

ABSTRACT
Synapses are very specialized compartments with high metabolic demand to maintain neurotransmission, an essential step for basic brain function. Neurons are post-mitotic and synapses need to stay functional over time—sometimes over decades. Given that synapses are often at a long distance from the cell body, they must use local mechanisms to regulate protein quality control. We show that macroautophagy/autophagy is one of these local processes and found that it is under strict control of the synapse-enriched protein EndoA/Endophilin-A, previously only implicated in endocytosis. Metabolic and neuronal stimulation induce synaptic autophagy and phosphorylation of EndoA by the Parkinson disease kinase Lrrk/LRRK2 is essential to promote the process. EndoA induces membrane curvature in vitro, and, mechanistically, phosphorylated EndoA creates curved membrane-protein docking sites that are capable of recruiting Atg3. Our work reveals a synapse-enriched branch of autophagy under the control of EndoA that may be deregulated in Parkinson disease.
KEYWORDS: ATG3, autophagic docking stations, autophagy, Drosophila, Endophilin-A, LRRK2, membrane curvature, neurodegeneration, Parkinson disease, synapses
Synapses are densely packed with proteins and organelles that are used many times during neurotransmission. High release rates and metabolic stress at synapses will more likely result in the accumulation of damaged proteins, impeding efficient synaptic function. This may also be relevant to many neurodegenerative diseases, including, for example, Parkinson disease that is thought to start from subtle synaptic defects that progress to blunt degeneration. However, how protein quality control occurs at synapses and how defects in these processes lead to neurodegeneration remain enigmatic.
Previous studies showed that autophagosomes can form at synaptiC-terminals and that defects in macroautophagy influence neurotransmitter release, but the molecular mechanisms of regulation and how autophagosomes form at synapses remained elusive. We showed that in addition to amino acid deprivation, neuronal stimulation also triggers autophagy at Drosophila synaptiC-terminals. Twenty min of electrical nerve stimulation or the activation of genetically encoded temperature-sensitive ion channels cause the formation of Atg8-positive structures: synaptic autophagosomes. These observations indicate that synapses contain the necessary machinery to create these structures. Indeed, axonal transport would be too slow to provide the necessary material within these time frames, and the motor neurons can also be severed without affecting autophagosome formation.
Autophagosomes display a very typical structure in most cell types, but at synapses, the morphology of Atg8-positive organelles had not been revealed. We used correlative light and electron microscopy to characterize the ultrastructure of synaptic autophagosomes. We found that Atg8-positive puncta usually contain only a single membrane and some of them accumulate smaller intralumenal vesicular structures that are electron dense in nature. Given the unique morphology of autophagosomes at synapses we wondered what are the molecular mechanisms that are at the origin of their formation.
It is known that several proteins needed for autophagy recognize specific lipids as well as the lipid packing defects in highly curved membranes. Interestingly, we identified a well-recognized membrane remodeling factor, EndoA, that is synapse specific and was already known to act during synaptic vesicle endocytosis. We showed that EndoA co-immunoprecipitates with Atg9-positive membranes, and the Milosevic group recently also found the mouse homolog of this protein to colocalize with autophagosomes. Recent work had shown that phosphorylation of the central amphiphatic helix of SH3GL2/EndophilinA, the human EndoA homolog, switches the function of the protein from creating shallow membrane deformations to very highly curved membranes. When not phosphorylated, the amphiphatic helix of SH3GL2/Endophilin-A inserts deep into the lipid layer, but phosphorylation of the amphiphilic region renders the helix more negatively charged such that it induces structural changes at the protein-lipid interphase leading to the stretching of the head groups. This activity results in the creation of highly curved membrane zones. Given that highly curved membrane zones are known to serve as docking sites for autophagic factors, we reasoned that EndoA is a good candidate to regulate the formation of autophagosomes at synapses. Using giant unilamellar vesicles we were able to test this model in vitro and showed that these vesicles, incubated with phosphomimetic EndoA, recruit mCherry-tagged Atg3. Binding of Atg3 to curved membranes is required for the Atg3-mediated lipidation of Atg8 and therefore is critical for autophagosome formation. While our data indicate that at synapses EndoA promotes this process, we currently do not know the nature of the mechanism that acts to recruit Atg3 at other cellular locations that are not enriched for EndoA.
In subsequent work we confirmed our model also in vivo using the Drosophila neuromuscular junction synapse. Without any stimulation, a phosphomimic EndoA is already sufficient to promote autophagosome formation. Conversely, fruit flies that only express phosphodead EndoA fail to induce the formation of autophagosomes, also when stimulated. In line with our in vitro results, we show that Atg3—and as a consequence Atg8—are no longer recruited during autophagy at synapses of flies that only express phosphodead EndoA. The data indicate that EndoA is needed during these early steps of synaptic autophagy, and our results suggest the protein creates docking sites for autophagic factors such as Atg3, but likely also other proteins. These proteins are then needed to create synaptic autophagosomes that are Atg8 positive (see Fig. 1). Given the unique morphology of synaptic autophagosomes, likely other synapse-enriched proteins are playing a role as well, but their identity is still concealed, and it will be interesting to identify them.
Figure 1.
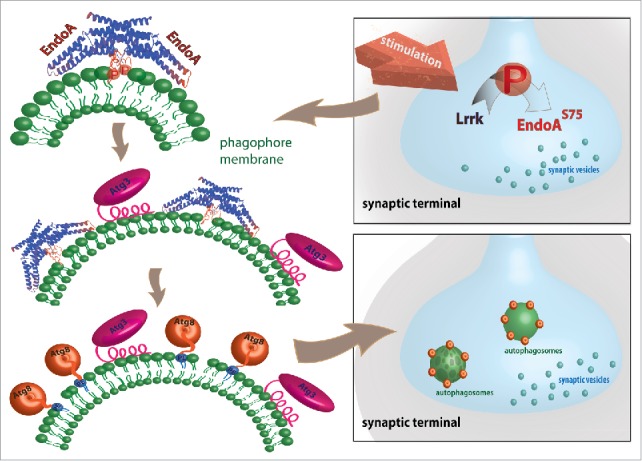
Speculative cartoon representation of EndoA function in autophagosome formation. In addition to neuronal and metabolic stimulation also expression of kinase active LRRK2 enhances autophagosome formation at the synapse. Lrrk/LRRK2 phosphorylates serine 75 in the central amphiphatic helix of the EndoA dimer leading to highly curved membranes by stretching lipid headgroups. The N-terminal domain of Atg3 can sense these highly curved membranes on phagophores (the precursors to autophagosomes). Atg3 insertion on highly curved membranes leads to lipidation (conjugation to phosphatidylethanolamine [PE]) of Atg8 on these membranes and promotes the progression of autophagosome formation.
Phosphorylation of the central amphiphatic helix in EndoA has been shown to be mediated by Lrrk and its human homolog LRRK2, a kinase that is the most commonly mutated protein in Parkinson disease. Similar to EndoA phosphodead mutants, Lrrk mutants do not support synaptic autophagy. Conversely, expression of a kinase hyperactive LRRK2 mutant (G2019S) is sufficient to induce autophagy, similar to the EndoA phosphomimetic mutant. Using fruit fly genetics, we establish that EndoA acts downstream of Lrrk to induce synaptic autophagosome formation. Given that neuronal stimulation induces synaptic autophagy (and likely EndoA phosphorylation), it will be interesting to elucidate how neuronal activity can activate Lrrk/LRRK2 activity. Perhaps the GTPase domain that is also contained in the Lrrk/LRRK2 protein plays a role here as well?
Autophagic defects have been linked to neurodegeneration, and mutations in ATG7 and ATG5 are risk factors for Parkinsonism. Interestingly, we observed that the balance in EndoA phosphorylation state is critical for photoreceptor and dopaminergic neuron survival. An interesting avenue to pursue would be to decipher if the correlation between dysregulation of EndoA phosphorylation and an imbalance in autophagy are also the direct cause of neurodegeneration. Nevertheless, experimental work on how autophagic defects at the synapse participate in the early onset of the disease are compelling. Especially so when considering that EndoA is functionally connected to at least 3 different proteins that when mutated cause Parkinson disease: LRRK2, PARK2/Parkin and SYNJ1 (synaptojanin 1). Noteworthy, all 3 proteins have independently been implicated in aspects of autophagy, and it will be interesting to connect them together into a common functional pathway that regulates the process at synaptiC-terminals.