

ABSTRACT
Macroautophagy/autophagy plays a key role in cellular quality control by eliminating protein aggregates and damaged organelles, which is essential for the maintenance of neuronal homeostasis. Defective autophagy has been implicated in the pathogenesis of Alzheimer disease (AD). In AD brains, autophagic vacuoles (AVs) accumulate massively within dystrophic neurites. This raises a fundamental question as to whether impaired autophagic clearance contributes to AD-associated autophagic stress. We recently revealed that AD neurons display defective retrograde transport and accumulation of amphisomes predominantly in axons and presynaptic terminals. Amyloid β (Aβ) oligomers are enriched in axons and interact with dynein motors. This interaction interferes with the coupling of the dynein motor with its adaptor SNAPIN. Such deficits disrupt dynein-driven retrograde transport of amphisomes, thus trapping them in distal axons and impairing their degradation in the soma. Therefore, our study provides new mechanistic insights into AD-linked autophagic pathology, and builds a foundation for developing potential AD therapeutic strategies by rescuing retrograde transport of amphisomes.
KEYWORDS: Alzheimer, amphisome, amyloid β, autophagic stress, autophagosome, autophagy, dynein, late endosome, retrograde transport, Snapin
Alzheimer disease (AD) is an age-related progressive neurodegenerative disease that affects a staggering percentage of the aging population and causes memory loss and cognitive decline. AD is characterized by the formation of senile plaques in patient brains. Amyloid β (Aβ) peptide, the principal component of senile plaques, is derived from serial proteolysis of APP (amyloid β precursor protein) by β-site-APP-cleaving enzyme 1 (BACE1) and γ secretase. Intracellular Aβ is enriched in the brain of both AD mouse models and human patients and associated with dystrophic neurites and abnormal synaptic morphology. Accumulation of intracellular Aβ is well correlated with the onset and progression of AD.
Neurons are highly polarized cells with a long axon. Recent studies have established that autophagosomes are formed in distal axons and move in a retrograde direction toward the soma where mature lysosomes are mainly located. Following fusion with somatic lysosomes, autophagic cargoes are degraded within autolysosomes by lysosomal hydrolases. Thus, neurons face the special challenge of transporting newly generated autophagosomes from distal axons to the soma. Such retrograde transport is initiated by recruitment of late endosome (LE)-loaded dynein-SNAPIN motor-adaptor complexes after fusion with LEs to form amphisomes. Ultrastructural analysis has revealed that AD brains exhibit a unique Aβ-associated autophagic pathology: autophagic vacuoles (AVs) massively accumulate within large swellings along dystrophic neurites. Aβ1–42 is associated with LEs or multivesicular bodies (MVBs) and AVs in AD brains. This raises a fundamental question: does intracellular Aβ accumulation impair autophagic clearance and thus augment autophagic stress in AD neurons?
To address this question, we first determined how autophagy is altered in an AD-related mutant human APP (HsAPP) transgenic (Tg) mouse model. We found that AVs accumulate predominantly in distal axons and presynaptic terminals of AD neurons. The majority of these AVs are co-labeled by autophagic markers LC3 or SQSTM1/p62 and the LE marker insulin-like growth factor 2 receptor/Cation-independent mannose-6-phosphate receptor (IGF2R/CI-MPR) suggesting that they are amphisomes in nature, following fusion with LEs. Using transmission electron microscopy (TEM), we detected massive accumulation of AV-like vesicles within dystrophic/swollen neurites in mutant HsAPP Tg mouse brains. Most of these vesicles show late-stage degradative AV (AVd)-like structures after fusion with late endocytic organelles, which contain electron-dense amorphous material and small internal vesicles and/or organelles at various degradation stages. Interestingly, a striking number of AVd-like structures are retained at presynaptic terminals of AD mice. These observations are consistent with increased levels of SQSTM1 and LC3-II in synaptosomal preparations from the brains of mutant HsAPP Tg mice and AD patients. These findings indicate aberrant AV retention in axons and at synapses of AD neurons and promote us to hypothesize that AD-linked autophagic stress may result from impaired retrograde transport of autophagosomes from distal AD axons toward the soma for degradation.
To test our hypothesis, we first examined the distribution and motility of AVs along the axon of AD neurons cultured from mutant HsAPP Tg mice. At basal conditions, LC3 is associated with vesicles as lipidated LC3-II within AD axons. The majority of these AVs colocalize with RAB7-labeled LEs, suggesting effective formation of amphisomes by fusion of these two organelles. The density of axonal AVs, particularly amphisomes, is robustly increased in AD neurons relative to that of wild-type (WT) controls. Moreover, while GFP-LC3 is mainly diffuse in WT axons, a significant portion of LEs move in a retrograde direction toward the soma along the same axon. Strikingly, LEs and amphisomes in AD neurons display a similar pattern of motility: reduced retrograde transport along the same axon (Fig. 1).
Figure 1.
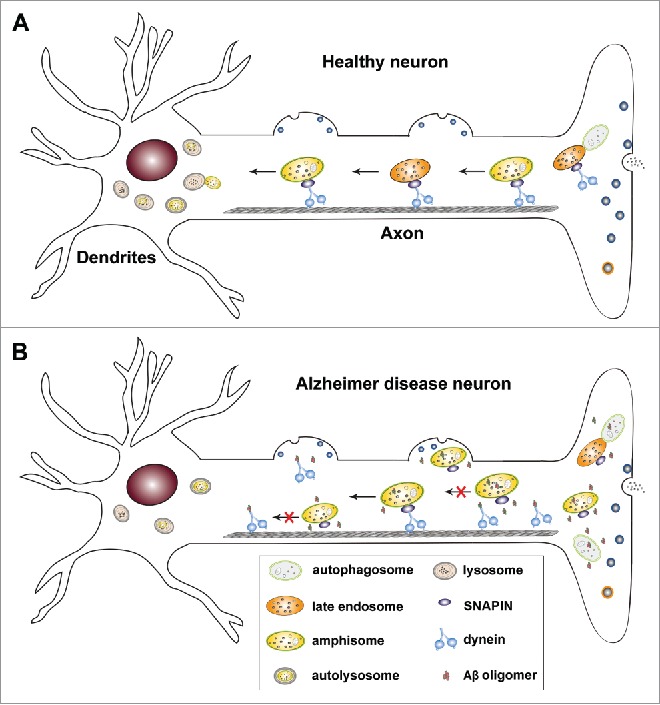
Dynein-SNAPIN-mediated retrograde transport plays a key role in the maintenance of axonal homeostasis by facilitating autophagic clearance in neurons. (A) In healthy neurons, autophagosomes are predominantly generated in distal axons. Through fusion with late endosomes (LEs) to form amphisomes, nascent autophagosomes gain long-distance retrograde motility by recruiting LE-loaded dynein-SNAPIN motor-adaptor transport machinery. Efficient retrograde transport rapidly moves amphisomes from distal axons to the soma where active lysosomes are relatively enriched. Such a mechanism enables neurons to facilitate cargo degradation within autolysosomes in the soma, thereby decreasing axonal stress. (B) In Alzheimer disease (AD) neurons, elevated cytoplasmic amyloid β (Aβ) oligomers interact with dynein motors and competitively interfere with dynein-SNAPIN coupling. Such deficits disrupt recruitment of dynein motors onto SNAPIN-loaded LEs and/or amphisomes, and reduce their retrograde transport toward the soma for lysosomal clearance. As a result, amphisomes are trapped in distal axons and at presynaptic terminals, augmenting autophagic stress in AD neurons.
Next, we asked whether these amphisomes are associated with Aβ oligomers, the toxic form of Aβ linked to AD pathogenesis. We demonstrated that oligomeric Aβ1–42 is enriched in distal axons and associated with amphisomes within dystrophic axons surrounding amyloid plaques in AD mouse brains. To confirm these light imaging results at the ultrastructural level, we performed immuno-EM analysis using the A11 antibody, which detects soluble Aβ oligomers. The anti-A11 immuno-gold grains are abundant in the cytoplasm of AD axons. More importantly, ∼57.3% of AVd-like structures are labeled with or surrounded by these oligomeric Aβ gold particles.
To provide mechanistic insight into impaired AV retrograde transport, we revealed that Aβ forms a complex with dynein motors, and recruitment of dynein motors to amphisomes is markedly reduced in mutant HsAPP mouse brains. Moreover, dynein-SNAPIN coupling is impaired in AD patient brains. We further determined whether soluble Aβ1–42 oligomers interfere with the loading of dynein motors onto AVs. We found that DNAIC (dynein, axonemal, intermediate chain) specifically interacts with Aβ1–42. Through this binding Aβ1–42 competitively interrupts the formation of dynein-SNAPIN motor-adaptor complexes. As a result, the recruitment of dynein motors to SNAPIN-loaded LEs and/or amphisomes is reduced. Our data provides compelling evidence that dynein-SNAPIN coupling, and thus motor loading onto their transport cargoes, is compromised by elevated levels of intracellular Aβ1–42 oligomers. Thus, cytoplasmic Aβ1–42 oligomers could impair dynein-driven retrograde transport of AVs in AD neurons, causing AD-linked autophagic pathology (Fig. 1).
Intriguingly, the Snapin-deficient mouse brain recapitulates AD-associated autophagic stress in axons. Amphisomes accumulate in the hippocampal mossy fibers of Snapin-mutant mice, which are composed of axons and presynaptic terminals. The loading of dynein motors onto LEs and/or amphisomes is reduced to ∼55% in Snapin-mutant mouse brains, suggesting that dynein-SNAPIN coupling is one of the major dynein-recruiting mechanisms for both LEs and amphisomes. Given that Snapin deficiency results in AD-like autophagic pathology, and if our hypothesis is correct, elevating SNAPIN expression in AD neurons would reverse axonal autophagic stress. We showed that overexpressing SNAPIN attenuates AV retention in axons and presynaptic terminals of AD neurons by enhancing AV retrograde transport under both basal conditions and upon autophagic activation. Therefore, these results indicate that SNAPIN-enhanced retrograde transport efficiently removes AVs from distal axons for somatic lysosomal clearance, reducing autophagic stress in AD axons.
In summary, our study provides new mechanistic insights into how intracellular Aβ impairs dynein-mediated retrograde transport of LEs and amphisomes, thereby leading to axonal autophagic pathology in AD neurons. Our study also establishes a foundation for future investigation into the regulation of dynein-SNAPIN coupling and thus retrograde transport to attenuate autophagy defects in AD brains.
Abbreviations
- Aβ
amyloid β
- AD
Alzheimer disease
- APP
amyloid precursor protein
- AV
autophagic vacuole
- AVd
degradative AV
- DNAIC
dynein, axonemal, intermediate chain
- IGF2R/CI-MPR
insulin-like growth factor 2 receptor/Cation-independent mannose-6-phosphate receptor
- LE
late endosome
- MVBs
multivesicular bodies
- TEM
transmission electron microscopy
- Tg
transgenic
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This research was supported by the National Institutes of Health (R00AG033658 and R01NS089737 to Q.C.); the Alzheimer Association (NIRG-14–321833 to Q.C.); and the Charles and Johanna Busch Biomedical Award (to Q.C.).