

ABSTRACT
The modulation of canonical macroautophagy/autophagy for therapeutic benefit is an emerging strategy of medical and pharmaceutical interest. Many drugs act to inhibit autophagic flux by targeting lysosome function, while others were developed to activate the pathway. Here, we report the surprising finding that many therapeutically relevant autophagy modulators with lysosomotropic and ionophore properties, classified as inhibitors of canonical autophagy, are also capable of activating a parallel noncanonical autophagy pathway that drives MAP1LC3/LC3 lipidation on endolysosomal membranes. Further, we provide the first evidence supporting drug-induced noncanonical autophagy in vivo using the local anesthetic lidocaine and human skin biopsies. In addition, we find that several published inducers of autophagy and mitophagy are also potent activators of noncanonical autophagy. Together, our data raise important issues regarding the interpretation of LC3 lipidation data and the use of autophagy modulators, and highlight the need for a greater understanding of the functional consequences of noncanonical autophagy.
KEYWORDS: autophagy, LAP, lipidation, lysosome, lysosomotropic, noncanonical
Introduction
The canonical autophagy pathway is an evolutionarily conserved lysosomal degradation pathway, through which intracellular proteins and organelles are degraded and recycled. Autophagy plays a major role in cell and organismal homeostasis and autophagic dysfunction contributes to the pathology of many diseases, including cancer and neurodegenerative disorders.1 Recent research efforts have focused on the development of drugs to either activate or inhibit the canonical autophagy pathway, with many high-throughput drugs screens having been performed to identify new targets.2-6
Canonical autophagy can be initiated by various cellular stresses, including nutrient withdrawal. The pathway is orchestrated by multiple proteins including an upstream complex (ULK1/2, RB1CC1/FIP200, ATG13 and ATG101) hereafter referred to as the ULK complex, a class III phosphatidylinositol 3-kinase (PIK3C3/VPS34) and a ubiquitin-like conjugation system (including ATG3, 4, 5, 7, 9, 10, 12 and 16L1). These autophagy proteins act in concert to form compartments termed phagophores that engulf intracellular constituents; completion and maturation into double-membrane autophagosomes allows them to be targeted for degradation.7 Upon fusion with lysosomes, autophagosomes and their contents are degraded and essential nutrients recycled. As such, the canonical autophagy pathway can be split into 2 stages; first, initiation of autophagosome formation and second, their flux through the lysosome. Autophagosome formation, and therefore autophagy itself, is often measured using the protein MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3). LC3 is a cytosolic ubiquitin-like protein that becomes conjugated to the membrane lipid phosphatidylethanolamine (PE) on forming autophagosomes. Therefore, the appearance of LC3 puncta by microscopy, or the intensity of faster-migrating lipidated LC3 (LC3-II) on SDS-PAGE gels, gives an indication of pathway activity, and are widely used to assay autophagy. Autophagosome number, and levels of lipidated LC3, can differ depending on the point at which the pathway is disrupted. Inhibition of early formation, for instance via inhibition of or depletion of the ULK complex, will reduce autophagosome numbers. Conversely, inhibition of flux, via lysosome inhibition, will accumulate lipidated LC3 on undegraded autophagosomes.8
It is now well established that autophagy proteins can also target and modify non-autophagosome compartments through a noncanonical pathway.9-11 This noncanonical autophagy pathway mediates the lipidation of LC3 to single-membrane intracellular compartments, including those of the endolysosomal system, following a range of macroendocytic engulfment events. The noncanonical autophagy pathway functions during the physiological process of LC3-associated phagocytosis (LAP);11-15 during which LC3 is lipidated to phagosomes housing a variety of pathogens or dead cells. Similar endolysosomal LC3 lipidation is observed on macropinosomes and during entosis,9 a live-cell engulfment process.10 LC3 lipidation in these processes is thought to regulate the fusion of vesicles with lysosomes. In the case of LAP this has important implications regarding the killing and clearance of pathogens, processing of antigens and macrophage cytokine responses following phagocytosis.11,14-17
Mechanisms underlying regulation of the noncanonical pathway are now being explored. Recently, we reported that noncanonical autophagy and endolysosomal LC3 lipidation can be promoted by certain lysosomotropic drugs, such as chloroquine, and the bacterial toxin VacA.18 Osmotic imbalances generated in endolysosomal compartments by these agents promote the recruitment of some autophagy proteins and lipidation of LC3. Indeed, simply altering extracellular tonicity induces an osmotic flux of water into endolysosomal compartments, which is sufficient to promote noncanonical autophagy. Physiological and drug-induced examples of noncanonical autophagy share the same features of being morphologically and genetically distinct from canonical autophagy. Indeed, noncanonical autophagy and endolysosomal LC3 lipidation proceed in the absence of autophagosomes.
Here we present evidence that activation of noncanonical autophagy is a surprisingly common feature of therapeutically relevant lysosomotropic agents and ionophores and provide evidence that this can occur in vivo. Importantly, we also show that carbonyl cyanide m-chlorophenylhydrazone (CCCP), a commonly used inducer of mitophagy, and amiodarone, an activator of autophagy with applications to anticancer therapies, are capable of activating noncanonical autophagy in parallel. This raises important issues in the interpretation of experiments where LC3 is used as a marker for autophagy, and highlights the need for greater understanding of the functional consequences of noncanonical autophagy.
Results
Ionophores and lysosomotropic drugs raise lysosomal pH
Lysosomes are acidic intracellular compartments where low pH is generated and maintained by the vacuolar-type H+-translocating ATPase (V-ATPase) proton pump and an array of ion channels.19 We tested the effect of a range of ionophores and lysosomotropic drugs (Table S1) for their effect on lysosomal pH by measuring LysoTracker Red fluorescence in MCF10A human breast epithelial cells. As expected, direct inhibition of V-ATPase activity using bafilomycin A1 (Baf) raised lysosomal pH and reduced LysoTracker Red staining (Fig. 1A and 1B). Nigericin (Nig) and monensin (Mon) are ionophores that drive the exchange of potassium and sodium, respectively, for hydrogen ions across membranes. In agreement with published work both nigericin and monensin dissipated lysosome pH (Fig. 1A and 1B).20 Lysosomotropic drugs are basic lipophilic compounds with certain physiochemical properties that promote their accumulation in acidic compartments.21 Some classic lysosomotropic drugs include chloroquine (CQ) and its derivative hydroxychloroquine (HCQ), which are weak base amines used therapeutically as antimalarials and more recently as anticancer drugs.22 Ammonium chloride (NH4Cl) is another weak base amine lysosomotropic drug. All 3 act to raise lysosome pH by sequestering hydrogen protons (Fig. 1A and 1B), and are extensively used to inhibit the canonical autophagy flux. Several pharmaceuticals also fall into the lysosomotropic drug category. Lidocaine hydrochloride (LH) and procainamide (ProA) are local anesthetics, also commonly used as antiarrhythmic drugs. Betahistine (BH) is a histamine receptor antagonist with lysosomotropic properties, used in the treatment of vertigo.23 Lidocaine hydrochloride, procainamide and betahistine all raised lysosomal pH (Fig. 1A and 1B). Alongside raising lysosomal pH, all of the drugs tested induced extensive formation of vacuoles (vacuolation) in cells. This observation has previously been reported and attributed to osmotic swelling of endolysosomal compartments driven by ionic imbalances or increased solute content of the compartments.24
Figure 1.
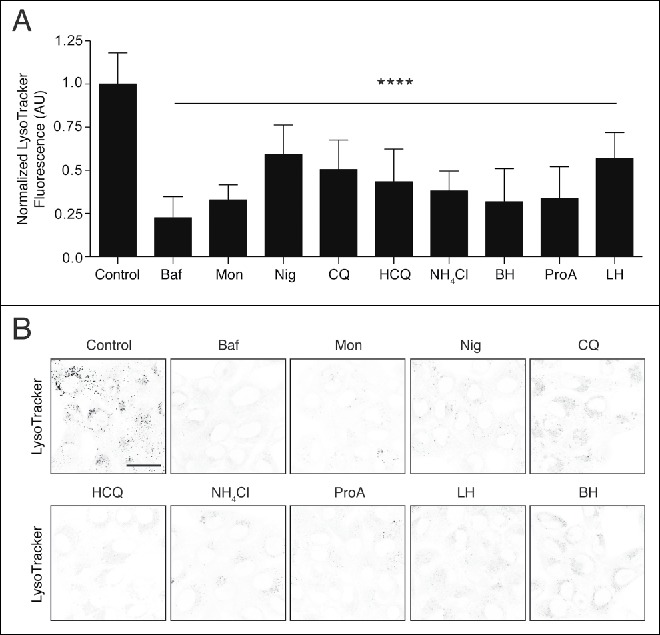
Effects of lysosomotropic and ionophore drugs on lysosome pH using LysoTracker. MCF10A cells were labeled with LysoTracker Red for 2 h in the presence of absence of bafilomycin A1 (Baf, 100 nM), monensin (Mon, 50 μM), nigericin (Nig, 10 μM), chloroquine (CQ, 100 μM), hydroxychloroquine (HCQ, 100 μM), NH4Cl (10 mM), betahistine (BH, 5 mM), procainamide (ProA, 5mM) or lidocaine hydrochloride (LH, 2.5 mM). (A) Quantification of LysoTracker fluorescence. Data are mean ± SD of 8 fields of view from 2 independent experiments. One-way ANOVA, P<0.0001. AU, arbitrary units. (B) Representative inverted grayscale images of LysoTracker staining in cells. Bar: 20 μm for all images.
These data demonstrate that the panel of commonly used drugs, of different classes, all raise lysosomal pH, and, importantly, achieve this without inhibiting the V-ATPase directly.
Ionophores and lysosomotropic drugs induce LC3 lipidation independently from canonical autophagy
Inhibition of lysosome function, by raising lysosomal pH, results in the inhibition of autophagic flux and accumulation of autophagosomes. Monitoring LC3 lipidation is one of the major research methodologies used to assess the canonical autophagy pathway. All of the drugs tested in Fig. 1, which raise lysosomal pH, have been implicated in inhibiting autophagic flux and increasing LC3 lipidation levels (Table S1). However, in light of recent work demonstrating that LC3 lipidation can be induced independently of canonical autophagy,18 we sought to test whether these drugs also activated a noncanonical autophagy pathway.
To differentiate between LC3 lipidation associated with autophagosomes and that associated with endolysosomal membranes during noncanonical autophagy, an autophagosome-deficient HEK293 cell line was generated using CRISPR/Cas9 to delete ATG13. ATG13 is part of the upstream ULK complex, which is essential for autophagosome formation.25,26 As shown by western blotting, wild-type cells displayed robust LC3 lipidation upon activation of canonical autophagy by starvation or inhibition of MTOR by PP242, whereas ATG13−/− cells did not respond (Fig. S1A and S1B). Using confocal microscopy, GFP-LC3-expressing wild-type cells displayed an increased LC3 puncta number following starvation, which represents autophagosome structures, and which was not seen in ATG13−/− cells. Importantly, adding back mCherry-tagged ATG13 in ATG13−/− cells restored GFP-LC3 puncta formation upon starvation (Fig. S1B), confirming this is a specific effect. Thus, ATG13−/− HEK293 cells provide an effective model to detect potential LC3 lipidation events not associated with autophagosomes.
To screen for noncanonical LC3 lipidation, dose responses were performed where wild-type and ATG13−/− HEK293 cells were treated for 2 h with drugs at the indicated concentrations, in full medium. Cell lysates were then probed for LC3 and GAPDH by western blotting. As expected, the V-ATPase inhibitor bafilomycin A1 induced an increase in lipidated LC3 (LC3-II) in wild-type cells, via inhibition of basal autophagic flux, while having no effect on LC3-II levels in autophagosome-deficient ATG13−/− cells (Fig. 2A and 2B and Fig. S2A and S2B). Monensin, nigericin, chloroquine, hydroxychloroquine, NH4Cl, procainamide, lidocaine hydrochloride and betahistine all induced LC3 lipidation in wild-type cells (Fig. 2A and Fig. S2A). Strikingly, all of these drugs promoted LC3 lipidation in ATG13−/− cells (Fig. 2B and Fig. S2B), indicating that they activate noncanonical autophagy.
Figure 2.
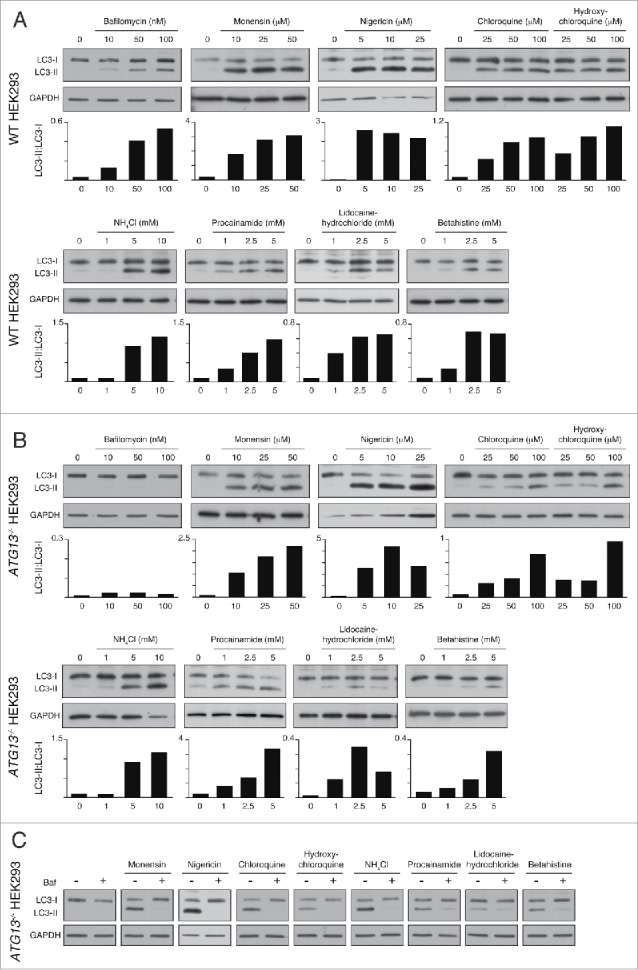
Lysosomotropic and ionophore dugs activate ATG13-independent, V-ATPase-dependent LC3 lipidation. Representative western blots for LC3 and GAPDH in (A) wild-type and (B) ATG13−/− HEK293 cells treated with drugs at the indicated concentrations for 2 h. Ratios of lipidated LC3-II:nonlipidated LC3-I were quantified and graphed. (C) ATG13−/− HEK293 cells were pretreated with bafilomycin A1 (Baf, 100nM) for 20 min before drug treatments. Representative western blots for LC3 and GAPDH are shown.
Together, these data indicate that a wide range of drugs activate noncanonical autophagy and promote LC3 lipidation in an ATG13-independent manner, in parallel to their known effects on inhibiting autophagic flux. These findings build on our previous studies of lysosomotropic drugs and extend this observation to include a wider range of drugs and ionophores. Interestingly, we note that LC3 lipidation in ATG13−/− cells tends to require higher drug concentrations as compared with wild-type cells, indicating that inhibition of autophagic flux and activation of noncanonical autophagy may be achieved at different concentrations.
Ionophore- and lysosomotropic drug-induced LC3 lipidation is dependent on V-ATPase activity
Inhibition of V-ATPase by bafilomycin A1 does not affect the LC3 lipidation step during autophagosome formation. In contrast, unconventional LC3 lipidation during noncanonical autophagy such as for LC3-associated phagocytosis (LAP) and entosis, are completely inhibited by bafilomycin A1.18 These data point to a more direct role for the V-ATPase in activating the LC3 lipidation machinery during noncanonical autophagy, although the mechanism of V-ATPase action in this process remains unknown.
To determine the effect of V-ATPase inhibition on LC3 lipidation induced by our panel of drugs we pretreated ATG13−/− HEK293 cells with bafilomycin A1 before treatment with the indicated drugs. Bafilomycin A1 pretreatment completely abolished drug-induced LC3 lipidation in all cases, supporting the conclusion that these drugs activate a noncanonical autophagy response (Fig. 2C).
Ionophores and lysosomotropic drugs activate noncanonical autophagy and promote LC3 lipidation to endolysosomal compartments
The data above clearly show that the panel of drugs tested induced ATG13-independent LC3 lipidation. To establish whether this LC3 lipidation was associated with noncanonical autophagy and endolysosomal membranes, we used confocal microscopy to image LC3 localization during drug treatment. Using Atg13 (atg13−/−) and Atg9 (atg9−/−) knockout mouse embryonic fibroblast (MEF) cell lines, both of which are deficient in autophagosome biogenesis,26,27 we stained endogenous LC3 and the lysosome marker LAMP1 following drug treatments. In both knockout cell lines, bafilomycin A1 has no obvious effect, whereas the other drug treatments induce dramatic LC3 relocalization (Fig. 3 and 4). LC3 was frequently colocalized with enlarged LAMP1 compartments (Fig. 3 and 4 crops). Similar results have been found in epithelial cells, using a GFP-LC3-expressing MCF10A cell line in which ATG13 has been deleted (Fig. S1C and S1D and Fig. S3A). These structures are unlikely to be autophagosomes in nature given their large size and the genetic backgrounds of the cell lines used. Corroborating this, there is no detectable increase in the early autophagosome marker WIPI2 following drug treatment (Fig. S4). Together, these findings indicate that a range of drugs induce LC3 lipidation of endolysosomal compartments distinct from autophagosomes. Interstingly, activation of lipidation did not appear to be restricted to LC3 family members as we also detected relocalization of GFP-GABARAPL2 in atg9−/− MEFs upon drug treatment (Fig. S5).
Figure 3.
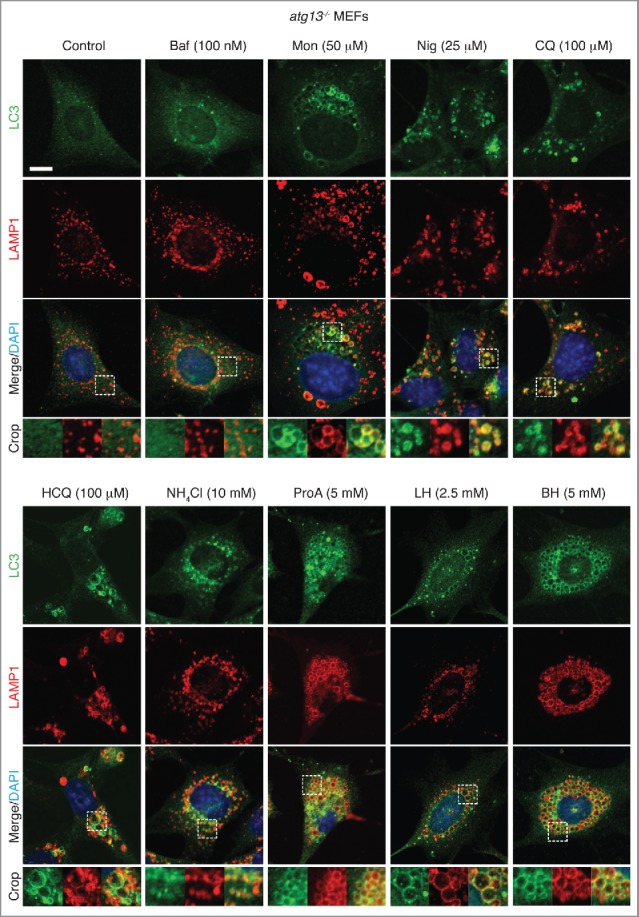
Activation of noncanonical autophagy and endolysosomal LC3 lipidation by lysosomotropic and ionophore drugs in atg13−/− MEFs. Confocal images of endogenous LC3 and LAMP1 immunostaining in atg13−/− MEFs treated with drugs at the indicated concentrations for 1–2 h. Inserts show zoomed regions highlighting colocalization of the LC3 and LAMP1 signal. Bar: 5 μm, for all images.
Figure 4.
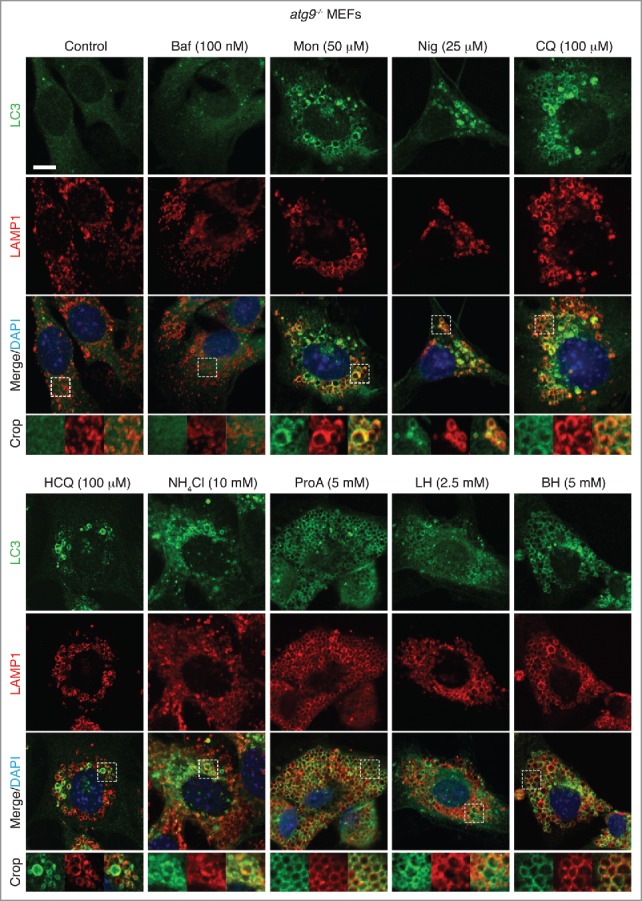
Activation of noncanonical autophagy and endolysosomal LC3 lipidation by lysosomotropic and ionophore drugs in atg9−/− MEFs. Confocal images of endogenous LC3 and LAMP1 immunostaining in atg9−/− MEFs treated with drugs at the indicated concentrations for 1–2 h. Inserts show zoomed regions highlighting colocalization of the LC3 and LAMP1 signal. Bar: 5 μm, for all images.
To confirm that the drug-induced LC3 lipidation and relocalization observed was associated with noncanonical autophagy, additional experiments were performed. Drug-induced effects on LC3 were lipidation dependent, as no lipidated LC3 was observed by western blot in ATG16−/− MCF10A cells upon drug treatment (Fig. S6A and S6B), and the lipidation-defective GFP-LC3G120A mutant showed no relocalization (Fig. S6C). In line with these results we also detected colocalization of the ATG12–ATG5-ATG16L1 complex to GFP-LC3-positive endomembranes in ATG13−/− cells upon drug treatment (Fig. S6C). Finally, GFP-LC3 was found to localize to V-ATPase-positive compartments following drug treatment, consistent with the proposed role that the V-ATPase functions in noncanonical autophagy (Fig. S6D).
Lysosomotropic drugs have the potential to damage lysosomes, which can then be targeted for engulfment by newly formed phagophores.28 To exclude the possibility that the observed LC3 response was due to autophagosomal targeting of damaged lysosomes, LGALS3/galectin-3 was used as a recently described marker of damaged endomembranes28 LGALS3 is a cytosolic protein that recognizes glycoproteins present on the extracellular side of the plasma membrane and lumenal side of intracellular vesicles. Damage of lysosome membranes allows LGALS3 to access lumenal glycoproteins. We monitored mCherry-LGALS3 and GFP-LC3 in autophagosome-deficient ATG13−/− MCF10A cells. Using glycyl-L-phenylalanine 2-naphthylamide (GPN) as a positive control for lysosome rupture,29 we detected potent induction of LGALS3 puncta (Fig. S7A). However, none of the lysosomotropic drugs used in this study induced any obvious LGALS3 translocation. Indeed, the GFP-LC3-positive vacuoles formed upon drug treatment were LGALS3 negative (Fig. S7B). These data rule out a role for autophagosome targeting of damaged vesicles in the observed LC3 response to the tested drugs.
Using multiple cell lines and genetic backgrounds, these data demonstrate that LC3 can be lipidated to endolysosomal membranes upon treatment with a range of ionophores and lysosomotropic drugs, independently of the canonical autophagy pathway. Colocalization of LC3 and LAMP1 is not complete suggesting that these drugs may also induce LC3 lipidation to other intracellular membranes in addition to endolysosomes. Taken together, the genetic requirements, morphological data and V-ATPase dependence of drug-induced LC3 lipidation demonstrate that these drugs are capable of activating a noncanonical autophagy pathway and endolysosomal LC3 lipidation.
Topical application of local anesthetics induces noncanonical autophagy in vivo
To determine whether lysosomotropic drug-induced noncanonical autophagy occurs in vivo, human skin biopsies were examined from control patients and those that had received a topical application of the local anesthetic EMLA before biopsy. EMLA cream is an anesthetic containing the active ingredients lidocaine and prilocaine, and has previously been reported to induce cellular vacuolation of subcutaneous keratinocytes and epithelial cells at the site of application.30,31 Using ATG13−/− MCF10A cells, we confirmed that lidocaine can activate noncanonical autophagy by promoting GFP-LC3 recruitment to swollen LAMP1-positive vesicles (Fig. 5A). We next stained control and EMLA-treated skin biopsies for LC3 by immunofluorescence. In agreement with published work, we found the most intense LC3 staining within the stratum granulosum (SG) layer.32,33 In 5/5 control samples, LC3 staining appeared as a diffusely grainy pattern. However, in 8/11 EMLA-treated samples LC3 localized to larger punctate compartments often observed as ring-like LC3 structures (Fig. 5B), reminiscent of those seen in lidocaine-treated MCF10A cells. To attempt to determine the identity of these EMLA-induced LC3-positive structures, we stained samples for CDSN (corneodesmosin), a marker for lamellar granules. Lamellar granules are acidic single-membrane lysosome-related organelles found in SG cells,34,35 and, as such, represent the type of compartment that can be targeted by noncanonical autophagy. Similar to LC3, EMLA treatment promoted a swelling of CDSN-positive structures (Fig. 5C). By costaining samples for LC3 and CDSN, we observed LC3 staining on some enlarged CDSN-positive structures following EMLA treatment (Fig. 5D). Finally, using transmission electron microscopy, we detected enlarged single-membrane compartments in EMLA-treated samples (Fig. 5E). Together these data support the idea that EMLA application promotes LC3 lipidation to non-autophagosome membranes, likely to represent lamellar granules in skin epidermal cells. These data are the first to propose that lysosomotropic drug-induced noncanonical autophagy and endolysosomal LC3 lipidation can be induced in vivo. This raises important questions regarding the consequences following activation of this pathway in vivo, which need to be addressed in the future.
Figure 5.

Local anesthetic EMLA promotes noncanonical autophagy in vivo. (A) Confocal images of GFP-LC3 and endogenous LAMP1 immunostaining in ATG13−/− MCF10A cells treated with lidocaine (2.5 mM) for 4 h. Inserts show zoomed regions highlighting colocalization of the LC3 and LAMP1 signal. Bar: 5 μm. Confocal imaging of (B) LC3 and (C) CDSN immunostaining in formalin-fixed paraffin embedded skin biopsies from (i) control patients and (ii) patients treated with the local anesthetic EMLA. Bar: 10 μm. Zoomed regions of representative maximal intensity Z-projections of stacks are shown (iii, iv). Bar: 4 μm. Swollen LC3- and CDSN-positive compartments (arrows) are observed in samples from patients treated with EMLA. (D) LC3 (green) and CDSN (white) and DAPI (blue) costaining of control and EMLA-treated skin biopsies. Colocalized signals (arrows) are observed in EMLA samples. Bar: 10 μm. (E) TEM images of an EMLA-treated biopsy. Arrows denote swollen compartments. N, nucleus. A cropped region is shown in the insert.
CCCP induces endolysosomal LC3 lipidation
Considering our data using inhibitors of canonical autophagy flux, we next sought to test whether published activators of canonical autophagy have any effect on the noncanonical pathway. CCCP is a proton uncoupler that acts as an ionophore for hydrogen.36 Treatment of cells with CCCP promotes mitochondrial dysfunction by dissipating proton gradients between inner and outer mitochondrial membranes. The E3 ubiquitin ligase PARK2/parkin is then recruited and regulates the engulfment and degradation of damaged mitochondria through a selective autophagic process termed mitophagy.37 Previously, LC3 lipidation observed following CCCP treatment has been attributed to ATG13-dependent autophagosome formation during mitophagy or bulk autophagy.38,39 However, in addition to eliminating proton gradients at mitochondria, CCCP will also dissipate proton gradients at lysosomes.40 In light of our results using the ionophores nigericin and monensin, we questioned whether CCCP could induce noncanonical autophagy and endolysosomal LC3 lipidation. We found that CCCP induced LC3 lipidation in a dose-dependent manner in both wild-type and ATG13−/− HEK293 cells, which could be inhibited by bafilomycin A1 pretreatment (Fig. 6A and 6B). Entotic corpse vacuoles are single-membrane acidic compartments formed following the live engulfment and killing of epithelial cells by the process entosis. These vacuoles represent the type of compartment that can be targeted by noncanonical autophagy.9,18 Using confocal microscopy we observed that CCCP promoted the robust recruitment of GFP-LC3 to LAMP1-positive entotic corpse vacuoles in wild-type and ATG13−/− MCF10A cells, in a bafilomycin A1-sensitive manner (Fig. 6C and 6B and Fig. S4B). We also noted a strong colocalization of GFP-LC3 with LAMP1-positive lysosomes upon CCCP treatment, which was resistant to a PIK3C3/VPS34 inhibitor (IN-1) able to inhibit starvation-induced GFP-LC3 puncta (Fig. 6E). Taken together, the ATG13- and PIK3C3/VPS34-independent lipidation of LC3 to non-autophagosome membranes clearly demonstrates that CCCP can activate noncanonical autophagy. CCCP did induce WIPI2 puncta in wild-type but not atg13−/− MEFs suggesting some activation of canonical autophagy (Fig. 6F). Bafilomycin A1 inhibited CCCP-induced LC3 lipidation (Fig. 6B and 6D) but not CCCP-mediated mitochondria depolarization and fragmentation (Fig. 6G). Thus, CCCP's ability to activate noncanonical autophagy is separate to its ability to promote mitochondria dysfunction. These data strengthen the conclusion that while CCCP does induce mitophagy, it is also a potent inducer of endolysosomal LC3 lipidation in parallel.
Figure 6.
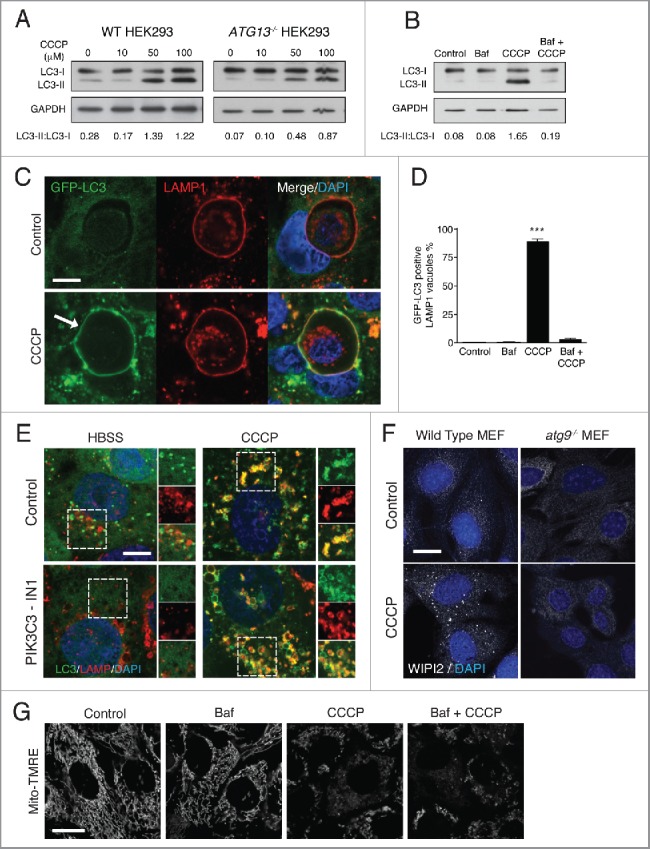
CCCP activates noncanonical autophagy. (A) Representative western blots of LC3 and GAPDH in wild-type and ATG13−/− HEK293 cells treated with increasing concentrations of CCCP. (B) ATG13−/− HEK293 cells were treated with Baf, CCCP or Baf + CCCP and lysates probed for LC3 and GAPDH. Ratios of lipidated LC3-II:nonlipidated LC3-I were quantified below. (C) Confocal images of GFP-LC3 and LAMP1 immunostaining on entotic corpse vacuoles in MCF10A cells treated with CCCP (100 μM) for 1 h. Arrow indicates lipidated LC3 on vacuoles. Bar: 10 μm for all images. (D) Quantification of GFP-LC3 recruitment to LAMP1-positive entotic vacuoles in cells treated with Baf (100 nM), CCCP (50 μM) or Baf + CCCP; data are mean ± SEM from 3 independent experiments, P<0.0003. (E) Confocal images of GFP-LC3 and LAMP1 immunostaining in MCF10A cells starved with HBSS or treated with CCCP (100 μM) for 2 h +/− PIK3C3 inhibitor IN-1 (5 μM) pre-treatment for 1 h. Zoomed regions highlight colocalization of LC3 and LAMP1 signals. Bar: 5 μm for all images. (F) Confocal images of WIPI2 staining in wild-type and atg9−/− MEFs +/− CCCP (100 μM) for 1 h. Bar: 20 μm for all images. (G) Confocal images of MCF10A cells labeled with Mito-TMRE treated with Baf (100 nM), CCCP (50 μM) or Baf + CCCP). Bar: 20 μm for all images
Amiodarone induces endolysosomal LC3 lipidation
Amiodarone is an FDA-approved antiarrhythmic drug used in the treatment of cardiac dysrhythmias. Amiodarone was identified via multiple microscopy-based screens as an inducer of GFP-LC3 puncta.6,41 Through its action as an L-type Ca2+ channel antagonist, amiodarone can decrease intracellular calcium levels, which inhibits the calcium-dependent cysteine proteases called calpains. This in turn activates autophagy, potentially through an MTOR-independent pathway.42 However, amiodarone also has lysosomotropic properties, accumulating in lysosomes and inducing their osmotic swelling.43 Considering our new data, we investigated whether noncanonical autophagy participates in amiodarone-induced LC3 lipidation.
In line with previous reports, we find that amiodarone raised lysosome pH as detected by loss of LysoTracker Red staining in MCF10A cells (Fig. 7A). We also noted swelling of intracellular LAMP1-positive vacuoles. Using wild-type and ATG13−/− HEK293 cells we found that amiodarone could induce a dose-dependent induction of LC3 lipidation in both cell lines by western blotting, which was inhibited by bafilomycin A1 pretreatment. (Fig. 7A-C). By immunofluorescence staining of endogenous LC3 we found that amiodarone could promote the relocalization of LC3 to swollen LAMP1 compartments in autophagy-deficient atg13−/− and atg9−/− MEFs (Fig. 7D). Amiodarone-induced GFP-LC3 relocalization was not inhibited by a PIK3C3/VPS34 inhibitor (Fig. 7E). These data provide clear and strong evidence that amiodarone can activate noncanonical autophagy. However, as with CCCP, we do detect amiodarone-induced WIPI2 puncta in wild-type, but not in atg9−/− MEFs, indicating that amiodarone induces both canonical and noncanonical autophagy in parallel (Fig. 7F).
Figure 7.

Amiodarone activates noncanonical autophagy. (A) Quantification of LysoTracker fluorescence intensity in MCF10A cells treated with amiodarone (Ami, 50 μM) for 2 h. Dashed line represents background fluorescence. Levels are mean +/− standard deviation. AU, arbitrary units. (B) Representative western blots of LC3 and GAPDH in wild-type and ATG13−/− HEK293 cells treated with increasing concentrations of amiodarone. (C) ATG13−/− HEK293 cells were treated with Baf (100 nM), amiodarone (Ami, 50 μM) or Baf + Ami and lysates probed for LC3 and GAPDH. Ratios of lipidated LC3-II:nonlipidated LC3-I were quantified below. (D) Confocal images of endogenous LC3 and LAMP1 immunostaining in atg13−/− and atg9−/− MEFs treated with amiodarone (50 μM) for 2 h. Inserts show zoomed regions highlighting colocalization of LC3 and LAMP1 signals. Bar: 10 μm for all images. (E) Confocal images of GFP-LC3 and LAMP1 immunostaining in MCF10A cells starved with HBSS or treated with amiodarone (50 μM) for 2 h +/− PIK3C3 inhibitor IN-1 (5 μM) pre-treatment for 1 h. Zoomed regions highlight colocalization of LC3 and LAMP1 signals. Bar: 5 μm for all images. (F) Confocal images of WIPI2 staining in wild-type and atg9−/− MEFs treated with +/− amiodarone (50 μM) for 2 h. Bar: 20 μm for all images.
Discussion
In this study, we report that a range of drugs with lysosomotropic or ionophore properties activate a noncanonical autophagy pathway, that directs LC3 lipidation to single-membrane endolysosomal compartments independently of the canonical autophagy pathway. Increased LC3 lipidation by these drugs has previously been attributed to inhibition of canonical autophagic flux or promotion of autophagosome formation (Table S1). However, our data clearly shows that these drugs can also induce LC3 lipidation in the absence of ATG13 and ATG9 or PIK3C3/VPS34 activity, all of which are required for autophagosome formation. It is highly unlikely that the observed LC3 lipidation is a result of alternative routes of autophagosome formation, such as BECN1/Beclin 1- or ULK1/2-independent pathways, as these mechanisms still require some autophagy machinery not necessary for the described noncanonical pathway. Instead our data indicate that LC3 is lipidated directly onto non-autophagosome membranes associated with the endolysosomal system.
In this study, we provide the first evidence supporting lysosomotropic drug-mediated activation of noncanonical autophagy in vivo. In human skin biopsy samples where the anesthetic cream EMLA containing lidocaine had been applied topically, we observed swollen LC3-positive vesicles in epidermal cells of the stratum granulosum layer, which bear markers of lamellar granules, lysosome-like organelles in skin. While we cannot exclude a role for canonical autophagy in these human samples, the single-membrane, low pH nature of the lamellar granules represents the characteristic type of compartment targeted by noncanonical autophagy. Indeed EMLA has previously been shown to induce vacuolation of single-membrane structures based on electron microscopy.30
We also report that CCCP, commonly used to induce mitophagy, can activate noncanonical autophagy in parallel. Interestingly, one study reported CCCP-induced LC3 lipidation occurring in the absence of RB1CC1/FIP200, ATG13 and ATG14; however, the authors did not determine what this lipidated LC3 represented.44 We now demonstrate that CCCP can promote LC3 lipidation to endolysosomal membranes completely independent from canonical autophagy. This is achieved at higher concentrations than those reported to induce mitophagy, but are still within levels commonly used in the study of autophagy. The requirement for higher concentrations may reflect the differences in sensitivity between mitochondria and lysosomes to proton permeability.
Amiodarone has been reported by several groups to be an activator of autophagosome formation.41-43,45,46 Amiodarone can promote autophagy via MTOR inhibition, possibly linked to its effect on lysosome function,42 and has also been proposed to activate an MTOR-independent autophagy pathway. As such it represents a drug of clinical interest where autophagy activation may be achieved, without further complicating effects of MTOR inhibition. For instance, induction of autophagy by amiodarone has been implicated in enhancing survival in mice following hepatectomy,47 and supressing hepatocellular carcinoma.48 In our study we clearly show that amiodarone is capable of activating noncanonical autophagy and endolysosomal LC3 lipidation in parallel to its effects on autophagosome formation, a parallel activity that will be important to consider.
We recently reported that osmotic imbalances within endolysosomal compartments can activate noncanonical autophagy and endolysosomal LC3 lipidation in a V-ATPase-dependent manner.18 This osmotic mechanism has been proposed to underlie physiological examples of endolysosmal LC3 lipidation such as LC3-associated phagocytosis and entosis. Considering that all of the drugs tested in our study have previously been shown to induce an osmotically driven swelling of single-membrane endolysosomal compartments, and our new data show that drug-induced LC3 lipidation is suppressed by inhibitors of the V-ATPase, we conclude that the same mechanism activating noncanonical autophagy is functional.
The results presented here raise important questions regarding the use and interpretation of LC3-based autophagy assays. Many genetic and pharmaceutical based high-throughput screens for autophagy modulators have been conducted with LC3 lipidation or puncta formation used as the readout. It should now be appreciated that LC3 is not a specific autophagosome marker and can be lipidated to non-autophagosome membranes. While not applicable to all known autophagy modulators, our results show that many drugs used to modulate the canonical autophagy pathway, primarily with lysosomotropic or ionophore properties, can also act as potent inducers of noncanonical autophagy and endolysosomal LC3 lipidation. This finding is an important consideration in the design and interpretation of future autophagy screens. Related to this, tandem RFP-GFP-LC3 constructs are now commonly used to measure autophagosome flux, whereby an increased dual signal is assumed to reflect a block in autophagosome-lysosome fusion. However, considering that LC3 associated with endolysosomal membranes will also retain both RFP and GFP signals, our findings on noncanonical autophagy further complicate the interpretation of such results. Therefore, we would recommend that in addition to assays measuring LC3 lipidation, autophagy screens should include other markers associated with autophagosome formation, such as autophagosome-associated phosphatidylinositol-3-phosphate effectors ZFYVE1/DFCP1 and WIPI2, to infer an effect on canonical autophagy induction. Similarly, measurement of autophagosome receptors such as SQSTM1/p62 or protein aggregates would give an accompanying indication on the degradation capacity of the autophagy pathway.
Our results highlight the need for further understanding of the functions and implications of noncanonical autophagy, which remain to be fully understood. LC3 lipidation to phagosomes during LAP is thought to modulate the fusion of lysosomes with the phagosome and degradation of its cargo. However, the exact mechanisms underlying this event are not known. Similarly, how LC3 on phagosomes directs the cytokine response in macrophages and dendritic cells remains to be determined. LC3 present on autophagosomes and endosomes have recently been shown to modulate MAPK/ERK signaling, enabling these compartments to act as a signaling platform.49,50 Whether noncanonical autophagy could generate similar endolysosomal signaling hubs would be of great interest and possibly inform on the efficacy and function of some autophagy-modulating drugs that possess endolysosomal LC3 lipidation properties.
Material and methods
Antibodies and reagents
The following primary antibodies were used at the indicated dilutions: anti-LC3A/B (1:1000 for western blotting and 1:100 for immunofluorescence; Cell Signaling Technology, 4108), anti-LC3A (1:100 for immunohistofluorescence; Abgent, 1805a), anti-ATG12 (1:100; Cell Signaling Technology, 2010), anti-ATG13 (1:1000; Cell Signaling Technology, 6940), anti-ATG16L1 (1:1000; Cell Signaling Technology, 8089), anti-human LAMP1 and anti-mouse LAMP1 (1:100; Becton Dickinson, 555798 and 553792), anti-WIPI2 (1:100; Bio-Rad, MCA5780GA), anti-CDSN/corneodesmosin (R&D Systems, AF5725), anti-GAPDH (1:2000; Santa Cruz Biotechnology, sc-25778), and anti-ATP6V0D1 (1:50; ABCAM, ab56441). The following inhibitors and reagents were used: Amiodarone hydrochloride (Sigma, A8423), bafilomycin A1 (Tocris Biosciences, 1334), betahistine dihydrochloride (Sigma, B4638), chloroquine (Sigma, C6628), CCCP (Sigma, C2759), HBSS (Gibco, 14025–092), hydroxychloroquine (Sigma, H0915), lidocaine (Sigma, L7757), lidocaine hydrochloride monohydrate (Sigma, L5647), monensin (Sigma, M5273), NH4Cl (Sigma, 213330), nigericin (Sigma, N7143), PP242 (Tocris, 4257), procainamide hydrochloride (Sigma, P9391). Mito-TMRE (abcam, 113852), PIK3C3/VPS34 inhibitor (IN-1) was kindly provided by Dr I. Ganley, University of Dundee.
Cell culture and transfection
MCF10A cells stably expressing GFP-LC3 were cultured in DMEM/F12 (Gibco, 11320–033) supplemented with 5% horse serum (Gibco, 16050–122), 20 ng/mL EGF (Peprotech, AF-100–15), 10 µg/mL insulin (Sigma, I9278), 0.5 µg/mL hydrocortisone (Sigma, H0888), 100 ng/µL cholera toxin (Sigma, C8052) and 100 U/mL penicillin and 100 µg/mL streptomycin (Gibco, 15140–122). MEF cell lines were maintained in DMEM (Gibco, 41966029) supplemented with 10% fetal bovine serum (Sigma, F9665) and 100 U/mL penicillin and 100 µg/mL streptomycin. Wild type, atg13−/− and atg9−/− MEFs and HEK293 cells stably expressing GFP-LC3 (kindly provided by N. Ktistakis, Babraham Institute) were cultured in complete DMEM supplemented with 0.4 mg/mL G418 (Melford Laboratories, G0175). Transfections of HEK293 GFP-LC3 cells were performed using the Lipofectamine® 2000 Transfection Reagent (Invitrogen, 11668–019). Transfections of MCF10A GFP-LC3 cells were performed using a Nucleofector II instrument (Lonza) and Lonza nucleofection kit V (Lonza, VCA-1003) following the manufacturer's guidelines. Electroporated cells were selected with 2.5 µg/mL puromycin (Sigma, P8833). The mCherry-LGALS3 expression plasmid was kindly provided by Dr. Felix Randow (MRC-LMB, Cambridge, UK). The pBabe.puro-DU-GFP GABARAPL2 (40072) expression plasmids were purchased from MRC-PPU Reagents. The pBabe.puro-GFP-LC3G120A expression plasmid was kindly provided by Dr. Noor Gammoh, University of Edinburgh, UK.
CRISPR/Cas9-mediated ATG13 and ATG16L1 knockout
To generate ATG13 knockout (KO) in HEK293 GFP-LC3 cells, CRISPR guide RNA (gRNA) forward and reverse sequences (Table S2) with overhangs for ligation into AflII restriction site were annealed and the resulting 100 base pair double-strand DNA fragment was cloned into the gRNA Cloning Vector (Addgene, 41824; deposited by Dr George Church) linearized with AflII restriction enzyme (NEB, R0520) using the Gibson Assembly® Master Mix (NEB, E2611). The recombinant ATG13 gRNA vector was co-transfected with a Cas9 expression vector (Addgene, 43861; deposited by Dr Keith Joung) in HEK293 GFP-LC3 cells. Single cell clones were generated following limiting dilution in 96-well plates.
To generate ATG13 and ATG16L1 KO in MCF10A GFP-LC3 cells, gRNA sequences with overhangs for containing a BpiI site were annealed and cloned into the pSpCas9(BB)-2A-GFP (Addgene, 48138; deposited by Dr Feng Zhang) digested with the BpiI restriction enzyme (Thermo Scientific, ER1011). The recombinant plasmid was electroporated in MCF10A GFP-LC3 cells along with a pBABE-puro construct (Addgene, 1764; deposited by Dr Hartmut Land). Cells were selected with 2.5 μg/ml puromycin (Sigma, P8833) for 48 h and single cells seeded into each well of 96-well plates.
After clonal expansion, KO clones were selected based on the absence of ATG13 or ATG16L1 protein detection by western blot. The rescue of a canonical autophagy response after MTOR inhibition and autophagic flux inhibition was verified in selected cells following transient transfection of an ATG13 expression vector (pmCherry-C1-ATG13 plasmid kindly provided by N. Ktistakis, Babraham Institute, UK).
Lysosomal pH measurement
To assess intracellular pH, cells were grown on 35-mm glass bottom dishes (MatTek, P35G-1.5–14C) and incubated in culture medium containing 67 nM LysoTracker Red DND-99 (Life Technologies, L-7528) for 2 h in the absence or presence of lysosomotropic and ionophore drugs. Confocal live-cell imaging was performed with the Confocal Zeiss LSM 780 (Carl Zeiss Ltd) equipped with a 40x oil immersion 1.40 numerical aperture (NA) objective and an incubation chamber set at 37°C and 5% CO2. Image acquisition was performed with Zen software (Carl Zeiss Ltd). All image processing (brightness and contrast) was performed on all pixels in each image and intracellular fluorescence intensity was measured using ImageJ software (NIH).
Immunofluorescence
For fluorescence immunocytology, cells were grown on glass coverslips, incubated as described in the figure legends, fixed in ice-cold methanol at −20°C for 5 min and washed in phosphate-buffered saline (PBS; Sigma, D8537). Samples were blocked in 5% bovine serum albumin (BSA; Sigma, A7906) in PBS for 1 h at room temperature (RT) before overnight incubation with primary antibodies in blocking buffer at 4°C. Following PBS washes samples were incubated with Alexa Fluor 488/568 goat anti-rabbit (H+L) or anti-mouse (H+L) secondary antibodies (1:500; Invitrogen, A-11034 and A-11031) for 1 h at RT. DNA was stained with DAPI (1 µg/mL; Sigma, D8417) before mounting coverslips with ProLong® Gold antifade mounting medium (Life Technologies, P36930). Image acquisition was performed with the Confocal Zeiss LSM 780 microscope (Carl Zeiss Ltd) equipped with a 40x oil immersion 1.40 numerical aperture (NA) objective using Zen software (Carl Zeiss Ltd). Images used in figures are representative of at least 3 independent repeats.
Immunofluorescence of skin biopsies
For fluorescence immunohistology, formalin-fixed paraffin embedded skin biopsy sections were dewaxed in xylene and rehydrated through descending ethanol concentrations. Antigen retrieval was performed by heating the sections at 100°C in citrate-based buffer, pH 6 (Vector laboratories, H-3300) for 20 min. Samples were then blocked in TBS-T (20 mM Tris, 140 mM NaCl, pH 7.6, 1% Tween 20 [Sigma, T9416]) supplemented with 5% BSA and 0.1 M glycine for 45 min at RT before overnight staining in primary antibodies diluted in blocking buffer at 4°C. Following washes in TBS-T, samples were incubated with Alexa Fluor 488 donkey anti-rabbit IgG (H+L) and/or TRITC donkey anti-sheep IgG (H+L) (Jackson ImmunoResearch, 711–545–152 and 713–025–147, respectively) secondary antibodies (1:400 in blocking buffer) for 45 min at RT. DNA was stained with DAPI (1 µg/mL) before mounting coverslips with ProLong® Gold antifade mounting medium. Confocal imaging of fixed samples was performed as above. All image processing (brightness and contrast) was performed on all pixels in each image.
TEM of skin biopsies
The skin biopsy sample was immersed in a fixative solution of 4% paraformaldehyde and 1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) for 48 h at 4°C. The skin biopsy slices were then washed in PBS, postfixed by incubation for 1 h with 2% osmium tetroxide and dehydrated in a graded series of ethanol solutions. Samples were embedded in Epon resin (Electron Microscopy Sciences, 14120), which was allowed to polymerize for 48 h at 60°C. Ultrathin sections were cut, stained with 2.5% uranyl acetate, 1% lead citrate, and deposited on EM grids for examination under a JEOL 1011 transmission electron microscope (TEM).
Western blotting
Cells were scraped into ice-cold RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1% Triton X-100 (Sigma, T8787), 0.1% SDS (Sigma, L3771), 0.5% sodium deoxycholate (Sigma, D6750) and lysed on ice for 10 min. Lysates were centrifuged for 10 min at 10,000 g at 4°C. Supernatants were then separated on 15% or 10% polyacrylamide SDS-PAGE gels and transferred to polyvinylidene difluoride membranes. Membranes were blocked in TBS-T supplemented with 5% BSA for 1 h at room temperature and incubated overnight at 4°C with primary antibodies diluted in blocking buffer. They were then incubated with a horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology, 7074S) and proteins were detected using enhanced chemiluminescence (GE Healthcare Life Sciences, RPN2209). Densitometry analysis was performed using ImageJ software.
Statistics
Indicated P values were obtained using the Student t test or one-way ANOVA using Graphpad Prism software.
Supplementary Material
Abbreviations
- ATG
autophagy-related
- Baf
bafilomycin A1
- BH
betahistine
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- CDSN
corneodesmosin
- CRISPR
clustered regularly interspaced short palindromic repeats
- CQ
chloroquine
- EMLA
eutectic mixture of local anesthetics
- GFP
green fluorescent protein
- GPN
glycyl-L-phenylalanine 2-naphthylamide
- HCQ
hydroxychloroquine
- LAMP1
lysosomal-associated membrane protein 1
- LAP
LC3-associated phagocytosis
- LD
lidocaine
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MEF
mouse embryonic fibroblast
- Mon
monensin
- LGALS3/galectin-3
lectin, galactose, soluble 3
- MTOR
mechanistic target of rapamycin
- Nig
nigericin
- ProA
procainamide
- RB1CC1
RB1-inducible coiled-coil 1
- V-ATPase
vacuolar-type H+-translocating ATPase
- WIPI2
WD repeat domain, phosphoinositide interacting 2
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank the members of the Florey laboratory for their comments on the manuscript and the imaging and genome targeting facilities at the Babraham Institute for their assistance. We thank Dr Nicholas Ktiskatis for sharing cell lines, antibodies and plasmids and Dr Ian Ganley for the VPS34 inhibitor IN-1. Electron Microscopy data were obtained with the technical assistance of C. Hayot and J. Rousseau at the IBiSA Electron Microscopy Facility of University François Rabelais of Tours.
Funding
This work was supported by a Cancer Research UK fellowship C47718/A16337.
References
- [1].Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. The New England journal of medicine 2013; 368:651-62; PMID: 23406030; http://dx.doi.org/ 10.1056/NEJMra1205406 [DOI] [PubMed] [Google Scholar]
- [2].Hale CM, Cheng Q, Ortuno D, Huang M, Nojima D, Kassner PD, Wang S, Ollmann MM, Carlisle HJ. Identification of modulators of autophagic flux in an image-based high content siRNA screen. Autophagy 2016; 12:713-26; PMID: 27050463; http://dx.doi.org/ 10.1080/15548627.2016.1147669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Orvedahl A, Sumpter R Jr., Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, et al.. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011; 480:113-7; PMID: 22020285; http://dx.doi.org/ 10.1038/nature10546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rubinsztein DC, Bento CF, Deretic V. Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J Exp Med 2015; 212:979-90; PMID: 26101267; http://dx.doi.org/ 10.1084/jem.20150956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709-30; PMID: 22935804; http://dx.doi.org/ 10.1038/nrd3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, et al.. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proceedings of the National Academy of Sciences of the United States of America 2007; 104:19023-8; PMID: 18024584; http://dx.doi.org/ 10.1073/pnas.0709695104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Research 2014; 24:24-41; PMID:24366339; https://dx.doi.org/26799652 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID: 26799652; http://dx.doi.org/ 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nature cell biology 2011; 13:1335-43; PMID: 22002674; http://dx.doi.org/ 10.1038/ncb2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Florey O, Overholtzer M. Autophagy proteins in macroendocytic engulfment. Trends in cell biology 2012; 22:374-80; PMID: 22608991; http://dx.doi.org/ 10.1016/j.tcb.2012.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al.. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450:1253-7; PMID: 18097414; http://dx.doi.org/ 10.1038/nature06421 [DOI] [PubMed] [Google Scholar]
- [12].Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, Glogauer M, Grinstein S, Brumell JH. Activation of antibacterial autophagy by NADPH oxidases. Proceedings of the National Academy of Sciences of the United States of America 2009; 106:6226-31; PMID: 19339495; http://dx.doi.org/ 10.1073/pnas.0811045106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ma J, Becker C, Lowell CA, Underhill DM. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. The Journal of biological chemistry 2012; 287:34149-56; PMID: 22902620; http://dx.doi.org/ 10.1074/jbc.M112.382812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proceedings of the National Academy of Sciences of the United States of America 2011; 108:17396-401; PMID: 21969579; http://dx.doi.org/ 10.1073/pnas.1113421108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Romao S, Gasser N, Becker AC, Guhl B, Bajagic M, Vanoaica D, Ziegler U, Roesler J, Dengjel J, Reichenbach J, et al.. Autophagy proteins stabilize pathogen-containing phagosomes for prolonged MHC II antigen processing. The Journal of cell biology 2013; 203:757-66; PMID: 24322427; http://dx.doi.org/ 10.1083/jcb.201308173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, Li QZ, Yan M, Janke L, Guy C, et al.. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 2016; 533:115-9; PMID: 27096368; http://dx.doi.org/ 10.1038/nature17950 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [17].Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J, et al.. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nature cell biology 2015; 17:893-906; PMID: 26098576; http://dx.doi.org/ 10.1038/ncb3192 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [18].Florey O, Gammoh N, Kim SE, Jiang X, Overholtzer M. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 2015; 11:88-99; PMID: 25484071; http://dx.doi.org/ 10.4161/15548627.2014.984277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol 2012; 74:69-86; PMID: 22335796; http://dx.doi.org/ 10.1146/annurev-physiol-012110-142317 [DOI] [PubMed] [Google Scholar]
- [20].Tapper H, Sundler R. Role of lysosomal and cytosolic pH in the regulation of macrophage lysosomal enzyme secretion. Biochem J 1990; 272:407-14; PMID: 2268269; http://dx.doi.org/ 10.1042/bj2720407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].de Duve C, de Barsy T, Poole B, Trouet A, Tulkens P, Van Hoof F. Commentary. Lysosomotropic agents. Biochem Pharmacol 1974; 23:2495-531; http://dx.doi.org/ 10.1016/0006-2952(74)90174-9 [DOI] [PubMed] [Google Scholar]
- [22].Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol 2009; 625:220-33; PMID: 19836374; http://dx.doi.org/ 10.1016/j.ejphar.2009.06.063 [DOI] [PubMed] [Google Scholar]
- [23].Ramos Alcocer R, Ledezma Rodriguez JG, Navas Romero A, Cardenas Nunez JL, Rodriguez Montoya V, Deschamps JJ, Liviac Ticse JA. Use of betahistine in the treatment of peripheral vertigo. Acta Otolaryngol 2015; 135:1205-11; PMID: 26245698; http://dx.doi.org/ 10.3109/00016489.2015.1072873 [DOI] [PubMed] [Google Scholar]
- [24].Marceau F, Bawolak MT, Lodge R, Bouthillier J, Gagne-Henley A, Gaudreault RC, Morissette G. Cation trapping by cellular acidic compartments: beyond the concept of lysosomotropic drugs. Toxicol Appl Pharmacol 2012; 259:1-12; PMID: 22198553; http://dx.doi.org/ 10.1016/j.taap.2011.12.004 [DOI] [PubMed] [Google Scholar]
- [25].Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. The Journal of biological chemistry 2009; 284:12297-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kaizuka T, Mizushima N. Atg13 Is Essential for Autophagy and Cardiac Development in Mice. Mol Cell Biol 2015; 36:585-95; PMID: 26644405; http://dx.doi.org/ 10.1128/MCB.01005-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. The Journal of cell biology 2010; 190:1005-22; PMID: 20855505; http://dx.doi.org/ 10.1083/jcb.200912089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, et al.. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. The EMBO journal 2013; 32:2336-47; PMID: 23921551; http://dx.doi.org/ 10.1038/emboj.2013.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012; 482:414-8; PMID: 22246324; http://dx.doi.org/ 10.1038/nature10744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cazes A, Prost-Squarcioni C, Bodemer C, Heller M, Brousse N, Fraitag S. Histologic cutaneous modifications after the use of EMLA cream, a diagnostic pitfall: review of 13 cases. Arch Dermatol 2007; 143:1074-6; PMID: 17709674; http://dx.doi.org/ 10.1001/archderm.143.8.1074 [DOI] [PubMed] [Google Scholar]
- [31].Vallance H, Chaba T, Clarke L, Taylor G. Pseudo-lysosomal storage disease caused by EMLA cream. J Inherit Metab Dis 2004; 27:507-11; PMID: 15303008; http://dx.doi.org/ 10.1023/B:BOLI.0000037352.98317.5a [DOI] [PubMed] [Google Scholar]
- [32].Chikh A, Sanza P, Raimondi C, Akinduro O, Warnes G, Chiorino G, Byrne C, Harwood CA, Bergamaschi D. iASPP is a novel autophagy inhibitor in keratinocytes. Journal of cell science 2014; 127:3079-93; PMID: 24777476; http://dx.doi.org/ 10.1242/jcs.144816 [DOI] [PubMed] [Google Scholar]
- [33].Rossiter H, Konig U, Barresi C, Buchberger M, Ghannadan M, Zhang CF, Mlitz V, Gmeiner R, Sukseree S, Fodinger D, et al.. Epidermal keratinocytes form a functional skin barrier in the absence of Atg7 dependent autophagy. J Dermatol Sci 2013; 71:67-75; PMID: 23669018; http://dx.doi.org/ 10.1016/j.jdermsci.2013.04.015 [DOI] [PubMed] [Google Scholar]
- [34].Ishida-Yamamoto A, Simon M, Kishibe M, Miyauchi Y, Takahashi H, Yoshida S, O'Brien TJ, Serre G, Iizuka H. Epidermal lamellar granules transport different cargoes as distinct aggregates. J Invest Dermatol 2004; 122:1137-44; PMID: 15140216; http://dx.doi.org/ 10.1111/j.0022-202X.2004.22515.x [DOI] [PubMed] [Google Scholar]
- [35].Chapman SJ, Walsh A. Membrane-coating granules are acidic organelles which possess proton pumps. J Invest Dermatol 1989; 93:466-70; PMID: 2550559; http://dx.doi.org/ 10.1111/1523-1747.ep12284032 [DOI] [PubMed] [Google Scholar]
- [36].Kasianowicz J, Benz R, McLaughlin S. The kinetic mechanism by which CCCP (carbonyl cyanide m-chlorophenylhydrazone) transports protons across membranes. J Membr Biol 1984; 82:179-90; PMID: 6096547; http://dx.doi.org/ 10.1007/BF01868942 [DOI] [PubMed] [Google Scholar]
- [37].Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology 2008; 183:795-803; PMID: 19029340; http://dx.doi.org/ 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, Zhang J, Iyengar R, Jung CH, Suen DF, et al.. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell 2011; 43:572-85; PMID: 21855797; http://dx.doi.org/ 10.1016/j.molcel.2011.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kwon KY, Viollet B, Yoo OJ. CCCP induces autophagy in an AMPK-independent manner. Biochem Biophys Res Commun 2011; 416:343-8; PMID: 22119190; http://dx.doi.org/ 10.1016/j.bbrc.2011.11.038 [DOI] [PubMed] [Google Scholar]
- [40].Padman BS, Bach M, Lucarelli G, Prescott M, Ramm G. The protonophore CCCP interferes with lysosomal degradation of autophagic cargo in yeast and mammalian cells. Autophagy 2013; 9:1862-75; PMID: 24150213; http://dx.doi.org/ 10.4161/auto.26557 [DOI] [PubMed] [Google Scholar]
- [41].Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, Nabi IR, Roberge M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One 2009; 4:e7124; PMID: 19771169; http://dx.doi.org/ 10.1371/journal.pone.0007124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, et al.. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4:295-305; PMID: 18391949; http://dx.doi.org/ 10.1038/nchembio.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Morissette G, Ammoury A, Rusu D, Marguery MC, Lodge R, Poubelle PE, Marceau F. Intracellular sequestration of amiodarone: role of vacuolar ATPase and macroautophagic transition of the resulting vacuolar cytopathology. Br J Pharmacol 2009; 157:1531-40; PMID: 19594752; http://dx.doi.org/ 10.1111/j.1476-5381.2009.00320.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen D, Chen X, Li M, Zhang H, Ding WX, Yin XM. CCCP-Induced LC3 lipidation depends on Atg9 whereas FIP200/Atg13 and Beclin 1/Atg14 are dispensable. Biochem Biophys Res Commun 2013; 432:226-30; PMID: 23402761; http://dx.doi.org/ 10.1016/j.bbrc.2013.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lee KY, Oh S, Choi YJ, Oh SH, Yang YS, Yang MJ, Lee K, Lee BH. Activation of autophagy rescues amiodarone-induced apoptosis of lung epithelial cells and pulmonary toxicity in rats. Toxicol Sci 2013; 136:193-204; PMID: 23912912; http://dx.doi.org/ 10.1093/toxsci/kft168 [DOI] [PubMed] [Google Scholar]
- [46].Renna M, Jimenez-Sanchez M, Sarkar S, Rubinsztein DC. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. The Journal of biological chemistry 2010; 285:11061-7; PMID: 20147746; http://dx.doi.org/ 10.1074/jbc.R109.072181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lin CW, Chen YS, Lin CC, Chen YJ, Lo GH, Lee PH, Kuo PL, Dai CY, Huang JF, Chung WL, et al.. Amiodarone as an autophagy promoter reduces liver injury and enhances liver regeneration and survival in mice after partial hepatectomy. Sci Rep 2015; 5:15807; PMID: 26515640; http://dx.doi.org/ 10.1038/srep15807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, Lin YJ, Wu CT, Liu HS. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology 2014; 59:505-17; PMID: 23913306; http://dx.doi.org/ 10.1002/hep.26659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Barrow-McGee R, Kishi N, Joffre C, Menard L, Hervieu A, Bakhouche BA, Noval AJ, Mai A, Guzman C, Robert-Masson L, et al.. Beta 1-integrin-c-Met cooperation reveals an inside-in survival signalling on autophagy-related endomembranes. Nature communications 2016; 7:11942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Martinez-Lopez N, Athonvarangkul D, Mishall P, Sahu S, Singh R. Autophagy proteins regulate ERK phosphorylation. Nature communications 2013; 4:2799; PMID: 24240988; http://dx.doi.org/ 10.1038/ncomms3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.