

ABSTRACT
Autophagy plays critical and complex roles in many human diseases, including diabetes and its complications. However, the role of autophagy in the development of diabetic retinopathy remains uncertain. Core histone modifications have been reported involved in the development of diabetic retinopathy, but little is known about the histone variants. Here, we observed increased autophagy and histone HIST1H1C/H1.2, an important variant of the linker histone H1, in the retinas of type 1 diabetic rodents. Overexpression of histone HIST1H1C upregulates SIRT1 and HDAC1 to maintain the deacetylation status of H4K16, leads to upregulation of ATG proteins, then promotes autophagy in cultured retinal cell line. Histone HIST1H1C overexpression also promotes inflammation and cell toxicity in vitro. Knockdown of histone HIST1H1C reduces both the basal and stresses (including high glucose)-induced autophagy, and inhibits high glucose induced inflammation and cell toxicity. Importantly, AAV-mediated histone HIST1H1C overexpression in the retinas leads to increased autophagy, inflammation, glial activation and neuron loss, similar to the pathological changes identified in the early stage of diabetic retinopathy. Furthermore, knockdown of histone Hist1h1c by siRNA in the retinas of diabetic mice significantly attenuated the diabetes-induced autophagy, inflammation, glial activation and neuron loss. These results indicate that histone HIST1H1C may offer a novel therapeutic target for preventing diabetic retinopathy.
KEYWORDS: autophagy, cell toxicity, diabetic retinopathy, H4K16Ac, histone HIST1H1C, inflammation
Introduction
Diabetic retinopathy is the most common microvascular complication of diabetes, and the leading cause of blindness in working-aged people.1 Diabetes induces cell death in most kinds of retinal cells, including endothelial cells,2 pericytes,3 ganglion cells and Müller cells.4,5 In diabetic retinas, several forms of cell death have been described, including apoptosis,6 pyroptosis (inflammation-driven cell death),7 necrosis,8 and autophagic death.9 All of these cell death forms may contribute to the development of the early lesions associated with diabetic retinopathy, including acellular capillaries, neuronal dysfunction and glial activation.10,11
Autophagy is an intracellular catabolic pathway that degrades cell components, toxic aggregates, and damaged organelles, then recycles them as basic building blocks to maintain cellular homeostasis, especially when cells under certain stresses.12 Autophagy is regulated by a set of ATG proteins, such as BECN1/Beclin 1, ATG3, ATG5 and ATG7, which contribute to the initiation and elongation of autophagosomes.12,13 Recent studies on retinopathy, nephropathy and cardiomyopathy in diabetic rodents have suggested a link between abnormal autophagy and diabetic complications.,9,14,15
The retina is part of the central nervous system with highly expressed ATG proteins, such as LC3B/MAP1LC3B and ATG9,16 making it a good organ model to study the functions of autophagy. However, there is controversy over the role of autophagy in the retina. Increased autophagy promotes the survival of retinal ganglion cells after the axotomy of the optic nerve.17 In contrast, activated autophagy promotes the death of ganglion cells, eventually leading to retinal degeneration in a rat model of glaucoma.18 Although autophagy is generally regarded as cell protective under short durations of stress, the observation of increased autophagosomes in dying cells suggests it also can be regarded as a nonapoptotic form of cell death which leads to cell damage.19 Enhanced autophagy has been observed in the retinas of diabetic mice,9 however, whether autophagy functions as a prosurvival or prodeath signaling pathway in diabetic retina remains unclear.
Histone HIST1H1C/H1.2 is a variant of linker histone H1, which is expressed in almost all somatic cells.20 Previously, the functions of histone H1 and its variants were thought to be limited to bringing the entering and exiting linker DNA segments together and stabilizing the nucleosome.21 Recently, growing evidence reveals that histone H1 and its variants have some unknown functions.22,23 Among them, histone HIST1H1C has attracted attention because it plays important roles in multiple cell processes, including apoptosis,24 gene transcription and protein folding.25-27 Histone HIST1H1C has the highest expression level among all histone H1 variants in the mouse retina and hippocampus,28 implicating its critical role in the neuronal system. To date, most studies on histone HIST1H1C have been performed in cultured cells, the role of this important histone H1 variant in disease animal models, including diabetic retinopathy, is largely unknown.
In the present study, using in vivo and in vitro approaches, we found that histone HIST1H1C and ATG proteins were both upregulated in the retinas of diabetic rodents and in high-glucose cultured cells. We further investigated the role of histone HIST1H1C in regulating autophagy and its effects on diabetes-induced cell death, inflammation, and retinopathy lesions. Our results suggest histone HIST1H1C may be a novel therapeutic target for diabetic retinopathy.
Results
Hyperglycemia induces histone HIST1H1C elevation and autophagy in the retinas and cultured cells
By analyzing the gene expression profile on mouse retina,28 we found that histone Hist1h1c has the highest mRNA level among all somatic histone H1 variants (Fig. S1). To our knowledge, little has been reported about histone HIST1H1C in the development of diabetic retinopathy. Thus, we used 2 diabetic rodent models to investigate whether this histone variant is altered in the diabetic retinas. The body weights and nonfasted blood glucose (NFBG) levels of the experimental rodents used in the present study are provided in Table S1.
We found that the protein level of histone HIST1H1C in the retinas of streptozotocin (STZ)-induced diabetic rats was increased approximately 1.8-fold compared with those of nondiabetic rats (Fig. 1A). Immunohistochemical staining further demonstrated that the increased histone HIST1H1C was localized to the ganglion cell layer (GCL) and inner nuclear layer (INL) in the retinas of STZ-induced diabetic rats (Fig. 1B). We also found that BECN1, the ATG12–ATG5 complex, ATG3, and the LC3B-I to LC3B-II conversion were significantly increased in the retinas of STZ-induced diabetic rats (Fig. 1C and D). Moreover, histone HIST1H1C was also increased in the retinas of Ins2+/− mice (Fig. 1E), while the increased histone HIST1H1C was mainly localized to the GCL and INL (Fig. 1F). Furthermore, a significant increase in ATG5 and LC3B-I to CL3B-II conversion was also observed in the retinas of Ins2+/− mice (Fig. 1G-H). These observations suggested histone HIST1H1C and autophagy may play important roles in the development of diabetic retinopathy.
Figure 1.

Diabetes increases histone HIST1H1C and autophagy in the retinas. (A) Representative western blots (upper panel) with the respective quantitative densitometric result (lower panel) of histone HIST1H1C in the rat retinas. (B) Representative images of histone HIST1H1C staining on the rat retinal sections. (C-D) Representative western blots (C) with the respective quantitative densitometric results (D) of indicated autophagy-related proteins in the rat retinas. (E) Representative western blots (upper panel) with the respective quantitative densitometric result (lower panel) of histone HIST1H1C in the mouse retinas. (F) Representative images of histone HIST1H1C staining on the mouse retinal sections. (G-H) Representative western blots (G) with quantitative densitometric results (H) of indicated autophagy-related proteins in the mouse retinas. n = 4–6 in each group; N, nondiabetes: D, diabetes; *p < 0.05 compared with nondiabetic or Ins2+/+ mice; brown, positively-stained cells; purple, hemotoxylin-stained nuclei; scale bar: 50 μm.
To confirm that hyperglycemia per se induces alterations in histone HIST1H1C and autophagy, a retinal Müller cell line (rMC-1) and a transformed human embryonic kidney cell line (293T) were treated with high glucose. Significantly increased histone HIST1H1C levels were observed in both rMC-1 and 293T cells after high glucose treatment (Fig. 2A-D). Furthermore, high glucose increased the levels of the ATG12–ATG5 complex, ATG5, ATG3 and LC3B-I to LC3B-II conversion in rMC-1 cells (Fig. 2A-B), as well as the levels of BECN1, ATG12–ATG5 complex, ATG3 and LC3B-I to LC3B-II conversion in 293T cells (Fig. 2C and D). To eliminate the possibility that these changes were due to high-glucose-induced osmotic effects, mannitol was used as an osmotic control. Increased LC3B-I to LC3B-II conversion and elevated HIST1H1C level were only found in the high glucose-treated, but not in the mannitol-treated rMC-1 cells (Fig. S2A). Moreover, the mRNA levels of Hist1h1c, Map1lc3b, Atg5, Atg7 and the percentage of autophagic cells were increased only in the high glucose-treated, but not in the mannitol-treated rMC-1 cells (Fig. S2B and C). Thus, the observed elevated histone HIST1H1C and autophagy were caused by high-glucose stress, but not by osmotic stress.
Figure 2.

Hyperglycemia increases histone HIST1H1C and autophagy in cultured cells. (A-B) Representative western blots (A) with the respective quantitative densitometric result (B) of the indicated proteins in the rMC-1 cells. (C-D) Representative western blots (C) with the respective quantitative densitometric results (D) of the indicated proteins in the 293T cells. n = 6 each group; NG, normal glucose; HG, high glucose; *p< 0.05 compared with the normal glucose.
Overexpression of histone HIST1H1C induces autophagy, inflammation and cell toxicity
To investigate the relationship between histone HIST1H1C and autophagy, we transfected a plasmid expressing human histone HIST1H1C into rMC-1 and 293T cells, respectively. Detection of the HA-tag and an exogenous histone HIST1H1C band indicated successful overexpression of histone HIST1H1C (Figs. 3A and S3A). In rMC-1 cells, overexpression of histone HIST1H1C induced upregulation of the ATG12–ATG5 complex, ATG7, ATG3 and LC3B-I to LC3B-II conversion (Fig. 3A and B). Furthermore, co-transfecting HIST1H1C with GFP-LC3 significantly increased the percentage of autophagic cells compared with the control group (from 8% to 21%; Fig. 3C and D). To investigate whether overexpression of histone HIST1H1C promotes autophagy flux, 2 autophagy inhibitors (chloroquine, CQ and bafilomycin A1, BafA1) were used. SQSTM1/p62 is a well-known substrate of autophagy,29 measurement of the cellular SQSTM1 level, together with the conversion of LC3B-I to LC3B-II, is regarded to be well correlated with autophagic flux.30 The level of SQSTM1 was dramatically reduced after overexpression of histone HIST1H1C in rMC-1 cells (Fig. 3E and F). Compared to the untreated histone HIST1H1C-overexpressing cells, CQ and BafA1 treatments significantly induced SQSTM1 accumulation and further enhanced LC3B-I to LC3B-II conversion (Fig. 3E and F), suggesting that overexpression of histone HIST1H1C promotes autophagy flux in cultured retinal cells. Similarly, overexpression of histone HIST1H1C in 239T cells also increased the levels of ATGs and cell autophagy (Fig. S3).
Figure 3.

Overexpression of histone HIST1H1C increases autophagy flux in rMC-1. (A-B) Representative western blots (A) with quantitative densitometric results (B) of the indicated proteins. (C-D) Representative images (C) with quantitative result (D) of autophagic cells. (E-F) Representative western blots (left panels) with quantitative densitometric results (right panels) of the indicated proteins following the CQ treatment (E) and the BafA1 treatment (F). n = 4–6 in each group; CQ, chloroquine; BafA1, bafilomycin A1; *p < 0.05 compared with pCI group; $ p < 0.05 compared with pH1.2 group; ex, exogenous, en, endogenous; scale bar: 10 μm.
Diabetes has been known to promote glial cell activation, a pathological markers of diabetic retinopathy.31 Inflammation and cell death are 2 key characteristics of diabetic retinopathy.31,32 Overexpression of histone HIST1H1C dramatically increased expression of GFAP (glial fibrillary acidic protein), a marker of glial cell activation,33 in rMC-1 cells (Fig. 4A and B). Further, overexpression of histone HIST1H1C induced transcription of inflammatory factors, such as Ccl2 and Il6, and significantly reduced cell viability in the rMC-1 cells (Fig. 4C and D). It has been reported that histone HIST1H1C acts as an apoptosis-mediator when released from the nucleus to the cytoplasm, and then transferred to mitochondria upon X-ray irradiation-induced DNA double-strand breaks.24 Thus, we investigated whether histone HIST1H1C translocation induced apoptosis contributes to the observed effects on cell viability by using cells overexpressing histone HIST1H1C or being treated with high glucose. Histone HIST1H1C was enriched in the nuclei of the rMC-1 cells under the conditions tested, as measured by immunofluorescence staining (Fig. S4A) and nuclear/cytoplasmic fractionation assay (Fig. S4B). Similar results were also observed in the 293T cells using nuclear/cytoplasmic fractionation assay (Fig. S4C). Together, these data suggested histone HIST1H1C may promote cell death in a different way from its previously reported function as an apoptosis-mediator in the conditions we investigated.24
Figure 4.
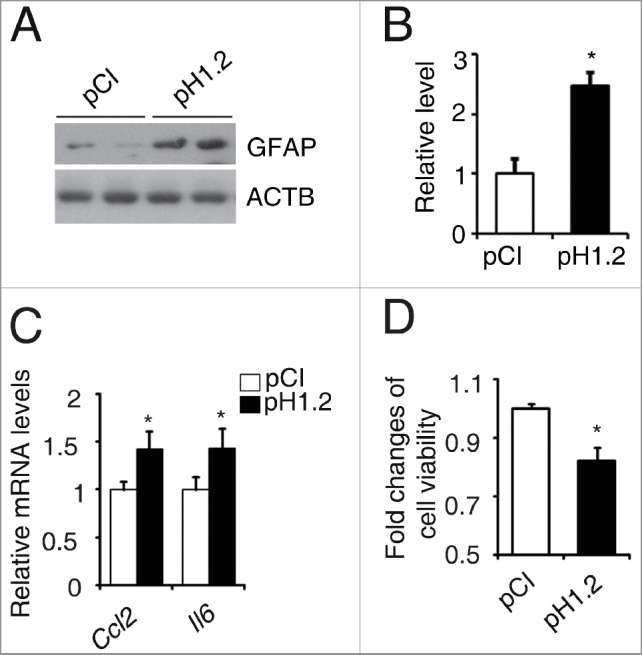
Overexpression of histone HIST1H1C promotes glial activation, inflammation and reduces cell viability in the rMC-1 cells. (A-B) Representative western blots (A) with the quantitative densitometric results (B) of GFAP. (C) qPCR results of Ccl2 and Il6. (D) Cell viability. n = 6 in each group; *p< 0.05 compared with the pCI group.
We also found that vitreous injection of rapamycin, an autophagy inducer, to the retina, induced increasing LC3B-I to LC3B-II conversion and decreased the protein levels of TUBB3/β-tubulin III and POU4F1/Brn3a, 2 retinal neuronal cell markers34 but no alteration on phosphorylated H2AFX/H2A.X (a marker of DNA damage35) (Fig. S5), indicating autophagy can promote retinal cell loss without causing DNA damage. To investigate whether the histone HIST1H1C overexpression induced autophagy contributes to cell death we observed, wild-type and atg7−/− MEF (a mouse embryonic fibroblast) cells were used. ATG7 is an E1 ubiquitin activating enzyme-like protein that promotes the formation of the ATG12–ATG5 complex, and plays an important role in autophagosome formation.29 Compared to the wild-type MEF cells, only very low levels of ATG12–ATG5 complex and LC3B-I to LC3B-II conversion were found in the atg7−/− MEF cells; consistently, very few autophagic cells were found in the rapamycin-treated or untreated atg7−/− MEF cells (Fig. S6A and B), which agrees with the previous reports that ATG7 is important for autophagosome formation.29 Importantly, Atg7 knockout significantly inhibited the histone HIST1H1C overexpression-induced autophagy (Fig. S6C and D); meanwhile, the reduced cell viability induced by histone HIST1H1C overexpression was partly normalized by the Atg7 knockout in the MEF cells (Fig. S6E).
Knockdown of histone HIST1H1C reduces both basal and stress-induced autophagy, inflammation and cell toxicity
To further investigate the role of histone HIST1H1C in regulating autophagy, shRNA was used to knock down the histone HIST1H1C level. Compared to the control (rMC-1Vec) cells, significant decreases in histone Hist1h1c mRNA and protein levels were observed in shHist1h1c rMC-1 cells (Fig. 5A and B). In histone HIST1H1C knockdown rMC-1 cells, significantly downregulated transcription of Atg genes (Becn1, Atg12, Atg7, Atg5, Atg3 and Map1lc3b), downregulated protein levels of ATGs, LC3B-I to LC3B-II conversion and decreased percentage of autophagic cells were observed (Fig. 5C-F).
Figure 5.

Knockdown of histone HIST1H1C reduces basal autophagy level in the rMC-1 cells. (A) qPCR results of histone HIST1H1C. (B) Representative western blots (left panel) with quantitative densitometric results (right panel) of histone HIST1H1C. (C) qPCR results of the indicated Atg genes. (D-E) Representative western blots (D) with quantitative densitometric results (E) of the indicated proteins. (F) Representative images (upper panel) with quantitative results (lower panel) of autophagic cells. n = 6 in each group; Vec, empty vector; *p < 0.05 compared with Vec; scale bar: 10 μm.
Next, we investigated whether knockdown of histone HIST1H1C could prevent cells from stress-induced autophagy. Histone HIST1H1C knockdown significantly reduced starvation- or rapamycin-induced increase of the percentage of autophagic cells in rMC-1 cells (Fig. 6A and B). In addition to these classic inducers of autophagy, knockdown of histone HIST1H1C also inhibited the high glucose-induced increase of the percentage of autophagic cells in rMC-1 cells (Fig. 6A and B).
Figure 6.
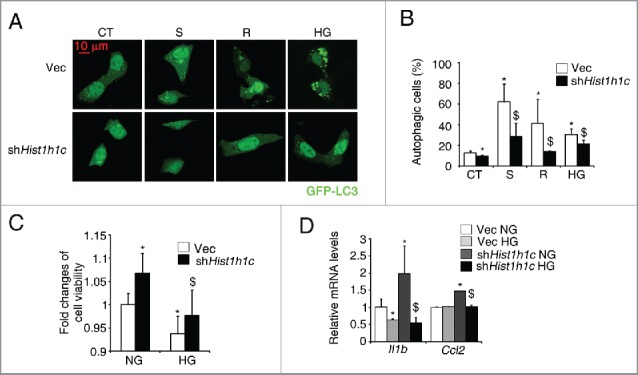
Knockdown of histone HIST1H1C suppresses stresses-induced autophagy levels in the rMC-1 cells. (A-B) Representative images (A) and quantitative results of autophagic cells (B) following indicated treatments. (C) Cell viability. (D) qPCR results of Il1b and Ccl2. n = 6 in each group. Vec, empty vector; CT, control; S, Starvation; R, rapamycin; HG, high glucose *p < 0.05 compared with Vec; $ p < 0.05 compared with Vec treated with indicated treatments; scale bar: 10 μm.
We further investigated whether histone HIST1H1C knockdown could rescue cells from high glucose-induced cell death and inflammation. In rMC-1 cells, histone HIST1H1C knockdown not only significantly reduced the high glucose-induced cell toxicity, but also improved cell survival rate in cells cultured under the normal glucose (Fig. 6C). Of great interest, knockdown of histone HIST1H1C reduced the high glucose-induced upregulation of proinflammatory factor, such as Il1b and Ccl2 in rMC-1 cells (Fig. 6D). Taken together, our results suggest knockdown of histone HIST1H1C, at least, partly inhibits the stresses induced autophagy and high glucose induced inflammation/cell toxicity in cultured retinal cells.
Overexpression of HIST1H1C reduces H4K16Ac level by upregulating HDACs
The MTOR (mechanistic target of rapamycin [serine/threonine kinase]) pathway is considered to be the primary regulatory pathway of autophagy.12 Usually, when the activity of MTOR is suppressed, as evidenced by reduced levels of phosphorylated MTOR and RPS6KB/p70S6K, a substrate of MTOR, autophagy initiates.29 However, the level of neither phosphorylated MTOR nor phosphorylated RPS6KB was changed in histone HIST1H1C-overexpressing rMC-1 cells (Fig. 7A and B).
Figure 7.
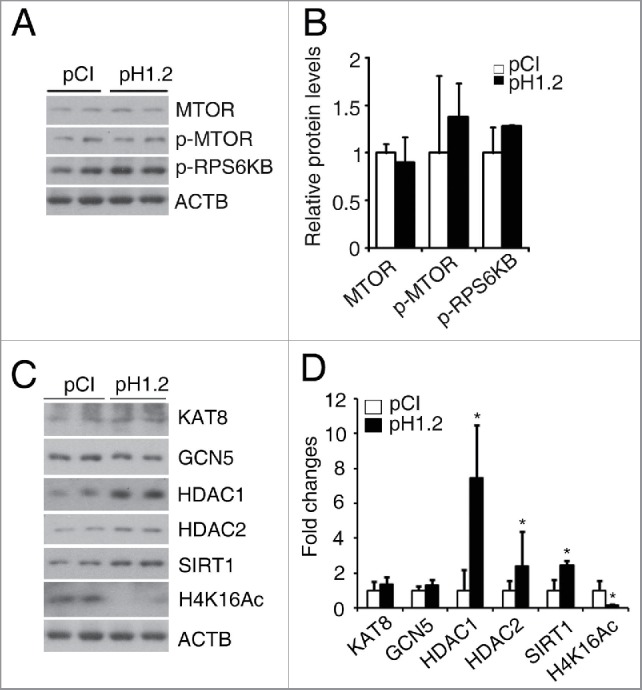
Overexpression of histone HIST1H1C reduces H4K16Ac level by regulating HDACs. (A-B) Representative western blots (A) of MTOR, p-MTOR, p-RPS6KB and ACTB with quantitative densitometric results (B) in the rMC-1 cells. (C-D) Representative western blots (C) of the indicated HATs, HDACs and H4K16Ac with quantitative densitometric results (D) in the rMC-1 cells. n = 6 in each group; *p < 0.05 compared with pCI group.
It has been reported that the deacetylation of H4K16 is directly linked to the upregulation of Atg genes.36 To investigate whether histone HIST1H1C regulates autophagy through H4K16 acetylation (H4K16Ac), we measured the levels of this mark. We also investigated histone acetyltransferases such as KAT8/MOF (K[lysine] acetyltransferase 8), KAT2A/GCN5 (K[lysine] acetyltransferase 2A), and histone deacetylases including HDAC1, HDAC2 and SIRT1, all of these have been reported to regulate acetylation of H4K16 and lead to DNA damage response, cell cycle arrest or autophagy.37,38 Overexpression of histone HIST1H1C significantly reduced H4K16Ac level in rMC-1 cells (Fig. 7C and D). Overexpression of histone HIST1H1C significantly increased the levels of HDAC1/2 and SIRT1, but not the histone acetyltransferases examined (Fig. 7C and D). These data suggest histone HIST1H1C may promote autophagy through regulating HDAC-mediated deacetylation of H4K16.
Overexpression of histone HIST1H1C in the retina induces lesions similar to those observed in diabetic retinopathy
Since histone HIST1H1C regulates autophagy, cell toxicity, and inflammation in retinal cell lines, we explored whether overexpression of histone HIST1H1C in the retinas induces autophagy and lesions of diabetic retinopathy. The schematic workflow of intravitreal injection of AAV-HIST1H1C and AAV2/2-GFP in the retinas is illustrated (Fig. 8A). 14 d post injection, histone HIST1H1C was successfully overexpressed in the retina (Fig. 8B). Overexpression of histone HIST1H1C increased the mRNA levels of Becn1, Atg12, Atg7, Atg5 and Atg3 (Fig. 8C), as well as the protein levels of BECN1, the ATG12–ATG5 complex, ATG5, ATG3 and LC3B-I to LC3B-II conversion (Fig. 8D), indicating that histone HIST1H1C per se increases autophagy in the retinas. Consistent with the in vitro results, overexpression of histone HIST1H1C in the retinas also dramatically reduced the acetylation of H4K16 by upregulation of HDAC1 and SIRT1 (Fig. 8E).
Figure 8.

Overexpression of histone HIST1H1C promotes autophagy, gliosis and loss of neuronal cells in the retina. (A) Experiment scheme of vitreous injection of rats with AAV-GFP/AAV-HIST1H1C. (B) Representative western blots of histone HIST1H1C and ACTB. (C) qPCR results of Atg genes. (D) Representative western blots of ATG proteins and HSP70 (left panel) with quantitative densitometric results (right panel). (E) Representative western blots of HDAC1, SIRT1 and H4K16Ac (left panel) with quantitative densitometric results (right panel) in the retinas. (F) Representative western blots of GFAP (left panel) with quantitative densitometric results (right panel) in the retinas. (G) Representative images of GFAP on the retinal sections. (H) qPCR results of Il6 and Ccl2. (I) Representative images of hematoxylin and eosin staining on retinal sections (left panel) with quantitative result of cell numbers in the GCL (right panel). (J) Quantitative results of retinal layer thickness. n = 4–5 in each group; *p < 0.05 compared with AAV-GFP group; red, GFAP-positive staining; purple, hemotoxylin stained nuclei; scale bar: 50 μm.
Furthermore, the level of GFAP was dramatically increased in the retinas of AAV-HIST1H1C group (Fig. 8F and G). Moreover, overexpression of histone HIST1H1C significantly increased the inflammatory response as demonstrated by increased mRNA levels of Il6 and Ccl2 (Fig. 8H). Meanwhile, overexpression of histone HIST1H1C significantly reduced the number of cells in the GCL (Fig. 8I), as well as the thickness of INL, IPL (inner plexiform player) and GCL (Fig. 8I and J), suggesting histone HIST1H1C overexpression contributes to the neuronal dysfunction in the retina. Together, these data demonstrated that overexpression of histone HIST1H1C in the retinas promotes autophagy, inflammation and neuron loss, pathological changes similar to what happens during the development of diabetic retinopathy.
Knockdown of histone HIST1H1C inhibits the lesions of diabetic retinopathy
To examine whether knockdown the diabetes-induced overexpression of histone HIST1H1C can rescue the lesions of diabetic retinopathy, siHist1h1c and the negative control siNC were intravitreously injected into Ins2+/+ and Ins2+/− mice at 6-mo-old (Fig. 9A). The diabetes-induced overexpression of histone HIST1H1C was successfully inhibited after siHist1h1c injection (Fig. 9B and C). The increased LC3B-I to LC3B-II conversion and decreased SQSTM1 in diabetic retinas were partly rescued by the knockdown of histone HIST1H1C (Fig. 9B and -C). Additionally, knockdown of histone HIST1H1C also downregulated the protein level of BECN1 in the diabetic retinas (Fig. 9B and C). Meanwhile, knockdown of histone HIST1H1C normalized H4K16Ac level through HDAC1 and SIRT1 levels in diabetic retinas (Fig. 9D and E). Importantly, knockdown of histone HIST1H1C partly rescued the diabetes-induced downregulation of POU4F1 level (Fig. 9F), and repressed the diabetes-induced transcription of proinflammatory factors, such as Il6, Ccl2, Vcam1 and Icam1, as well as glial activation marker Gfap (Fig. 9G). These data suggest that knockdown of histone HIST1H1C partly rescues the diabetes-induced autophagy, inflammation and some pathological lesions in the retinas.
Figure 9.

Knockdown of histone HIST1H1C rescues the diabetic-induced autophagy, gliosis and loss of ganglion cells. (A) Experiment scheme for mouse vitreous injection with siNC/siHist1h1c. (B-C) Representative western blots of the indicated ATG proteins, histone HIST1H1C and ACTB (B) and quantitative densitometric results (C). (D-E) Representative western blots of SIRT1, HDAC1 and H4K16Ac (D) with quantitative densitometric results (E). (F) Representative western blots (left panel) of POU4F1 with quantitative densitometric result (right panel). (G) qPCR results of indicated inflammatory genes and Gfap. (H) Proposed mechanism for histone HIST1H1C regulation of autophagy in the development of diabetic retinopathy. n = 4–5 in each group; *p < 0.05 compared with Ins2+/+ mice injected with siNC; $ p < 0.05 compared with Ins2+/− mice injected with siNC; # p = 0.08 or p = 0.07 compared with Ins2+/− mice injected with siNC.
Discussion
Autophagy plays critical roles in cell homeostasis. Dysfunction in autophagy is associated with multiple diseases including diabetes.9,14,15 In STZ-induced diabetic mice, diastolic dysfunction is accompanied with a significant increase in autophagy.14 Meanwhile, increased autophagy is observed in both podocytes of diabetic mice and podocytes cultured under diabetic-like conditions.15 Conversely, autophagy is suppressed in db/db mouse, a type 2 diabetes mouse model,14 possibly due to high intracellular nutrient energy status and activated MTOR pathway.14,39 Here, we found increased autophagy in the retinas of type 1 diabetic rodents, as well as in retinal cells and 239T cells cultured under hyperglycemic conditions (Fig. 1C and D, G and H, Fig. 2), which is consistent with a recent finding in the retinas of the STZ-induced diabetic mice.9 More interestingly, we also found increased histone HIST1H1C, an important histone variant, under the above conditions (Fig. 1A and B, E and F, Fig. 2).
Histone HIST1H1C is originally regarded as a repressor of gene transcription.26 However, growing studies have proven that histone HIST1H1C has multiple functions in cell cycle, DNA-damage induced apoptosis, and fibrillar stabilization.24,27,40 Furthermore, histone HIST1H1C can interact with a phosphorylated form of RNA polymerase II to potentiate gene transcription,25 indicating histone HIST1H1C also acts as a transcription activator. In this study, we demonstrated that histone HIST1H1C overexpression significantly upregulated most of ATGs we investigated, while histone HIST1H1C knockdown had the opposite effects both in vivo and in vitro (Fig. 3A and B, Fig. 5C-E, Fig. 8C and D and Fig. 9B and C), demonstrating histone HIST1H1C acts as an activator in the regulation of autophagy.
We further identified that overexpression of histone HIST1H1C promoted the autophagy through regulating H4K16 acetylation both in the retina and in the retinal cell lines (Fig. 7C and D and Fig. 8D and E). To our knowledge, this is the first report to identify a histone H1 variant regulates acetylation level of core histone both in mammalian cells and in animal model, although a previous study has demonstrated that acetylation of H4K16 is augmented in the histone H1-depleted germline stem cell of Drosophila ovary which promotes the self-renewal of these cells.41 However, in Drosophila, a nonmammalian system, no histone H1 variant has been found so far;42 while at least 6 variants of histone H1 exist in mammalian somatic cells, each has unique functions in different cell types,21 suggesting the nonequivalent function of histone H1 and HIST1H1C (especially between Drosophila histone H1 and mammalian histone variant HIST1H1C). It has been reported that MOF is the key enzyme that responsible for deacetylation of H4K16 and promotes the transcription of Atg genes.36 However, we found histone HIST1H1C mainly upregulates HDACs (primary SIRT1 and HDAC1), while KAT8 is unchanged, to maintain the deacetylation of H4K16 (Fig. 7C and D). How does histone HIST1H1C regulate the levels of SIRT1 and HDAC1, and whether H4K16Ac is the only core histone modification regulated by histone HIST1H1C, await further investigation.
Previously studies found that neuronal dysfunction is important in the development of diabetic retinopathy,10,43 and diabetic retinopathy is also regarded as a neurodegenerative disease.44 Since most neurodegenerative diseases are associated with accumulation of reactive oxygen species and mitochondrial dysfunction in neurons, some evidence has suggested that upregulation of autophagy may protect against neurodegeneration.29 In contrast, mouse-specific depletion of Atg7 in substantia nigra neurons has a lasting and robust protection of the axon in an acute axon injury model.45 Furthermore, autophagosomes can be a source of reactive oxygen species under some circumstances, which aggravates neurotoxicity.46 These reports suggest that inhibition of autophagy may be beneficial for neural cells after certain types of injuries. In the present study, we found that neuron loss in the retinas overexpressing histone HIST1H1C (Fig. 8I and J), and knockdown of histone HIST1H1C partly inhibited the diabetes induced neuronal cell death (Fig. 9F). Our results suggested that histone HIST1H1C promotes cell death, at least partly via HIST1H1C-induced autophagy, since knockout of Atg7 significantly reduced histone HIST1H1C overexpression induced autophagy and cell death (Fig. S6C-E). It has been reported that histone HIST1H1C can trigger cell apoptosis in response to the X-ray irradiation-induced DNA double-strand breaks, depending on its release from nuclear to cytoplasmic compartments.24 However, in the present study, histone HIST1H1C is still enriched in the nuclei of cells we used under either high glucose treatment or histone HIST1H1C overexpression (Fig. S4), suggesting histone HIST1H1C may not act as a direct apoptosis mediator upon the stresses used in these cell lines. However, we certainly cannot exclude the possibility that histone HIST1H1C may translocate to cytoplasm in other cell types or under other stimuli. Furthermore, we demonstrated that rapamycin induces autophagy and neuron loss without causing change of DNA damage marker in the retina (Fig. S5). All these data indicate that histone HIST1H1C regulates the survival of neurons by autophagy, which contributes to the development of diabetic retinopathy.
In this study, we also noticed a few inconsistencies between high-glucose treated cell lines and diabetic retinas. In vivo data suggested increased BECN1 in STZ-induced diabetic retinas and AAV-HIST1H1C infected retinas (Fig. 1C and Fig. 8D), whereas knockdown of histone HIST1H1C in retinas of Akita mice significantly reduced the protein level of BECN1 (Fig. 9B), indicating that histone HIST1H1C regulates BECN1 level in in vivo studies. On the other hand, BECN1 level was unchanged in rMC-1 cells under high glucose stress (Fig. 2A). We also noticed that histone HIST1H1C enriched in nuclei in rMC-1 cells under the high glucose stress or upon histone HIST1H1C overexpression (Fig. S4), however, immunohistochemical studies demonstrated there was cytoplasmic staining of histone HIST1H1C in some cells on the retinal sections of diabetic rodents (Fig. 1B and F). The difference between in vivo and in vitro results may due to the mixed retinal cell types in GCL and INL of the retinas, and more complicated nature of diabetic conditions than high glucose per se.
As little is known about the roles of histone variants in autophagy, especially in autophagy-related diseases, our finding that histone HIST1H1C is upregulated in diabetic retina and high glucose cultured cells, and that increased histone HIST1H1C promotes autophagy and lesions in the early development of diabetic retinopathy (Fig. 9H), suggests histone HIST1H1C as a novel factor to regulate autophagy level in tissues, and may thus be further considered as a novel therapeutic target for treating or preventing diabetic retinopathy.
Methods
Experimental animals
Male Sprague-Dawley rats were obtained from the ABSL-III laboratory of Wuhan University. Diabetes was induced as described previously.11 One wk after STZ (Amresco, N407–1G) injection, rats with NFBG above 250 mg/dl (measured with a OneTouch blood glucose meter, LifeScan, Milpitas, CA, USA) were regarded as diabetic. Age-matched rats, citrate buffer injected, were used as nondiabetic controls. Retinas were collected 2 mo after the onset of diabetes. Breeding pairs of Ins2+/− (Insulin2+/−; Akita) mice, a spontaneous type 1 diabetes mouse model, were obtained from the Model Animal Research Center of Nanjing University, and bred/genotyped as described previously.45,47 Six-mo-old male Ins2+/− mice and their nondiabetic (Ins2+/+) littermates were used. Animals were handled according to the Guidelines of the China Animal Welfare Legislation and approved by the Committee on Ethics in the Care and Use of Laboratory Animals of College of Life Sciences, Wuhan University.
Overexpression and knockdown of histone HIST1H1C in the retina
Human histone HIST1H1C/H1.2 cDNA amplified from a human cDNA library purchased from ABclonal Technology (Oxfordshire, UK) was packed into AAV2/2 viral vectors and purified by SBO Medical Biotechnology (Shanghai, China). AAV-GFP or AAV- HIST1H1C (1 × 1012 vg/ml) was injected into the vitreous of Sprague-Dawley rats (5 μl/eye). 14 d after injection, retinas were collected for qPCR and western blot analysis. 28 d after injection, retinas were collected for pathological analysis.
Mouse histone Hist1h1c siRNA (siHist1h1c, 5′ GGTCAAGAGCGCGTCTAAA 3′) and a scrambled siRNA (siNC) were designed and synthesized by RiBobio Co. (Guangzhou, China). siNC or siHist1h1c (5 μg) was injected into the vitreous of Ins2+/+ and Ins2+/− mice at 6-mo-old. 7 d after injection, retinas were harvested for qPCR and western blot analysis.
Histological examination and immunohistochemical/fluorescence staining of the retinas
Retinal sections were deparaffinized and rehydrated as we previously reported.48,49 For histological examination, sections were stained with hematoxylin and eosin (Beyotime, C0107/C0109). High resolution (× 400 magnification) pictures were taken with an Olympus BX60 microscope. The nuclei in the GCL (not including nuclei in the vessels) and the thicknesses of different retinal layers were measured using the Image-Pro Plus Software (Media Cybernetics, Rockville, MD, USA) in 5 different areas per retinal sample.
For immunohistochemical staining, retinal sections were incubated overnight with a rabbit anti-histone HIST1H1C/H1.2 antibody (Abcam, 17677; 1:1000 dilution). After extensive washing, sections were further incubated with a biotinylated anti-rabbit antibody (Vector Laboratories, BA-1000). Positive staining was visualized by DAB substrate reaction (Vector laboratories, SK-4100) following the ABC kit protocol (Vector laboratories, PK-6100), and costaining with hematoxylin. High-resolution pictures were taken with an Olympus BX60 microscope.
For immuno-fluorescence staining, retinal sections were incubated with a rabbit anti-GFAP antibody (Abcam, 7260; 1:1000 dilution) followed by an anti-rabbit antibody conjugated with TRITC (tetramethyl rhodamine isothiocyanate) (Life Technologies, A11010; 1:200 dilution). Sections were then covered with DAPI (4,6-diamidino-2-phenylindole) (Sigma, D9542) and anti-fading medium (Beyotime, P0126), and imaged as described above.
Cell culture
rMC-1 (a rat Müller cell line), 293T (a large T antigen transformed human embryonic kidney cell line), and Atg7+/+ or atg7−/− MEF (mouse embryonic fibroblast) cells were cultured in DMEM media (Hyclone, SH30021.FS) supplemented with 10% FBS (Hyclone, SV30087.02) and 1% sodium pyruvate (Hyclone, SV30010). All cells were incubated at 37°C in 5% CO2. For high glucose treatment, additional 20 mM D-glucose (Amresco, 50–99–7) or 20 mM mannitol (Sigma, M9546) as the osmotic control were added to respective cultured media for 48 h. For starvation treatment, media was replaced with HBSS (Hyclone, SH30030.02B) for 4 h. For rapamycin treatment, cells were cultured in media containing 0.5 μg/ml rapamycin (LC laboratories, R-500G) for 6 h. To inhibit autophagy, 50 μM CQ (Sigma, 6628) or 100 nM BafA1 (Santa Cruz Biotechnology, 88899–55–2) was added to respective culture media for 12 h.
In vitro transfection and establishment of stable knockdown cell lines
HIST1H1C cDNA was cloned with NotI and EcoRI restriction sites into a pIRES-Neo vector (pCI; a gift from Dr. X. Zhang, Wuhan University), which contains Flag and HA tags, named pH1.2. For overexpression experiments, pH1.2, pCI, GFP-LC3 plasmids were transfected using Lipofectamine LTX (Invitrogen, 11668–019). Cells were collected for qPCR and western blot analysis at 48 h after transfection.
Stable Hist1h1c knockdown cell lines were established as described previously.50 Briefly, rMC-1 cells were transfected with either blank pSuper vector (OligoEngine, VEC-PRT-0002) or vector containing shRNA targeting Hist1h1c. Stable Hist1h1c knockdown cell lines were selected using puromycin (Amresco, J593) at the concentration of 1 μg/ml.
Autophagy analysis
To assess autophagy, cells were cotransfected with plasmid encoding GFP-LC3 and pH1.2 or pCI and treated as indicated above. Cells were imaged with an Olympus FV1000 Viewer confocal microscope. A cell containing more than 10 cytoplasmic GFP dots was counted as an autophagic cell.29 At least 200 cells were analyzed per treatment. Each experiment was repeated at least 3 times.
To assess autophagy flux, cells were transfected with pH1.2 or pCI. At 36 h after transfection, cells were treated with or without 50 μM CQ and 100 nM Baf A1 for 12 h.
Immunofluorescence staining for cultured cells
The cultured cells were fixed with 4% formaldehyde and blocked with 2% BSA (Amresco, E588). A primary antibody against histone HIST1H1C/H1.2 (Abcam, 17677; 1:2000 dilution) or LC3B (Sigma, L7543; 1:500) was applied overnight. The cells were then incubated with an anti-rabbit antibody conjugated with TRITC (Life Technologies, A11010), followed by DAPI costaining, and imaged with an Olympus FV1000 Viewer confocal microscope.
MTT assay
An MTT assay was performed as described previously to determine cell viability.51 Briefly, cells were plated in 96-well plates at a density of 5000 cells/well and incubated at 37°C in 5% CO2 for 12 h. After treated as indicated for 48 h, cells were co-incubated with 10 μl MTT (Amresco, 298–93–1; 5 mg/ml) per well for 4 h. The media was removed and DMSO (Amresco, 0231) was added to each well. Absorbance was measured at 490 nm with a MUITISKAN FC plate reader (Thermo Scientific, Waltham, MA). Cell viability was reported as the fold change of OD value, the value of the respective control group was set up as one-fold.
Western blot analysis
Freshly isolated retinas or cultured cells were sonicated in ice-cold RIPA buffer (Beyotime, P0013B) and protein concentrations were determined. Proteins were fractionated by SDS-PAGE, electroblotted onto PVDF membrane (Millipore, IPVH00010) and probed with primary antibodies (Table S2). Protein bands detected by the antibodies were visualized by enhanced chemiluminescence (Beyotime, P0018) and evaluated using Quantity One 1-D Analysis Software (Bio-Rad, Hercules, CA) as we described previously.52 The expression levels of target proteins and the ratio of LC3B-II: I were first quantified relative to ACTB/β-actin in the same sample, then the relative protein expression levels in different groups were normalized to the respective control group, which was arbitrarily set as 1.
Quantitative real-time PCR (qPCR)
RNA was extracted from retinas or cells using RNAiso Plus (Takara Biotechnology, 9109) as described previously.49,53 cDNA synthesis was performed using the M-MLV First Strand Kit (Invitrogen, 28025–021). Primer sequences for the target genes are provided in Table S3. qPCR were performed as described previously.54 The formation of a single product for each primer set was confirmed by observing only one peak in the melting curve for each reaction. Αctb or Rn18s were used as internal controls. The relative difference is expressed as the fold change calculated using the 2−ΔΔCT method.
Nuclear and cytoplasmic extraction
After the indicated treatments, cells were freshly collected. Cytoplasmic and nuclear fractions were separated by a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, P0027) according to the manufacturer's instruction.
Statistical analysis
All results were expressed as the mean ± SD (standard deviation). Data were analyzed using the nonparametric Kruskal-Wallis test followed by the Mann-Whitney test for more than 2 group comparison, while the Mann-Whitney test was used for 2 group comparison. Differences were considered statistically significant at p < 0.05.
Abbreviations
- AAV
adeno-associated virus
- ATG
autophagy related
- BafA1
bafilomycin A1
- CQ
chloroquine
- GCL
ganglion cell layer
- GFAP
glial fibrillary acidic protein
- HAT
histone acetyltranferase
- HDAC
histone deacetylase
- INL
inner nuclei layer
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NFBG
nonfasted blood glucose
- SIRT1
sirtuin 1
- STZ
streptozotocin
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
pSuper plasmid, pGFP-LC3 plasmid, pCI plasmid and Atg7+/+ and atg7−/− MEF cells are gifts from Drs. H. Shu, W. Li, X. Zhang and M. Chen (Wuhan University).
Funding
This work was supported by the National Basic Research Program of China (2012CB524901), the Natural Science Foundation of China (No. 31271370, 81222043, 31471208, 31500941 and 31671195), the Natural Science Foundation of Hubei Province (2016CFA012) and the Front Youth Program of HUST.
References
- [1].Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet 2010; 376:124-36; PMID:20580421; http://dx.doi.org/ 10.1016/S0140-6736(09)62124-3 [DOI] [PubMed] [Google Scholar]
- [2].Leal EC, Manivannan A, Hosoya K, Terasaki T, Cunha-Vaz J, Ambrosio AF, Forrester JV. Inducible nitric oxide synthase isoform is a key mediator of leukostasis and blood-retinal barrier breakdown in diabetic retinopathy. Invest Ophthalmol Vis Sci 2007; 48:5257-65; PMID:17962481; http://dx.doi.org/ 10.1167/iovs.07-0112 [DOI] [PubMed] [Google Scholar]
- [3].Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes 2002; 51:2241-8; PMID:12086956; http://dx.doi.org/ 10.2337/diabetes.51.7.2241 [DOI] [PubMed] [Google Scholar]
- [4].Asnaghi V, Gerhardinger C, Hoehn T, Adeboje A, Lorenzi M. A role for the polyol pathway in the early neuroretinal apoptosis and glial changes induced by diabetes in the rat. Diabetes 2003; 52:506-11; PMID:12540628; http://dx.doi.org/ 10.2337/diabetes.52.2.506 [DOI] [PubMed] [Google Scholar]
- [5].Kusner LL, Sarthy VP, Mohr S. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase: a role in high glucose-induced apoptosis in retinal Muller cells. Invest Ophthalmol Vis Sci 2004; 45:1553-61; PMID:15111614; http://dx.doi.org/ 10.1167/iovs.03-1294 [DOI] [PubMed] [Google Scholar]
- [6].Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci 2011; 52:1156-63; PMID:21357409; http://dx.doi.org/ 10.1167/iovs.10-6293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vincent JA, Mohr S. Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes 2007; 56:224-30; PMID:17192486; http://dx.doi.org/ 10.2337/db06-0427 [DOI] [PubMed] [Google Scholar]
- [8].Babel J, Leuenberger P. A long term study on the ocular lesions in streptozotocin diabetic rats. Albrecht Von Graefes Arch Klin Exp Ophthalmol 1974; 189:191-209; PMID:4360703; http://dx.doi.org/ 10.1007/BF00414781 [DOI] [PubMed] [Google Scholar]
- [9].Piano I, Novelli E, Della Santina L, Strettoi E, Cervetto L, Gargini C. Involvement of Autophagic Pathway in the Progression of Retinal Degeneration in a Mouse Model of Diabetes. Front Cell Neurosci 2016; 10:42; PMID:26924963; http://dx.doi.org/ 10.3389/fncel.2016.00042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zheng L, Howell SJ, Hatala DA, Huang K, Kern TS. Salicylate-based anti-inflammatory drugs inhibit the early lesion of diabetic retinopathy. Diabetes 2007; 56:337-45; PMID:17259377; http://dx.doi.org/ 10.2337/db06-0789 [DOI] [PubMed] [Google Scholar]
- [11].Wang LL, Chen H, Huang K, Zheng L. Elevated histone acetylations in Muller cells contribute to inflammation: a novel inhibitory effect of minocycline. Glia 2012; 60:1896-905; PMID:22915469; http://dx.doi.org/ 10.1002/glia.22405 [DOI] [PubMed] [Google Scholar]
- [12].Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124-31; PMID:20034776; http://dx.doi.org/ 10.1016/j.ceb.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al.. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383-435; PMID:20959619; http://dx.doi.org/ 10.1152/physrev.00030.2009 [DOI] [PubMed] [Google Scholar]
- [14].Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, Watanabe T, Morishita K, Okada H, Kawasaki M, et al.. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy 2015; 11:1146-60; PMID:26042865; http://dx.doi.org/ 10.1080/15548627.2015.1051295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lenoir O, Jasiek M, Henique C, Guyonnet L, Hartleben B, Bork T, Chipont A, Flosseau K, Bensaada I, Schmitt A, et al.. Endothelial cell and Podocyte Autophagy Synergistically protect from Diabetes-induced Glomerulosclerosis. Autophagy 2015; 11:1130-45; PMID:26039325; http://dx.doi.org/ 10.1080/15548627.2015.1049799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mitter SK, Rao HV, Qi X, Cai J, Sugrue A, Dunn WA Jr., Grant MB, Boulton ME. Autophagy in the retina: a potential role in age-related macular degeneration. Adv Exp Med Biol 2012; 723:83-90; PMID:22183319; http://dx.doi.org/ 10.1007/978-1-4614-0631-0_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rodriguez-Muela N, Germain F, Marino G, Fitze PS, Boya P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ 2012; 19:162-9; PMID:21701497; http://dx.doi.org/ 10.1038/cdd.2011.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Park HY, Kim JH, Park CK. Activation of autophagy induces retinal ganglion cell death in a chronic hypertensive glaucoma model. Cell Death Dis 2012; 3:e290; PMID:22476098; http://dx.doi.org/ 10.1038/cddis.2012.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest 2005; 115:2679-88; PMID:16200202; http://dx.doi.org/ 10.1172/JCI26390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Meergans T, Albig W, Doenecke D. Varied expression patterns of human H1 histone genes in different cell lines. DNA Cell Biol 1997; 16:1041-9; PMID:9324306; http://dx.doi.org/ 10.1089/dna.1997.16.1041 [DOI] [PubMed] [Google Scholar]
- [21].Carruthers LM, Bednar J, Woodcock CL, Hansen JC. Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: mechanistic ramifications for higher-order chromatin folding. Biochemistry 1998; 37:14776-87; PMID:9778352; http://dx.doi.org/ 10.1021/bi981684e [DOI] [PubMed] [Google Scholar]
- [22].Zhao X, Sidoli S, Wang L, Wang W, Guo L, Jensen ON, Zheng L. Comparative proteomic analysis of histone post-translational modifications upon ischemia/reperfusion-induced retinal injury. J Proteome Res 2014; 13:2175-86; PMID:24628298; http://dx.doi.org/ 10.1021/pr500040a [DOI] [PubMed] [Google Scholar]
- [23].Harshman SW, Young NL, Parthun MR, Freitas MA. H1 histones: current perspectives and challenges. Nucleic Acids Res 2013; 41:9593-609; PMID:23945933; http://dx.doi.org/ 10.1093/nar/gkt700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Konishi A, Shimizu S, Hirota J, Takao T, Fan Y, Matsuoka Y, Zhang L, Yoneda Y, Fujii Y, Skoultchi AI, et al.. Involvement of histone H1.2 in apoptosis induced by DNA double-strand breaks. Cell 2003; 114:673-88; PMID:14505568; http://dx.doi.org/ 10.1016/S0092-8674(03)00719-0 [DOI] [PubMed] [Google Scholar]
- [25].Kim K, Lee B, Kim J, Choi J, Kim JM, Xiong Y, Roeder RG, An W. Linker Histone H1.2 cooperates with Cul4A and PAF1 to drive H4K31 ubiquitylation-mediated transactivation. Cell Rep 2013; 5:1690-703; PMID:24360965; http://dx.doi.org/ 10.1016/j.celrep.2013.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim K, Choi J, Heo K, Kim H, Levens D, Kohno K, Johnson EM, Brock HW, An W. Isolation and characterization of a novel H1.2 complex that acts as a repressor of p53-mediated transcription. J Biol Chem 2008; 283:9113-26; PMID:18258596; http://dx.doi.org/ 10.1074/jbc.M708205200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Roque A, Sortino R, Ventura S, Ponte I, Suau P. Histone H1 Favors Folding and Parallel Fibrillar Aggregation of the 1–42 Amyloid-beta Peptide. Langmuir 2015; 31:6782-90; PMID:26023729; http://dx.doi.org/ 10.1021/la504089g [DOI] [PubMed] [Google Scholar]
- [28].Carter TA, Greenhall JA, Yoshida S, Fuchs S, Helton R, Swaroop A, Lockhart DJ, Barlow C. Mechanisms of aging in senescence-accelerated mice. Genome Biol 2005; 6:R48; PMID:15960800; http://dx.doi.org/ 10.1186/gb-2005-6-6-r48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; http://dx.doi.org/ 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:2852113; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res 2011; 30:343-58; PMID:21635964; http://dx.doi.org/ 10.1016/j.preteyeres.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Feenstra DJ, Yego EC, Mohr S. Modes of Retinal Cell Death in Diabetic Retinopathy. J Clin Exp Ophthalmol 2013; 4:298; PMID:3963519; http://dx.doi.org/2155-9570.1000298 PMID:24672740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol 2011; 93:421-43; PMID:21219963; http://dx.doi.org/ 10.1016/j.pneurobio.2011.01.005 [DOI] [PubMed] [Google Scholar]
- [34].Sippl C, Tamm ER. What is the nature of the RGC-5 cell line? Adv Exp Med Biol 2014; 801:145-54.PMID:24664692; http://dx.doi.org/ 10.1007/978-1-4614-3209-8_19 [DOI] [PubMed] [Google Scholar]
- [35].Talbert PB, Henikoff S. Environmental responses mediated by histone variants. Trends Cell Biol 2014; 24:642-50; PMID:25150594; http://dx.doi.org/ 10.1016/j.tcb.2014.07.006 [DOI] [PubMed] [Google Scholar]
- [36].Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, Hermanson O, Rosenfeld MG, Klionsky DJ, Joseph B. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 2013; 500:468-71; PMID:23863932; http://dx.doi.org/ 10.1038/nature12313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Spedale G, Timmers HT, Pijnappel WW. ATAC-king the complexity of SAGA during evolution. Genes Dev 2012; 26:527-41; PMID:22426530; http://dx.doi.org/ 10.1101/gad.184705.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rea S, Xouri G, Akhtar A. Males absent on the first (MOF): from flies to humans. Oncogene 2007; 26:5385-94; PMID:17694080; http://dx.doi.org/ 10.1038/sj.onc.1210607 [DOI] [PubMed] [Google Scholar]
- [39].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/ 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sancho M, Diani E, Beato M, Jordan A. Depletion of human histone H1 variants uncovers specific roles in gene expression and cell growth. PLoS Genet 2008; 4:e1000227; PMID:18927631; http://dx.doi.org/ 10.1371/journal.pgen.1000227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sun J, Wei HM, Xu J, Chang JF, Yang Z, Ren X, Lv WW, Liu LP, Pan LX, Wang X, et al.. Histone H1-mediated epigenetic regulation controls germline stem cell self-renewal by modulating H4K16 acetylation. Nat Commun 2015; 6:8856; PMID:26581759; http://dx.doi.org/ 10.1038/ncomms9856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Perez-Montero S, Carbonell A, Moran T, Vaquero A, Azorin F. The embryonic linker histone H1 variant of Drosophila, dBigH1, regulates zygotic genome activation. Dev Cell 2013; 26:578-90; PMID:24055651; http://dx.doi.org/ 10.1016/j.devcel.2013.08.011 [DOI] [PubMed] [Google Scholar]
- [43].Kern TS, Barber AJ. Retinal ganglion cells in diabetes. J Physiol 2008; 586:4401-8; PMID:2614025; http://dx.doi.org/jphysiol.2008.156695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cuenca N, Fernandez-Sanchez L, Campello L, Maneu V, De la Villa P, Lax P, Pinilla I. Cellular responses following retinal injuries and therapeutic approaches for neurodegenerative diseases. Prog Retin Eye Res 2014; 43:17-75; PMID:25038518; http://dx.doi.org/ 10.1016/j.preteyeres.2014.07.001 [DOI] [PubMed] [Google Scholar]
- [45].Cheng HC, Kim SR, Oo TF, Kareva T, Yarygina O, Rzhetskaya M, Wang C, During M, Talloczy Z, Tanaka K, et al.. Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J Neurosci 2011; 31:2125-35; PMID:21307249; http://dx.doi.org/ 10.1523/JNEUROSCI.5519-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kubota C, Torii S, Hou N, Saito N, Yoshimoto Y, Imai H, Takeuchi T. Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J Biol Chem 2010; 285:667-74; PMID:19850931; http://dx.doi.org/ 10.1074/jbc.M109.053058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chen H, Li J, Jiao L, Petersen RB, Peng A, Zheng L, Huang K. Apelin inhibits the development of diabetic nephropathy by regulating histone acetylation in Akita mouse. J Physiol 2014; 592:505-21; PMID:24247978; http://dx.doi.org/ 10.1113/jphysiol.2013.266411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sun Y, Xue W, Song Z, Huang K, Zheng L. Restoration of Opa1-long isoform inhibits retinal injury-induced neurodegeneration. J Mol Med (Berl) 2016; 94:335-46; PMID:26530815; http://dx.doi.org/ 10.1007/s00109-015-1359-y [DOI] [PubMed] [Google Scholar]
- [49].Cai R, Xue W, Liu S, Petersen RB, Huang K, Zheng L. Overexpression of glyceraldehyde 3-phosphate dehydrogenase prevents neurovascular degeneration after retinal injury. FASEB J 2015; 29:2749-58; PMID:25805836; http://dx.doi.org/ 10.1096/fj.14-265801 [DOI] [PubMed] [Google Scholar]
- [50].Huang J, Wan D, Li J, Chen H, Huang K, Zheng L. Histone acetyltransferase PCAF regulates inflammatory molecules in the development of renal injury. Epigenetics 2015; 10:62-72; PMID:25496441; http://dx.doi.org/ 10.4161/15592294.2014.990780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Liu X, Zhou Y, Peng A, Gong H, Huang L, Ji K, Petersen RB, Zheng L, Huang K. MPHOSPH1: a potential therapeutic target for hepatocellular carcinoma. Cancer Res 2014; 74:6623-34; PMID:25269478; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-1279 [DOI] [PubMed] [Google Scholar]
- [52].Li J, Huang J, Li JS, Chen H, Huang K, Zheng L. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol 2012; 56:900-7; PMID:22173165; http://dx.doi.org/ 10.1016/j.jhep.2011.10.018 [DOI] [PubMed] [Google Scholar]
- [53].Chen H, Wan D, Wang L, Peng A, Xiao H, Petersen RB, Liu C, Zheng L, Huang K. Apelin protects against acute renal injury by inhibiting TGF-beta1. Biochim Biophys Acta 2015; 1852:1278-87; PMID:25748499; http://dx.doi.org/ 10.1016/j.bbadis.2015.02.013 [DOI] [PubMed] [Google Scholar]
- [54].Li JS, Wang WJ, Sun Y, Zhang YH, Zheng L. Ursolic acid inhibits the development of nonalcoholic fatty liver disease by attenuating endoplasmic reticulum stress. Food Funct 2015; 6:1643-51; PMID:25892149; http://dx.doi.org/ 10.1039/c5fo00083a [DOI] [PubMed] [Google Scholar]