

Abstract
Background
Ethanol (EtOH) neurotoxicity can result in devastating effects on brain and behavior by disrupting homeostatic signaling cascades and inducing cell death. One such mechanism involves double-stranded RNA activated protein kinase (PKR), a primary regulator of protein translation and cell viability in the presence of a virus or other external stimuli. EtOH-mediated up-regulation of interferon-gamma (IFNγ; the oxidative stress-inducible regulator of PKR), PKR, and its target, p53, are still being fully elucidated.
Methods
Using western blot analysis, immunofluorescence and linear regression analyses, changes in the IFNγ-PKR-p53 pathway following chronic ethanol treatment in the frontal cortex of rodents were examined. The role of PKR on cell viability was also assessed in EtOH-treated cells using PKR overexpression vector and PKR inhibitor.
Results
In rats chronically-fed ethanol, PKR, phosphorylated PKR (p-PKR), IFNγ, and p53 were significantly increased following chronic ethanol exposure. Linear regression revealed a significant correlation between IFNγ and p-PKR protein levels, as well as p-PKR expression and age of EtOH exposure. Overexpression of PKR resulted in greater cell death, while use of PKR inhibitor enhanced cell viability in EtOH-treated cells.
Conclusions
Chronic EtOH exposure activates the IFNγ-PKR-p53 pathway in the frontal cortex of rodents. p-PKR expression is greater in brains of rodents exposed to EtOH at earlier ages compared to later life, suggesting a mechanism by which young brains could be more susceptible to EtOH-related brain injury. PKR and p-PKR were also colocalized in neurons and astrocytes of rats. This study provides additional insight into biochemical mechanisms underlying alcohol use related neuropathology and warrants further investigation of PKR as a potential pharmacotherapeutic target to combat EtOH-related neurotoxicity, loss of protein translation and brain injury.
Alcohol use disorders (AUDs) are the fifth leading risk factor globally for premature death and disability, and are associated with neuropsychiatric impairment which affects physical health, mood, memory, executive function and cognition (Tarter and Alterman, 1984, Nicolas et al., 1993, Dupont et al., 1996). Several molecular consequences of ethanol (EtOH)-induced brain injury have been identified, including an increase in apoptotic signaling, oxidative stress production, transcriptional or translational dysregulation and inhibition of protein synthesis, leading to cell death (Moonat et al., 2010, Ou et al., 2010, Luo, 2012, Trudell et al., 2014). Moreover, EtOH appears to be especially deleterious to young brains. Studies have illustrated that adolescent-aged brains are more vulnerable to EtOH-induced damage and cell death compared with EtOH exposure in adult-aged brains (Crews et al., 2000; Hermens et al., 2013; Kane et al., 2014). However, the biochemical mechanisms underlying these differential effects in young brains are not clearly understood.
We recently noted that expression of double stranded RNA-activated protein kinase (PKR) is higher in the postmortem brain of AUD patients who had early onset AUD compared to individuals who began drinking in later life (Johnson et al., 2015). PKR, an interferon-induced kinase which contains an N-terminal dsRNA-binding regulatory domain and C-terminal catalytic domain (Williams, 1999), is most widely known for counteracting viral infiltration of the host cell (Garcia et al., 2006). In the presence of dsRNA or other cellular stressors, PKR is activated through autophosphorylation on multiple serine/threonine residues leading to subsequent phosphorylation of target substrates, the most characterized of which being the alpha subunit of eukaryotic initiation factor 2 (eIF2apha)(Garcia et al., 2006). Activated PKR (phosphorylated PKR; [p-PKR]) impairs eIF2A activity, which thereby inhibits protein synthesis (Hovanessian, 1989, Williams, 1999) and induces cell death (Srivastava et al., 1998, Gil et al., 1999, Donze et al., 2004). In addition, PKR activates p53 via interaction with the C-terminal which may initiate apoptotic signaling cascades (Williams, 1999; Garcia et al., 2006). Several factors/pathways are responsible for regulating PKR expression and activity. One of these PKR regulators, interferon-gamma (IFN-γ), is a cytokine which induces transcription of PKR through JAK-STAT signaling and IFN-γ activated sequence elements (GAS) in the PKR promoter (Hovanessian, 1989; Tanaka and Samuel, 1994, Garcia et al., 2006, Pyo et al., 2008). IFN-γ also activates PKR catalytic activity directly through mRNA binding to PKR regulatory subunit (Cohen-Chalamish et al., 2009). In addition, SP1-like transcription factors are reported to regulate PKR transcription through Sp1-binding sites in the promoters (Das et al., 2006, Bin et al., 2011).
PKR mediates neuronal cell death in several neurodegenerative disorders apart from viral infection, including Alzhiemer’s, Huntington’s and Parkinson’s diseases (Onuki et al., 2004; Peel, 2004; Bando et al., 2005; Morel et al., 2009). Additionally, PKR and IFNγ are induced by EtOH and involved in the subsequent neurotoxic consequences of EtOH exposure (Lanzke et al., 2007, Ke et al., 2011), and mediate neural cell death from EtOH exposure (Chen et al., 2006). Furthermore, IFNγ is up-regulated by reactive oxygen species (ROS) and EtOH exposure (Lanzke et al., 2007), and is a central player in oxidative stress-induced PKR expression and subsequent apoptosis (Pyo et al., 2008). EtOH has been documented to increase p53-induced oxidative stress and cell death in neurons as well (Kuhn and Miller, 1998; Chu et al., 2007). Previous studies also note increased expression of PKR/p-PKR/p53 in fetal alcohol syndrome and that EtOH-induced PKR expression decreases cell survival in the central nervous system (CNS) (Chen et al., 2006; Chu et al., 2007). Up-regulation of PKR has also been identified as a mechanism of EtOH-induced death of cerebellar granule cells (Luo, 2012). Moreover, EtOH-induced stress of endoplasmic reticulum, as well as EtOH-induced thiamine deficiency increases PKR and PKR-related neuronal cell death (Ke et al., 2009, Ke et al., 2011). Interestingly, PKR inhibitors (PKRIs) have been demonstrated to assuage cellular stress, leading to increased cell viability (Shimazawa and Hara, 2006). Because the IFNγ-PKR pathway has been linked to EtOH-induced neurodegeneration, and our previous report indicated that this pathway is more enhanced in early-onset AUD (Johnson et al., 2015), we hypothesize that alcohol consumption at a younger age induces greater IFNγ-PKR signaling which could result in greater neurodegeneration in later life.
In this study, we extended our investigation of EtOH-induced PKR regulation by evaluating IFNγ, PKR, p-PKR, and p53 protein expression following exposure of rats to chronic EtOH in the frontal cortex. The Lieber-Decarli liquid EtOH diet used in this study is a well-established, chronic EtOH rodent model which closely mimics many EtOH-related consequences of AUD in humans (Lieber et al., 2008). The liquid diet is also ideal to produce blood EtOH concentrations less than 50mM, which is commonly found among alcoholics and result in the physiological effects of chronic EtOH use (Henriksen et al., 1997; Ou et al., 2010; Olson et al., 2013). The frontal cortex was examined in this study because it is a highly vulnerable region to EtOH-induced brain damage and subsequent neurocognitive dysfunction (Moselhy et al., 2001; Moriyama et al., 2002). In addition, we further examined the relationship between age-of-onset of EtOH exposure and the expression of p-PKR in mice. Understanding these age-specific changes in EtOH-related PKR induction and consequential signaling aberrations may provide new pharmacotherapeutic strategies to combat EtOH-related brain injury. Last, the degree to which PKR is involved in EtOH-induced cell death and drugs which could rescue EtOH-induced PKR ugpregulation is still obscure. Therefore, we manipulated PKR using an overexpression vector and a PKRI in neural cells exposed to EtOH to determine the significance of PKR on EtOH-mediated cell death.
MATERIALS AND METHODS
Animal Studies
The rodent experiments used in this study were conducted according to the Ethical Guidelines on Animal Experimentation and approved by the Institutional Animal Care and Usage Committee at the University of Mississippi Medical Center. Male Wistar rats used in the chronic EtOH feeding studies were obtained from the Indiana University Alcohol Research Center only. C57/6J mice at ages of 1, 4, or 9 months were bread at UMMC and used in the age-of-onset study. All rodents were individually housed in a temperature- and humidity-controlled room with a 12:12-h light/dark cycle with ad libitum access to food and water until the start of EtOH treatments.
Chronic EtOH Rodent Model
Male adult, EtOH-preferring Wistar rats (weighing 180 to 220 g) were fed either the Lieber-DeCarli EtOH diet or a control diet (Dyets Inc., Bethlehem, PA). After being acclimatized for three days, rats were then allowed free access to the control liquid diet without EtOH for three days and randomly assigned to the EtOH-fed (n = 9) or control (n = 9) groups. For age-of-onset analysis, male adult, C57 mice were randomly divided into groups which received the Lieber-DeCarli EtOH (n = 18) or control (n = 18) diets beginning at ages of 1 month (n=6), 4 months (n = 8), and 9 months (n =4). The liquid EtOH diet consisted of increasing amounts of EtOH over 28 days which resulted in a final diet containing 36% of calories from EtOH (6.4%) (Ou et al., 2011). The specific diet regimen was as follows: no EtOH for three days, followed by 2.5% for three days, then 5.0% for five days, and finally 6.4% for 17 days. The final EtOH diet consisted of ~14 g EtOH/day/kg which resulted in a blood EtOH concentration (BAC) of less than 50 mM (Ou et al., 2011), well within the range leading to physiological effects observed in AUD (Brinkmann et al., 2002). The EtOH diet used in this study was designed based of the well-documented Lieber-Decarli diet with a final diet composed of 36% EtOH-derived caloric intake. This paradigm reproduces many aspects of human alcohol dependence in animal models such as fatty liver, hyperlipidemia, various metabolic and endocrine disorders, tolerance to EtOH and other drugs, physical dependence and withdrawal, the fetal alcohol syndrome and liver changes (Lieber et al., 2008). The control rats were fed an isocaloric glucose liquid diet without EtOH (Dyets, Inc.) (Ou et al., 2011). All rats, in either EtOH-fed or control groups, were sacrificed by decapitation on day 29. Immediately following decapitation, one hemisphere containing the entire half of the brain was directly placed in 4% paraformaldehyde to be fixed for immunohistochemical studies, while the frontal cortex of the remaining hemisphere was dissected out and stored at −80°C until further use (Ou et al., 2011).
Western Blot Analysis
Forty micrograms of total protein from each rat was separated on 10.5% SDS-polyacrylamide gels by electrophoresis and transferred to PVDF membranes in a BioRad mini tank apparatus (Ou et al., 2011). After transfer, the membranes were blocked at room temperature for 1h with 5% nonfat dry milk. Membranes were then incubated with either rabbit polyclonal anti-PKR (Santa Cruz, Dallas, TX), rabbit monoclonal anti-p-PKR (Abcam, Cambridge, UK), rabbit monoclonal IFNγ (Abcam), or mouse monoclonal p53 (Santa Cruz) primary antibodies overnight at 4 °C. Following incubation with respective HRP-conjugated secondary antibodies, goat anti-mouse IgG or goat anti-rabbit IgG (Santa Cruz), bands were visualized by horseradish peroxidase reaction using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Following band visualization, the membrane was stripped for 20 min at room temperature in Restore Western Blot Stripping Buffer (Thermo Scientific), blocked, washed, then re-probed with mouse anti-actin primary antibody (1:10,000; Millipore) and HRP-conjugated goat anti-mouse IgG (Santa Cruz) as a control for sample loading. Protein bands were visualized by the ChemiDoc XRS+ Imaging System (BioRad). The optical density of the autoradiographic bands was calculated and normalized to those of actin using Quantity One Plus analysis software (Ou et al., 2011).
Triple-Labeling Immunofluorescence
The brains from control rats and rats that were exposed to chronic EtOH feeding were embedded into a Multibrain® gelatin block and cut into 40 μm coronal free-floating sections by Neuroscience Associates (Knoxville, TN). Sections were then stored at −20° C in antigen preservative (polyvinyl pyrrolidone, phosphate-buffered saline, ethylene glycol) until further use. After removal from antigen preservative, sections were washed (3x for 5min/each) with phosphate-buffered saline (PBS) and blocked with 5% normal goat serum for 1h. Sections were incubated with rabbit anti-p-PKR antibody (1:100; Sigma, St. Louis, MO) or rabbit anti-PKR antibody (K-17, 1:150; Santa Cruz, CA). Sections were additionally incubated with mouse anti-NeuN antibody (a neuronal marker; 1:400; Millipore, Billerica, MA) or chicken anti-GFAP antibody (glial fibrillary acidic protein; an astrocytic marker; 1:1000; Abcam, Cambridge, MA) overnight at 4° C followed by PBS washing (3x for 5 min/each). Secondary antibody incubation was performed with a Cy3-conjugated goat anti-rabbit IgG (red, for p-PKR and PKR), Cy5-conjugated goat anti-mouse IgG (magenta, for NeuN; Jackson ImmunoResearch Inc, West Grove, PA), or Alexa-488-conjugated goat anti-chicken IgG (green, for GFAP; Jackson ImmunoResearch Inc., West Grove, PA) at room temperature for 2h. Following antibody incubations, sections were rinsed in PBS (3x for 5 min/each), counterstained and mounted using Vectashield mounting medium with 4,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA) to analyze under a Zeiss Observer Z1 deconvolution fluorescent microscope system (Oberkochen, Germany) with Slidebook software (Intelligent Imaging Innovations, Inc. Denver CO).
Cell Culture and MTT Assay
Human neuroblastoma SH-SY5Y and glioblastoma UM 1242-G were purchased from The American Type Culture Collection (Rockville, Maryland) and seeded to 6-well plates. Culture media contained MEM or DMEM, and F12 (1:1) supplemented with 10% HyClone™ fetal bovine serum (Thermo Scientific, Waltham, MA) and Pen-Strep antibiotics (Life Technologies, Carlsbad, CA). Cells were cultured in an incubator at 37 °C and 5% CO2 throughout. All cells were seeded 1 day prior to EtOH treatment or transfections. To examine the effect of PKR on EtOH-induced cell death, the PKR expression vector and the empty expression control vector pCMV3.1 (Thermo Fisher Scientific, Waltham, MA) were transiently transfected (2μg/well) using Superfect transfection reagent (Qiagen, Venlo, Limburg) according to manufacturer’s instructions. In addition, PKR inhibitor (500nM; Millipore, Bellerica, MA) was administered to EtOH-treated and control cells. At the start of EtOH treatments, fresh medium containing 150mM of EtOH, a concentration within the standard range of in vitro studies (Ku et al., 2006, Johnson et al., 2007), was dispersed to cells 1x daily for 2 days. Control cells which did not receive EtOH received fresh medium without EtOH 1x daily for 2 days. Following 2 days of EtOH treatment, cell viability was measured by the metabolism of 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), which was incubated with cells under normal culturing conditions for 4 hours, followed by solubilization of formazan crystals by the addition of 1 mL of DMSO into each well (Johnson et al., 2007). Optical density was then measured at 572nm using a microplate reader (Biorad, Hercules, CA).
Statistical Analysis
For multiple comparisons, a 2-way ANOVA followed by Tukey’s post hoc analysis was used. For two group comparisons, a Student’s t-test was used. Pearson correlation was used for the correlation of p-PKR and age. A value of p < 0.05 was considered significant. All data are reported as mean ± SEM.
RESULTS
Chronic EtOH Exposure Increases the Protein Expression of PKR, p-PKR, IFN-γ, and p53 in the Frontal Cortex of Rats
To further elucidate the putative neurotoxic effects of chronic alcohol use on brain tissue, the frontal cortex from rats exposed to chronic EtOH treatment (four weeks) and EtOH-preferring matched controls were subjected to Western blot analysis. To fully evaluate our working hypothesis, namely that EtOH elicits activation of the IFNγ-PKR-p53 signaling cascade, the protein expression levels of PKR, activated PKR (p-PKR), IFNγ, and p53 were determined from the frontal cortex of 9 EtOH-treated rats and 9 age- and sex-matched controls. Rats that were chronically exposed to EtOH revealed a 48% increase (t(16) = 2.43; p<0.05) in PKR protein expression compared to control rats (Fig. 1). Because p-PKR is the activated, functioning form of PKR in humans and animals, we also determined p-PKR protein expression. Similarly, p-PKR protein expression was increased by 53% (t(16) = 3.12; p<0.04) in EtOH-fed rats compared to control rats (Fig. 1), suggesting a close match between the up-regulation of PKR and subsequent PKR activity following chronic EtOH exposure.
Figure 1. Chronic ethanol (EtOH) exposure increased the expression of PKR and p-PKR in the frontal cortex of rats.

Rats were fed EtOH or control liquid diets over 28 days and the protein expression of PKR and p-PKR were evaluated. (A) Representative immunoblot depicting PKR, p-PKR, and actin bands from control and EtOH-treated rats. (B) Expression of PKR was significantly increased in EtOH-treated rats compared to EtOH-preferring control rats. (C) Expression of p-PKR was significantly increased in EtOH-treated rats compared to EtOH-preferring control rats. Data presented as mean±SEM; *p<.05.
The protein expression of IFNγ was increased also by 54% (t(16) = 2.12; p<0.05) in rats exposed to chronic EtOH compared to control rats (Fig. 2A). Furthermore, to assess the relationship of IFNγ and p-PKR protein, linear regression analysis was performed from western blot data of EtOH-treated Wistar rats. Indeed, a significant positive correlation was found among EtOH-treated rats between the protein expression levels of IFNγ and p-PKR (Fig. 2B; R2 = 0.61; p<0.05), suggesting activation of IFNγ by EtOH leads to an increase in p-PKR protein expression. This indicates that the EtOH-dependent mechanism of PKR induction is tightly coupled with IFNγ transcriptional effects which is also upregulated following chronic alcohol exposure. Last, because p53 is a major downstream signaling component of PKR activity (Williams, 1999), p53 protein expression was evaluated. Rats that were exposed chronically to EtOH displayed a 58% increase in p53 compared to control rats (Figure 2C; t(16) = 2.83; p< 0.02). These results suggest that, indeed, the IFNγ-PKR-p53 signaling cascade is activated by ~50% following chronic exposure to EtOH in rats and provides evidence of a potential mechanism involving PKR on cell death that is associated with exposure to EtOH.
Figure 2. Interferon-gamma and p53 are increased by chronic ethanol (EtOH) exposure in the frontal cortex of rats.

Rats were fed EtOH or control liquid diets over 28 days and the protein expression of IFNγ and p53 were evaluated. (A) Expression of IFNγ was significantly elevated in EtOH-treated rats compared to control rats. (B) The protein expression of IFNγ is positively correlated with p-PKR (C) Expression of p53 was significantly elevated in EtOH-treated rats compared to control rats. Upper Panels in A and C: Representative western blot showing immunolabeling in control and EtOH-treated rats. Lower Panels in A and C: Quantitative graph of average optical density ratios in the EtOH-preferring control (n=9) and EtOH-fed (n=9) groups. Data presented as mean±SEM; *p<.05.
Cellular and Sub-cellular Localization of PKR and p-PKR in Neuronal and Glial Cells in the Rat Prefrontal Cortex
The expression of PKR/p-PKR in neuronal and/or glial cells in rat brain has not been previously investigated with immunohistochemistry. Triple-labeling immunofluorescence was performed (Fig. 3) using rat prefrontal cortex sections (from normal control rats and EtOH-treated rats). Figure 3A depicts cytoplasmic and nuclear localization of PKR (red) in PFC of control rats. PKR signal is enhanced in PFC of EtOH-treated rats (Fig. 3B). By contrast, Fig. 3C illustrates subcellular localization of p-PKR (red) to nuclear compartments of neurons (magenta) and astrocytes (green) in a punctate-like fashion, which is much more abundant in EtOH-treated rats compared to control rats. This suggests a non-viral, alternative function for the EtOH-induced nuclear translocation of p-PKR in neural cells, a neuropathological feature of which has been documented in several neurodegenerative disorders (Bando et al., 2005).
Figure 3. Cellular and subcellular localization of PKR and p-PKR in neurons and astrocytes.

Representative photomicrographs obtained from the pre-frontal cortex (PFC) to demonstrate the cellular distribution of PKR/p-PKR (red) immunoreactivity. The cell nucleus was counterstained by DAPI (blue). The astrocytes were labeled by GFAP immunoreactivity (IR) (green) and neurons were labeled by NeuN IR (magenta). (A) PKR IR distribution in PFC of rats fed with control diet depicting PKR localization to the cell nucleus; (B) PKR IR distribution in PFC of EtOH-treated rats depicting enhanced PKR localization to both nuclear and cytosolic compartments compared to control rats; (C) p-PKR IR distribution in PFC of rats fed with EtOH diet depicting p-PKR localization to the nucleus of neurons and both cytosol and nucleus in astrocytes.
The Protein Expression of p-PKR is Elevated in EtOH-Exposed Mice and Correlated with Age of Onset of EtOH Exposure
Previously, our group reported a negative correlation between age-of-onset of AUD and p-PKR expression in human brain (Johnson et al., 2015). To determine whether this effect also occurred in a rodent model of chronic EtOH exposure, we subjected mice at different ages to chronic EtOH treatments and determined p-PKR expression. At all ages investigated, EtOH increased the expression of p-PKR in homogenates of the frontal cortex of the C57/6J mice (p < 0.05, Fig. 4), which is consistent with results from rats (Fig. 1). A significant negative correlation was found for the age at which mice were exposed to chronic EtOH treatment and the protein expression of p-PKR (R2 = 0.6513; p < 0.001). Interestingly, mice exposed to control diet displayed a correlation coefficient R2 of 0.2626 (p < 0.03). These data imply that p-PKR expression decreases with age in rodents, while EtOH exposure augments greater p-PKR in younger rodents according to age-of-onset. Similar to our finding in humans (Johnson et al., 2014), a conserved feature of EtOH-mediated p-PKR expression is dependent upon the age of onset of exposure to EtOH, where p-PKR expression is greater in younger life and decreases with age This finding may explain why younger brains are more vulnerable to EtOH-induced neuropathology compared to mature brains in later life.
Figure 4. Phosphorylated PKR protein expression is correlated with age of onset of chronic ethanol (EtOH) exposure in rodents.

Control mice and EtOH-treated mice were assessed for p-PKR expression at various ages. Mice were exposed to EtOH beginning at 1, 4, or 9 months and were fed EtOH according to the Lieber-DeCarli diet for 28 days. p-PKR expression was measured thereafter and was negatively correlated with age of onset of EtOH exposure, with the youngest mice (1 mo. old) having the greatest p-PKR expression, which decreased with age at the time of EtOH exposure. Control mice also showed a negative correlation with respect to age and p-PKR expression. *p < 0.05 vs. control
PKR Overexpression Enhances EtOH-Induced Cell Death, but Inhibition of PKR Activity Provides Neuroprotection Against EtOH Treatment in SH-SY5Y and U-1242MG Cells
Because EtOH has been demonstrated to up-regulate the PKR and this effect is thoroughly documented to induce cell death apart from viral defense, we investigated the role of PKR on cell viability in cells exposed to EtOH. To this end, cells were exposed to either a 2-day EtOH treatment (150mM) only, EtOH treatment plus transfection of PKR expression vector, or EtOH treatment plus PKRI administration, after which all cells were subjected to MTT assay to determine the degree of viable cells following EtOH treatment compared to control, non-treated cells. Following EtOH exposure, cell viability was reduced in SH-SY5Y (61.9±3.77 vs 100±1.67; p<.001) and U-1242MG (67.1±3 vs 100±2.87; p<.001) cells by 39% and 33%, respectively, versus control cells (Fig. 5). However, EtOH-exposed cells treated with PKRI enhanced cell viability in SH-SY5Y cells (77.6±3.44 vs 61.93±3.77; p=.005) by 25% and in U-1242MG cells (81.1±2.85 vs 67.1±-3; p=.002) by 21% compared to EtOH-exposed cells without PKRI treatment (Fig. 5A). Conversely, PKR overexpression significantly decreased cell viability in EtOH-exposed SH-SY5Y cells (Fig. 5B; 31.4±10.8 vs 67.2±6.5; p=.006) by 53%, and in U-1242MG cells (Fig. 5C; 36.3±5 vs 69.5±2.36; p<.001) by 37% compared to the EtOH-exposed cells, which were transfected with the empty control vector (pCMV3.1). These findings highlight a role of PKR on cell viability under conditions of EtOH exposure, which suggests that PKR and downstream signaling components have a potent effect on cellular homeostasis when insulted. Moreover, because PKRI provided significant neuroprotection against EtOH, it may be considered a viable candidate pharmacotherapuetic intervention to prevent EtOH-related brain injury.
Figure 5. PKR is involved in ethanol (EtOH)-induced cell death.
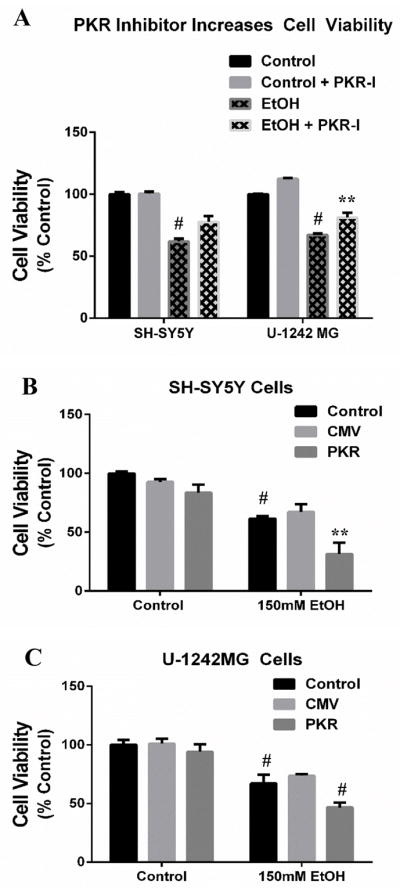
SH-SY5Y and U-1242MG cells were exposed to EtOH for 2 days. Subgroups of EtOH-exposed cells were transfected to overexpress PKR, or treated with a PKRI for the duration of EtOH exposure. (A) EtOH decreases cell viability in both SH-SY5Y and U-1242 cells, but concomitant PKRI administration significantly alleviates loss of cell viability induced by EtOH. (B) SH-SY5Y cells which overexpress PKR result in a significant loss of cell viability compared to EtOH-treated cells. pCMV empty vector was used as a transfection control vector. (C) Similarly, U-1242MG cells which overexpress PKR result in a significant loss of cell viability compared to EtOH-treated cells. pCMV empty vector was used as a transfection control vector. All data presented as mean±SEM; p*<.05; **p<.01; #p<.001.
DISCUSSION
EtOH exposure elicits a variety of mechanisms which culminate in aberrant, EtOH-inducible signaling cascades leading to loss of cellular homeostasis and cell death. One of the key molecular consequences of EtOH exposure is the production of ROS which occurs indirectly and through the metabolism of EtOH itself (Wu and Cederbaum, 2003; Das and Vasudevan, 2007; Ou et al., 2010). As a high oxidative stress load is capable of inducing IFNγ (Lanzke et al., 2007), which subsequently promotes the transcription of PKR (Pyo et al., 2008) and results in cell death, we propose that EtOH-induced ROS accumulation is the driving force underlying EtOH-induced upregulation of PKR (Fig. 6). Several studies have documented elevated PKR expression in neurodegenerative diseases such as Huntington’s, Parkinson’s, and Alzheimer’s diseases (Onuki et al., 2004; Peel, 2004; Bando et al., 2005; Morel et al., 2009). The present study offers further evidence for a conserved EtOH-responsive feature invovling PKR up-regulation in rodent models of chronic EtOH exposure. This study, combined with our previous work demonstrating elevated PKR in human postmortem brain of alcohol-dependent subjects, suggests that PKR is involved, to some degree, with the neuropathological consequences of AUD. PKR may contribute to a neurobiological insult in AUD in a manner comparable with other neurodegenerative diseases. Together, these results justify further examination of PKR and PKR-related signaling pathways, which may contribute to alcohol-induced brain injury in humans. As such, PKR is a possible candidate for pharmacological intervention in cases of alcohol misuse.
Figure 6. Proposed ethanol (EtOH)-induced PKR signaling.

The accumulation of EtOH-induced reactive oxygen species (ROS) elevates IFN-γ expression which drives the transcription of PKR at IFN-γ Activating Sequence (GAS) elements. IFN-γ also phosphorylates PKR through direct mRNA binding to the regulatory subunit. PKR transcription can also be controlled by Sp1-like transcription factors through Sp1 binding sites. Activated p-PKR can translocate to the nucleus and also induces phosphorylation of eukaryotic translation initiation factor 2α, which inhibits protein translation, leading to cell death. pPKR also increases p53 which causes increased oxidative stress and cell death.
This is the first study to investigate changes in PKR regulation in response to chronic EtOH exposure in rats. The protein expression of PKR, p-PKR, p53, and the PKR-inducer, IFNγ, was markedly increased in the frontal cortex of rats exposed to a liquid EtOH diet for 28 days, a model for chronic use of EtOH. Moreover, a positive correlation was found between expression levels of p-PKR and IFNγ, indicating that the transcriptional activator of PKR is tightly coupled to its active isoform. These findings also further elucidate chronic EtOH-responsive PKR signaling pathways, one of which subsequently activates p53. p53 is well documented to induce multiple downstream pathways resulting in apoptosis and is also a canonical EtOH-induced signaling regulator, which has been documented in rodents (Kuhn and Miller, 1998; Chu et al., 2007) and cells (de la Monte et al., 2000). A major function of p53 signaling is its ability to regulate ROS levels through activation of a third of H2O2-inducible genes, suggesting that p53 has an imperative influence over the ROS response (Desaint et al., 2004). Therefore, p53 up-regulation, mediated by EtOH-induced PKR signaling, may serve as a key contributor to ROS production associated with EtOH exposure and may likely elicit many other ROS-related secondary effects, which could collectively result in cell death.
The role of PKR on EtOH-mediated neural cell death was further characterized in this study by utilizing PKR overexpression vector and PKR inhibitor in both neuroblastoma (dopaminergic, SH-SY5Y) and glioblastoma (oligodendroglia, U-1242 MG) cell lines. In both SH-SY5Y and U-1242MG cell lines, PKR overexpression decreased cell viability following EtOH exposure compared to EtOH-only treated cells. Conversely, EtOH-treated cells with concomitant administration of PKR inhibitor (0.5μM) ameliorated viability and neuroprotection from EtOH, compared to EtOH-only treated cells. These data are supported by previous studies indicating that treatment with PKRI yielded substantial neuroprotection against EtOH-induced cell death in SK-N-MC and cerebellar granule cells (Chen et al., 2006, Ke et al., 2009).
To further characterize EtOH-induced PKR expression, we performed a series of triple-immunofluorescence labeling of PKR and p-PKR in rats exposed to chronic EtOH. PKR was confined largely to the cytosol in both neurons and astrocytes in the frontal cortex of EtOH-exposed rats. Furthermore, in EtOH-exposed rats, p-PKR-immunoreactivity was more pronounced in the nuclei of neurons, and both nuclei and cytosol of astrocytes, as compared to control rats. PKR expression displayed a uniform pattern of distribution, while p-PKR appeared punctate-like within the nucleus. p-PKR displays punctate nuclear expression in grey matter of individuals with neurodegenerative diseases (Peel, 2004; Bando et al., 2005), suggesting that EtOH-induced nuclear p-PKR expression may be a preceding indication of future neuropathology. Interestingly, PKR has also been detected endogenously in the nucleus of cells and suggested to be involved in ribosome biosynthesis (Jeffrey et al., 1995). Although PKR has been identified in both nuclear and cytoplasmic fractions, most functions belonging to PKR occur in the cytoplasm; hence, the role of nuclear PKR after exposure to EtOH remains unclear. However, several studies have attributed clinical and pathological significance to nuclear PKR, which has been documented in tissues from patients with Alzheimer and other neurodegenerative diseases (Peel, 2004). This may be of significance, since nuclear translocation of p-PKR has been identified as a critical factor of neural cell death in various neuropathologies (Bando et al., 2005). It also implicates other possible functions of PKR, such as transcriptional regulation, outside of its known cytosolic roles.
A major finding from this study is that greater expression of p-PKR protein is associated with an early age of onset of EtOH exposure in rodents. Previously, we discovered that p-PKR protein was elevated to a higher level in human subjects with AUD who began drinking chronically at younger vs. older ages (Johnson et al., 2015). Thus, mechanisms leading to higher p-PKR expression may become exacerbated with early life exposure to EtOH, leading to higher p-PKR expression and greater risk of brain injury in later life. Interestingly, p-PKR expression was also significantly correlated with younger age in control rodents, suggesting that p-PKR expression naturally declines with age. This may explain why younger brains are more vulnerable to the pejorative biological effects of EtOH (Crews et al., 2000; De Bellis et al., 2000), which are further augmented by PKR up-regulation in early life.
PKR has emerged as a novel pharmacotherapuetic target to prevent EtOH-related brain injury, as determined in the current study and by the work done by many other laboratories. The current study further demonstrates a conserved mechanism of PKR whereby EtOH exposure elicits the up-regulation of the IFNγ-PKR-p53 pathway in rodents in a manner similar to AUD subjects (Johnson et al., 2015). Since the IFNγ-PKR can be activated by oxidative stress, it is a possibility that EtOH-related oxidative stress is a mechanism driving the changes we have observed in this study (Fig. 6). In addition, several studies have documented dysfunctions in protein synthesis associated with EtOH exposure (Hong-Brown et al., 2001; Lang et al., 2001). Due to its predominant role in inhibiting protein synthesis, PKR could be culpable for the effects seen in these studies. Although further studies to outline the intricacies of the EtOH-responsive effect of PKR and subsequent signaling aberrations are justified, PKR may serve as a potential pharmacological target to disengage downstream molecular events from the pathological effects of EtOH.
Acknowledgments
The authors appreciate the support provided by the Laboratory Animal Facilities and the Animal Behavior Core (ABC) at the University of Mississippi Medical Center. This study was supported by Public Health Service Grants R01 AA020103, P30 GM103328, an Alcohol Research Resource Award Grant (R24 AA015512-02), a Carraway Foundation Grant, and a MIND center subcontract to JMW. The authors have no interest conflict for this publication.
References
- Bando Y, Onuki R, Katayama T, Manabe T, Kudo T, Taira K, Tohyama M. Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson’s disease and Huntington’s disease. Neurochemistry international. 2005;46:11–18. doi: 10.1016/j.neuint.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Bin L, Howell MD, Kim BE, Streib JE, Hall CF, Leung DY. Specificity protein 1 is pivotal in the skin’s antiviral response. The Journal of allergy and clinical immunology. 2011;127:430–438. e431–432. doi: 10.1016/j.jaci.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann B, Beike J, Kohler H, Heinecke A, Bajanowski T. Incidence of alcohol dependence among drunken drivers. Drug and alcohol dependence. 2002;66:7–10. doi: 10.1016/s0376-8716(01)00177-6. [DOI] [PubMed] [Google Scholar]
- Chen G, Ma C, Bower KA, Ke Z, Luo J. Interaction between RAX and PKR modulates the effect of EtOH on protein synthesis and survival of neurons. The Journal of biological chemistry. 2006;281:15909–15915. doi: 10.1074/jbc.M600612200. [DOI] [PubMed] [Google Scholar]
- Chu J, Tong M, de la Monte SM. Chronic EtOH exposure causes mitochondrial dysfunction and oxidative stress in immature central nervous system neurons. Acta neuropathologica. 2007;113:659–673. doi: 10.1007/s00401-007-0199-4. [DOI] [PubMed] [Google Scholar]
- Cohen-Chalamish S, Hasson A, Weinberg D, Namer LS, Banai Y, Osman F, Kaempfer R. Dynamic refolding of IFN-gamma mRNA enables it to function as PKR activator and translation template. Nature chemical biology. 2009;5:896–903. doi: 10.1038/nchembio.234. [DOI] [PubMed] [Google Scholar]
- Crews FT, Braun CJ, Hoplight B, Switzer RC, III, Knapp DJ. Binge EtOH consumption causes differential brain damage in young adolescent rats compared with adult rats. Alcoholism, clinical and experimental research. 2000;24(11):1712–1723. [PubMed] [Google Scholar]
- Das S, Ward SV, Tacke RS, Suske G, Samuel CE. Activation of the RNA-dependent protein kinase PKR promoter in the absence of interferon is dependent upon Sp proteins. The Journal of biological chemistry. 2006;281:3244–3253. doi: 10.1074/jbc.M510612200. [DOI] [PubMed] [Google Scholar]
- Das SK, Vasudevan DM. Alcohol-induced oxidative stress. Life sciences. 2007;81:177–187. doi: 10.1016/j.lfs.2007.05.005. [DOI] [PubMed] [Google Scholar]
- De Bellis MD, Clark DB, Beers SR, Soloff PH, Boring AM, Hall J, Kersh A, Keshavan MS. Hippocampal Volume in adolescent-onset alcohol use disorders. The American Journal of Psychiatry. 2000;157(5):737–744. doi: 10.1176/appi.ajp.157.5.737. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Ganju N, Banerjee K, Brown NV, Luong T, Wands JR. Partial rescue of EtOH-induced neuronal apoptosis by growth factor activation of phosphoinositol-3-kinase. Alcoholism, clinical and experimental research. 2000;24:716–726. [PubMed] [Google Scholar]
- Desaint S, Luriau S, Aude JC, Rousselet G, Toledano MB. Mammalian antioxidant defenses are not inducible by H2O2. The Journal of biological chemistry. 2004;279:31157–31163. doi: 10.1074/jbc.M401888200. [DOI] [PubMed] [Google Scholar]
- Donze O, Deng J, Curran J, Sladek R, Picard D, Sonenberg N. The protein kinase PKR: a molecular clock that sequentially activates survival and death programs. The EMBO journal. 2004;23:564–571. doi: 10.1038/sj.emboj.7600078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont RM, Rourke SB, Grant I, Lehr PP, Reed RJ, Challakere K, Lamoureux G, Halpern S. Single photon emission computed tomography with iodoamphetamine-123 and neuropsychological studies in long-term abstinent alcoholics. Psychiatry Res. 1996;67:99–111. doi: 10.1016/0925-4927(96)02769-2. [DOI] [PubMed] [Google Scholar]
- Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiology and molecular biology reviews: MMBR. 2006;70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J, Alcami J, Esteban M. Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the alpha subunit of eukaryotic translation initiation factor 2 and NF-kappaB. Molecular and cellular biology. 1999;19:4653–4663. doi: 10.1128/mcb.19.7.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen JH, Gronbaek M, Moller S, Bendtsen F, Becker U. Carbohydrate deficient transferrin (CDT) in alcoholic cirrhosis: a kinetic study. J Hepatol. 1997;26:287–292. doi: 10.1016/s0168-8278(97)80043-8. [DOI] [PubMed] [Google Scholar]
- Hermens DF, Lagopoulos J, Tobias-Webb J, De Regt T, Dore G, Juckes L, Latt N, Hickie IB. Pathways to alcohol-induced brain impairment in young people: A review. Cortex. 2013;49(1):3–17. doi: 10.1016/j.cortex.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Hong-Brown LQ, Frost RA, Lang CH. Alcohol impairs protein synthesis and degradation in cultured skeletal muscle cells. Alcoholism, clinical and experimental research. 2001;25:1373–1382. [PubMed] [Google Scholar]
- Hovanessian AG. The double stranded RNA-activated protein kinase induced by interferon: dsRNA-PK. Journal of interferon research. 1989;9:641–647. doi: 10.1089/jir.1989.9.641. [DOI] [PubMed] [Google Scholar]
- Jeffrey IW, Kadereit S, Meurs EF, Metzger T, Bachmann M, Schwemmle M, Hovanessian AG, Clemens MJ. Nuclear localization of the interferon-inducible protein kinase PKR in human cells and transfected mouse cells. Experimental cell research. 1995;218:17–27. doi: 10.1006/excr.1995.1126. [DOI] [PubMed] [Google Scholar]
- Johnson S, Duncan J, Hussain SA, Chen G, Luo J, McLaurin C, May W, Rajkowska G, Ou XM, Stockmeier CA, Wang JM. The IFNgamma-PKR pathway in the prefrontal cortex reactions to chronic excessive alcohol use. Alcoholism, clinical and experimental research. 2015;39:476–484. doi: 10.1111/acer.12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Z, Wang X, Liu Y, Fan Z, Chen G, Xu M, Bower KA, Frank JA, Li M, Fang S, Shi X, Luo J. EtOH induces endoplasmic reticulum stress in the developing brain. Alcoholism, clinical and experimental research. 2011;35:1574–1583. doi: 10.1111/j.1530-0277.2011.01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke ZJ, Wang X, Fan Z, Luo J. EtOH promotes thiamine deficiency-induced neuronal death: involvement of double-stranded RNA-activated protein kinase. Alcoholism, clinical and experimental research. 2009;33:1097–1103. doi: 10.1111/j.1530-0277.2009.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn PE, Miller MW. Expression of p53 and ALZ-50 immunoreactivity in rat cortex: effect of prenatal exposure to EtOH. Experimental neurology. 1998;154:418–429. doi: 10.1006/exnr.1998.6907. [DOI] [PubMed] [Google Scholar]
- Lang CH, Kimball SR, Frost RA, Vary TC. Alcohol myopathy: impairment of protein synthesis and translation initiation. The international journal of biochemistry & cell biology. 2001;33:457–473. doi: 10.1016/s1357-2725(00)00081-9. [DOI] [PubMed] [Google Scholar]
- Lanzke N, Kleinwachter R, Kerschischnik S, Sargsyan L, Groneberg DA, Kamradt T, Liesenfeld O, Krenn V, Sander M, Spies C. Differential effects of EtOH on IFN-gamma- and TNF-alpha-producing splenic T lymphocytes in a murine model of gram-negative pneumonia. Addiction biology. 2007;12:59–68. doi: 10.1111/j.1369-1600.2006.00042.x. [DOI] [PubMed] [Google Scholar]
- Li TK, Lumeng L, McBride WJ, Waller MB. Progress toward a voluntary oral consumption model of alcoholism. Drug Alcohol Depend. 1979;4:45–60. doi: 10.1016/0376-8716(79)90040-1. [DOI] [PubMed] [Google Scholar]
- Lieber CS, Leo MA, Wang X, Decarli LM. Effect of chronic alcohol consumption on Hepatic SIRT1 and PGC-1alpha in rats. Biochemical and biophysical research communications. 2008;370:44–48. doi: 10.1016/j.bbrc.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Luo J. Mechanisms of EtOH-induced death of cerebellar granule cells. Cerebellum. 2012;11:145–154. doi: 10.1007/s12311-010-0219-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonat S, Starkman BG, Sakharkar A, Pandey SC. Neuroscience of alcoholism: molecular and cellular mechanisms. Cellular and molecular life sciences: CMLS. 2010;67:73–88. doi: 10.1007/s00018-009-0135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel M, Couturier J, Lafay-Chebassier C, Paccalin M, Page G. PKR, the double stranded RNA-dependent protein kinase as a critical target in Alzheimer’s disease. Journal of cellular and molecular medicine. 2009;13:1476–1488. doi: 10.1111/j.1582-4934.2009.00849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama Y, Mimura M, Kato M, Yoshino A, Hara T, Kashima H, Kato A, Watanabe A. Executive dysfunction and clinical outcome in chronic alcoholics. Alcoholism, clinical and experimental research. 2002;26:1239–1244. doi: 10.1097/01.ALC.0000026103.08053.86. [DOI] [PubMed] [Google Scholar]
- Moselhy HF, Georgiou G, Kahn A. Frontal lobe changes in alcoholism: a review of the literature. Alcohol and alcoholism. 2001;36:357–368. doi: 10.1093/alcalc/36.5.357. [DOI] [PubMed] [Google Scholar]
- Nicolas JM, Catafau AM, Estruch R, Lomena FJ, Salamero M, Herranz R, Monforte R, Cardenal C, Urbano-Marquez A. Regional cerebral blood flow-SPECT in chronic alcoholism: relation to neuropsychological testing. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 1993;34:1452–1459. [PubMed] [Google Scholar]
- Olson KN, Smith SW, Kloss JS, Ho JD, Apple FS. Relationship between blood alcohol concentration and observable symptoms of intoxication in patients presenting to an emergency department. Alcohol and Alcoholism. 2013;48(4):386–389. doi: 10.1093/alcalc/agt042. [DOI] [PubMed] [Google Scholar]
- Onuki R, Bando Y, Suyama E, Katayama T, Kawasaki H, Baba T, Tohyama M, Taira K. An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer’s disease. The EMBO journal. 2004;23:959–968. doi: 10.1038/sj.emboj.7600049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou XM, Johnson C, Lu D, Johnson S, Paul IA, Austin MC, Iyo AH, Miguel-Hidalgo JJ, Luo J, Bell RL, Grunewald M, Wang J, Sittman DB. EtOH increases TIEG2-MAO B cell death cascade in the prefrontal cortex of EtOH-preferring rats. Neurotoxicity research. 2011;19:511–518. doi: 10.1007/s12640-010-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou XM, Stockmeier CA, Meltzer HY, Overholser JC, Jurjus GJ, Dieter L, Chen K, Lu D, Johnson C, Youdim MB, Austin MC, Luo J, Sawa A, May W, Shih JC. A novel role for glyceraldehyde-3-phosphate dehydrogenase and monoamine oxidase B cascade in EtOH-induced cellular damage. Biological psychiatry. 2010;67:855–863. doi: 10.1016/j.biopsych.2009.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel AL. PKR activation in neurodegenerative disease. Journal of neuropathology and experimental neurology. 2004;63:97–105. doi: 10.1093/jnen/63.2.97. [DOI] [PubMed] [Google Scholar]
- Pyo CW, Lee SH, Choi SY. Oxidative stress induces PKR-dependent apoptosis via IFN-gamma activation signaling in Jurkat T cells. Biochemical and biophysical research communications. 2008;377:1001–1006. doi: 10.1016/j.bbrc.2008.10.103. [DOI] [PubMed] [Google Scholar]
- Shimazawa M, Hara H. Inhibitor of double stranded RNA-dependent protein kinase protects against cell damage induced by ER stress. Neuroscience letters. 2006;409:192–195. doi: 10.1016/j.neulet.2006.09.074. [DOI] [PubMed] [Google Scholar]
- Srivastava SP, Kumar KU, Kaufman RJ. Phosphorylation of Eukaryotic Translation Initiation Factor 2 Mediates Apoptosis in Response to Activation of the Double-stranded RNA-dependent Protein Kinase. Journal of Biological Chemistry. 1998;273:2416–2423. doi: 10.1074/jbc.273.4.2416. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Samuel CE. Mechanism of interferon action: structure of the mouse PKR gene encoding the interferon-inducible RNA-dependent protein kinase. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:7995–7999. doi: 10.1073/pnas.91.17.7995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarter RE, Alterman AI. Neuropsychological deficits in alcoholics: etiological considerations. Journal of studies on alcohol. 1984;45:1–9. doi: 10.15288/jsa.1984.45.1. [DOI] [PubMed] [Google Scholar]
- Trudell JR, Messing RO, Mayfield J, Harris RA. Alcohol dependence: molecular and behavioral evidence. Trends in pharmacological sciences. 2014;35:317–323. doi: 10.1016/j.tips.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch KA, Carson A, Lawrie SM. Brain structure in adolescents and young adults with alcohol problems: Systematic review of imaging studies. Alcohol and Alcoholism. 2013;48(4):433–444. doi: 10.1093/alcalc/agt037. [DOI] [PubMed] [Google Scholar]
- Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- Wu D, Cederbaum AI. Alcohol, oxidative stress, and free radical damage. Alcohol research & health: the journal of the National Institute on Alcohol Abuse and Alcoholism. 2003;27:277–284. [PMC free article] [PubMed] [Google Scholar]