441).
Box 2 Figure.
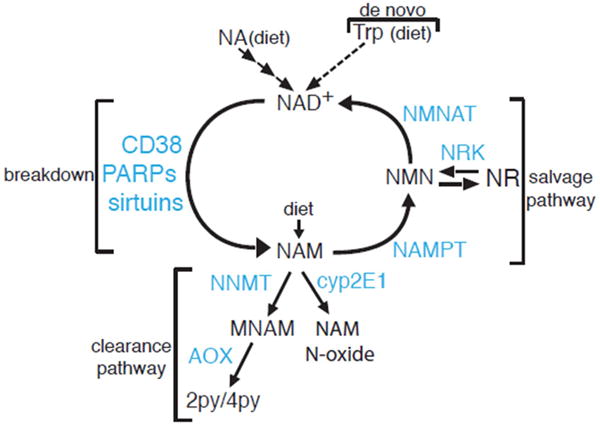
NAD+ Synthesis, Recycling and Clearance. Schematic representation of enzymatic reactions involved in the metabolism of NAD+, including NAD+ synthesis from dietary precursors, breakdown, recycling and clearance of metabolic intermediates.
NNMT and MNAM in C.elegans
Sirtuins are NAD+-dependent enzymes linked to improved metabolic health [39,40]. Although originally described as deacetylases, Sirt4 has mono-ADP-ribosyltransferase activity, while Sirt5 and Sirt6 also remove longer acyl groups from lysines (succinyl, malonyl, myristoyl and others) [41,42]. Sirt1 is by far the best studied member of the family, and deacetylates several substrates involved in glucose, lipid and energy metabolism, such as PGC1α, FoxO1, LXR, SREBPs and many others [43–49]. A recent study linked the consumption of NAD+ by Sirt1 to increased lifespan extension in C.elegans through NNMT and MNAM [13]. The worm NNMT orthologue Anmt-1 methylates NAM to MNAM, which is subsequently oxidized by Gad-3, the worm orthologue of Aox. Aox is known to produce reactive oxygen species (ROS), and oxidation of MNAM causes a transient increase in ROS [13]. This increase appears to be necessary for C.elegans life-span extension. This model is supported by several pieces of evidence; life-span extension by Anmt-1 overexpression or MNAM is blocked by Gad-3 knockdown or by treatment with general antioxidants, such as butylated hydroxyanisole (BHA) or N-acetylcysteine [13]. Whether MNAM oxidation increases ROS in mammalian tissues is currently unknown. Treatment of the mammalian neuroblastoma SH-SY5Y cells with MNAM did not affect complex I activity, the main site of ROS production, but rescued the cells from the toxicity of complex I inhibitors, such as 1-methyl-4-phenylpyridinium (MPP+) and rotenone, while also increasing cellular ATP levels [50]. Similarly, treatment with MNAM protected renal tubular kidney cells from lipotoxicity and cell death [51]
NNMT and MNAM in liver
Hepatocytes express the highest levels of NNMTiii,iv [1], suggesting a potential role for NNMT in the liver. Consistent with this idea, NNMT expression is associated with lower serum cholesterol, triglycerides and other parameters, both in mice and in humans [20]. The metabolic effects of NNMT in the liver are mediated by MNAM [20]. MNAM increases Sirt1 protein levels and activity by interfering with its ubiquitination. Consistent with this model, NNMT knockdown in hepatocytes decreased intracellular MNAM levels, lowered Sirt1 protein and increased acetylation of FoxO1, a well-characterized Sirt1 target. The opposite is also true; NNMT overexpression or treatment of hepatocytes with MNAM increased Sirt1 protein while decreasing FoxO1 acetylation. Functionally, NNMT and MNAM increased glucose 6 phosphatase (G6Pc) and phosphoenolpyruvate carboxykinase (Pck1) expression and glucose production, and these effects were prevented by pharmacological and genetic inhibition of Sirt1 [20]. The precise molecular target of MNAM is not known but it could involve a component involved in Sirt1 degradation, possibly an E3-ligase, such as MDM2 or others [52]. Consistent with the in vitro effects, dietary supplementation of MNAM prevented some of metabolic dysregulation of mice fed HFD. MNAM increased liver Sirt1 protein levels, suppressed hepatocyte fatty acid and cholesterol synthesis and decreased liver triglyceride and cholesterol content, and liver inflammation [20]. These improvements occurred without changes in body weight and adiposity in the MNAM-supplemented groups, suggesting a primary effect of MNAM on the liver. Thus, both NAD+ biosynthesis and clearance pathways use Sirt1 as a downstream effector. Elevated NAD+ levels increase Sirt1 enzymatic activity and NAM methylation by NNMT increases Sirt1 protein.
MNAM and endothelial function
Recent studies suggest that the product of NNMT, MNAM possesses biological activity and is not merely a terminal product of NAM clearance. Pharmacological doses of MNAM acutely increase secretion of prostacyclin (PGI2) from endothelial cells and may regulate thrombotic as well as inflammatory processes in rodent models of thrombosis [53]. MNAM also enhances nitric oxide release from endothelial cells and vasodilation of human blood vessels, in response to a calcium ionophore and acetylcholine [54]. The mechanism of action of MNAM in endothelium is incompletely understood but it may involve cyclooxygenase 2 (COX2) and endothelial nitric oxide synthase (eNOS). Thus, MNAM might be a therapeutically relevant target for functional disorders of the endothelium, such as thrombosis, high blood pressure and atherosclerosis.
NNMT in inflammation and injury
Increased NNMT expression has been observed in human and rodent models of injury associated with inflammation. NNMT expression is significantly increased in the lungs and skeletal muscle of patients with chronic obstructive pulmonary disease (COPD) with muscle wasting [55,56]. Significant upregulation of NNMT expression is also seen in the skeletal muscle of patients with various forms of dystrophies and can also be modeled in the mdx mouse, a model Duchenne ‘s muscular dystrophy [57]. Furthermore, experimental liver injury with concavalin A and induction of pulmonary hypertension also increase NNMT expression and activity [58,59]. The increased inflammation in these disease states and injury models is thought to drive the increase in NNMT expression. This is supported by in vitro experiments showing that direct stimulation of human skeletal muscle myoblasts with IL-6, TNFα and TGFβ all increase NNMT expression [55]. The role of NNMT and MNAM in these inflammatory states needs to be more fully elucidated but is thought to represent a protective compensatory response to injury [55,60].
NNMT and methyl donor balance
Cancer cells
With advancements in microarray and proteomic techniques, it became apparent that NNMT expression is increased in a wide variety of cancers. NNMT mRNA is increased in primary glioblastoma tumors compared to normal brain samples [61], human papillary thyroid cancer cell lines [62], in renal clear cell carcinoma [63] and bladder cancer [64]. The NNMT protein is increased in gastric cancers [65,66], human colorectal cancers [67] and oral squamous cell carcinoma [68]. One notable exception is hepatocellular carcinoma, where NNMT expression is suppressed [69]. Soon after the connection between NNMT and cancer was described, a causal role for NNMT in various cancer phenotypes was demonstrated. NNMT knockdown decreased proliferation and migration of 253J laval bladder cancer cells [64]. Similarly, NNMT knockdown inhibited the proliferation and/or metastasis of renal clear cell carcinoma cells, pancreatic cancer cells Panc-1, oral squamous carcinoma, and others [70–72]. Collectively, these data support the notion that NNMT overexpression enhances the aggressiveness of various cancers. The factors driving NNMT overexpression in cancers are not known, but are likely at the level of transcription, as NNMT gene amplification has not been reported. These studies established the functional role of NNMT in various cancers but the proximal mechanism of action was not elucidated. A plausible pathway was provided by Ulanovskaya and colleagues, which proposed that NNMT acts as a methyl donor sink [73]. SAM is a required co-substrate for all methyltransferases including NNMT and histone methyltransferases. Consistent with NNMT enzymatic activity, NNMT overexpression in the renal clear cell carcinoma 769P cells lowered SAM/SAH ratio and altered the epigenetic state of the cells. Global H3 methylation on lysine 9 (H3K9) and lysine 27 (H3K27) was decreased (major epitopes associates with transcriptional repression), while expression of some pro-oncogenic gene products was increased [73]. On the other hand, knockdown of NNMT in the ovarian carcinoma cell SKOV3 had the opposite effect as it increased SAM/SAH ratio, and global H3K9 and H3K27 trimethylation. Similar changes in methylation were also observed in one non-histone protein, protein phosphatase 2, suggesting that other cellular targets apart from histones might be differentially methylated by changes in NNMT expression and methyl donor levels [73,74]. These data provided a direct mechanistic link between NNMT and the regulation of epigenetic landscape of certain cancer cells.
Embryonic Stem cells
Recent work expanded the link between NNMT expression and global histone methylation to human embryonic stem cells (hESCs). Naive pluripotent hESCs express high levels of NNMT [75]. During the transition to a primed, more committed state, NNMT expression is downregulated. At the same time, histone methylation of repressive marks, such as H3K27me3, increase on developmental and key metabolic genes that regulate the switch between naive-to-primed hESCs; hypoxia-inducible factor (HIF) is activated and Wnt signaling is repressed. Consistent with published studies, the authors proposed that NNMT is involved in the maintenance of the naive ESC state because its activity consumes methyl donors to produce SAH and MNAM, and thus limits SAM availability for other methyltransferases. This in turn contributes to low levels of DNA and histone methylation, a known feature of the naive ESC state. Then during transition to the primed state, NNMT expression decreases and allows for more SAM accumulation and increased methylation of epigenetic marks [75].
Adipose tissue
As mentioned previously, NNMT is strongly expressed in the adipose tissue of humans and rodents. NNMT mRNA, protein and enzymatic activity increases during differentiation of 3T3L1 cells [76]. Accordingly, treatment of mouse adipose tissue explants and 3T3-L1 cells with excess NAM, the substrate of NNMT, significantly increased homocysteine secretion into the media, a byproduct of NNMT activity, whereas treatment with the NNMT product and competitive inhibitor, MNAM, reduced homocysteine release [76].
Human adipose tissue NNMT expression correlates positively with adiposity and insulin resistance [18,19]. Consistent with these correlations, antisense NNMT knockdown (NNMT ASO) in vivo protected C57BL6 mice from fat accumulation on a high fat diet (HFD) and improved their glucose tolerance compared with controls [38]. NNMT knockdown occurred both in the liver and fat, but the lean metabolic phenotype appears to be mediated by increased energy expenditure of the adipose tissue. Because NNMT is a major methyltransferase in adipocytes, the authors measured the SAM/SAH ratio. Indeed, NNMT knockdown increased SAM/SAH ratio and global histone H3K4 methylation, the latter associated with transcriptional activation [38]. The authors linked the lean phenotype to the activation of the polyamine cycle, arguing that increased substrate (SAM) and increased expression of the polyamine synthesis genes ornithine decarboxylase (ODC) and spermidine acetyltransferase (SAT1), due to epigenetic histone modifications, wastes carbons (acetyl-CoA) and energy, and protects mice from diet-induced obesity [38].
Two recent studies extend these findings to humans and show that adipose tissue NNMT and its circulating product MNAM correlates positively with insulin resistance and body mass index (BMI) [18,19]. In the study by Kannt and colleagues, NNMT expression is increased ~2-fold in subcutaneous and omental adipose tissue of type 2 diabetic subjects (men and women), compared to healthy controls. In addition, the circulating product of NNMT, MNAM, correlates positively with adipose tissue NNMT expression but only in T2D subjects. Omental adipose tissue NNMT expression and MNAM also correlate positively with insulin resistance, as measured by the euglycemic-hyperinsulinemic clamp (r2 = 0.41, p < 0.001 for NNMT; r2 = 0.44, p < 0.001 for MNAM). Importantly, interventions that increase insulin sensitivity, such as exercise and bariatric surgery, also decreased expression of NNMT in adipose tissue [18]. No association between NNMT expression and BMI was found in this study, as has been suggested by rodent data [38]. In contrast, Liu and colleagues reported a positive correlation between circulating MNAM and BMI, waist and hip circumference (BMI r = 0.22, p < 0.001 in men; r = 0.13, p = 0004 in women) in a larger cohort of 1160 Chinese individuals [19]. Curiously, the average BMI in the study by Kaant and colleagues was in between 33 and 47 for control and T2D individuals, respectively. On the other hand, the average BMI in the study by Liu and colleagues was only 23–24. One would expect that the contribution of adipose tissue NNMT to the circulating MNAM would be higher in more obese individuals and a positive correlation between MNAM and BMI would be easier to establish, but that was not the case. The reason for this discrepancy is not clear and could be attributed to the different ethnic makeup and size of the cohorts. Overall, these observations suggest that NAM methylation by NNMT could be involved in the development of insulin resistance. Given the global changes in methyl donor balance and epigenetic histone methylation in NNMT ASO mice, it is highly plausible that the polyamine pathway is only one of the pathways that contribute to the metabolic phenotype (obesity and glucose intolerance), and more detailed analysis of the epigenetic and transcriptional profiles regulated by NNMT in adipose tissue is warranted. Whether NNMT inhibitors could be used for the treatment of obesity and diabetes remains to be seen. The apparent beneficial role of NNMT and MNAM in other tissues such as the liver and endothelium raise some questions for the long-term use of NNMT inhibitors as therapeutics.
Liver
NNMT is not considered a major methyltransferase activity in the liver. The majority of methyl donors in the liver, where most of the methylation in the body occurs, are used for the methylation of guanidinoacetic acid (GAA) to creatine by the guanidininoacetate N-methyltransferase (GAMT), and for the methylation of phosphatidylethanolamine (PE) to phosphatidylcholine (PC) by the phosphatidylethanolamine N-methyltransferase (PEMT) [77,78]. Consistent with that, NNMT knockdown did not change SAM/SAH ratio in the livers of mice or primary hepatocytes [20]. As mentioned previously, high doses of the NNMT substrate NAM caused liver SAM depletion but only on diets with limiting methyl donors [17]. Along the same lines, it has recently been reported that exogenous NAM lowers liver SAM content in mice null for the enzyme glycine N-methyltransferase (GNMT, GNMT-KO mice). GNMT is a major liver methyltransferase that methylates glycine to form sarcosine, a metabolite currently without known physiological function. GNMT activity protects liver from changes in SAM fluxes. During periods of high SAM consumption, GNMT activity declines, restoring SAM levels and vice versa [78]. This homeostatic mechanism is lacking in the livers of the GNMT-KO mice, which accumulate massive amounts of SAM that eventually causes DNA and protein hypermethylation and metabolic disturbances [79]. Exogenous NAM used by NNMT lowers liver SAM content of the GNMT-KO mice, thus improving their phenotype, but does not change SAM levels in the control mice [80]. Collectively, these results suggest that NNMT activity is not sufficient to alter liver methyl donor balance under normal conditions, but can do so in instances of substrate overdose and compromised methyl donor recycling.
Box 3. The methionine cycle.
The methionine cycle is a collection of enzymatic activities that catabolize and recycle methionine (Met) and its products. The principal function of the methionine cycle is to dispose of excess Met and to generate SAM, the universal methyl donor [77]. Liver occupies a special position in methylation reactions. More than 80% of all transmethylation reactions in the body occur in liver [77]. Three reactions account quantitatively for the majority of methyl donor consumption, the synthesis of the high energy phosphate donor, creatine by guanidinoacetic acid N-methyltransferase (GAMT), the synthesis of the major body phospholipid, phosphatidylcholine by phosphatidylethanolamine N-methyltransferase (PEMT), and the synthesis of sarcosine (methylglycine), a metabolite with no assigned activity, by glycine N-methyltransferase (GNMT) (see Box 3 Figure). The liver has specialized enzymes to deal with the high methionine flux. GNMT is responsive to intracellular SAM/SAH ratio and its methylation of glycine slows down to restore falling methyl donors, suggesting that GNMT is an integral part of the regulation of the methionine cycle [78]. In addition, the liver expresses high levels of the enzymes cystathionine beta synthase (CBS) and cystathionine gamma lyase (CGL) in the transulfuration pathway (TSS) that convert excess hcy to cysteine and a-ketoglutarate. The liver also expresses high levels of betaine homocysteine methyltransferase (BHMT), which helps to recycle hcy back to Met (Box 3 Figure). Mutations in methionine cycle enzymes can change the balance between SAM and SAH, and cause hypermethionemia and hyperhomocysteinemia. Methyltransferases have high affinity for both SAM and SAH, so it is the intracellular SAM/SAH ratio rather than their absolute concentrations that determines the fraction of the active enzymes (occupied with SAM rather than SAH). Changes in SAM/SAH ratio can affect the function of multiple enzymes and have global effects on protein and DNA methylation, leading to metabolic dysfunction and cancer [77,79].
Box 3 Figure.
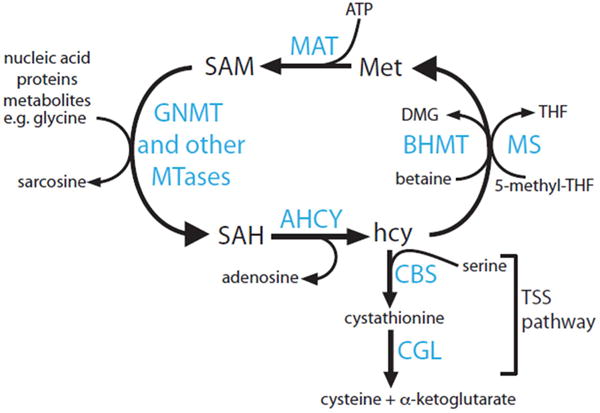
The Methionine Cycle. Schematic representation of enzymatic reactions involved in the catabolism of methionine, synthesis and utilization of S-adenosylmethionine (SAM) by methyltransferases, and recycling and disposal of homocysteine (hcy).
Concluding Remarks and Future Perspectives
Recent work has substantially increased our understanding of the role of NNMT in physiology. It became apparent that NNMT is more than a vitamin B3 clearance enzyme. Its enzymatic activity regulates global epigenetic histone profiles and its product MNAM, regulates posttranslational protein modifications (acetylation) through Sirt1. The functional consequences of altered NNMT expression are cell-specific and range from cancer cell proliferation, to altered metabolic rate of adipocytes, to changes in lipid and cholesterol metabolism of hepatocytes (Figure 3).
Figure 3. NNMT-centric view of the intracellular pathways regulated by NNMT.
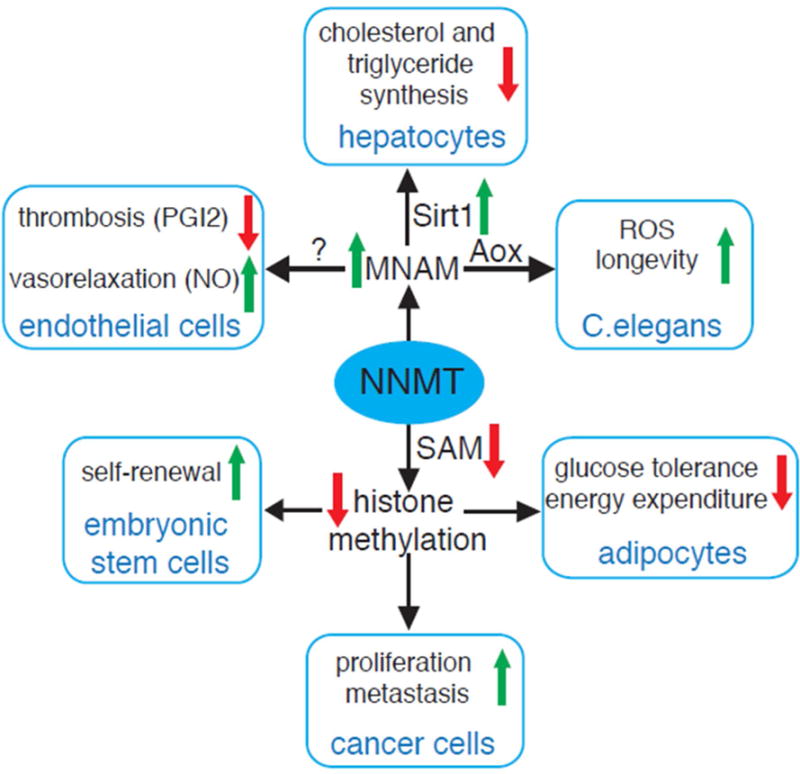
NNMT consumes SAM to methylate NAM, a form of vitamin B3. Consumption of SAM in adipose tissue, some cancer cells and hECSs decreases SAM levels and global methylation of selected epigenetic marks. The biological effects are cell specific and range from fat accumulation in adipocytes, to increased proliferation and invasiveness of cancer cells to maintenance of hESCs ‘stemness’. The product of NNMT, MNAM interferes with Sirt1 degradation leading to deacetylation of Sirt1 targets and suppression of triglyceride and cholesterol synthesis in hepatocytes. MNAM exerts antothrombotic and vasodillatory effects in endothelial cells. Oxidation of MNAM by the C.elegans homolog of Aox, increases ROS production and life span in the nematode.
As is true with any emerging area, there are many outstanding questions regarding the function of NNMT. Although widely assumed, the basic function of NNMT as a vitamin B3 clearance enzyme has not been rigorously tested. If NNMT is indeed the main clearance enzyme, then NAM may accumulate in the tissues of people with low NNMT activity to levels that may cause dysfunction, i.e. inhibition of sirtuins and PARPs. Proof of concept should come from the whole body NNMT-KO mouse, when available. This may be important as pharmacological doses of nicotinamide riboside (NR), NAD+ precursor, are being tested in preclinical models and in humans as a treatment for a variety of dysfunctions associated with obesity and aging (ClinicalTrials.gov Identifiers: NCT02303483, NCT02950441) [81,82]. Efficient removal of these precursors over time will be essential. Further, a high dose regimen of NAD+ precursors will generate increased levels of MNAM in the tissues [83]. Thus, NNMT activity may have a role in the metabolic benefits of the NAD+ supplement therapy by increasing Sirt1 protein, which is then being activated by increased tissue NAD+.
It is not known whether other related methyltransferase activities are capable of methylating NAM. Analyzing NNMT-KO mice may provide some answers for the presence of other NAM methylating activities. Given that MNAM itself is metabolically active, identifying these activities would be of obvious interest. Regarding the mechanism of action, discovering downstream targets of MNAM is critical in understanding its physiological function. Affinity purifications could be used to find proteins that bind MNAM directly. Quantitative proteomics approaches combined with expression profiling could be used to identify additional proteins increased by MNAM without changes in their mRNA expression, and uncover novel biological roles for this metabolite.
It is currently assumed that consumption of methyl donors by NNMT lowers the SAM/SAH ratio and drives the changes in histone methylation. Although quite plausible, estimates suggest that less than 1% of the total urinary loss of methyl groups can be attributed to NNMT activity (75 micromols/day for MNAM+2py+4py versus ~15 mmol total methyl group loss/day) [78,84], so the findings that NNMT can alter global methyl donor balance in several cell types could not have been anticipated based on its contribution to whole body methyl group loss, and requires further experimentation. This is an important point to clarify, since epigenetic marks can be modified independent of changes in methyl donors. A recent report showed that Sirt1 regulates the activity of MLL1, a histone methyltransferase for H3K4, one of the methyl lysine epitopes regulated by NNMT [85]. A careful comparison between the amount of methyl donors consumed by NNMT and the total methyl donors used in a cell type would provide additional evidence that NNMT is a major methyltransferase in that cell type. Along the same lines, it is not known whether NAM is the major intracellular substrate that is responsible for the methyl donor depletion by NNMT or whether there are others. Our overall understanding of NNMT and methyl donor regulation is still in its infancy.
Current concepts suggest that NNMT activity in the liver and endothelium is beneficial for the host but unfavorable in adipose tissue and in neoplasias. This apparent contradiction has raised questions regarding the endogenous role of NNMT in physiology and disease [86]. Only future work can sort out these broader questions. The pace of NNMT research is accelerating and recent advances have already substantially changed our understanding of the contribution of NNMT to NAD+ and methyl donor metabolism, with more discoveries certainly to come.
Trends Box.
NNMT expression is associated with multiple metabolic parameters in mice and in humans suggesting a functionally conserved metabolic role.
Emerging data show that the activity of NNMT can regulate posttranslational protein modifications: acetylation by affecting the protein expression of the Sirt1 deacetylase, and histone methylation by changing the global levels of the methyl donor SAM.
NNMT may be a novel effector of the metabolic function of vitamin B3. Vitamin B3 supplements may have previously unrecognized effects on epigenetics through NNMT that remain to be fully characterized.
Outstanding Questions Box.
How important is NNMT for whole body vitamin B3 metabolism? Does NAM accumulate in NNMT-KO mice or humans with low NNMT expression to levels that inhibit activity of sirtuins and PARPs?
What are the intracellular targets of MNAM? How does MNAM inhibit the ubiquitination of Sirt1? Is it through direct binding to components of the proteasome machinery or indirectly through other effectors (e.g. kinases)?
Is NAM the main NNMT substrate? Can NAM methylation account for a large fraction of SAM consumption that is consistent with the change in SAM/SAH ratio and epigenetics? If not, what other substrates can NNMT methylate? What is the impact of the global epigenetic changes (histone, DNA methylation) regulated by NNMT on the underlying transcriptome and how it affects cellular function?
Acknowledgments
This work was supported by a grant from Eli Lilly and the National Institute of Health DK083694.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Resources
Assembly GRCh38.p2
Hybrid Mouse Diversity Panel (HMDP) https://systems.genetics.ucla.edu/
References
- 1.Aksoy S, et al. Human liver nicotinamide N-methyltransferase. cDNA cloning, expression, and biochemical characterization. J Biol Chem. 1994;269:14835–14840. [PubMed] [Google Scholar]
- 2.Felsted RL, Chaykin S. N1-Methylnicotinamide Oxidation in a Number of Mammals. J Biol Chem. 1967;242:1274–1279. [PubMed] [Google Scholar]
- 3.Real AM, et al. Nicotinamide N-oxidation by CYP2E1 in human liver microsomes. Drug Metab Dispos Biol Fate Chem. 2013;41:550–553. doi: 10.1124/dmd.112.049734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaykin S, et al. The fate of nicotinamide in the mouse. Urinary metabolites. J Biol Chem. 1965;240:932–938. [PubMed] [Google Scholar]
- 5.Shibata K, et al. Niacin catabolism in rodents. J Nutr Sci Vitaminol (Tokyo) 1990;36:87–98. doi: 10.3177/jnsv.36.87. [DOI] [PubMed] [Google Scholar]
- 6.Najjar VA, Holt LE. The Excretion of Specific Fluorescent Substances in the Urine in Pellagra. Science. 1941;93:20–21. doi: 10.1126/science.93.2401.20. [DOI] [PubMed] [Google Scholar]
- 7.Sarett HP, et al. Studies in Nicotinic Acid Metabolism I. The Fate of Nicotinic Acid in Man. J Nutr. 1942;23:23–34. [Google Scholar]
- 8.Huff JW, Perlzweig WA. N1-Methylnicotinamide, a Metabolite of Nicotinic Acid in the Urine. J Biol Chem. 1943;150:395–400. [Google Scholar]
- 9.Cantoni GL. Methylation of Nicotinamide with a Soluble Enzyme System from Rat Liver. J Biol Chem. 1951;189:203–216. [PubMed] [Google Scholar]
- 10.Cantoni GL. THE NATURE OF THE ACTIVE METHYL DONOR FORMED ENZYMATICALLY FROM L-METHIONINE AND ADENOSINETRIPHOSPHATE1,2. J Am Chem Soc. 1952;74:2942–2943. [Google Scholar]
- 11.Alston TA, Abeles RH. Substrate specificity of nicotinamide methyltransferase isolated from porcine liver. Arch Biochem Biophys. 1988;260:601–608. doi: 10.1016/0003-9861(88)90487-0. [DOI] [PubMed] [Google Scholar]
- 12.Yan L, et al. Mouse liver nicotinamide N-methyltransferase: cDNA cloning, expression, and nucleotide sequence polymorphisms. Biochem Pharmacol. 1997;54:1139–1149. doi: 10.1016/s0006-2952(97)00325-0. [DOI] [PubMed] [Google Scholar]
- 13.Schmeisser K, et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat Chem Biol. 2013;9:693–700. doi: 10.1038/nchembio.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang-Lee YA, et al. Metabolic effects of nicotinamide administration in rats. J Nutr. 1983;113:215–221. doi: 10.1093/jn/113.2.215. [DOI] [PubMed] [Google Scholar]
- 15.Handler P, Dann WJ. The Inhibition of Rat Growth by Nicotinamide. J Biol Chem. 1942;146:357–368. [Google Scholar]
- 16.RIKANS LL, et al. FATTY LIVERS PRODUCED IN ALBINO RATS BY EXCESS NIACIN IN HIGH FAT DIETS. II. EFFECT OF CHOLINE SUPPLEMENTS. J Nutr. 1965;85:107–112. doi: 10.1093/jn/85.1.107. [DOI] [PubMed] [Google Scholar]
- 17.Henning SM, et al. Hepatic content of S-adenosylmethionine, S-adenosylhomocysteine and glutathione in rats receiving treatments modulating methyl donor availability. J Nutr. 1989;119:1478–1482. doi: 10.1093/jn/119.10.1478. [DOI] [PubMed] [Google Scholar]
- 18.Kannt A, et al. Association of nicotinamide-N-methyltransferase mRNA expression in human adipose tissue and the plasma concentration of its product, 1-methylnicotinamide, with insulin resistance. Diabetologia. 2015 doi: 10.1007/s00125-014-3490-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu M, et al. Serum N(1)-Methylnicotinamide Is Associated With Obesity and Diabetes in Chinese. J Clin Endocrinol Metab. 2015;100:3112–3117. doi: 10.1210/jc.2015-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong S, et al. Nicotinamide N-methyltransferase regulates hepatic nutrient metabolism through Sirt1 protein stabilization. Nat Med. 2015;21:887–894. doi: 10.1038/nm.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J, et al. Activation of nicotinamide N-methyltransferase gene promoter by hepatocyte nuclear factor-1beta in human papillary thyroid cancer cells. Mol Endocrinol Baltim Md. 2005;19:527–539. doi: 10.1210/me.2004-0215. [DOI] [PubMed] [Google Scholar]
- 22.Tomida M, et al. Stat3 up-regulates expression of nicotinamide N-methyltransferase in human cancer cells. J Cancer Res Clin Oncol. 2008;134:551–559. doi: 10.1007/s00432-007-0318-6. [DOI] [PubMed] [Google Scholar]
- 23.Ghazalpour A, et al. Hybrid mouse diversity panel: a panel of inbred mouse strains suitable for analysis of complex genetic traits. Mamm Genome Off J Int Mamm Genome Soc. 2012;23:680–692. doi: 10.1007/s00335-012-9411-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torgerson DG, et al. Meta-analysis of Genome-wide Association Studies of Asthma In Ethnically Diverse North American Populations. Nat Genet. 2011;43:887–892. doi: 10.1038/ng.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seshadri S, et al. Genetic correlates of brain aging on MRI and cognitive test measures: a genome-wide association and linkage analysis in the Framingham study. BMC Med Genet. 2007;8:S15. doi: 10.1186/1471-2350-8-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bromberg A, et al. Nicotinamide-N-methyltransferase (NNMT) in schizophrenia: genetic association and decreased frontal cortex mRNA levels. Int J Neuropsychopharmacol Off Sci J Coll Int Neuropsychopharmacol CINP. 2012;15:727–737. doi: 10.1017/S1461145711001179. [DOI] [PubMed] [Google Scholar]
- 27.Wang G, et al. Female specific association between NNMT gene and schizophrenia in a Han Chinese population. Int J Med Sci. 2014;11:1234–1239. doi: 10.7150/ijms.9426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sazci G, et al. Association of Nicotinamide-N-Methyltransferase Gene rs694539 Variant with Epilepsy. Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9364-2. [DOI] [PubMed] [Google Scholar]
- 29.Sazci A, et al. Association of nicotinamide-N-methyltransferase gene rs694539 variant with patients with nonalcoholic steatohepatitis. Genet Test Mol Biomark. 2013;17:849–853. doi: 10.1089/gtmb.2013.0309. [DOI] [PubMed] [Google Scholar]
- 30.Souto JC, et al. A genomewide exploration suggests a new candidate gene at chromosome 11q23 as the major determinant of plasma homocysteine levels: results from the GAIT project. Am J Hum Genet. 2005;76:925–933. doi: 10.1086/430409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115–130. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- 32.Cantó C, et al. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y, Sauve AA. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta. 2016;1864:1787–1800. doi: 10.1016/j.bbapap.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bitterman KJ, et al. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 35.Clark JB, et al. Inhibition of nuclear NAD nucleosidase and poly ADP-ribose polymerase activity from rat liver by nicotinamide and 5′-methyl nicotinamide. Biochim Biophys Acta. 1971;238:82–85. doi: 10.1016/0005-2787(71)90012-8. [DOI] [PubMed] [Google Scholar]
- 36.Revollo JR, et al. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 37.Burgos ES, Schramm VL. Weak coupling of ATP hydrolysis to the chemical equilibrium of human nicotinamide phosphoribosyltransferase. Biochemistry (Mosc) 2008;47:11086–11096. doi: 10.1021/bi801198m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraus D, et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature. 2014;508:258–262. doi: 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang HC, Guarente L. SIRT1 and other sirtuins in Metabolism. Trends Endocrinol Metab TEM. 2014;25:138–145. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He W, et al. Mitochondrial sirtuins: regulators of protein acylation and metabolism. Trends Endocrinol Metab TEM. 2012;23:467–476. doi: 10.1016/j.tem.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Houtkooper RH, et al. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodgers JT, et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 44.Nemoto S, et al. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306:2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 45.Nemoto S, et al. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 46.Liu Y, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456:269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li X, et al. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 48.Ponugoti B, et al. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J Biol Chem. 2010;285:33959–33970. doi: 10.1074/jbc.M110.122978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walker AK, et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010;24:1403–1417. doi: 10.1101/gad.1901210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parsons RB, et al. The expression of nicotinamide N-methyltransferase increases ATP synthesis and protects SH-SY5Y neuroblastoma cells against the toxicity of Complex I inhibitors. Biochem J. 2011;436:145–155. doi: 10.1042/BJ20101685. [DOI] [PubMed] [Google Scholar]
- 51.Tanaka Y, et al. 1-Methylnicotinamide ameliorates lipotoxicity-induced oxidative stress and cell death in kidney proximal tubular cells. Free Radic Biol Med. 2015;89:831–841. doi: 10.1016/j.freeradbiomed.2015.10.414. [DOI] [PubMed] [Google Scholar]
- 52.Peng L, et al. Ubiquitinated Sirtuin 1 (SIRT1) Function Is Modulated during DNA Damage-induced Cell Death and Survival. J Biol Chem. 2015;290:8904–8912. doi: 10.1074/jbc.M114.612796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chlopicki S, et al. 1-Methylnicotinamide (MNA), a primary metabolite of nicotinamide, exerts anti-thrombotic activity mediated by a cyclooxygenase-2/prostacyclin pathway. Br J Pharmacol. 2007;152:230–239. doi: 10.1038/sj.bjp.0707383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Domagala TB, et al. Nitric oxide production and endothelium-dependent vasorelaxation ameliorated by N1-methylnicotinamide in human blood vessels. Hypertension. 2012;59:825–832. doi: 10.1161/HYPERTENSIONAHA.111.183210. [DOI] [PubMed] [Google Scholar]
- 55.Kim HC, et al. Expression and functional significance of nicotinamide N-methyl transferase in skeletal muscles of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181:797–805. doi: 10.1164/rccm.200906-0936OC. [DOI] [PubMed] [Google Scholar]
- 56.Savarimuthu Francis SM, et al. Genes and gene ontologies common to airflow obstruction and emphysema in the lungs of patients with COPD. PloS One. 2011;6:e17442. doi: 10.1371/journal.pone.0017442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang H, et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436–1443. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- 58.Sternak M, et al. Nicotinamide N-methyltransferase (NNMT) and 1-methylnicotinamide (MNA) in experimental hepatitis induced by concanavalin A in the mouse. Pharmacol Rep PR. 2010;62:483–493. doi: 10.1016/s1734-1140(10)70304-2. [DOI] [PubMed] [Google Scholar]
- 59.Fedorowicz A, et al. Activation of the nicotinamide N-methyltransferase (NNMT)-1-methylnicotinamide (MNA) pathway in pulmonary hypertension. Respir Res. 2016;17:108. doi: 10.1186/s12931-016-0423-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jakubowski A, et al. 1-Methylnicotinamide protects against liver injury induced by concanavalin A via a prostacyclin-dependent mechanism: A possible involvement of IL-4 and TNF-α. Int Immunopharmacol. 2016;31:98–104. doi: 10.1016/j.intimp.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 61.Markert JM, et al. Differential gene expression profiling in human brain tumors. Physiol Genomics. 2001;5:21–33. doi: 10.1152/physiolgenomics.2001.5.1.21. [DOI] [PubMed] [Google Scholar]
- 62.Xu J, et al. Enhanced expression of nicotinamide N-methyltransferase in human papillary thyroid carcinoma cells. J Clin Endocrinol Metab. 2003;88:4990–4996. doi: 10.1210/jc.2002-021843. [DOI] [PubMed] [Google Scholar]
- 63.Yao M, et al. Gene expression analysis of renal carcinoma: adipose differentiation-related protein as a potential diagnostic and prognostic biomarker for clear-cell renal carcinoma. J Pathol. 2005;205:377–387. doi: 10.1002/path.1693. [DOI] [PubMed] [Google Scholar]
- 64.Wu Y, et al. Overlapping gene expression profiles of cell migration and tumor invasion in human bladder cancer identify metallothionein 1E and nicotinamide N-methyltransferase as novel regulators of cell migration. Oncogene. 2008;27:6679–6689. doi: 10.1038/onc.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jang JS, et al. The differential proteome profile of stomach cancer: identification of the biomarker candidates. Oncol Res. 2004;14:491–499. doi: 10.3727/0965040042380441. [DOI] [PubMed] [Google Scholar]
- 66.Lim BH, et al. Overexpression of nicotinamide N-methyltransferase in gastric cancer tissues and its potential post-translational modification. Exp Mol Med. 2006;38:455–465. doi: 10.1038/emm.2006.54. [DOI] [PubMed] [Google Scholar]
- 67.Roessler M, et al. Identification of nicotinamide N-methyltransferase as a novel serum tumor marker for colorectal cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2005;11:6550–6557. doi: 10.1158/1078-0432.CCR-05-0983. [DOI] [PubMed] [Google Scholar]
- 68.Sartini D, et al. Nicotinamide N-methyltransferase upregulation inversely correlates with lymph node metastasis in oral squamous cell carcinoma. Mol Med Camb Mass. 2007;13:415–421. doi: 10.2119/2007-00035.Sartini. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim J, et al. Expression of nicotinamide N-methyltransferase in hepatocellular carcinoma is associated with poor prognosis. J Exp Clin Cancer Res CR. 2009;28:20. doi: 10.1186/1756-9966-28-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu T, et al. Effects of nicotinamide N-methyltransferase on PANC-1 cells proliferation, metastatic potential and survival under metabolic stress. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2015;35:710–721. doi: 10.1159/000369731. [DOI] [PubMed] [Google Scholar]
- 71.Tang SW, et al. Nicotinamide N-methyltransferase induces cellular invasion through activating matrix metalloproteinase-2 expression in clear cell renal cell carcinoma cells. Carcinogenesis. 2011;32:138–145. doi: 10.1093/carcin/bgq225. [DOI] [PubMed] [Google Scholar]
- 72.Pozzi V, et al. RNA-mediated gene silencing of nicotinamide N-methyltransferase is associated with decreased tumorigenicity in human oral carcinoma cells. PloS One. 2013;8:e71272. doi: 10.1371/journal.pone.0071272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ulanovskaya OA, et al. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat Chem Biol. 2013;9:300–306. doi: 10.1038/nchembio.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Palanichamy K, et al. NNMT silencing activates tumor suppressor PP2A, inactivates oncogenic STKs and inhibits tumor forming ability. Clin Cancer Res Off J Am Assoc Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sperber H, et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat Cell Biol. 2015;17:1523–1535. doi: 10.1038/ncb3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Riederer M, et al. Adipose tissue as a source of nicotinamide N-methyltransferase and homocysteine. Atherosclerosis. 2009;204:412–417. doi: 10.1016/j.atherosclerosis.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 77.Mato JM, et al. Methionine metabolism and liver disease. Annu Rev Nutr. 2008;28:273–293. doi: 10.1146/annurev.nutr.28.061807.155438. [DOI] [PubMed] [Google Scholar]
- 78.Mudd SH, et al. Methyl balance and transmethylation fluxes in humans. Am J Clin Nutr. 2007;85:19–25. doi: 10.1093/ajcn/85.1.19. [DOI] [PubMed] [Google Scholar]
- 79.Martínez-Chantar ML, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatol Baltim Md. 2008;47:1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Varela-Rey M, et al. Fatty liver and fibrosis in glycine N-methyltransferase knockout mice is prevented by nicotinamide. Hepatol Baltim Md. 2010;52:105–114. doi: 10.1002/hep.23639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cantó C, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Trammell SAJ, et al. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci Rep. 2016;6:26933. doi: 10.1038/srep26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trammell SAJ, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. doi: 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Okamoto H, et al. Diurnal variations in human urinary excretion of nicotinamide catabolites: effects of stress on the metabolism of nicotinamide. [Online] doi: 10.1093/ajcn/77.2.406. Available: http://ajcn.nutrition.org. [Accessed: 15-Jun-2016] [DOI] [PubMed]
- 85.Aguilar-Arnal L, et al. NAD+-SIRT1 control of H3K4 trimethylation through circadian deacetylation of MLL1. Nat Struct Mol Biol. 2015;22:312–318. doi: 10.1038/nsmb.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Trammell SAJ, Brenner C. NNMT: A Bad Actor in Fat Makes Good in Liver. Cell Metab. 2015;22:200–201. doi: 10.1016/j.cmet.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brown KD, et al. Activation of SIRT3 by the NAD+ precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 2014;20:1059–1068. doi: 10.1016/j.cmet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yoshino J, et al. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fang EF, et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016;24:566–581. doi: 10.1016/j.cmet.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bi HC, et al. N-methylnicotinamide and nicotinamide N-methyltransferase are associated with microRNA-1291-altered pancreatic carcinoma cell metabolome and suppressed tumorigenesis. Carcinogenesis. 2014;35:2264–2272. doi: 10.1093/carcin/bgu174. [DOI] [PMC free article] [PubMed] [Google Scholar]