

ABSTRACT
Selectivity of autophagy is achieved by target recognition; however, the number of autophagy receptors identified so far is limited. In this study we demonstrate that a subset of tripartite motif (TRIM) proteins mediate selective autophagy of key regulators of inflammatory signaling. MEFV/TRIM20, and TRIM21 act as autophagic receptors recognizing their cognate targets and delivering them for autophagic degradation. MEFV recognizes the inflammasome components NLRP3, CASP1 and NLRP1, whereas TRIM21 specifically recognizes the activated, dimeric from of IRF3 inducing type I interferon gene expression. MEFV and TRIM21 have a second activity, whereby they act not only as receptors but also recruit and organize key components of autophagic machinery consisting of ULK1, BECN1, ATG16L1, and mammalian homologs of Atg8, with a preference for GABARAP. MEFV capacity to organize the autophagy apparatus is affected by common mutations causing familial Mediterranean fever. These findings reveal a general mode of action of TRIMs as autophagic receptor-regulators performing a highly-selective type of autophagy (precision autophagy), with MEFV specializing in the suppression of inflammasome and CASP1 activation engendering IL1B/interleukin-1β production and implicated in the form of cell death termed pyroptosis, whereas TRIM21 dampens type I interferon responses.
KEYWORDS: autophagy receptors, BEC1, familial Mediterranean fever, inflammasome, interferon, interleukin-1β, IRF3, tripartite motif class of proteins (TRIMs), ULK1
The tripartite motif proteins (TRIMs) comprise a large protein family in metazoans, with 82 members encoded by the human genome excluding pseudogenes. Since recent evidence suggests a link between TRIMs and autophagy, we examined the role of TRIM-mediated autophagy in immunological processes, an area where both TRIMs and autophagy play a major role. We started the study by addressing a still incompletely understood process of how IFNG/interferon-γ, a major regulator of immunological responses and a key player in defense against intracellular pathogens, induces autophagy. We carried out an siRNA screen and found that 24 TRIMs positively affect autophagy induction in response to IFNG.
One of the TRIMs identified in the screen was MEFV/TRIM20, which is strongly inducible by INFG and is a risk locus for familial Mediterranean fever (FMF). We found that MEFV regulates autophagy by forming protein complexes with multiple key autophagic factors. MEFV assembles autophagic regulators, ULK1 and BECN1, simultaneously. AMPK is also present in MEFV protein complexes, and MEFV preferentially enriches phosphorylated forms of ULK1 at Ser317 and Ser555, residues which are targets for activating phosphorylation by AMPK in the induction of autophagy. ATG16L1, a component of the autophagy E3-like complex that regulates LC3 conjugation and autophagosome formation, is also present in MEFV protein complexes. This fact may reflect direct association or reinforcement of known links between ULK1 and ATG16L1. MEFV also interacts with mammalian Atg8 paralogs (mAtg8s); MEFV interacts with MAP1LC3A, MAP1LC3C, GABARAP, GABARAPL1 and GABARAPL2, with a preference for GABARAP and conspicuous absence of LC3B binding. The interaction between MEFV and mAtg8s occurs through 3 LIR-like sequences with each of them capable of interacting with GABARAP. Thus, MEFV regulates autophagy by forming an activating platform for sets of autophagy regulators and effector factors.
MEFV has more than 300 FMF-associated variants mainly in its C-terminal PRY/SPRY domain, where MEFV binds inflammasome components such as NLRP3, NLRP1, and CASP1. We found that MEFV acts as a receptor for selective autophagy of inflammasome components. MEFV recognizes inflammasome components, brings ULK1 to NLRP3-MEFV receptor-target recognition complexes, and directs assembly of autophagosomes to sequester and degrade the target (inflammasome components). As a result, inflammasome components, and MEFV itself, are subjected to autophagic degradation. Through this process, MEFV suppresses CASP1 activation and IL1B production. Of note, the presence of MEFV's cognate autophagic target NLRP3 in MEFV protein complexes activates ULK1, indicating that substrate recognition activates the entire MEFV autophagy platform. MEFV's role in autophagy plays a critical role in the disease process of FMF. FMF patients suffer from a prolonged period of fever due to the hyperactivation of the NLRP3 inflammasome. FMF-associated variants of MEFV reduce autophagic activity due to a diminished ability to form protein complexes with ULK1. As a result, FMF-associated variants of MEFV display a reduced capacity to downregulate inflammasome components, which results in excessive IL1B production as seen in FMF patients.
Another TRIM found to be a positive factor in our screen was TRIM21. TRIM21, which is also known as Ro52/SSA, is associated with autoimmune diseases including systemic lupus erythematosus (SLE) and Sjögren syndrome. In these autoimmune diseases, type I interferon (IFN) response is hyperactivated. TRIM21 interacts through its C-terminal SPRY domain with the transcriptional factor IRF3, which is a critical transcriptional regulator of the type I IFN response. TRIM21, as an autophagy receptor, interacts with IRF3. Furthermore, TRIM21 colocalizes with IRF3 in ULK1- or MAP1LC3-positive puncta in the cytoplasm. We found that TRIM21 also interacts with ULK1, BECN1, a subset of mAtg8s, and SQSTM1/p62. Finally, IRF3 is found in the same protein complexes with ULK1 in the presence of TRIM21. Thus, we wondered if TRIM21 is a bona fide autophagic receptor for IRF3, analogous to what we observed with MEFV and NLRP3. During viral infection with HIV, cytosolic DNA induces a type I interferon response through endogenous second messenger (cyclic GMP-AMP) by utilizing its adaptor protein TMEM173/STING that results in IRF3 dimerization. Indeed, we found that TRIM21 targets the dimerized form of IRF3 for degradation by autophagy and suppresses the type I IFN response.
The above findings exemplify how autophagy exerts its effects on immunological processes beyond the previously described autophagic receptors, and explain in part how IFNG induces autophagy and suppresses excessive inflammasome or type I IFN activation. These findings show furthermore how autophagy through receptor-regulators acts as a smart balancer of key innate immunity responses, potentially serving as a guardian against excessive inflammation, which in turn causes pathology during autoimmune processes or in infections causing cytokine storms.
In general, our study shows that TRIMs, as receptor-regulators, orchestrate several aspects of selective autophagy: TRIMs as receptors directly recognize their targets, whereas TRIMs as regulators assemble autophagy machinery (Fig. 1). We termed this process ‘precision autophagy’ to indicate the fact that the receptors act both as organizers and activators of autophagy machinery to deliver a highly selective autophagic degradation of targets, and to emphasize a distinct feature that precision autophagy does not require ubiquitination of targets but rather relies on direct recognition via specific protein-protein interactions and domains in TRIMs. However, we cannot exclude a modulatory role of ubiquitin in these processes (e.g., in stabilization of the recognition or activation complexes or both); since TRIMs are E3 ubiquitin ligases, this needs to be explored. The above phenomena and features are common to the TRIM family of proteins. Precision autophagy targets both endogenous (inflammasome components by MEFV, and activated, dimeric IRF3 by TRIM21) and exogenous (HIV1 by TRIM5) targets. TRIMs possess a variety of recognition domains and motifs that may expand the scope of autophagic targeting to different types of molecules and molecular patterns. TRIMs can homo- and hetero-oligomerize and may provide a combinatorial power in recognition of complex autophagic targets. Our study shows that the large family of TRIMs endows cells with the ability to deploy autophagy with great precision and greatly increases the repertoire of currently known autophagic receptors.
Figure 1.
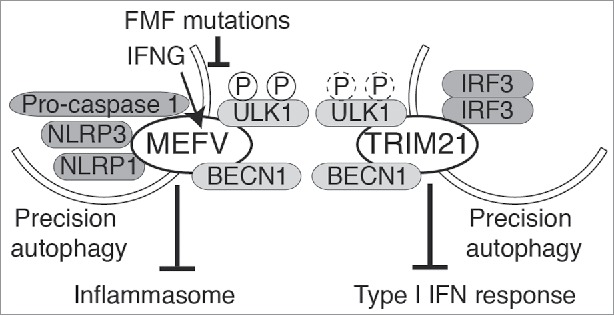
Precision autophagy prevents excessive inflammation. TRIM20/MEFV and TRIM21, both contributing to autophagy stimulated in response to IFNG, directly bind their respective cargo and recruit autophagic machinery (including ULK1 and BECN1) to execute sequestration and degradation. MEFV/TRIM20, mutated in familial Mediterranean fever, targets the inflammasome components for autophagic degradation, whereas TRIM21, associated with autoimmune diseases, targets dimerized IRF3, to suppress inflammasome activity and type I IFN response, respectively. ULK1 in the MEFV autophagic platform gets activated upon recognition of its specific targets.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.