

Abstract
Smooth muscle cell (SMC) proliferation and fibrosis contribute to the development of advanced atherosclerotic lesions. Oxidative stress caused by increased production or unphysiological location of reactive oxygen species (ROS) is a known major pathomechanism. However, in atherosclerosis, in particular under hyperglycaemic/diabetic conditions, the hydrogen peroxide-producing NADPH oxidase type 4 (NOX4) is protective. Here we aim to elucidate the mechanisms underlying this paradoxical atheroprotection of vascular smooth muscle NOX4 under conditions of normoand hyperglycaemia both in vivo and ex vivo. Following 20-weeks of streptozotocin-induced diabetes, Apoe−/− mice showed a reduction in SM-alpha-actin and calponin gene expression with concomitant increases in platelet-derived growth factor (PDGF), osteopontin (OPN) and the extracellular matrix (ECM) protein fibronectin when compared to non-diabetic controls. Genetic deletion of Nox4 (Nox4−/− Apoe−/− ) exacerbated diabetes-induced expression of PDGF, OPN, collagen I, and proliferation marker Ki67. Aortic SMCs isolated from NOX4-deficient mice exhibited a dedifferentiated phenotype including loss of contractile gene expression, increased proliferation and ECM production as well as elevated levels of NOX1-associated ROS. Mechanistic studies revealed that elevated PDGF signalling in NOX4-deficient SMCs mediated the loss of calponin and increase in fibronectin, while the upregulation of NOX1 was associated with the increased expression of OPN and markers of proliferation. These findings demonstrate that NOX4 actively regulates SMC pathophysiological responses in diabetic Apoe−/− mice and in primary mouse SMCs through the activities of PDGF and NOX1.
Keywords: Vascular smooth muscle cell, NOX4, Diabetes, Atherosclerosis
1. Introduction
Reactive oxygen species (ROS) produced by NADPH oxidase (NOX) enzymes are major contributors to oxidative stress and redox signalling in the vasculature [1]. The NOX4 isoform constitutively generates hydrogen peroxide (H2O2) that under non-disease conditions is produced at tightly controlled levels in discrete subcellular locations to mediate various physiological processes in vascular cells including the maintenance of the contractile phenotype in smooth muscle cells (SMCs) [2]. Pathological activation of NOX enzymes, particularly NOX4 has been shown to affect differentiation such as preadipocytes [3] and promote inflammation in various disease models, including osteoporosis and vascular disease [4,5]. In the context of diabetes NOX enzymes are known to play central roles in vascular disease progression [6]. Chronic hyperglycaemia as seen in type 1 diabetes accelerates the progression of atherosclerosis from early to advanced lesions with an increasing importance of cell types capable of extracellular matrix (ECM) synthesis and remodelling. We and others have demonstrated that atherosclerosis development can be modulated in response to NOX4 deletion or overexpression. Whereby overexpression of NOX4 results in a reduction in atherosclerosis [7], while genetic deletion has been shown to increase atherosclerosis development [8–10]. The high degree of plasticity exhibited by vascular SMCs allows for a diversity of phenotypes in response to chronic exposure to the diabetic milieu; however, the potential role of NOX4 in this process remains relatively unexplored.
The significant contribution of vascular SMCs to atherosclerosis plaque development has gained recent attention with the refinement of approaches to identify [11] and track [12] phenotypically modulated SMCs. SMC phenotype switching is characterised by a loss of smooth muscle markers and a gain of proliferative and synthetic activity mediated by factors such as PDGF and OPN [13–15]. Interestingly, SMCs isolated and cultured from streptozotocin (STZ)-diabetic rats show decreased SM-α-actin, elevated superoxide production, increased OPN expression and enhanced PDGF-mediated proliferation [16,17], consistent with such a switch in the SMC phenotype. Few studies have directly tested the effects of high glucose (HG) on PDGF-BB expression; however, HG has been shown to increase PDGF-BB receptor binding and downstream signalling events [18]. Increased expression of OPN has also been demonstrated in the medial layer of arteries from diabetic humans and rodents [19]. Previous work by Lyle et al. [20] identified a link between H2O2 and OPN expression in SMCs; however, this has not yet been investigated in the context of diabetes.
Better understanding the role of NOX4 in SMC pathobiology would provide greater insight into the controversial, and potentially vasoprotective, role of NOX4 in diabetes-associated atherosclerotic disease. Therefore, we investigated the role of NOX4 in vascular SMC fibrosis and proliferation in Apoe−/− mice 20-weeks after STZ-induced diabetes and complemented these in vivo studies with additional mechanistic experiments in mouse aortic SMCs with and without HG treatment.
2. Materials and methods
2.1. Generation of experimental mice
All animal procedures were approved by the Alfred Medical Research and Education Precinct (AMREP) Animal Ethics Committee in accordance with the National Health and Medical Research Council of Australia guidelines. The animals were maintained on a 12-hour light/dark cycle with unrestricted access to water and standard chow (Specialty Feeds, Glen Forrest, WA, Australia) under pathogen-free conditions in the Precinct Animal Centre of the Baker IDI Heart and Diabetes Institute. Nox4−/− [21] mice were backcrossed with Apoe−/− mice on the C57BL/6J background (B6.129P2-Apoetm1Unc /J, Jax Labs, Bar Harbor, ME, USA) for 10 generations to generate Nox4−/− Apoe−/− double knockout animals (Transnetyx, USA); as previously described in [9]. Diabetes was induced in six-week-old Nox4−/− Apoe−/− and Nox4−/− Apoe−/− wildtype male mice by five daily i.p. injections of streptozotocin [55 mg/kg dissolved in citrate buffer (Sigma-Aldrich, St Louis, MO, USA)] [22]. Mice with a blood glucose level ≥15 mmol/L were deemed diabetic and included in the study. After 20 weeks, the mice were killed by an i.p. injection of sodium pentobarbital (100 mg/kg; Euthatal, Sigma-Aldrich, Castle Hill, NSW, Australia).
2.2. Isolation and culture of primary aortic smooth muscle cells (SMCs)
SMCs from mouse thoracic aortas (stripped of adventitia) were isolated by enzymatic digestion and grown in DMEM with 10% fetal bovine serum (FBS) as described previously [23]. Cells from passages 4–8 were grown to 70–80% confluence for experiments, unless otherwise specified.
2.3. Gene expression analysis by Quantitative RT-PCR
Total RNA was extracted from frozen aortas and SMCs using TRIzol reagent (Invitrogen Australia, Mt Waverly, VIC, Australia) and reversed transcribed to cDNA using the Superscript First Strand Synthesis System (Life Technologies BRL, Grand Island, NY, USA). Quantitative real-time PCRs were performed using a Taqman system (ABI Prism 7500; Perkin-Elmer, Poster City, CA, USA) for OPN, PDGFbb, fibronectin, collagen I, CTGF, TGFβ, PCNA and Nox1 and genes. The SYBR Green PCR Master Mix (Applied Biosystems, Darmstadt, Germany) system was used for transgelin, calponin and Ki67 genes. Probes and primers were designed using a Primer Express program (primer sequences in Supplemental Table 1) and purchased from Applied Biosystems (ABI, Foster City, CA, USA). Gene expression was normalised relative to the housekeeping gene, 18S ribosomal RNA (18S rRNA Taqman Control Reagent kit) and expressed as fold inductions relative to wild-type non-diabetic mice that were arbitrarily assigned a value of 1.
2.4. Western blot analyses
Protein lysates of whole aortas (n=4–6/group) and whole cells (average of 3–4 independent experiments) were prepared in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, 150 mM NaCl, 5 mM EDTA, 0.05% SDS, 0.5% deoxycholate) with fresh protease and phosphatase inhibitors (PMSF, aprotinin, leupeptin) as described previously [9,24]. For proteins secreted into culture supernatant (fibronectin and collagen) proteins, SMC were grown in DMEM 0.1% FBS supplemented with L-ascorbic acid and beta-aminopropionitrile (50 μg /ml of each) for three days. Supernatant proteins were concentrated via sequential centrifugation in Amicon Ultra Centrifugal Filters as per the manufacturers instructions (Merck Millipore, Billerica, MA, USA). Concentration of protein in all extracts was determined by Bradford Assay. Equal amounts of protein diluted in Laemmli buffer were separated by SDS-PAGE and transferred to a nitrocellulose membrane. All blots were incubated with primary antibodies overnight at 4 °C and secondary antibodies for 1 h at room temperature. Antibody details are outlined in Supplemental Table 2. SMC proteins were visualised by enhanced chemiluminescence (ECL, GE) and detected with Amersham Hyperfilm ECL (GE), with band densitometry performed using ImageJ software (National Institutes of Health, Bethesda, MD). Aortic proteins were scanned by the Odyssey Infrared Imaging System (LI-COR Biosciences) using the appropriate channels and band intensity quantified using Quantity-One software (Bio-Rad).
2.5. Circulating biomarkers
The concentrations of PDGF-BB and OPN were measured in serum collected from mice 1 week prior to the experimental endpoint. Frozen samples were sent to Aushion BioSystems (Billerica, MA, USA) for quantification using the Circa™ Immunoassay platform.
2.6. Immunohistochemical staining of atherosclerotic lesions
Paraffin-fixed sections (4 μm) of aorta were stained for markers of fibrosis and proliferation: OPN, PDGF, SM-alpha-actin, fibronectin, collagen I, and Ki67. Antibody dilutions and details are outlined in Supplemental Table 3. De-paraffinized sections were quenched in 3% H2O2 in TRIS-buffered saline (pH 7.6) to inhibit endogenous peroxidase activity. Collagen I was unmasked by pepsin digest. For this, sections were incubated in dH2O for 10 min at 37 °C then transferred to a pepsin (1 mg/ml; #P6887, Sigma)-0.2 N HCl solution for 10 min at for 37 °C followed by washes in dH2O and PBS at room temperature. Sections were processed for immunohistochemistry as described previously [25]. Non-specific binding was blocked by incubation with 2.5% Normal Horse Serum (Vector Laboratories, Burlingame, CA, USA) for 30 min at room temperature. Primary antibodies were applied overnight incubation at 4 °C in a humidified chamber. Sequential blocks of Avidin and Biotin (Vector Laboratories) were followed by incubation with the appropriate biotinylated secondary antibody (Vector Laboratories) for 30 min. All sections were incubated with horseradish peroxidase-conjugated streptavidin (Vectastain Elite ABC Staining Kit; Vector Laboratories), visualised with 3,3′-diamino-benzidine tetrahydrochloride/H2O2 (DAB; Sigma-Aldrich, St Louis, MO, USA) and counterstained with Mayer’s haematoxylin prior to mounting with DePex. Images were captured on a light microscope and digitized with a high-resolution camera (Olympus BX-50; Olympus Optical) at ×10 magnification and quantified in a blinded manner (Image-Pro Plus; v6.0). Results represent positively stained area as a percentage of the total aorta area. Positive staining for Ki67 was expressed per aortic medial area (excluding plaque).
2.7. Immunocytochemical staining of MASMs
Vascular SMCs were grown on glass coverslips at 100,000 cells/well (in a 6-well plate), with each condition in triplicate. Cells were processed as previously described [26]. In short, cell were quickly washed with PBS, fixed with 10% formaldehyde for 10 min, rinsed with dH2O and permeabilized with 0.5% Triton-X-100 in PBS for 5 min. After blocking in filtered 3% bovine serum albumin for 1 h, cells were incubated with anti-OPN (1:100; see Supplemental Table III) overnight at 4 °C and then incubated with fluorophore-conjugated secondary antibody for 1 h at room temperature. After repeated washes in dH2O, stained cells were mounted in ProLong Gold Antifade Reagent (InVitrogen #P-36931) containing 4′, 6-diamidino-2-phenylindole (DAPI) for nuclei staining. Images were acquired with a Zeiss LSM 510 META Laser Scanning Confocal Microscope System using a 40 × oil objective lens and Zeiss ZEN acquisition software. Threshold settings of the confocal microscope remained constant. Controls with no primary or secondary antibody showed no fluorescence. The images are maximum intensity projections of Z-series from the base through the top of the cell.
2.8. Growth curve
MASM cells at passage 6 were seeded at 30,000 cells per well into a 6 well plate; n=3 per condition. At 24, 48, 72 and 96 h after seeding cells were trypsinised, stained with trypan blue and live cells counted by haemocytometer. Data from the 72-hr time point are presented.
2.9. Reactive oxygen species measurements
2.9.1. Amplex red (hydrogen peroxide)
Hydrogen peroxide was measured in SMCs using the Amplex red commercial kit following the manufacturer’s instructions (Molecular Probes, Eugene, OR, USA). In short, SMCs seeded in 6 well plates were exposed to high glucose (25 mM) or normal glucose DMEM (10% FBS) for 2 days prior to protein harvest. Twenty microliters of whole cell isolate, standards and blank were assayed in triplicate in black 96-well plates after addition of prewarmed (37 °C) working solution containing 100 μM Amplex red reagent and 0.2 U/ml horseradish peroxidase. Fluorescence intensity was measured in 30-minute intervals at 37 °C on a Victor fluorescence microplate reader at 544 nm excitation/590 nm emission. Data from the 120-min time point are presented as mmol of hydrogen peroxide standardized to protein concentration.
2.9.2. L-012 (superoxide)
MASM cells were seeded at 20,000 cells per well into black 96-well microplates; n=6 per condition. After 24 h, wells were washed with pre-warmed Krebs-HEPES and incubated with 100 μl of Krebs-HEPES supplemented with L-012 (Wako Chemicals, Virginia, USA) at a concentration of 100 μM at 37 °C for 15 min. Chemiluminescence was recorded every 5 min for 60 min at 37 °C on a luminometer (Berthold Technologies, Germany). Data from the 45-minute time point are presented. In experiments with NOX inhibition, Nox4−/− Apoe−/− SMCs were prepared in 96-well plates (as outlined above) and treated with the NOX1/NOX4 dual inhibitor GKT137831 (1 μM/ml) prior to the addition of L-012.
2.10. PDGF-BB treatment and inhibition studies
Cells were seeded at 50,000 cells/well in 6 well plates and allowed to adhere overnight before serum deprivation for 48 h in 0.1% FBS DMEM. Prior to treatment, PDGF and anti-PDGF neutralising antibody were incubated for 1 h at 37 °C. Nox4−/− ApoE−/− SMCs were treated with the PDGF-BB (10 ng/ml; Sigma #P 4306, Saint Louis, MO, USA) and inhibitor (67 μM/ml; Merck-Millipore #06-127, Temecula, CA, USA) cocktail while Nox4−/− Apoe−/− SMCs were treated with PDGF inhibitor (67 μg/ml) alone for 24 h. In experiments with PDGF-BB and PDGF inhibitor treatments, subconfluent MASMs were made quiescent in DMEM with 0.1% fetal bovine serum for 48 h prior to treatment with PDGF (10 ng/ml) with or without PDGF neutralising antibody (67 μM/ml) for 24 h.
2.11. Nox1 silencing
The knockdown of Nox1 in primary mouse aortic SMCs was achieved by transfection with siRNA directed against mouse Nox1 (NM_172203) (Mm_Nox1_3 FlexiTube siRNA, Qiagen, Valencia, CA, USA). SMCs were seeded at 200,000 cells/60-mm dish. The following day, non-target control (AllStars Negative Control siRNA, Qiagen) or siRNA against Nox1 were incubated separately with Lipofectamine RNAiMAX reagent (7.5 μl/well; Life Technologies, Carlsbad, CA) in Opti-MEM (Life Technologies) for 10 min. Cells were equilibrated with warmed Opti-MEM prior to the addition of non-target or Nox1 siRNA mixture (final concentration of 10 nM/well). Treatment conditions were replicated in triplicate. After overnight of incubation with siRNA, the medium was replaced with fresh DMEM with 0.1% FBS. Cells were harvested for RNA isolation 2 days after transfection. The knockdown efficiency in Nox4−/− Apoe−/− SMCs was verified by qRT-PCR to be greater than 50% for Nox1 mRNA.
2.12. H2O2 treatment
SMCs were treated with H2O2 to assess the H2O2-dependent effects of Nox4 deletion. After 48 h of exposure to normal or high glucose media in T75 flasks, cells were trypsinized and seeded at 200,000 cells/well in 6 well plate. The following day, SMCs were treated with H2O2 (100 μM; BSPA5.500, LabServ Thermo Fisher Scientific Australia) for 4 h and harvested immediately for RNA extraction. Each condition was repeated in triplicate.
2.13. Statistical analysis
All data are presented as mean ± SEM. Groups were analysed by one-way and two-way ANOVA with an LSD post hoc test for multiple comparison of the means using SPSS Statistics version 22 (IBM, St Leonards, NSW, Australia). A p value of <0.05 was considered statistically significant.
3. Results
3.1. Altered SMC phenotypic marker expression in the aortas from diabetic NOX4-deficient mice
The effects of diabetes and NOX4 on characteristic markers of the SMC phenotype were first assessed in vivo using aortas from non-diabetic and diabetic wildtype (Nox4−/− Apoe−/− ) and double knockout (Nox4−/− Apoe−/− ) mice. Diabetes was associated with reduced aortic gene expression of the hallmark SMC contractility markers, SM-α-actin and calponin (Fig. 1A, left panels). Lack of NOX4 also reduced SM-α-actin and calponin in non-diabetic mice while further reducing SM-α-actin in diabetic mice when compared to Nox4−/− Apoe−/− counterparts. The mediators of SMC phenotype switching, PDGF-BB and OPN, were elevated in diabetic mouse aorta (Fig. 1A, right panels). Furthermore, NOX4 deficiency markedly increased both PDGF-BB gene and protein expression in the diabetic setting (Fig. 1A and B). In contrast, only diabetic Nox4−/− Apoe−/− mice showed increased aortic OPN gene (Fig. 1A, right panel) and protein levels (Fig. 1D). Consistent with observations in the aorta, NOX4-deficient mice displayed increased circulating levels of PDGF (Fig. 1E, left panel). Serum OPN levels were increased in response to diabetes in Nox4−/− Apoe−/− mice and basally by NOX4 deficiency in non-diabetic mice (Fig. 1E, right panel and Supplementary Fig. 1). These data suggest that both diabetes and NOX4 modulate the expression of contractility genes and PDGF-BB in the aorta and circulation. In contrast, regulation of aortic OPN expression by NOX4 is only apparent in the context of diabetes.
Fig. 1.
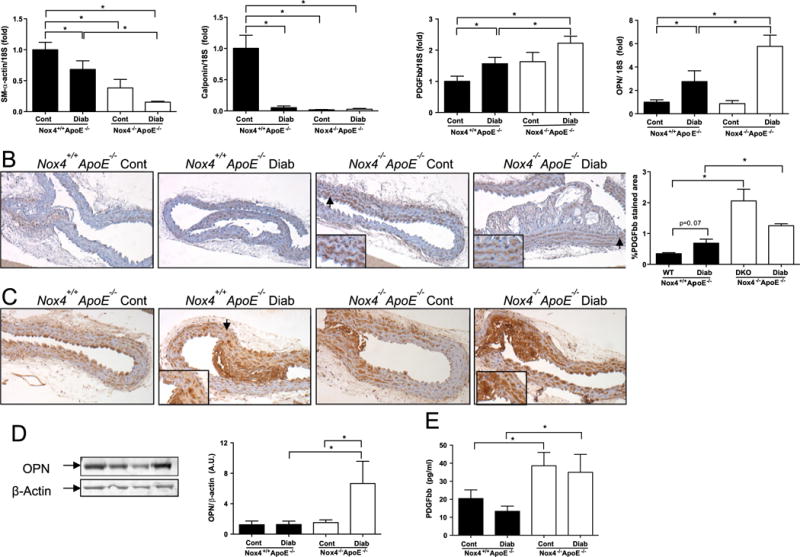
Downregulation of contractile SMC markers with concomitant upregulation of PDGF-BB and osteopontin (OPN) in aortas from diabetic NOX4-deficient mice. (A) mRNA levels of SM-α-actin, calponin, PDGF-BB, and OPN relative to 18S in aortas from NOX4-deficient (Nox4+/+ Apoe−/− , filled bars) and control (NOX4−/− Apoe−/− , open bars) mice measured by quantitative real time polymerase chain reaction (qRT-PCR). (B) Representative immunohistochemistry images and analysis showing PDGF-BB staining in medial layer of aortas from Nox4+/+ Apoe−/− and NOX4−/− Apoe−/− mice; Brown: PDGF-BB, blue: nuclear counterstaining with Hematoxylin. OPN protein expression in aortas from Nox4+/+ Apoe−/− and NOX4−/− Apoe−/− mice as assessed by (C) immunohistochemistry and (D) immunoblotting with fold changes normalised to the β-actin level. (E) Circulating levels of PDGF-BB protein in the serum from NOX4-deficient mice compared to control mice. Values are mean±SEM (n=6–8). *p<0.05.
3.2. Expression of fibrosis and proliferation is increased in NOX4-deficient SMCs
We next investigated responses downstream of PDGF and OPN signalling in SMCs involved in atherosclerotic plaque development, fibrosis and proliferation. Diabetes induced expression of the extracellular matrix (ECM) protein fibronectin in the aortas of Nox4−/− Apoe−/− and Nox4−/− Apoe−/− mice (Fig. 2A). An additional fibrosis marker Collagen I was only upregulated in Nox4−/− Apoe−/− mice with diabetes (Fig. 2B). Similarly, expression of the proliferative marker Ki67 followed a trend to increase in diabetic mice that lacked NOX4 (Fig. 2C). These findings suggest a role for NOX4 in the suppression of Collagen I and potentially Ki67 expression in the vasculature exposed to the diabetic milieu.
Fig. 2.
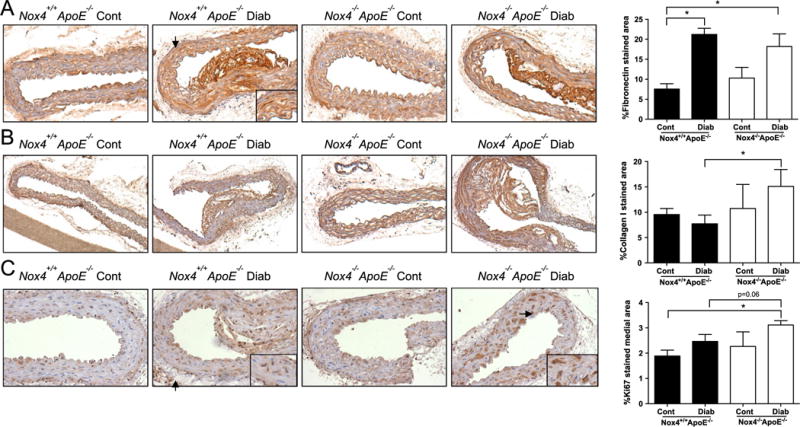
Elevated expression of fibrosis and proliferation markers in the aortas from diabetic NOX4-deficient mice. Representative images and analysis (right) of immunohistochemical staining for (A) Fibronectin, (B) Collagen I and (C) Ki67 in the aortas of Nox4+/+ Apoe−/− (filled bars) and Nox4−/− Apoe−/− (open bars) mice. Values are mean positively stained area±SEM (n=6). *p<0.05.
3.3. Enhanced PDGF-BB and osteopontin (OPN) expression in NOX4-deficient SMCs
To study the potential mechanisms underpinning NOX4- and diabetes- mediated SMC phenotype modulation, we isolated aortic SMCs from Nox4−/− Apoe−/− and Nox4−/− Apoe−/− mice and cultured these cells under normal glucose (NG; 5.5 mM) and high glucose (HG; 25 mM) conditions. In contrast to the reduced contractile gene expression seen in whole aortas from diabetic mice, treatment of SMCs with HG did not alter SM-alpha-actin expression and led to a slight elevation in both transgelin and calponin mRNA levels (Fig. 3A). Consistent with the in vivo studies, NOX4 deficiency significantly decreased contractile gene expression (Fig. 3A) as well as calponin protein expression (Fig. 3B), further supporting a prominent role for NOX4 in the maintenance of SMC phenotype. Treatment of Nox4−/− Apoe−/− SMCs with HG did not mirror the effects of diabetes on aortic PDGF and OPN gene expression. However, consistent with our in vivo findings in Nox4−/− Apoe−/− aortas, SMCs from these Nox4−/− Apoe−/− mice displayed heightened expression of PDGF and OPN (Fig. 3C). Moreover, only Nox4−/− Apoe−/− SMCs cultured under HG conditions demonstrated a small increase in OPN protein expression (Fig. 3D) and a distinct intracellular OPN distribution pattern when compared to Nox4−/− Apoe−/− SMCs (Fig. 3E). These observations further support a unique function of NOX4 on OPN protein expression in the setting of HG.
Fig. 3.
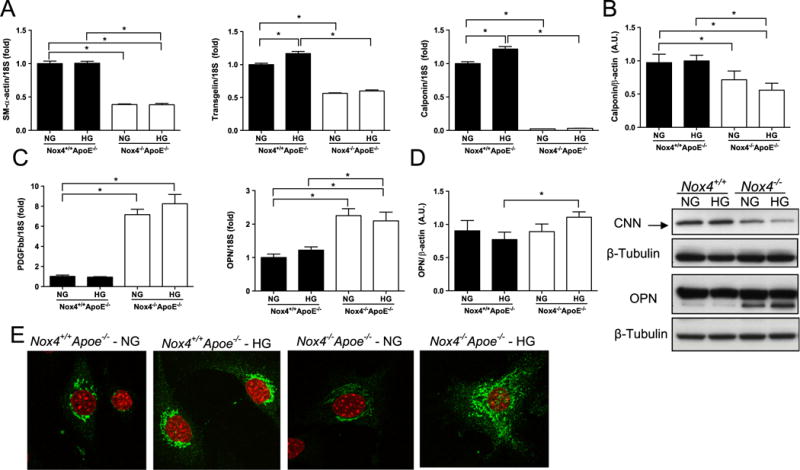
Lack of NOX4 decreases expression of SMC contractility markers in association with marked increases in PDGF-BB and osteopontin (OPN) in mouse aorta SMCs. (A) Relative SM-α-actin, transgelin and calponin mRNA levels in SMCs derived from Nox4+/+ Apoe−/− (filled bars) and Nox4−/− Apoe−/− (open bars) mouse aortas cultured in normal (NG) and high glucose (HG) conditions. (B) Immunoblotting of calponin protein in Nox4+/+ Apoe−/− (denoted Nox4+/+ ) and Nox4−/− Apoe−/− (denoted Nox4+/+) SMCs. (C) PDGF-BB and OPN gene expression as well as OPN protein expression as shown by immunoblotting (D) and immunocytochemistry (E). Analysis of immunoblots represents fold changes from Nox4+/+ Apoe−/− SMCs cultured under NG conditions normalised to the β-tubulin level (n=4). Immunocytochemistry images show Red: nuclear DAPI staining and Green: OPN, magnification ×40. *p<0.05.
3.4. Expression of fibrosis and proliferation markers in NOX4-deficient SMCs
To explore the functional consequences of increased PDGF-BB and OPN in NOX4-deficient SMCs, we evaluated markers of fibrosis and proliferation. Irrespective of the glucose concentration, SMCs lacking NOX4 showed increased expression of genes encoding the ECM proteins fibronectin and Collagen I in addition to the important pro-fibrotic factors TGFβ and CTGF (Fig. 4A). HG treatment induced Collagen I gene expression in Nox4−/− Apoe−/− and Nox4−/− Apoe−/− cells to a similar extent. Analysis of ECM proteins secreted into the culture media revealed a trend for increased fibronectin production (Fig. 4B) combined with significantly enhanced collagen production in Nox4−/− Apoe−/− SMCs (Fig. 4C). HG induced gene expression of the proliferation marker Ki67, which was further elevated in Nox4−/− Apoe−/− SMCs (Fig. 4D, left panel). The additional proliferative marker PCNA was unaffected by HG but markedly increased in SMCs lacking NOX4 (Fig. 4D, right panel). Growth curve analysis revealed an increase in SMC proliferation under HG conditions, which was further increased in those cells lacking NOX4 (Fig. 4E). Thus, aortic Ki67 mRNA levels mirrored the pro-proliferative effects of HG on SMCs whereas PCNA more closely reflected the effect of NOX4. Taken together, these findings demonstrate an important role for NOX4 in preventing SMC fibrosis and HG-mediated proliferation.
Fig. 4.
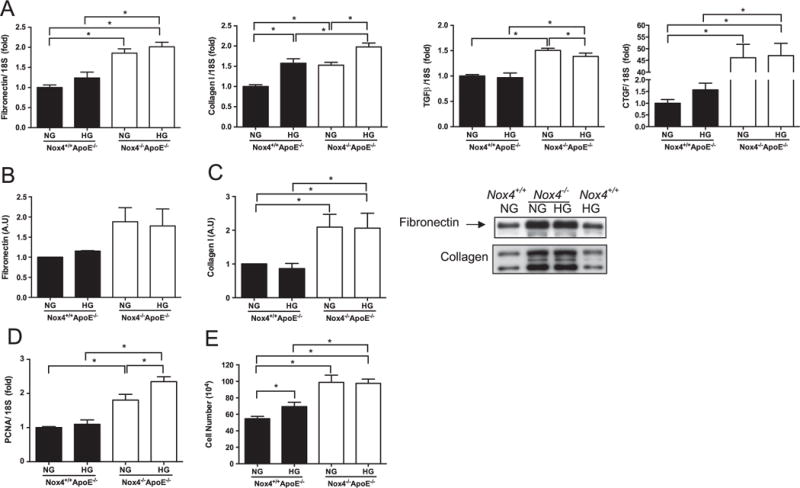
Upregulation of fibrosis and proliferation markers in SMCs from NOX4-deficient mouse aortas. (A) Relative mRNA levels of extracellular matrix proteins fibronectin and collagen I as well as profibrotic mediators transforming growth factor (TGF)-β and connective tissue growth factor (CTGF) in SMCs derived from Nox4−/− Apoe−/− (filled bars) and Nox4−/− Apoe−/− (open bars) aortas cultured in normal (NG) and high glucose (HG) conditions. Extracellular fibronectin (B) and collagen I (C) protein expression measured by immunoblotting culture media of Nox4−/− Apoe−/− (denoted Nox4−/− ) and Nox4−/− Apoe−/− (denoted Nox4−/− ) SMCs with fold changes normalised to the NG Nox4−/− Apoe−/− SMCs. (D) Expression of proliferation genes PCNA and (E) growth curve assessment of proliferation. Values are mean±SEM (n=4–6). *p<0.05.
3.5. NOX4 deficiency reduces hydrogen peroxide (H2O2) production and increases superoxide (O2− ) in high glucose-treated SMCs in association with marked increases in NOX1
The effect of NOX4 deletion on H2O2 and O2•− production by SMCs was measured via Amplex Red and LO12 chemiluminescence, respectively. Nox4−/− Apoe−/− SMCs cultured under HG conditions showed decreased H2O2 levels when compared to HG treated Nox4−/− Apoe−/− SMCs (Fig. 5A). HG induced O2•− production in Nox4−/− Apoe−/− cells and to a greater extent in Nox4−/− Apoe−/− SMCs (Fig. 5B). We investigated whether the increase in O2•− -was associated with compensatory upregulation of the other prominent NOX isoform in vascular SMCs, NOX1. Indeed SMCs from Nox4−/− Apoe−/− mice showed dramatic increases in Nox1 gene expression compared to Nox4−/− Apoe−/− SMCs (Fig. 5C and Supplemental Fig. 2A). mRNA levels of the NOX1 cytosolic organizer subunit NOXO1 were unchanged while activator subunit NOXA1 was only detectable in Nox4−/− Apoe−/− SMCs (Supplemental Fig. 2B and C). Lastly, treatment of Nox4−/− Apoe−/− SMCs with the NOX1/NOX4 dual inhibitor GKT137831 significantly reduced O2•− levels (Fig. 5D) suggesting a role for NOX1 in O2•− production in NOX4-deficient SMCs.
Fig. 5.
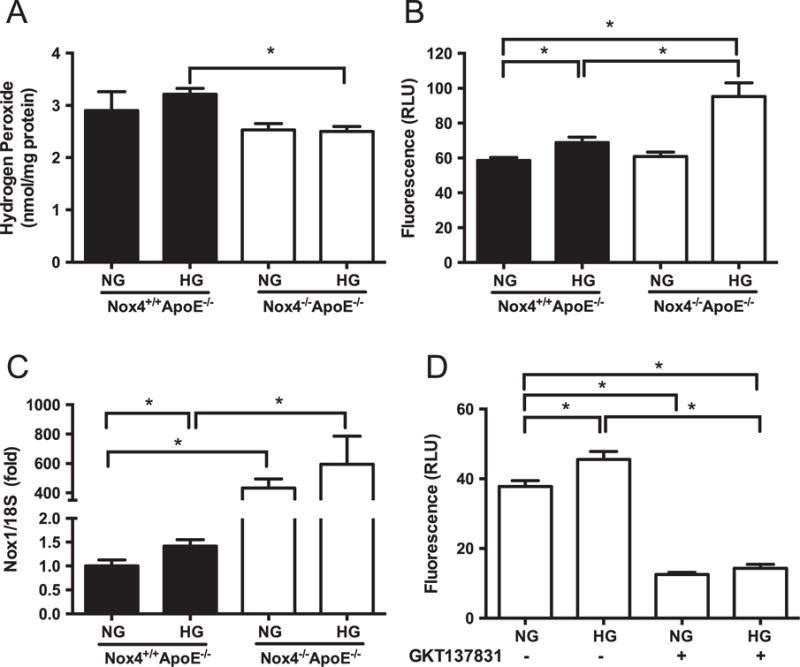
Reduced hydrogen peroxide and increased superoxide production associated with elevated Nox1 gene expression in high glucose treated NOX4-deficient SMCs. (A) Hydrogen peroxide (A) and superoxide (B) production in SMCs from Nox4+/+ Apoe−/− (filled bars) and Nox4−/− Apoe−/− (open bars) aortas cultured under normal (NG) and high glucose (HG) conditions as measured by Amplex Red and L012 chemiluminescence, respectively. (C) Expression of Nox1 gene relative to 18S. (D) Superoxide levels in DKO cells treated with or without the NOX1/NOX4 dual inhibitor GKT137831. Values are mean±SEM (n=6). *p<0.05.
Next we conducted a series of experiments to dissect the relative contributions of upregulated NOX1 and PDGF as well as the loss of NOX4-derived H2O2 to the observed phenotypic phenomena. According to our working hypothesis, the most upstream perturbation of the signalling cascade was NOX4 deficiency followed by elevated PDGF that acts as an upstream inducer of NOX1 expression and activity.
3.6. Silencing of NOX1 attenuates the aberrant induction of PDGF-BB, osteopontin (OPN), and pro-proliferative gene expression in NOX4-deficient SMCs
NOX1 siRNA decreased gene expression of NOX1 in Nox4+/+ Apoe−/− SMCs by 50% (Fig. 6A; 1.0±0.008 to 0.49±0.09). The dramatic increase in NOX1 mRNA levels in NOX4-deficient SMCs was significantly, albeit only modestly (12%), reduced by siRNA treatment. Silencing of NOX1 reduced expression of PDGF-BB (Fig. 6B) and OPN (Fig. 6C) in both Nox4+/+ Apoe−/− and Nox4−/− Apoe−/− cells. In Nox4−/− Apoe−/− SMCs, silencing of Nox1 significantly increased the expression of SM-α-actin in high glucose conditions only, with Nox4+ /+ Apoe−/− SMCs being unaffected (Fig. 6D). In Nox4−/− Apoe−/− SMCs, silencing of NOX1 reduced fibronectin and collagen I (Supplemental Fig. 3A and B) expression but was unable to modulate the elevated expression of these ECM genes observed in the Nox4−/− Apoe−/− SMCs. In contrast, proliferation genes Ki67 and PCNA (Supplemental Fig. 3C and D) were reduced in both Nox4+/+ Apoe−/− and Nox4−/− Apoe−/− cells silenced for NOX1. Therefore, NOX1-derived ROS are important mediators of PDGF and OPN expression as well as downstream proliferation pathways in SMCs that lack NOX4.
Fig. 6.
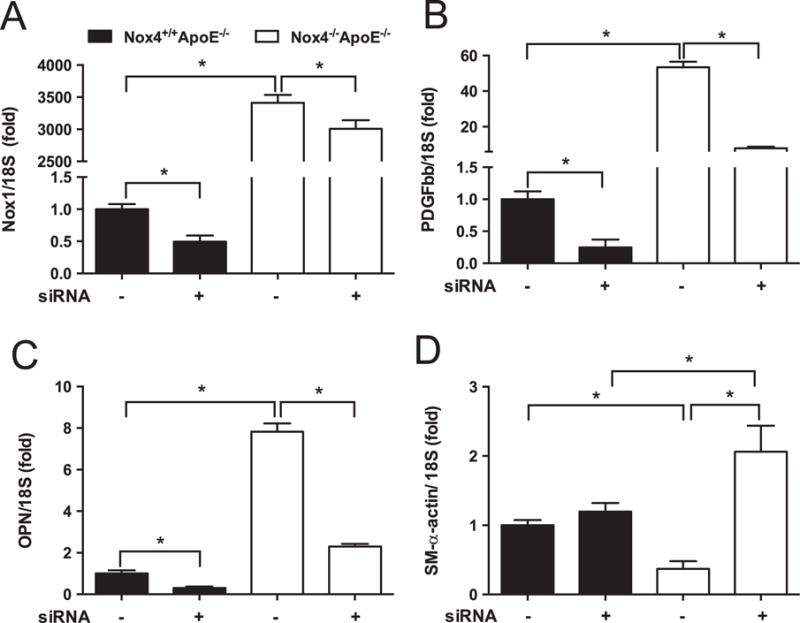
Silencing of NOX1 attenuates the aberrant expression of NOX1, PDGF-BB, osteopontin (OPN), and SM-α-actin genes in NOX4-deficient SMCs. (A) NOX1, (B) PDGF-BB, (C) OPN, and (D) SM-α-actin mRNA levels relative to 18S in Nox4+/+ Apoe−/− (filled bars) and Nox4−/− Apoe−/− (open bars) SMCs following knockdown with siRNA against NOX1. Values are mean±SEM (n=6). *p<0.05.
3.7. Inhibition of PDGF reverses aberrant expression of calponin, PDGF-BB and fibronectin in NOX4-deficient SMCs
The potent and pleiotropic effects of PDGF on vascular SMC pathophysiology are well characterised. In order to elucidate the role of PDGF in the NOX4-mediated phenotypic switch of SMCs, we performed inhibition experiments using a PDGF neutralising antibody (67 μg/ml, for 24 h). To confirm efficacy of the neutralising antibody, we treated Nox4+/+ Apoe−/− cells with PDGF or with PDGF neutralising antibody. In all cases, neutralising antibody reversed the effect of PDGF, except for the contractility marker calponin, which was elevated in Nox4+/+ Apoe−/− SMCs treated with inhibitor when compared to treatment with PDGF alone (Fig. 7A). In Nox4−/− Apoe−/− cells, inhibition of PDGF normalised calponin (Fig. 7A) and PDGF expression levels (Fig. 7B). PDGF treatment dramatically induced OPN expression in Nox4+/+ Apoe−/− SMCs, which was reversed by PDGF inhibition (Fig. 7C). Conversely, the heightened expression of OPN in Nox4−/− Apoe−/− SMCs was unaffected by PDGF inhibition (Fig. 7C). In Nox4+/+ Apoe−/− SMCs, PDGF-induced fibronectin (Fig. 7D) and Collagen I (Supplemental Fig. 4A) gene expression was prevented by inhibitor treatment. In Nox4−/− Apoe−/− SMCs, only fibronectin expression was dampened by anti-PDGF (Fig. 7D). PDGF induced proliferative gene expression in Nox4+/+ Apoe−/− SMCs; however, treatment with anti-PDGF was ineffective at reducing heightened Ki67 and PCNA expression levels in Nox4−/− Apoe−/− SMCs (Supplemental Fig. 4B and C).
Fig. 7.
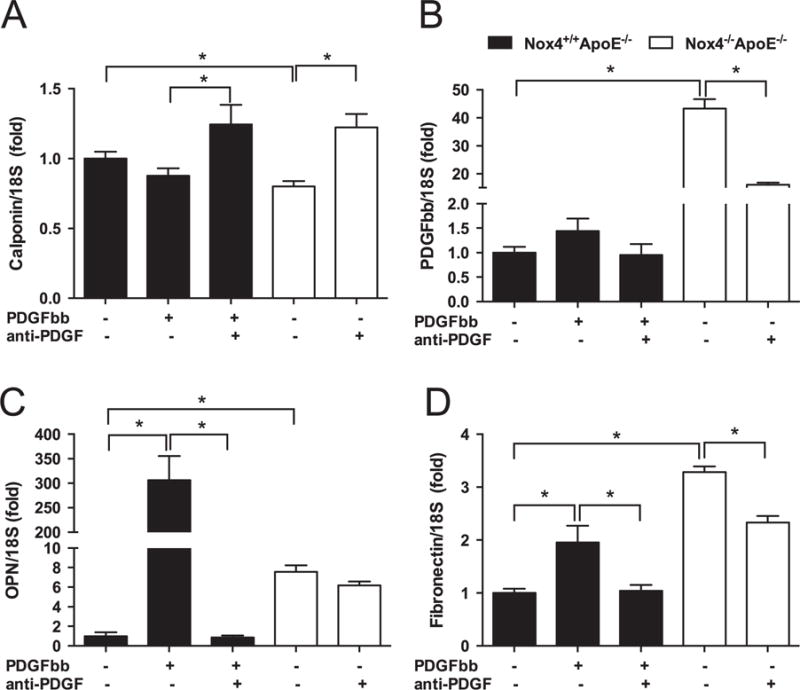
Inhibition of PDGF reverses aberrant expression of calponin, PDGF-BB and fibronectin in NOX4-deficient SMCs. Gene expression of (A) Calponin, (B) PDGF-BB, (C) osteopontin (OPN), and (D) Fibronectin in SMCs derived from Nox4+/+ Apoe−/− (filled bars) and Nox4−/− Apoe−/− (open bars) following treatment with PDGF-BB (10 ng/ml for 24 h, Nox4+/+ Apoe−/− SMCs only) or PDGF neutralising antibody (anti-PDGF; 67 μg/ml for 24 h). Values are mean±SEM (n=3). *p<0.05.
Previous studies have shown that ECM production and proliferation of SMCs during neointimal formation is associated with PDGF-induced ROS production by NOX1. Here we also observed induction of Nox1 gene expression by PDGF in Nox4+/+ Apoe−/− SMCs cultured under HG conditions (Supplemental Fig. 4C). Treatment of Nox4−/− Apoe−/− SMCs with HG further increased Nox1 mRNA levels, an effect which could be inhibited by the PDGF neutralising antibody (Supplemental Fig. 4D). Importantly, basal expression of NOX1 in Nox4+/+ Apoe−/− or Nox4−/− Apoe−/− SMCs cultured under NG conditions was unaffected by anti-PDGF treatment (Supplemental Fig. 4D).
3.8. Treatment of NOX4-deficient SMCs with hydrogen peroxide (H2O2) normalises aberrant expression of PDGF-BB, OPN and fibronectin genes
Treatment of Nox4+/+ Apoe−/− SMCs with HG reduced PDGF expression whereas treatment with H2O2 had no effect. In contrast, treatment of Nox4−/− Apoe−/− SMCs with H2O2 reduced expression of PDGF in the setting of HG (Supplementary Fig. 5A). Expression of OPN in Nox4+/+ Apoe−/− cells was increased by HG and reduced by H2O2 treatment (Supplementary Fig. 5B). Similarly, heightened OPN levels in Nox4−/− Apoe−/− cells were dramatically reduced by H2O2 (Supplementary Fig. 5B). Treatment of Nox4+/+ Apoe−/− or Nox4−/− Apoe−/− SMCs with H2O2 also reduced expression of fibronectin under HG conditions (Supplementary Fig. 5C). These effects of H2O2 on Nox4−/− Apoe−/− cells cultured under HG conditions were also observed for Collagen I, Ki67, PCNA, and Nox1 gene expression levels (Supplemental Fig. 5D–G).
4. Discussion
4.1. Downregulation of contractile SMC markers and concomitant upregulation of PDGF-BB and osteopontin (OPN) in aortas from diabetic NOX4-deficient mice
Vascular NADPH oxidase (NOX) enzymes play a central role in the normal and pathological responses of vascular smooth muscle cells (SMCs) that underscore the development and progression of vascular disease. Previous studies have identified NOX4-derived ROS as important mediators of the contractile, differentiated, ‘healthy’ SMC phenotype [2]. Few studies have explored SMC differentiation in vivo in response to changes in local environmental cues including NOX-derived ROS, hyperglycaemia and growth factors [15]. In this study, a marked reduction in the expression of the contractility markers SM-α-actin and calponin was observed in the aortas of diabetic Apoe−/− mice, which were further decreased by genetic deletion of NOX4. Furthermore, SMCs isolated from the aortas of NOX4-deficient Apoe−/− mice showed reductions in SM-α-actin, calponin and transgelin expression thus supporting a clear role for NOX4 in the maintenance of the differentiated SMC phenotype, particularly in the setting of hyperglycaemia.
SMC phenotypic transition is associated with a loss of smooth muscle markers and a gain of markers of proliferative and synthetic activity, including PDGF and OPN [15]. Increased expression of PDGF and OPN was seen in the aortas from diabetic Apoe−/− mice that was exacerbated by deletion of NOX4. In contrast, HG treatment of Nox4+/+ Apoe−/− or Nox4−/− Apoe−/− SMCs did not increase PDGF expression. Interestingly, HG treatment decreased PDGF expression in NOX4-expressing cells and only in NOX4-deficient cells treated with H2O2. Redox regulation of growth factor signalling has been shown previously for epidermal growth factor (EGF) where oxidation of the extracellular environment activates metalloproteinase-mediated shedding of EGF-like ligands [27]. Silencing of NOX1 attenuated PDGF expression in Nox4−/− Apoe−/− SMCs cultured under NG conditions. PDGF is a known inducer of NOX1 activity [28] and in these studies it was shown that inhibition of PDGF blunted basal and HG-induced Nox1 gene expression in Nox4−/− Apoe−/− SMCs. Similarly, EGF stimulation of the EGF receptor has been shown to be upstream of NOX1 activation in vascular SMCs [29]. Taken together, these data suggest a positive feedback mechanism between NOX1 and PDGF expression. Interestingly, HG induced NOX1 gene expression in Nox4−/− Apoe−/− but not Nox4+/+ Apoe−/− cells, suggesting that NOX4-derived ROS suppress the pathway through which glucose regulates NOX1. In line with this finding, Gray et al. [9] also demonstrated that genetic deletion of Nox4 prevented diabetes-induced Nox1 gene expression in the aortas of Apoe−/− mice. Overall, these findings suggest that diabetes-induced PDGF gene expression is a combined effect of hyperglycaemia and other factors, such as ROS.
4.2. NOX1-associated ROS regulate diabetes-induced expression of osteopontin (OPN) and proliferation markers in Nox4-deficient SMCs
Lee et al. [23] have shown that PDGF-induced ROS production by NOX1 controls SMC proliferation. In this study we showed that elevated Nox1 gene expression in NOX4-deficient SMCs correlated with heightened expression of proliferation markers PCNA and Ki67 as well as accelerated cell growth under HG conditions. OPN has also been reported to stimulate vascular SMC proliferation [30] and increase in expression in response to diabetes [19]. Here we show that deletion of NOX4 leads to enhanced diabetes-mediated expression of OPN in the aortas of Apoe−/− mice. Interestingly, NOX4-deficient SMCs displayed dramatic increases in OPN gene expression while OPN protein was only elevated in the setting of high glucose. Redox regulation of OPN was first described by Lyle et al. [20] where H2O2 (50 μM for 6–18 h)-treated rat SMCs stimulated OPN gene and protein expression. In these studies we demonstrated that acute treatment of Apoe−/− mouse-derived SMCs with H2O2 (100 μM for 4 h) or siRNA against NOX1 decreased OPN gene expression. These findings suggest that NOX1-derived superoxide when converted to H2O2 drives OPN expression. Interesting work by Arnold and colleagues [31] showed that H2O2 formed from the dismutation of superoxide produced by NOX1 induces the expression of PDGF in addition to other cell cycle and growth-related genes We propose that in the setting of NOX4 deficiency, HG treatment elevates levels of superoxide leading to localized increases of H2O2 sufficient to increase OPN transcription and translation.
The OPN protein in rodents is frequently resolved by western blot as a doublet [20]; however, it is not yet known whether this due to transcriptional or post-translational modification of OPN. In dendritic cells, these 2 bands of higher and lower molecular weight proteins represent the secreted and intracellular forms, respectively [32]. We have demonstrated by western blot and immunocytochemistry that NOX4-deficient SMCs expressed more of the lower molecular weight intracellular OPN. Furthermore, global NOX4 deletion in vivo prevented the diabetes-mediated release of OPN into the circulation. Further studies are required to discern whether the two OPN forms have distinct biological properties in vascular SMCs, with studies in immune cells proposing a role for intracellular OPN in cytoskeletal rearrangement and in signal transduction pathways [33]. Our data indicate that NOX4 regulates the production and retention of OPN protein in SMCs in part via upregulation of NOX1, which promotes a proliferative and synthetic phenotype.
4.3. Increased expression of pro-fibrotic factors and extracellular matrix (ECM) proteins in the NOX4-deficient SMCs
Phenotypically modulated SMCs are prominent synthesisers of ECM in atherosclerotic lesions [34], but few studies have examined the role of NOX4 in vascular SMC fibrotic responses. Previous studies by Schurmann et al. [8] identified that upon deletion of NOX4 there was increased fibrotic response within the aorta. Additionally, Babelova et al. [35] demonstrated that deletion of NOX4 in various models of renal disease increased fibrotic responses. In this present study, we identified deposition of the fibrillar collagen, Collagen I, in the aorta was significantly greater in diabetic mice lacking NOX4 when compared to their Nox4+/+ Apoe−/− counterparts. Similarly, NOX4-deficient SMCs displayed not only increased expression of collagen I and fibronectin but also key profibrogenic factors, TGFβ and CTGF when compared to Nox4+/+ Apoe−/− controls. Elevated fibronectin expression in Nox4−/− Apoe−/− SMCs was reduced by PDGF inhibition but not by silencing of NOX1. Interestingly, H2O2 treatment of Nox4−/− Apoe−/− SMCs cultured under HG conditions suppressed the secretion of fibronectin and collagen I into the extracellular space. In contrast to our findings, studies in cardiac [36] and lung fibroblasts [37] showed that NOX4 induced TGFβ-mediated fibrosis. While it is known that TGFβ increases NOX4 expression and activity [38] in VSMCs, few studies have examined the reciprocal effect of NOX4-derived ROS on fibrotic gene expression in this cell type; especially in the context of diabetes. Here we have demonstrated NOX4-mediated suppression of fibrotic responses in SMCs via PDGF and H2O2. However, as NOX4 has been shown to be a driver of fibrosis in epithelial models such as lung fibrosis models [39–41], one cannot discount that NOX4 deletion is acting through different mechanisms within this model.
Recent studies by Tong et al. [42] have explored the effects of SMC-NOX4 on atherosclerosis induced by western diet and partial left common carotid artery (LCA) ligation in FVB/N Apoe−/− mice that overexpress a human dominant negative (DN) form of NOX4 specifically in SMCs. Contrary to the atherogenic effects previously observed in C57/Bl6J Apoe−/− mice with genetic deletion of Nox4 and rendered diabetic [9] or subjected to Western-diet with subsequent partial LCA ligation [8], these authors showed an attenuation of atherosclerosis in DN mice linked to reduced inflammation, proliferation and migration via downregulation of soluble epoxide hydrolase 2. Of note, the proliferation assays presented in Schurmann et al., were restricted to SMCs isolated from FVB/N mice with the authors citing difficulties obtaining data from Apoe−/− SMCs, which was possible in the current study. Fur- thermore, the effects of the human DN construct on the endogenous NOX1 isoform or major pro-proliferative factors, such as Ki67, were not reported. Intriguingly, the reduction in atherosclerosis seen in DN mice was not mirrored by significant changes in lesion SM-α-actin-positive cells and, if anything, the percentage of ‘non-macrophage’ SM-α-actin-stained cells trended towards reduction. This data may provide some continuity across the studies, whereby lack of NOX4 leads to a loss of expression of characteristic SMC markers. It is increasingly apparent that the role of NOX4 in the development of atherosclerosis is far more complex than anticipated and that careful interpretation of results in the context of animal model and experimental methodology is required.
4.4. Diabetes and NOX4 modulate SMC phenotypic changes of direct relevance to atherosclerotic disease
Based on these studies, we propose a novel mechanism for NOX4-mediated SMC responses in normal and high glucose conditions. SMCs lacking NOX4 no longer produce H2O2 in response to HG. Reduced NOX4-dependent H2O2 levels result in increased expression of PDGF, which subsequently reduces calponin and increases fibronectin gene expression. Under NG conditions, NOX4-deficient SMCs showed increased PDGF independent of changes in H2O2 and increased Nox1 expression independent of PDGF but regulated in part by H2O2. Under HG conditions, PDGF induces NOX1 expression that is associated with elevated superoxide levels and downstream induction of osteopontin expression in addition to the proliferation genes Ki67 and PCNA. Taken together, these findings demonstrate a central role for NOX4 in the suppression of vascular SMC proliferation and fibrosis, responses that define the de-differentiated SMC phenotype associated with the accelerated progression of atherosclerosis, as seen in diabetes.
Supplementary Material
Acknowledgments
This work was supported by the National Health & Medical Research Council (NHMRC) project grant of Australia (APP1005851), the Juvenile Diabetes Research Foundation (JDRF 2SRA-259) and the Diabetes Australia Research Trust. SPG is supported by the Australian Diabetes Society Skip Martin Early Career Fellowship (SM101). EDM is supported by the National Heart Foundation Postgraduate Scholarship (PB12M6943). HHHWS is supported by the EU Marie Curie International Reintegration Grant and the ERC Advanced Grant 30983347E RADMED. KKG is supported by the National Institute of Health Grants HL038206 and HL095070. KJD and MC are supported by NHMRC Senior Research and Senior Principal Fellowships (KJD - APP1059124; MC - APP1078808), respectively. Supported in part by the Victorian Government’s Operational Infrastructure Support Program.
The authors would like to thank Ms. Bonnie Seidel-Rogol, Ms. Maryann Arnstein, and Ms. Kylie Gilbert (Baker IDI Heart & Diabetes Institute, Melbourne, Australia) for technical assistance.
Abbreviations
- Apoe
Apolipoprotein e
- CTGF
Connective tissue growth factor
- CNN
Calponin
- DN
Dominant negative
- ECM
Extracellular matrix
- FBS
Foetal bovine serum
- HG
High glucose
- LCA
Left common carotid artery
- mRNA
Messenger ribonucleic acid
- MASM
Mouse Aortic Smooth Muscle Cells
- NG
Normal glucose
- NOX
NADPH oxidase
- OPN
Osteopontin
- PCNA
Proliferating cell nuclear antigen
- PDGF
Platelet-derived growth factor
- qRT-PCR
Quantitative real-time polymerase chain reaction
- ROS
Reactive oxygen species
- siRNA
Small interfering ribonucleic acid
- SMC
Smooth muscle cell
- STZ
Streptozotocin
- TGF
Transforming growth factor
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed.2016.07.013.
Footnotes
Disclosure
CS is a paid employee and own shares in Genkyotex SA, Geneva, Switzerland. The other authors report no conflicts.
References
- 1.Lassègue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol – Regul Integr Comp Physiol. 2003;285(254–2):R277–R297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 2.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Schmidt HH, Lassegue B, Griendling KK. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2007;27(1):42–48. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schroder K, Wandzioch K, Helmcke I, Brandes RP. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol. 2009;29(2):239–245. doi: 10.1161/ATVBAHA.108.174219. [DOI] [PubMed] [Google Scholar]
- 4.Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res. 2012;110(9):1217–1225. doi: 10.1161/CIRCRESAHA.112.267054. [DOI] [PubMed] [Google Scholar]
- 5.Goettsch C, Babelova A, Trummer O, Erben RG, Rauner M, Rammelt S, Weissmann N, Weinberger V, Benkhoff S, Kampschulte M, Obermayer-Pietsch B, Hofbauer LC, Brandes RP, Schroder K. NADPH oxidase 4 limits bone mass by promoting osteoclastogenesis. J Clin Investig. 2013;123(11):4731–4738. doi: 10.1172/JCI67603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Marco E, Jha JC, Sharma A, Wilkinson-Berka JL, Jandeleit-Dahm KA, de Haan JB. Are reactive oxygen species still the basis for diabetic complications? Clin Sci. 2015;129(2):199–216. doi: 10.1042/CS20150093. [DOI] [PubMed] [Google Scholar]
- 7.Craige SM, Kant S, Reif M, Chen K, Pei Y, Angoff R, Sugamura K, Fitzgibbons T, Keaney JF., Jr Endothelial NADPH oxidase 4 protects ApoE−/−mice from atherosclerotic lesions. Free Radic Biol Med. 2015;89:1–7. doi: 10.1016/j.freeradbiomed.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schurmann C, Rezende F, Kruse C, Yasar Y, Lowe O, Fork C, van de Sluis B, Bremer R, Weissmann N, Shah AM, Jo H, Brandes RP, Schroder K. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur Heart J. 2015 doi: 10.1093/eurheartj/ehv460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gray SP, Di Marco E, Kennedy K, Chew P, Okabe J, El-Osta A, Calkin AC, Biessen EA, Touyz RM, Cooper ME, Schmidt HH, Jandeleit-Dahm KA. Reactive oxygen species can provide atheroprotection via NOX4-dependent inhibition of inflammation and vascular remodeling. Arterioscler Thromb Vasc Biol. 2015 doi: 10.1161/ATVBAHA.115.307012. [DOI] [PubMed] [Google Scholar]
- 10.Langbein H, Brunssen C, Hofmann A, Cimalla P, Brux M, Bornstein SR, Deussen A, Koch E, Morawietz H. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J. 2016;37(22):1753–1761. doi: 10.1093/eurheartj/ehv564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95(2):156–164. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21(6):628–637. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89(12):1147–1154. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 14.Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, Frutkin A, Dichek D, Giachelli CM. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res. 2009;104(6):733–741. doi: 10.1161/CIRCRESAHA.108.183053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 16.Pandolfi A, Grilli A, Cilli C, Patruno A, Giaccari A, Di Silvestre S, De Lutiis MA, Pellegrini G, Capani F, Consoli A, Felaco M. Phenotype modulation in cultures of vascular smooth muscle cells from diabetic rats: association with increased nitric oxide synthase expression and superoxide anion generation. J Cell Physiol. 2003;196(2):378–385. doi: 10.1002/jcp.10325. [DOI] [PubMed] [Google Scholar]
- 17.Mori S, Takemoto M, Yokote K, Asaumi S, Saito Y. Hyperglycemia-induced alteration of vascular smooth muscle phenotype. J Diabetes Complicat. 2002;16(1):65–68. doi: 10.1016/s1056-8727(01)00189-1. [DOI] [PubMed] [Google Scholar]
- 18.Inaba K, Zhou D, Yang B, Vacek I, Sun AM. Normalization of diabetes by xenotransplantation of cryopreserved microencapsulated pancreatic islets. Application of a new strategy in islet banking. Transplantation. 1996;61(2):175–179. doi: 10.1097/00007890-199601270-00001. [DOI] [PubMed] [Google Scholar]
- 19.Takemoto M, Yokote K, Nishimura M, Shigematsu T, Hasegawa T, Kon S, Uede T, Matsumoto T, Saito Y, Mori S. Enhanced expression of osteopontin in human diabetic artery and analysis of its functional role in accelerated atherogenesis. Arterioscler Thromb Vasc Biol. 2000;20(3):624–628. doi: 10.1161/01.atv.20.3.624. [DOI] [PubMed] [Google Scholar]
- 20.Lyle AN, Remus EW, Fan AE, Lassegue B, Walter GA, Kiyosue A, Griendling KK, Taylor WR. Hydrogen peroxide regulates osteopontin expression through activation of transcriptional and translational pathways. J Biol Chem. 2014;289(1):275–285. doi: 10.1074/jbc.M113.489641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, Schmidt HHHW. Post-stroke inhibition of induced nadph oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010;8(9):e1000479. doi: 10.1371/journal.pbio.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsueh W, Abel ED, Breslow JL, Maeda N, Davis RC, Fisher EA, Dansky H, McClain DA, McIndoe R, Wassef MK, Rabadán-Diehl C, Goldberg IJ. Recipes for creating animal models of diabetic cardiovascular disease. Circ Res. 2007;100(10):1415–1427. doi: 10.1161/01.RES.0000266449.37396.1f. [DOI] [PubMed] [Google Scholar]
- 23.Lee MY, Martin A San, Mehta PK, Dikalova AE, Garrido AM, Datla SR, Lyons E, Krause KH, Banfi B, Lambeth JD, Lassegue B, Griendling KK. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. 2009;29(4):480–487. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown DI, Lassegue B, Lee M, Zafari R, Long JS, Saavedra HI, Griendling KK. Poldip2 knockout results in perinatal lethality, reduced cellular growth and increased autophagy of mouse embryonic fibroblasts. PLoS One. 2014;9(5):e96657. doi: 10.1371/journal.pone.0096657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, De Haan JB, Koulis C, El-Osta A, Andrews KL, Chin-Dusting JPF, Touyz RM, Wingler K, Cooper ME, Schmidt HHHW, Jandeleit-Dahm KA. NADPH Oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127(18):1888–1902. doi: 10.1161/CIRCULATIONAHA.112.132159. [DOI] [PubMed] [Google Scholar]
- 26.Williams HC, Martin A San, Adamo CM, Seidel-Rogol B, Pounkova L, Datla SR, Lassegue B, Bear JE, Griendling K. Role of coronin 1B in PDGF-induced migration of vascular smooth muscle cells. Circ Res. 2012;111(1):56–65. doi: 10.1161/CIRCRESAHA.111.255745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stanic B, Katsuyama M, Miller FJ., Jr An oxidized extracellular oxidation-reduction state increases Nox1 expression and proliferation in vascular smooth muscle cells via epidermal growth factor receptor activation. Arterioscler Thromb Vasc Biol. 2010;30(11):2234–2241. doi: 10.1161/ATVBAHA.110.207639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91phox homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88(9):888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 29.Seshiah PN, Kereiakes DJ, Vasudevan SS, Lopes N, Su BY, Flavahan NA, Goldschmidt-Clermont PJ. Activated monocytes induce smooth muscle cell death: role of macrophage colony-stimulating factor and cell contact. Circulation. 2002;105(2):174–180. doi: 10.1161/hc0202.102248. [DOI] [PubMed] [Google Scholar]
- 30.Gadeau AP, Campan M, Millet D, Candresse T, Desgranges C. Osteopontin overexpression is associated with arterial smooth muscle cell proliferation in vitro. Arterioscler Thromb. 1993;13(1):120–125. doi: 10.1161/01.atv.13.1.120. [DOI] [PubMed] [Google Scholar]
- 31.Arnold RS, Shi J, Murad E, Whalen AM, Sun CQ, Polavarapu R, Parthasarathy S, Petros JA, Lambeth JD. Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc Natl Acad Sci USA. 2001;98(10):5550–5555. doi: 10.1073/pnas.101505898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shinohara ML, Kim HJ, Kim JH, Garcia VA, Cantor H. Alternative translation of osteopontin generates intracellular and secreted isoforms that mediate distinct biological activities in dendritic cells. Proc Natl Acad Sci USA. 2008;105(20):7235–7239. doi: 10.1073/pnas.0802301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inoue M, Shinohara ML. Intracellular osteopontin (iOPN) and immunity. Immunol Res. 2011;49(1–3):160–172. doi: 10.1007/s12026-010-8179-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacLeod DC, Strauss BH, de Jong M, Escaned J, Umans VA, van Suylen RJ, Verkerk A, de Feyter PJ, Serruys PW. Proliferation and extracellular matrix synthesis of smooth muscle cells cultured from human coronary atherosclerotic and restenotic lesions. J Am Coll Cardiol. 1994;23(1):59–65. doi: 10.1016/0735-1097(94)90502-9. [DOI] [PubMed] [Google Scholar]
- 35.Babelova A, Avaniadi D, Jung O, Fork C, Beckmann J, Kosowski J, Weissmann N, Anilkumar N, Shah AM, Schaefer L, Schroder K, Brandes RP. Role of Nox4 in murine models of kidney disease. Free Radic Biol Med. 2012;53(4):842–853. doi: 10.1016/j.freeradbiomed.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 36.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97(9):900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 37.Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax. 2010;65(8):733–738. doi: 10.1136/thx.2009.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin-Garrido A, Brown DI, Lyle AN, Dikalova A, Seidel-Rogol B, Lassegue B, Martin A San, Griendling KK. NADPH oxidase 4 mediates TGF-beta-induced smooth muscle alpha-actin via p38MAPK and serum response factor. Free Radic Biol Med. 2011;50(2):354–362. doi: 10.1016/j.freeradbiomed.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jarman ER, Khambata VS, Cope C, Jones P, Roger J, Ye LY, Duggan N, Head D, Pearce A, Press NJ, Bellenie B, Sohal B, Jarai G. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am J Respir Cell Mol Biol. 2014;50(1):158–169. doi: 10.1165/rcmb.2013-0174OC. [DOI] [PubMed] [Google Scholar]
- 40.Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, Guichard C, Arbiser JL, Banfi B, Pache JC, Barazzone-Argiroffo C, Krause KH. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal. 2011;15(3):607–619. doi: 10.1089/ars.2010.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rhyu DY, Park J, Sharma BR, Ha H. Role of reactive oxygen species in transforming growth factor-beta1-induced extracellular matrix accumulation in renal tubular epithelial cells. Transplant Proc. 2012;44(3):625–628. doi: 10.1016/j.transproceed.2011.12.054. [DOI] [PubMed] [Google Scholar]
- 42.Tong X, Khandelwal AR, Wu X, Xu Z, Yu W, Chen C, Zhao W, Yang J, Qin Z, Weisbrod RM, Seta F, Ago T, Lee KS, Hammock BD, Sadoshima J, Cohen RA, Zeng C. Pro-atherogenic role of smooth muscle Nox4-based NADPH oxidase. J Mol Cell Cardiol. 2016 doi: 10.1016/j.yjmcc.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.