

ABSTRACT
SPHK1 (sphingosine kinase 1), a regulator of sphingolipid metabolites, plays a causal role in the development of hepatocellular carcinoma (HCC) through augmenting HCC invasion and metastasis. However, the mechanism by which SPHK1 signaling promotes invasion and metastasis in HCC remains to be clarified. Here, we reported that SPHK1 induced the epithelial-mesenchymal transition (EMT) by accelerating CDH1/E-cadherin lysosomal degradation and facilitating the invasion and metastasis of HepG2 cells. Initially, we found that SPHK1 promoted cell migration and invasion and induced the EMT process through decreasing the expression of CDH1, which is an epithelial marker. Furthermore, SPHK1 accelerated the lysosomal degradation of CDH1 to induce EMT, which depended on TRAF2 (TNF receptor associated factor 2)-mediated macroautophagy/autophagy activation. In addition, the inhibition of autophagy recovered CDH1 expression and reduced cell migration and invasion through delaying the degradation of CDH1 in SPHK1-overexpressing cells. Moreover, the overexpression of SPHK1 produced intracellular sphingosine-1-phosphate (S1P). In response to S1P stimulation, TRAF2 bound to BECN1/Beclin 1 and catalyzed the lysine 63-linked ubiquitination of BECN1 for triggering autophagy. The deletion of the RING domain of TRAF2 inhibited autophagy and the interaction of BECN1 and TRAF2. Our findings define a novel mechanism responsible for the regulation of the EMT via SPHK1-TRAF2-BECN1-CDH1 signal cascades in HCC cells. Our work indicates that the blockage of SPHK1 activity to attenuate autophagy may be a promising strategy for the prevention and treatment of HCC.
KEYWORDS: autophagy, BECN1/Beclin 1, HepG2 cells, invasion and metastasis, TRAF2
Introduction
Hepatocellular carcinoma (HCC) is a complication of chronic liver disease and is considered to be the most lethal type of cancer.1,2 Various factors contribute to HCC, such as oxidative/metabolic stress, growth factors and inflammatory factors.1,3 SPHK1 (sphingosine kinase 1), a regulator of sphingolipid metabolites, plays a role similar to that of growth factors in causing the development of HCC.2 Sphingosine suppresses tumor progression by mediating cell cycle arrest and inducing apoptosis.4 However, SPHK1 converts sphingosine to sphingosine-1-phosphate (S1P), which promotes cell growth, proliferation and survival.5 SPHK1 protein expression and mRNA level are both increased in HCC tissues.6 SPHK1 overexpression promotes the invasion and metastasis of SMMC-7721 cells.7 Our previous studies revealed that the inhibition of SPHK1 suppressed the invasion of HepG2 cells.2 These studies suggest that SPHK1 could augment the invasion and metastasis of HCC. However, the precise regulatory mechanisms and signaling pathways underlying the SPHK1-mediated promotion of HCC invasion and metastasis remain unknown.
The epithelial-mesenchymal transition (EMT) enables polarized epithelial cells to undergo multiple biochemical changes to exhibit characteristics of mesenchymal cell phenotypes, including enhanced migration and invasion capacities.8 The EMT converts benign tumors into invasive, metastatic tumors and plays a critical role in the regulation of tumor progression and metastasis.9 The loss of CDH1/E-cadherin expression is the foundation for the activation of the EMT,10 and the downregulated expression of CDH1 in HCC tissues contributes to tumor progression and development.11 In metastatic HCC tissue, the level of CDH1 decreases and the expression of other EMT markers (e.g., FN1 [fibronectin 1], and VIM [vimentin]) changes,12 suggesting that the EMT process is activated in metastatic HCC tissue. Because SPHK1 augments HCC invasion and metastasis and EMT stimulation promotes HCC progression and development, it is possible that SPHK1 promotes HCC invasion and metastasis via regulating the EMT.
In this study, we hypothesized that SPHK1 might play a role in the regulation of tumor metastasis through inducing the EMT. We found a correlation between the levels of SPHK1 and CDH1 expression in HCC tissues and studied the potential mechanism responsible for the SPHK1-mediated metastatic effect. Our findings suggest that SPHK1 can function as an endogenous inducer of tumor metastasis by regulating a critical autophagy modulator to maintain autophagy activation and promoting the lysosomal degradation of CDH1, the master suppressor of the EMT.
Results
SPHK1 stimulates the EMT in both HCC tissues and cells
We previously reported that the SPHK1 inhibitor SKI II suppresses the proliferation and migration of HepG2 cells.2,5 Here, we further determined the exact relationship between SPHK1 and malignancy grade in HCC tissues. We found that the SPHK1 expression and malignancy grade are positively correlated in HCC tissues (Fig. 1A and B). The loss of CDH1 expression was correlated with increased invasiveness through EMT induction,10 and we found that the expression of CDH1 was lower in HCC higher grades of malignancy (Fig. 1A and B). These results suggest that SPHK1 and CDH1 are negatively correlated and that SPHK1 might regulate the EMT process in malignant HCC tissues.
Figure 1.

SPHK1 stimulates the EMT in both HCC tissues and cells. (A, B) The expression of SPHK1 and CDH1 was correlated with malignancy grade in HCC tissues. The expression of SPHK1 was analyzed by staining (IHC) as indicated in Materials and Methods. (C, D) SPHK1 promotes cell migration and invasion. HepG2 cells stably expressing vector or MYC-SPHK1 were plated in the upper chamber of the filters for 24 h. Then, cells that had migrated to the underside of the transwell insert were counted. (E) SPHK1 overexpression decreased the levels of epithelial markers and increased the level of mesenchymal markers. HepG2 cells stably expressing vector or MYC-SPHK1 were harvested and the level of epithelial or mesenchymal markers was detected by western blot analysis. ACTB served as the loading control. The value for the vector was set to 1.0 and the other values were normalized. (F) SPHK1 overexpression decreased the expression of CDH1 and increased the expression of VIM. HepG2 cells stably expressing vector or MYC-SPHK1 were fixed in 4% paraformaldehyde and stained for CDH1 or VIM, and imaged by confocal microscopy, scale bar: 20 μm. Data are presented as the mean ± SE (n = 4).
We further investigated whether the expression of SPHK1 promotes the invasion and metastasis of HCC cells by regulating the EMT. Because both the expression of SPHK16 and mobility13 of HepG2 cells are lower than those of other HCC cells, we prepared HepG2 cells stably expressing either vector or MYC-SPHK1. The data showed that the overexpression of SPHK1 resulted in enhanced migration, as indicated by transwell migratory assay, as well as increased invasion, as indicated by Martrigel invasion assay (Fig. 1C and D). Then, we examined changes in the EMT markers. SPHK1 overexpression decreased the expression of the epithelial marker CDH1 and increased the expression of mesenchymal markers FN1, VIM and CDH2/N-cadherin (Fig. 1E and F). Similarly, we found that SPHK1 promoted the expression of EMT markers and induced the migration and invasion in other HCC cell lines (e.g. Huh7 and HCCLM3, Fig. S1A-1F). These results suggest that the overexpression of SPHK1 induces the EMT in both HCC tissues and cells.
SPHK1 accelerates the degradation of CDH1 through lysosomal pathways
Because the loss of CDH1 expression is the foundation of the EMT10 and SPHK1 decreased the expression of CDH1 (Fig. 1E and F), we speculated that SPHK1 stimulated the EMT through the downregulation of CDH1. The upregulation of CDH1 reduced the migration and invasion of SPHK1-overexpressing cells and decreased the expression of mesenchymal markers (e.g., FN1, VIM and CDH2), which suggests that SPHK1 stimulated the EMT through decreasing the expression of CDH1 in HepG2 cells (Fig. S2). We next investigated the exact regulatory effect of SPHK1-reduced CDH1 expression in HepG2 cells. We found that the overexpression of SPHK1 reduced the protein but not the mRNA expression of CDH1 in a concentration-dependent manner (Fig. 2A and B). Therefore, we hypothesized that SPHK1 might decrease the CDH1 level by regulating the stability of CDH1. We found that the cells overexpressing SPHK1 accelerated CDH1 degradation in the presence of the protein synthesis inhibitor cycloheximide (CHX) (Fig. 2C).
Figure 2.

SPHK1 promotes the lysosomal degradation of CDH1. (A) SPHK1 decreased the expression of CDH1. Cells were transfected with the indicated concentrations of MYC-SPHK1 or vector and protein lysates were analyzed by immunoblotting with the indicated antibodies. (B) SPHK1 did not affect the level of CDH1 mRNA in HepG2 cells. Cells were transfected with the indicated concentrations of MYC-SPHK1 or vector, and total RNA was isolated. CDH1 mRNA was analyzed by fluorescent quantitative RT-PCR, as indicated in Materials and Methods. (C) SPHK1 inhibits the degradation of CDH1 in HepG2 cells. HepG2 cells stably expressing vector or MYC-SPHK1 were transfected with pCMV6-CDH1 for 24 h and then treated with CHX (20 μmol/L) for the indicated times. The cell lysates were detected by western blotting using an anti-CDH1 antibody. (D) SPHK1 did not affect the proteasomal degradation of CDH1. HepG2 cells were transfected with pCMV6-CDH1 for 24 h. Cells were treated with MG132 (10 μmol/L) for 2 h, and then also treated with CHX for the indicated times. Immunoblotting was performed with the indicated antibody. (E) SPHK1 accelerated the lysosomal degradation of CDH1. Cells were transfected with pCMV6-CDH1 for 24 h. HepG2 cells were treated with CQ (100 μmol/L) for 12 h, and then CHX was added for the indicated times. Immunoblotting was performed with the indicated antibody. Data are presented as the mean ± SE (n = 4). NS, nonsignificant; CHX, cycloheximide; CQ, chloroquine.
Most proteins are degraded either by the proteasome or lysosomal pathways. We next examined which pathway participates in the regulation of SPHK1-induced CDH1 degradation. The inhibition of the proteasome by MG132 did not affect the degradation of CDH1 in SPHK1-overexpressing cells (Fig. 2D). However, the inhibition of lysosome function in the presence of chloroquine (CQ) delayed the degradation of CDH1 in SPHK1-overexpressing cells (Fig. 2E). Taken together, these data suggest that SPHK1 accelerates the degradation of CDH1 through lysosomal pathways.
SPHK1 stimulates autophagy in HCC cells
Lysosomal pathways include autophagic and endocytic lysosomal pathways. Previous studies have shown that CDH1 can be degraded by the endocytic lysosomal pathway.14 However, because SPHK1 stimulates autophagy in MCF-7 cells,15 we speculated that autophagy might participate in the lysosomal degradation of CDH1, and we investigated the effects of SPHK1 on the regulation of autophagy in HCC cells. We found that SPHK1 increased the number of autophagosomes; electron microscopy revealed the presence of double-membraned vacuolar structures with the morphological features of autophagosomes in SPHK1-overexpressing HepG2 cells (Fig. 3A and B). The conversion of the soluble form of MAP1LC3/LC3 (MAP1LC3-I) to a lipidated form (MAP1LC3-II) is a marker of autophagy, and SQSTM1/p62, a “cargo” protein, is recognized as a marker of autophagy flux.16 SPHK1 overexpression increased the expression of MAP1LC3-II and decreased the level of SQSTM1 in HepG2 cells (Fig. 3C). Furthermore, our results showed that SPHK1 augmented MAP1LC3 foci in HepG2 cells (Fig. 3D), and CQ enhanced the SPHK1-induced accumulation of MAP1LC3-II (Fig. 3E). Taken together, our data indicate that SPHK1 upregulates the autophagy activity in HCC cells.
Figure 3.

SPHK1 stimulates autophagy in HepG2 cells. (A, B) SPHK1 increased the number of autophagosomes (APs) in HepG2 cells. Electron microscopy revealed typical autolysosomes as observed in SPHK1-overexpressing cells (indicated by the red arrowhead). Typical mitochondrion is indicated by the green arrowhead. Magnification x 10,000–50,000. The number of autophagosomes was quantified as described in Materials and Methods. (C) SPHK1 overexpression upregulated the expression of autophagy-related proteins. HepG2 cells stably expressing vector or MYC-SPHK1 were harvested and autophagy-related proteins were detected by western blot analysis. (D) SPHK1 overexpression aggregated MAP1LC3 foci. HepG2 cells stably expressing vector or MYC-SPHK1 were fixed in 4% paraformaldehyde, stained with fluorochromes, and imaged by confocal microscopy, scale bar: 20 μm. MAP1LC3 foci per cell were quantified as described in Materials and Methods. (E) Treatment with CQ augmented the expression of MAP1LC3-II. HepG2 cells expressing either vector or MYC-SPHK1 were treated with or without CQ (100 μmol/L) for 12 h, and the expression of MAP1LC3 was measured by western blotting. Data are presented as the mean ± SE of 4 independent assays. N, nucleus; CQ, chloroquine.
SPHK1 induces the EMT in HCC cells by stimulating autophagy
Autophagy promotes HCC cell invasion through EMT activation.17 Thus, we hypothesized that SPHK1 induces the EMT by activating autophagy. However, we found that the inhibition of autophagy by MHY1485, an agonist of MTOR (mechanistic target of rapamycin), or by SBI-0206965, an autophagy kinase ULK1 inhibitor, did not affect the expression of EMT-related markers in SPHK1-overexpressing cells (Fig. 4A). In addition, suppression of autophagy by 3-MA, an inhibitor of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, or by CQ recovered the expression of CDH1 and decreased the expression of FN1, VIM and CDH2 in SPHK1-overexpressing cells (Fig. 4A). These data suggested that SPHK1 stimulated autophagy by affecting the autophagic PtdIns3K complex. Furthermore, the suppression of autophagy by 3-MA or CQ reduced migration (Fig. 4B and D) and invasion (Fig. 4C and E) in SPHK1-overexpressing cells. These data suggest that SPHK1 induces the EMT in HCC cells by stimulating autophagy.
Figure 4.
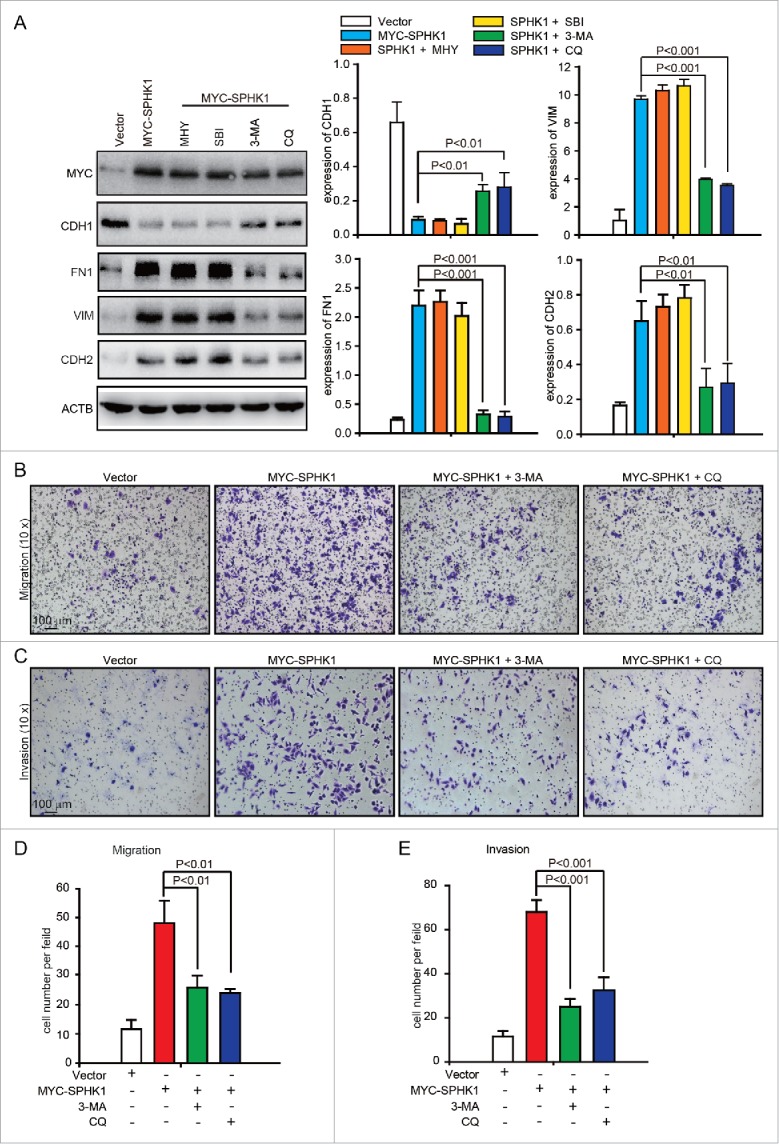
SPHK1 induces the EMT by stimulating autophagy. (A) The inhibition of autophagy recovered the expression of epithelial markers and mesenchymal markers in SPHK1-overexpressing HCC cells. HepG2 cells stably expressing vector or MYC-SPHK1 were treated with autophagic inhibitors MHY1485 (2 μmol/L, 6 h), SBI-0206965 (10 μmol/L, 2 h), 3-MA (10 mmol/L, 6 h) and CQ (100 μmol/L, 12 h) and then harvested. EMT-related protein expression was detected by western blot analysis. (B) Suppression of autophagy reduced cell migration in SPHK1-overexpressing HCC cells. HepG2 cells stably expressing vector or MYC-SPHK1 were plated in the upper chamber of transwell filters for 24 h and then treated with 3-MA (10 mmol/L, 6 h) or CQ (100 μmol/L, 12 h). Then cells migrating to the underside of the transwell insert were measured. (C) Suppression of autophagy reduced the invasion of SPHK1-overexpressing cells. The transwell invasion assay was performed as described in (B), except that the chambers were coated with basement membrane Matrigel. (D, E) Statistical analysis of cells per field in (B) and (C). The cells per field were quantified as described in Materials and Methods. MHY, MHY1485; SBI, SBI-0206965; CQ, chloroquine.
SPHK1 promotes the activation of autophagy through TRAF2
The overexpression of SPHK1 upregulated the intracellular and extracellular levels of S1P in HepG2 cells and the intracellular S1P content was greater (Fig. S1G). Hence, we considered the probability that SPHK1 regulated the lysosomal degradation of CDH1 through TRAF2 (TNF receptor associated factor 2), a downstream signaling factor of intracellular S1P.18 We found that the silencing of TRAF2 decreased the number of MAP1LC3 foci in SPHK1-overexpressing HepG2 cells (Fig. 5A and B). Furthermore, SPHK1 neither increased the expression of MAP1LC3-II nor decreased the level of SQSTM1 after TRAF2 silencing (Fig. 5C). These data suggest that SPHK1 stimulates autophagy through TRAF2.
Figure 5.

SPHK1 activates autophagy through TRAF2. (A, B) Silencing TRAF2 decreased the dots of MAP1LC3 in SPHK1-overexpressing cells. HepG2 cells were transfected with control-siRNA or si-TRAF2 and then fixed in 4% paraformaldehyde, stained with fluorochromes, and imaged by confocal microscopy; scale bar: 20 μm. (C) TRAF2 is necessary for the activation of autophagy when SPHK1 is overexpressed. The HepG2 cells were transfected with control-siRNA or si-TRAF2. Cell lysates were collected, and the expression of autophagy-related proteins was analyzed by western blotting. Data are presented as the mean ± SE of 4 independent assays. Con-siRNA, control-siRNA.
SPHK1 induces lysine 63-linked BECN1 ubiquitination through TRAF2
We further investigated the exact mechanism of how SPHK1 regulates the autophagic machinery to activate autophagy. TRAF6, an E3 ubiquitin ligase, stimulates autophagy through inducing the lysine 63-linked ubiquitination of BECN1/Beclin 1.19 SPHK1 was found to stimulate autophagy through TRAF2 (Fig. 5), and TRAF2 is known to induce the lysine 63-linked ubiquitination of substrates.18 Hence, we hypothesized that SPHK1 stimulates autophagy through the TRAF2-induced lysine 63-linked ubiquitination of BECN1. SPHK1 overexpression increased ubiquitination (Fig. 6A), especially lysine 63-linked ubiquitination (Fig. 6B), and did not affect the lysine 48-linked ubiquitination of BECN1 (Fig. 6C). In addition, silencing TRAF2 decreased the lysine 63-linked ubiquitination of BECN1 induced by SPHK1 (Fig. 6D). These data suggest that SPHK1 promotes the lysine 63-linked ubiquitination of BECN1 through TRAF2.
Figure 6.

SPHK1 induces the lysine 63-linked ubiquitination of BECN1 through TRAF2. (A-C) SPHK1 induced the lysine 63-linked ubiquitination of BECN1 and did not affect the lysine 48-linked ubiquitination of BECN1. HepG2 cells stably expressing vector or MYC-SPHK1 were transfected with a plasmid encoding HA-BECN1. The cell lysates were extracted and immunoprecipitated using an anti-HA antibody. The precipitates were then examined with anti-ubiquitin (A), anti-K63-specific ubiquitin (B) or anti-K48-specific ubiquitin (C). (D) TRAF2 was imperative for SPHK1 to induce the lysine 63-linked ubiquitination of BECN1. HepG2 cells were cotransfected with a plasmid encoding HA-BECN1 and control-siRNA (or si-TRAF2); then, the cell lysates were extracted and immunoprecipitated using an anti-HA antibody. The precipitates were then examined with anti-K63-specific ubiquitin antibody. WT, wild type; Ub, ubiquitin; Con-siRNA, control-siRNA.
SPHK1 stimulates lysine 63-linked BECN1 ubiquitination and triggers autophagy by binding to TRAF2
Ubiquitin-protein ligases, e.g., TRAF6 and AMBRA1, bind to BECN1 and then catalyze lysine 63-linked ubiquitination of BECN1 to trigger autophagy.19,20 In addition, TRAF2 induces the lysine 63-linked ubiquitination of substrates by binding to substrates, such as RIPK1/RIP1 (receptor interacting serine/threonine kinase 1) and TICAM1 (toll like receptor adaptor molecule 1).18,21 Hence, we speculated that TRAF2 may interact with BECN1 to catalyze its lysine 63-linked ubiquitination. Using coimmunoprecipitation and glutathione S-transferase (GST) affinity isolation assays, we found that TRAF2 could bind to BECN1 in vivo and in vitro (Fig. 7A and Fig. S3). The interaction of SPHK1 and TRAF2 is essential for lysine 63-linked poly-ubiquitination of RIPK1.18 We found that SPHK1 bound to both TRAF2 and BECN1 (Fig. 7B). Furthermore, the silencing of TRAF2 decreased not only the interaction of SPHK1 and BECN1 (Fig. 7C) and but also the lysine 63-linked ubiquitination of BECN1 in SPHK1-overexpressing cells (Fig. 6D). These results suggested that SPHK1 bound to BECN1 through TRAF2 and that this interaction between SPHK1 and TRAF2 was essential for the TRAF2-inducing lysine 63-linked poly-ubiquitination of BECN1. In addition, SPHK1 generates intracellular S1P (Fig. S1G), which binds to the RING domain of TRAF2 and this binding is also necessary for TRAF2-induced lysine 63-linked poly-ubiquitination.18
Figure 7.

SPHK1 stimulates the lysine 63-linked ubiquitination of BECN1 to trigger autophagy by binding to TRAF2. (A) BECN1 interacted with TRAF2. Whole cell lysates were extracted from HepG2 cells and then immunoprecipitated with anti-BECN1 antibody or an equal amount of mouse IgG and blotted with anti-TRAF2 antibody. (B) SPHK1 bound to TRAF2 and BECN1. Whole cell lysates were extracted from HepG2 cells stably expressing MYC-SPHK1 and then immunoprecipitated with anti-MYC (SPHK1) antibody or an equal amount of mouse IgG and blotted with anti-BECN1 or anti-TRAF2 antibody. (C) Silencing TRAF2 inhibited the binding of SPHK1 and BECN1. HepG2 cells stably expressing MYC-SPHK1 were cotransfected with a plasmid encoding HA-BECN1 and control-siRNA (or si-TRAF2). Whole cell extracts were immunoprecipitated with anti-MYC (SPHK1) antibody and blotted with an anti-HA (BECN1) antibody. (D) Deletion of the RING domain of TRAF2 reduced the lysine 63-linked ubiquitination of BECN1 and blocked the interaction of BECN1 with TRAF2 in SPHK1-overexpressing cells. HepG2 cells were cotransfected with a plasmid encoding HA-BECN1 and the indicated constructs of Flag-TRAF2. The cell lysates were extracted and immunoprecipitated using an anti-HA antibody. The precipitates were then analyzed with anti-K63-specific ubiquitin or anti-Flag antibody. (E) The deletion of the RING domain of TRAF2 did not induce autophagy. HepG2 cells were transfected with the indicated constructs of Flag-TRAF2 and the cell lysates were extracted. Autophagy-related proteins were detected by western blot analysis. Data are presented as the mean ± SE of 4 independent assays. Con-siRNA, control-siRNA; FL, full-length.
We further investigated whether S1P was necessary for the TRAF2-induced lysine 63-linked poly-ubiquitination of BECN1. We found that a treatment with S1P induced the lysine 63-linked poly-ubiquitination of BECN1 in cells stably expressing vector (Fig. S4). However, the deletion of the RING domain decreased the lysine 63-linked poly-ubiquitination of BECN1 and inhibited the interaction of BECN1 and TRAF2 in SPHK1-overexpressing cells (Fig. 7D), and then reduced the levels of autophagy-related proteins (Fig. 7E). These data suggested that S1P, which is generated by SPHK1, bound to the RING domain of TRAF2 and that this binding was essential for TRAF2 to induce the lysine 63-linked poly-ubiquitination of BECN1 as well as subsequent autophagy activation in HepG2 cells. Together, our data indicated that SPHK1 induced the lysine 63-linked poly-ubiquitination of BECN1 by interacting with TRAF2 and stimulating autophagy in HepG2 cells.
SPHK1 induces the EMT by stimulating BECN1 in HepG2 cells
We next investigated whether SPHK1 promoted the EMT through regulating BECN1-mediated autophagy activation. Overexpressing SPHK1 promoted the acceleration of migration and invasion (Fig. 1C and D), whereas silencing BECN1 interrupted the acceleration of migration and invasion in SPHK1-overexpressing cells (Fig. 8A and B). Overexpressing SPHK1 decreased the expression of CDH1 and increased the expression of FN1, VIM and CDH2 (Fig. 1E and F), whereas silencing BECN1 induced the expression of CDH1 and reduced the expression of FN1, VIM and CDH2 in SPHK1-overexpressing cells (Fig. 8C). These data suggested that SPHK1 induced the EMT and promoted cell migration and invasion through stimulating BECN1. Moreover, silencing BECN1 delayed the degradation of CDH1 in SPHK1-overexpressing cells (Fig. 8D). Together, our data indicated that SPHK1 activated BECN1-mediated autophagy through TRAF2 and then promoted the lysosomal degradation of CDH1 to induce the EMT in HepG2 cells (Fig. 8E).
Figure 8.
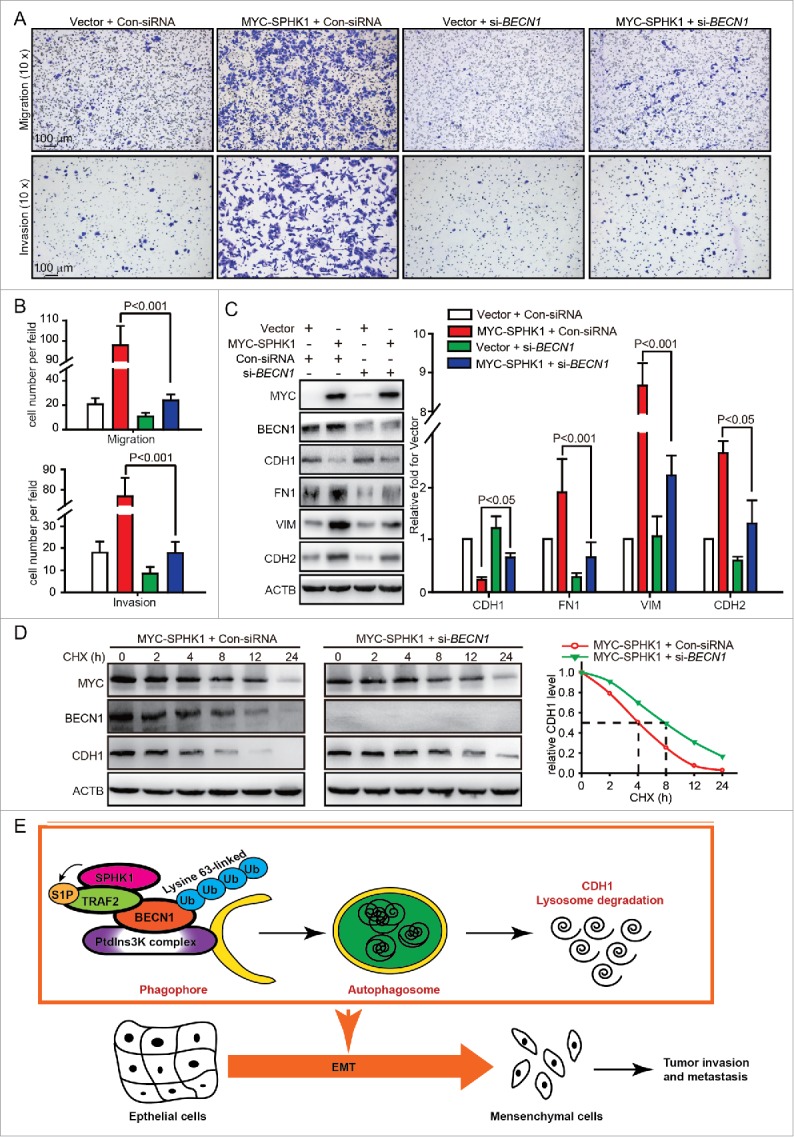
SPHK1 induces the EMT by stimulating BECN1 in HepG2 cells. (A, B) Silencing BECN1 blocked cell migration and invasion in SPHK1-overexpressing cells. HepG2 cells stably expressing vector or MYC-SPHK1 were transfected with control-siRNA or si-BECN1 and plated in the upper chamber of the filters for 24 h. Then cells migrating to the underside of the transwell insert were counted. (C) SPHK1 overexpression regulated the expression of EMT-related markers through stimulating BECN1. HepG2 cells stably expressing vector or MYC-SPHK1 were transfected with control-siRNA or si-BECN1 for 24 h and then harvested for cell lysate extraction. The level of epithelial or mesenchymal markers was detected by western blot analysis. (D) Silencing BECN1 blocked the degradation of CDH1 in SPHK1-overexpressing cells. HepG2 cells stably expressing MYC-SPHK1 were cotransfected with pCMV6-CDH1 and si-BECN1 (or control-siRNA) for 24 h and then treated with CHX (20 μmol/L) for the indicated times. The cell lysates were analyzed by western blotting using an anti-CDH1 antibody. (E) Schematic diagram of the mechanism of SPHK1-mediated autophagy and lysosomal CDH1 degradation that stimulates the EMT in HepG2 cells. Con-siRNA, control-siRNA.
Discussion
SPHK1 promotes tumor metastasis and invasiveness by multiple pathways. For example, SPHK1 promotes the metastasis of esophageal cancer by activating the EGFR (epidermal growth factor receptor) pathway22 and confers malignant phenotypes in colon cancer by regulating the PTK2/FAK (protein tyrosine kinase 2) pathway.23 EGFR activation leads to the EMT,24 and PTK2 is necessary for the process of TGFB/TGFβ (transforming growth factor β)-induced EMT.25 In this study, we found that SPHK1 promoted the EMT through stimulating TRAF2-mediated lysine 63-linked ubiquitination of BECN1 directly, the intracellular S1P-stimulated pathway, and then induced the lysosomal degradation of CDH1 in HepG2 cells. Bao et al. reported that SPHK1 promotes the migration and invasion of SMMC-7721 cells, and the S1PR1 (sphingosine-1-phosphate receptor 1) inhibitor VPC23019 effectively blocks it.7 However, the inhibition of S1PR1 by VPC23019 did not affect SPHK1 induction of the lysine 63-linked ubiquitination of BECN1 to stimulate autophagy and promotion of the EMT in HepG2 cells (Fig. S5), which seems to be contradictory with the above report. Probably, the expression of S1PR1 in HepG2 cells is much lower than in other HCC cells26 and also lower than SMMC-7721 cells (Fig. S5D). Hence, it is possible that the extracellular S1P-S1PR1 signal pathway does not affect the migration and invasion of HepG2 cells, whereas the extracellular S1P-S1PR1 signal pathway promotes the migration and invasion of SMMC-7721 cells. Moreover, the extracellular S1P stimulates the E3 ligase activity of TRAF2 by increasing the intracellular level of S1P in a S1PR1-independent manner.18 Hence, it is possible that the exogenous S1P induces the poly-ubiquitination of BECN1 by increasing the intracellular level of S1P (Fig. S4), which enhances the action of intracellular S1P.
Autophagy plays a dual role in the regulation of the EMT. The induction of autophagy leads to the loss of the metastatic phenotype by promoting the autophagy-mediated degradation of SNAI1/Snail and TWIST, 2 major EMT inducers, in breast cancer cells.9 The induction of autophagy suppresses the EMT and then impairs migration and invasion in glioblastoma cells.27 Autophagy would appear to be a negative regulator of the EMT process. However, autophagy promotes HCC invasion by activating the EMT.17 Additionally, our current observations showed that the attenuation of autophagy impairs the EMT and reduces the migration and invasion of HepG2 cells. Hence, we speculate that the function of autophagy in the regulation of the EMT is tissue-type dependent. Sun et al. found that BCL2/Bcl−2, an inhibitor of BECN1-dependent autophagy, promotes the EMT by binding to TWIST and leading to the transcriptional activation of TWIST in HCC;28 however, no changes in autophagy were detected in the process. As the dissociation of BCL2 and BECN1 is a trigger for autophagy,29 it is possible that BCL2 dissociates from its complex with BECN1 and then binds to TWIST.
The loss of CDH1 expression is the foundation of the EMT10 and we found that SPHK1 promotes the degradation of CDH1 by stimulating autophagy (Fig. 8D). Janda et al. reported that CDH1 is degraded by the endocytic lysosomal pathway,14 which is the same pathway by which the extracellular and plasma membrane proteins are degraded.30 However, signaling related to the autophagic inhibitor MTOR controls the exocyst complex to regulate the endocytosis pathway, and the exocyst complex also coordinates the PtdIns3K complex to initiate autophagy,31 which suggests mutual effects on autophagy and endocytosis. Moreover, it has been recently reported that the attenuation of autophagy halts the degradation of collagen, while the stimulation of autophagy by rapamycin accelerates the degradation of collagen,32 suggesting that autophagy participates in the regulation of the endocytic lysosomal pathway. Our finding showed that SPHK1 decreased the expression of CDH1 and did not change the RNA level of CDH1 in HCC cells (Fig. 2, Fig. S6A and Fig. S6C), and that the inhibition of autophagy by 3-MA or CQ recovered the expression of CDH1 in SPHK1-overexpressing cells (Fig. 4A, Fig. S6B and Fig. S6D) and the attenuation of autophagy by BECN1 silencing delayed the lysosomal degradation of CDH1 (Fig. 8D). These data suggested that SPHK1 induces the EMT by promoting the autophagy-linked lysosomal degradation of CDH1, at least in HCC cells.
SPHK1 stimulates autophagy in breast cancer cells,15 but the detailed regulatory mechanism is unclear. Our study indicated that SPHK1 activated autophagy through the TRAF2-mediated lysine 63-linked ubiquitination of BECN1 (Fig. 6). TRAF2, an established downstream component of the SPHK1 intracellular signaling pathway, regulates a variety of cellular activities facilitating the lysine 63-linked ubiquitination of such signaling proteins as RIPK1 and TICAM1.18,21 TRAF2 binds to RIPK1 and catalyzes the lysine-63-linked ubiquitination of RIPK1 for NFKB/NF-κB activation.18 Similarly, TICAM1 must be bound by TRAF2 to trigger the lysine-63-linked ubiquitination of TICAM1 for the activation of the TLR3 (toll-like receptor 3)-TLR4 pathway.21 Our study indicated that TRAF2 also bound to BECN1 and catalyzed the lysine 63-linked ubiquitination of BECN1 to activate autophagy and degrade CDH1. Additionally, previous work has indicated that the E3 ubiquitin ligase TRAF619 and AMBRA120 induce the lysine 63-linked ubiquitination of BECN1 for autophagy activation. Similarly, we found that the lysine 63-linked ubiquitination of BECN1 catalyzed by TRAF2 was essential for SPHK1-induced autophagy. However, silencing TRAF6 or AMBRA1 did not affect the activity of autophagy in SPHK1-overexpressing cells (Fig. S7A), which suggested that the stimulation of autophagy in HepG2 cells by SPHK1 relied on TRAF2, not TRAF6 or AMBRA1, and TRAF2 is a new component of the previously described pathways converging on BECN1. Thus, our study details a signaling pathway responsible for the regulation of the EMT in HCC cells via SPHK1-TRAF2-BECN1-CDH1 signaling cascades.
This study suggests that SPHK1 promotes the EMT by stimulating autophagy and then accelerating the lysosomal degradation of the epithelial marker CDH1 in HepG2 cells. SPHK1 catalyzes the lysine 63-linked ubiquitination of BECN1 by TRAF2 for autophagy activation. Our findings define a novel signaling pathway and the molecular mechanism by which SPHK1 regulates invasiveness and metastasis in HCC cells. Our work indicates that blockage of SPHK1 activity to attenuate autophagy may be a promising strategy for the prevention and treatment of HCC.
Materials and methods
Reagents and antibodies
CHX (01810), MG132 (C2211), anti-MAP1LC3 (L8918) and anti-SQSTM1 (P0067) antibodies were obtained from Sigma. Anti-human SPHK1 (ab71700) antibody was purchased from Abcam. Anti-CDH1 antibody (610181) was purchased from BD Biosciences. Anti-human VIM (2870), CDH2 (5741), TRAF2 (4724), BECN1 (3495), HA (3724), MYC (2278), Flag (2908) and ACTB/β-actin (4970) antibodies were obtained from Cell Signaling Technology. TRAF2 siRNA (sc-29509), BECN1 siRNA (sc-29797) and control siRNA (sc-37007) were purchased from Santa Cruz Biotechnology. Other materials were purchased from commercial sources.
Tissue microarray slides and immunohistochemistry
In vivo SPHK1 or CDH1 expression was detected by immunohistochemistry (IHC) using tissue microarrays (LV20810, US Biomax), which contained 169 HCC tissue specimens. IHC was performed according to standard protocols9 and then evaluated semiquantitatively as described previously.11
Plasmid construction and cell culture
MYC-tagged SPHK1 was created in the pCMV6-AN-MYC (PS100012, Origene) vector by standard subcloning. CDH1 was constructed in the pCMV6 vector (PS100001, Origene) by standard subcloning. HA-tagged BECN1 was constructed in the pCMV6-AC-HA vector (PS100004, Origene) by standard subcloning. Flag-tagged TRAF2 and its mutant (ΔRING) were cloned into the pFLAG-CMV-2 expression plasmid (E7033, Sigma).
Human liver carcinoma HepG2 cells were purchased from American Type Culture Collection (ATCC, HB-8065) and maintained according to their instructions. Transient transfection was performed using Lipofectamine 2000 (Invitrogen, 11668019) or Lipofectamine RNAiMax (Invitrogen, 13778150) according to the manufacturer's instructions. To generate HepG2 cells stably expressing SPHK1, the pCMV6-AN-MYC and the MYC-tagged SPHK1 plasmids were both transfected into HepG2 cells. After 24 h of transfection, stable transfectants were selected using neomycin, and a single clone was amplified using a dilution-cloning technique.
Cell migration and invasion assays
As described previously,8 migration was measured according to the ability of cells to migrate across a transwell filter (8-μm pores, Merck Millipore, PIEP12R48), invasion capability was measured according to the ability of the cells to migrate across a transwell filter coated with Matrigel (Corning, 354480). Cells suspended in serum-free Eagle's minimum essential medium (ATCC, 30–2003) were added to the upper chamber, and Eagle's minimum essential medium containing 20% fetal bovine serum was added to the lower chamber. After the cells were incubated at 37°C for 24 h, the cells that had migrated to the lower side of the upper chamber were stained with crystal violet, and the cells per microscopy field (× 10) were counted under a microscope.
Immunoprecipitation and western blot analysis
Cells were washed 3 times with phosphate-buffered saline (Invitrogen, 10010023) and then harvested. The cell lysates were extracted in co-immunoprecipitation buffer.16 As described previously,16,33 total cell lysates (5 mg of protein) were subjected to immunoprecipitation with the indicated antibodies overnight at 4°C and then incubated with protein A/G Plus-agarose (Santa Cruz Biotechnology, sc-2003) for 24 h at 4°C. After being washed 3 times, the immunocomplexes were mixed with 2×SDS loading buffer and boiled for 5 min. For the western blot analysis, co-precipitates or total cell lysates were electrophoresed by SDS-PAGE and transferred to PVDF membranes (Millipore, ISEQ00010). The membranes were immunoblotted with the appropriate antibodies and then developed using a ChemiImager 5500 imaging system (α Innotech Co., USA).
Real-time PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, 15596026). Total RNA (100 ng) was detected by using a SYBR Green One-Step qRT-PCR Kit (Invitrogen, 11736059) as described in the manual. The following primers were used: ACTB, 5′-CAA CTG GGA CGA CAT GGA GAA AAT-3′ and 5′-CCA GAG GCG TAC AGG GAT AGC AC-3′; CDH1, 5′- TTC CCA ACT CCT CTC CTG −3′ and 5′- AAA CCT TGC CTT CTT TGT C-3′.
Electron microscopy
As described previously,16 HepG2 cells were promptly fixed with 2.5% glutaraldehyde containing 0.1 mol/L sodium cacodylate and stored at 4°C. The samples were then embedded, and ultrathin (50–60 nm) sections were cut using an ultramicrotome (LEICA EM UC7). The samples were examined with a JEM-1400 electron microscope at 80 kV, and the images were captured with a Veleta TEM camera. The number of autophagosomes was quantified as described previously.16
Confocal assay
Standard protocols for immunofluorescence microscopy were used as described previously.16 The cells were plated on coverglass-bottom dishes (NEST, 801008), washed twice with phosphate-buffered saline, fixed in 4% paraformaldehyde for 20 min, and then washed 3 times. The cells on the dishes (0.5-μm thick) were prepared and stained with the primary antibodies overnight at 4°C. The sections were washed twice with phosphate-buffered saline, incubated with fluorochrome-labeled secondary antibodies (1: 200, Invitrogen, A-21206 and A-21202) for 30 min, and then washed 3 times. Images were obtained using a Leica SP2 confocal microscope (Leica Microsystems, Exton, PA) and analyzed with Leica confocal software.
To quantify the number of MAP1LC3 foci, a total of 100 cells were recorded with the Leica SP2 confocal microscope and analyzed using ImageJ analysis software. MAP1LC3 puncta with a diameter between 0.3 and 1 μm were scored as positive.16
Statistics
Data are represented as the mean ± SE. The statistical analyses were performed using the Student t test to compare 2 groups. The P values less than 0.05 were considered significant. All statistical analyses were performed using SPSS 17.0 software.
Supplementary Material
Abbreviations
- CHX
cycloheximide
- Con-siRNA
control-siRNA
- CQ
chloroquine
- EGFR
epidermal growth factor receptor
- EMT
epithelial-mesenchymal transition
- FL
full-length
- GST
glutathione S-transferase
- HCC
hepatocellular carcinoma
- IHC
immunohistochemistry
- MTOR
mechanistic target of rapamycin
- N
nucleus
- NS
nonsignificant
- PtdIns3K
class III phosphatidylinositol 3-kinase
- PTK2/FAK
protein tyrosine kinase 2
- RIPK1/RIP1
receptor interacting serine/threonine kinase 1
- SPHK1
sphingosine kinase 1
- S1P
sphingosine-1-phosphate
- S1PR1
sphingosine-1-phosphate receptor 1
- TGFB/TGFβ
transforming growth factor β
- TICAM1
toll like receptor adaptor molecule 1
- TRAF2
TNF receptor-associated factor 2
- Ub
ubiquitin
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Nature Scientific Foundation (31401186, 81473249, 81321004 and 81673471), National Mega-project for Innovative Drugs (2014ZX09201042) and the CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-2–002).
References
- [1].Lin H, Yan J, Wang Z, Hua F, Yu J, Sun W, Li K, Liu H, Yang H, Lv Q, et al.. Loss of immunity-supported senescence enhances susceptibility to hepatocellular carcinogenesis and progression in Toll-like receptor 2-deficient mice. Hepatology 2013; 57:171-82; PMID:22859216; http://dx.doi.org/ 10.1002/hep.25991 [DOI] [PubMed] [Google Scholar]
- [2].Zhang C, He H, Zhang H, Yu D, Zhao W, Chen Y, Shao R. The blockage of Ras/ERK pathway augments the sensitivity of SphK1 inhibitor SKI II in human hepatoma HepG2 cells. Biochem Biophys Res Commun 2013; 434:35-41; PMID:23545258; http://dx.doi.org/ 10.1016/j.bbrc.2013.03.070 [DOI] [PubMed] [Google Scholar]
- [3].Fuchs BC, Hoshida Y, Fujii T, Wei L, Yamada S, Lauwers GY, McGinn CM, DePeralta DK, Chen X, Kuroda T, et al.. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 2014; 59:1577-90; PMID:24677197; http://dx.doi.org/ 10.1002/hep.26898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Huang YL, Huang WP, Lee H. Roles of sphingosine 1-phosphate on tumorigenesis. World J Biol Chem 2011; 2:25-34; PMID:21537487; http://dx.doi.org/ 10.4331/wjbc.v2.i2.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Liu H, Zhang CX, Ma Y, He HW, Wang JP, Shao RG. SphK1 inhibitor SKI II inhibits the proliferation of human hepatoma HepG2 cells via the Wnt5A/beta-catenin signaling pathway. Life Sci 2016; 151:23-29; PMID:26944438; http://dx.doi.org/ 10.1016/j.lfs.2016.02.098 [DOI] [PubMed] [Google Scholar]
- [6].Zhang Z, Yan Z, Yuan Z, Sun Y, He H, Mai C. SPHK1 inhibitor suppresses cell proliferation and invasion associated with the inhibition of NF-kappaB pathway in hepatocellular carcinoma. Tumour Biol 2015; 36:1503-39; PMID:25537088; http://dx.doi.org/ 10.1007/s13277-014-2665-7 [DOI] [PubMed] [Google Scholar]
- [7].Bao M, Chen Z, Xu Y, Zhao Y, Zha R, Huang S, Liu L, Chen T, Li J, Tu H, et al.. Sphingosine kinase 1 promotes tumour cell migration and invasion via the S1P/EDG1 axis in hepatocellular carcinoma. Liver Int 2012; 32:331-38; PMID:22098666; http://dx.doi.org/ 10.1111/j.1478-3231.2011.02666.x [DOI] [PubMed] [Google Scholar]
- [8].Zhang H, Ma Y, Zhang S, Liu H, He H, Li N, Gong Y, Zhao S, Jiang JD, Shao RG. Involvement of Ras GTPase-activating protein SH3 domain-binding protein 1 in the epithelial-to-mesenchymal transition-induced metastasis of breast cancer cells via the Smad signaling pathway. Oncotarget 2015; 6:17039-53; PMID:25962958; http://dx.doi.org/ 10.18632/oncotarget.3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H, Yan J, Lv X, Chen X, Hu ZW. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res 2012; 72:3238-50; PMID:22719072; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3832 [DOI] [PubMed] [Google Scholar]
- [10].Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 2008; 68:3645-54; PMID:18483246; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-2938 [DOI] [PubMed] [Google Scholar]
- [11].Zhai B, Yan HX, Liu SQ, Chen L, Wu MC, Wang HY. Reduced expression of E-cadherin/catenin complex in hepatocellular carcinomas. World J Gastroenterol 2008; 14:5665-73; PMID:18837082; http://dx.doi.org/ 10.3748/wjg.14.5665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fransvea E, Angelotti U, Antonaci S, Giannelli G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology 2008; 47:1557-66; PMID:18318443; http://dx.doi.org/ 10.1002/hep.22201 [DOI] [PubMed] [Google Scholar]
- [13].Hsieh S, L PY, Hsieh CW, Li IT, Hsieh SL, Wu CC, Y SH, Ys Huang, Huay MW, Tu LW, et al.. Probing the Adhesion of Hepatocellular Carcinoma HepG2 and SK-Hep-1 Cells. J Chin Chem Soc 2012; 59:929-33; http://dx.doi.org/ 10.1002/jccs.201200129 [DOI] [Google Scholar]
- [14].Janda E, Nevolo M, Lehmann K, Downward J, Beug H, Grieco M. Raf plus TGFbeta-dependent EMT is initiated by endocytosis and lysosomal degradation of E-cadherin. Oncogene 2006; 25:7117-30; PMID:16751808; http://dx.doi.org/ 10.1038/sj.onc.1209701 [DOI] [PubMed] [Google Scholar]
- [15].Lavieu G, Scarlatti F, Sala G, Carpentier S, Levade T, Ghidoni R, Botti J, Codogno P. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem 2006; 281:8518-27; PMID:16415355; http://dx.doi.org/ 10.1074/jbc.M506182200 [DOI] [PubMed] [Google Scholar]
- [16].Liu H, Ma Y, He HW, Wang JP, Jiang JD, Shao RG. SLC9A3R1 stimulates autophagy via BECN1 stabilization in breast cancer cells. Autophagy 2015; 11:2323-34; PMID:26218645; http://dx.doi.org/ 10.1080/15548627.2015.1074372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y, Song Z, Zheng Q, Xiong J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013; 34:1343-51; PMID:23430956; http://dx.doi.org/ 10.1093/carcin/bgt063 [DOI] [PubMed] [Google Scholar]
- [18].Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, et al.. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010; 465:1084-88; PMID:20577214; http://dx.doi.org/ 10.1038/nature09128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal 2010; 3:ra42; PMID:20501938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xia P, Wang S, Du Y, Zhao Z, Shi L, Sun L, Huang G, Ye B, Li C, Dai Z, et al.. WASH inhibits autophagy through suppression of Beclin 1 ubiquitination. EMBO J 2013; 32:2685-96; PMID:23974797; http://dx.doi.org/ 10.1038/emboj.2013.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sasai M, Tatematsu M, Oshiumi H, Funami K, Matsumoto M, Hatakeyama S, Seya T. Direct binding of TRAF2 and TRAF6 to TICAM-1/TRIF adaptor participates in activation of the Toll-like receptor 3/4 pathway. Mol Immuno 2010; 47:1283-91; http://dx.doi.org/ 10.1016/j.molimm.2009.12.002 [DOI] [PubMed] [Google Scholar]
- [22].Pan J, Tao YF, Zhou Z, Cao BR, Wu SY, Zhang YL, Hu SY, Zhao WL, Wang J, Lou GL, et al.. An novel role of sphingosine kinase-1 (SPHK1) in the invasion and metastasis of esophageal carcinoma. J Transl Med 2011; 9:157; PMID:21936950; http://dx.doi.org/ 10.1186/1479-5876-9-157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu SQ, Su YJ, Qin MB, Mao YB, Huang JA, Tang GD. Sphingosine kinase 1 promotes tumor progression and confers malignancy phenotypes of colon cancer by regulating the focal adhesion kinase pathway and adhesion molecules. Int J Oncol 2013; 42:617-26; PMID:23232649 [DOI] [PubMed] [Google Scholar]
- [24].Holz C, Niehr F, Boyko M, Hristozova T, Distel L, Budach V, Tinhofer I. Epithelial-mesenchymal-transition induced by EGFR activation interferes with cell migration and response to irradiation and cetuximab in head and neck cancer cells. Radiother Oncol 2011; 101:158-64; PMID:21665310; http://dx.doi.org/ 10.1016/j.radonc.2011.05.042 [DOI] [PubMed] [Google Scholar]
- [25].Cicchini C, Laudadio I, Citarella F, Corazzari M, Steindler C, Conigliaro A, Fantoni A, Amicone L, Tripodi M. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res 2008; 314:143-52; PMID:17949712; http://dx.doi.org/ 10.1016/j.yexcr.2007.09.005 [DOI] [PubMed] [Google Scholar]
- [26].Heo MJ, Kim YM, Koo JH, Yang YM, An J, Lee SK, Lee SJ, Kim KM, Park JW, Kim SG. microRNA-148a dysregulation discriminates poor prognosis of hepatocellular carcinoma in association with USP4 overexpression. Oncotarget 2014; 5:2792-806; PMID:24798342; http://dx.doi.org/ 10.18632/oncotarget.1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Catalano M, D'Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, Faienza F, Esposito V, Limatola C, Cecconi F, Di Bartolomeo S. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol 2015; 9:1612-25; PMID:26022108; http://dx.doi.org/ 10.1016/j.molonc.2015.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sun T, Sun BC, Zhao XL, Zhao N, Dong XY, Che N, Yao Z, Ma YM, Gu Q, Zong WK, et al.. Promotion of tumor cell metastasis and vasculogenic mimicry by way of transcription coactivation by Bcl-2 and Twist1: a study of hepatocellular carcinoma. Hepatology 2011; 54:1690-706; PMID:21748764; http://dx.doi.org/ 10.1002/hep.24543 [DOI] [PubMed] [Google Scholar]
- [29].Liu H, Mi S, Li Z, Hua F, Hu ZW. Interleukin 17A inhibits autophagy through activation of PIK3CA to interrupt the GSK3B-mediated degradation of BCL2 in lung epithelial cells. Autophagy 2013; 9:730-42; PMID:23514933; http://dx.doi.org/ 10.4161/auto.24039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861-73; PMID:18006683; http://dx.doi.org/ 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- [31].Lamb CA, Dooley HC, Tooze SA. Endocytosis and autophagy: Shared machinery for degradation. BioEssays 2013; 35:34-45; PMID:23147242; http://dx.doi.org/ 10.1002/bies.201200130 [DOI] [PubMed] [Google Scholar]
- [32].Mi S, Li Z, Yang HZ, Liu H, Wang JP, Ma YG, Wang XX, Liu HZ, Sun W, Hu ZW. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Immunol 2011; 187:3003-14; PMID:21841134; http://dx.doi.org/ 10.4049/jimmunol.1004081 [DOI] [PubMed] [Google Scholar]
- [33].Liu H, Mi S, Li Z, Lv XX, Li K, Hua F, Hu ZW. SB216763, a selective small molecule inhibitor of glycogen synthase kinase-3, improves bleomycin-induced pulmonary fibrosis via activating autophagy. Acta Pharmaceutica Sinica B 2013; 4:226-33; http://dx.doi.org/ 10.1016/j.apsb.2013.05.004 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.