

ABSTRACT
Chaperone-mediated autophagy (CMA), a selective form of protein lysosomal degradation, is maximally activated in stress situations to ensure maintenance of cellular homeostasis. CMA activity decreases with age and in several human chronic disorders, but in contrast, in most cancer cells, CMA is upregulated and required for tumor growth. However, the role of CMA in malignant transformation remains unknown. In this study, we demonstrate that CMA inhibition in fibroblasts augments the efficiency of MYC/c-Myc-driven cellular transformation. CMA blockage contributes to the increase of total and nuclear MYC, leading to enhancement of cell proliferation and colony formation. Impaired CMA functionality accentuates tumorigenesis-related metabolic changes observed upon MYC-transformation. Although not a direct CMA substrate, we have found that CMA regulates cellular MYC levels by controlling its proteasomal degradation. CMA promotes MYC ubiquitination and degradation by regulating the degradation of C330027C09Rik/KIAA1524/CIP2A (referred to hereafter as CIP2A), responsible for MYC stabilization. Ubiquitination and proteasomal degradation of MYC requires dephosphorylation at Ser62, and CIP2A inhibits the phosphatase responsible for this dephosphorylation. Failure to degrade CIP2A upon CMA blockage leads to increased levels of phosphorylated MYC (Ser62) and to stabilization of this oncogene. We demonstrate that this phosphorylation is essential for the CMA-mediated effect, since specific mutation of this site (Ser62 to Ala62) is enough to normalize MYC levels in CMA-incompetent cells. Altogether these data demonstrate that CMA mitigates MYC oncogenic activity by promoting its proteasomal degradation and reveal a novel tumor suppressive role for CMA in nontumorigenic cells.
KEYWORDS: Autophagy, cancer, CIP2A, lysosomes, oncogene, proteolysis
INTRODUCTION
Autophagy is a highly conserved cellular process that mediates targeting and degradation of intracellular components (proteins and organelles) in lysosomes.1 This continuous clearance contributes to maintenance of cellular homeostasis by controlling levels and quality of intracellular constituents and ensures cell viability under stress conditions.2 In addition, the recycling ability of the autophagic process contributes to preserve a positive energetic balance by providing important metabolic precursors.3 Out of the 3 different types of autophagy that coexist in mammalian cells, microautophagy, macroautophagy and chaperone-mediated autophagy (CMA),2,4 only the interplay of macroautophagy and the oncogenic processes has been extensively studied. Thus, it is well accepted that macroautophagy plays a dual role on cancer, depending of tumor type and disease stage.5,6 Although macroautophagy displays tumor-suppressive functions, in different contexts, it can also facilitate tumor development and progression.5,6 In contrast, the interplay of other forms of autophagy with cancer has been less studied.7
In this work we focus on CMA, a form of autophagy in which selectivity is conferred by the presence in substrate proteins of a motif (KFERQ-like motif) that once recognized by the chaperone HSPA8 mediates their targeting for lysosomal degradation.4,8,9 The targeted substrate binds to lysosomes through the cytosolic portion of LAMP2A (lysosomal-associated membrane protein 2A).10 Substrate binding triggers LAMP2A multimerization into a high molecular order complex through which, upon unfolding, the substrate is translocated across the lysosomal membrane and readily degraded in the lysosomal lumen.4,7,11–14
Basal levels of CMA are detectable in almost all mammalian cell types but this process is maximally upregulated in response to stressors such as proteotoxicity, nutritional stress, hypoxia or genotoxic insults.7 Decreases of CMA activity have been implicated in neurodegeneration, metabolic abnormalities and aging.15–18 In contrast, CMA seems to be constitutively upregulated in multiple types of cancer cells and in tumors and its blockage in transformed cells has anticancer potential.7 Thus, CMA inhibition in malignant cells lowers cellular proliferation and reduces tumorigenic and metastatic capacity.7,19 Later reports have confirmed this dependence on CMA for cancer cell growth and attribute it to a diversity of mechanisms including: protection against oxidative damage,19 removal of negative regulators of cell proliferation20 and antioncogenes21 or maintenance of the metabolic switch favorable for cancer cell growth.22,23 However, the role of CMA on early stages of tumor development such as transformation of normal cells into cancerous cells has not been elucidated.
In this work, we have analyzed CMA requirement during MYC (myelocytomatosis viral oncogene homolog)-mediated fibroblasts malignant transformation. The transcription factor MYC/c-Myc, cellular ortholog of the oncogene v-myc; (avian myelocytomatosis virus), is key for malignant transformation of fibroblasts by inducing cell cycle progression and metabolic changes required for neoplastic transformation.24,25 Genetic alterations of the MYC gene are frequently found in human cancers26,27 where the MYC proto-oncogene can be activated through several mechanisms, including gene amplification and point mutations in its coding sequence.27 Phosphorylation events control proteasome-mediated degradation of MYC, explaining why residues such as threonine 58 (Thr58) and serine 62 (Ser62) (major sites of MYC phosphorylation) are often mutated in cancer.28
We demonstrate here that CMA prevents MYC-driven malignant transformation of fibroblasts by regulating its degradation. However, although MYC bears 2 KFERQ-like motifs (267VEKRQ271 and 361VLERQ365), which could make it amenable for CMA degradation and, in fact, it accumulates in CMA-defective cells, we found that in the conditions analyzed in this study, MYC is not a direct target for lysosomal degradation, and it is mostly degraded by the ubiquitin-proteasome system. We have discovered that CMA exerts an indirect control of MYC levels by degrading the recently described cancerous inhibitor of PPP2/PP2A, CIP2A (please note that current human [HGNC/HUGO] nomenclature for the commonly termed “CIP2A” is “KIAA1524," mouse [MGI] nomenclature is “C330027C09Rik” for the same gene and rat [RGD] nomenclature leads to “RGD1310335." Therefore, for the sake of simplicity we use “CIP2A” in subsequent instances). Blockage of CMA increases CIP2A levels, thus preventing PPP2-mediated-MYC dephosphorylation on the Ser62 residue, required for its ubiquitination and proteasomal degradation. In fact, CIP2A knockdown was able to revert the MYC accumulation observed in CMA-incompetent cells and their transformation advantage. Therefore, this work highlights an anticancerous role of CMA in untransformed cells mediated by its ability to tightly control MYC levels.
Results
CMA deficiency facilitates MYC-driven malignant transformation of fibroblasts
To gain insights into the potential role of CMA on early stages of carcinogenesis, we analyzed the effect of CMA blockage on cellular transformation induced by MYC proto-oncogene overexpression in mouse fibroblasts (NIH-3T3 cells). For this purpose, we blocked CMA using stable short hairpin RNA (shRNA) interference against Lamp2a (Fig. S1A and B) and then transduced control (ctrl) and LAMP2A knockdown cells (LAMP2A [-]) with lentivirus carrying a vector for MYC expression or an empty one. Despite a similar increase in Myc mRNA levels (about 60-fold increase) in ctrl or LAMP2A (-) cells (Fig. S1C), we found that MYC protein levels in CMA-deficient cells were significantly (P ≤ 0.01) higher than in ctrl cells (Fig. 1A and B). Using subcellular fractioning of MYC-transduced cells we confirmed that the nuclear levels of this transcription factor were also higher in LAMP2A (-) than in ctrl cells (Fig. 1C).
Figure 1.

Blockage of CMA potentiates MYC protein accumulation, proliferation and transformation driven by MYC overexpression. (A) Immunoblot for MYC and ACTB of total protein lysates from NIH-3T3 cells, control (ctrl) or LAMP2A knockdown (LAMP2A [-]), transduced with MYC or the empty vector. (B) Densitometry analysis of immunoblots as the one shown in A. Values are expressed relative to the ctrl empty-vector cells, normalized to Ponceau S staining (N = 6). (C) Immunoblot for the indicated proteins of cytoplasmic and nuclear subcellular fractions from MYC-transduced ctrl or LAMP2A (-) cells (N = 2). HSP90 and LMNA/Lamin A/C expression were used as cytoplasmatic and nuclear controls, respectively. (D) Growth curves (N = 9), (E) soft-agar transformation assay (N = 6) and (F) focus formation assay (N = 6) of the same cells from (A). (E and F) Above: representative images from each transformation assay; below quantification of colony and focus formation capacity, respectively. In all graphs values are presented as mean ± SEM. Two-way ANOVA and the Bonferroni post-hoc test were used and differences were considered significant for *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001
We next confirmed that the elevated MYC protein levels in CMA deficient cells potentiated several transformation-related phenotypes in these cells. Thus, growth curve analysis showed significantly (P ≤ 0.01) higher proliferation rates for the LAMP2A (-) MYC-transduced cells when compared with the other ones (Fig. 1D). Combination of LAMP2A knockdown and MYC protein overexpression also resulted in significantly higher cellular transformation capacity (Fig. 1E and F). CMA blockage exerted a synergic effect on MYC-driven malignant transformation both in the soft-agar colony formation assay (Fig. 1E) and in foci formation assay (Fig. 1F). Immunoblot for LAMP2A confirmed that the knockdown for LAMP2A remained stable through multiple cell division cycles (Fig. S1D). We also tested for any possible direct detrimental effect of the double manipulation in these cells (LAMP2A knockdown and MYC transfection) but did not find significant differences between treated and untreated cells in their levels of oxidized proteins (Fig. S1E) or overall cellular viability (Fig. S1F).
Differences in MYC abundance upon CMA blockage were not a peculiarity of this mouse cell line since mouse embryonic fibroblasts (MEFs) from lamp2a knockout mice (lamp2a KO) also displayed higher MYC levels (Fig. S2A, B) and proliferative rates (Fig. S2C) when compared with MEFs from wild-type littermate mice.
Changes in cellular metabolism, characterized by a switch from oxidative phosphorylation to aerobic glycolysis (known as the Warburg effect) are classically associated with tumor transformation.26 To investigate whether CMA blockage also cooperates in the acquisition of this key cancer metabolic phenotype by the MYC-transduced cells, we measured oxygen consumption (OCR) and extracellular acidification (ECAR) rates using the Seahorse Bioscience extracellular flux analyzer (Fig. 2). In agreement with the transformation assay data (Fig. 1E and F), CMA blockage potentiated the MYC-induced changes in OCR. Thus, LAMP2A (-) MYC cells presented a significant decrease in basal (P ≤ 0.05) and maximal respiration (P ≤ 0.001) when compared with the already reduced values observed upon expression of MYC in CMA proficient cells (Fig. 2A and B). In the case of aerobic glycolysis (measured by ECAR), we have previously reported that although blockage of CMA in untransformed cells leads to increased glycolytic flux due to failure to degrade glycolytic enzymes,17,29 blockage of CMA in transformed cells has a completely opposed effect as it reduces their glycolytic flux through transcriptional downregulation of the same enzymes.7 Consistent with those findings, untransformed LAMP2A (-) cells displayed a very pronounced increase in glycolysis and glycolytic capacity (P ≤ 0.01), that although, markedly reduced upon MYC transformation, still remained elevated relative to MYC transformed CMA competent cells (Fig. 2C and D). In summary, our results reveal that reduction of CMA activity in nontumorigenic cells made them more susceptible to MYC-driven malignant transformation. We propose that the significantly higher levels of MYC protein in CMA-incompetent cells are behind the synergistic effect of CMA blockage on MYC mediated cell proliferation, clonogenicity and tumor-favorable metabolic changes.
Figure 2.
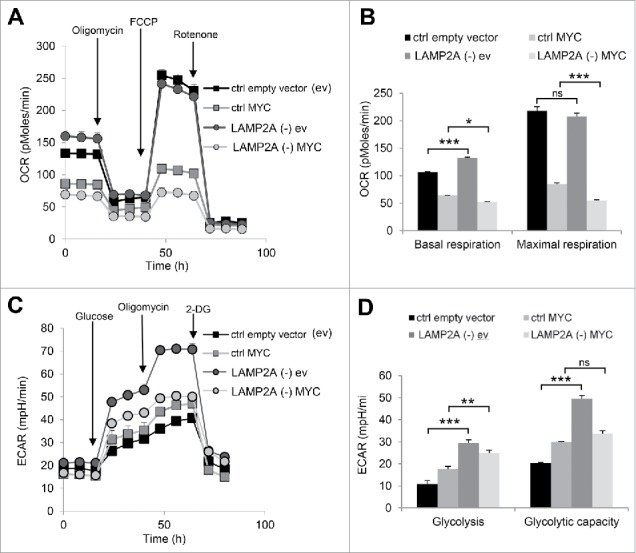
Blockage of CMA in MYC-transduced cells enhances changes in cellular metabolism associated with a transformed phenotype. (A) Seahorse Bioscience analyzer was used to measure oxygen consumption rate (OCR) in NIH-3T3 cells, control (ctrl) or LAMP2A knockdown (LAMP2A [-]), transduced with lentiviral-delivered MYC or empty vector (N = 3). (B) Quantification of basal and maximal respiration from OCR data (A). (C) A Seahorse Bioscience analyzer was used to measure extracellular acidification rate (ECAR) in the same cells as in (A) (N = 3). (D) Glycolysis and glycolytic capacity quantification form ECAR data (C). In all graphs values are presented as mean ± SEM. Two-way ANOVA and the Bonferroni post-hoc test were used and differences were considered significant for *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. ns represents nonsignificant changes.
MYC in transforming cells is not degraded through CMA, but by the proteasome
Given the pronounced increase in MYC levels in LAMP2A (-) cells (Fig. 1A and B), we investigated whether a fraction of cellular MYC is degraded by CMA. Analysis of the effect of lysosomal proteolysis inhibition on endogenous MYC levels (Fig. 3A and B) revealed that treatment with ammonium chloride and leupeptin to block lysosomal degradation did not lead to any significant increase on MYC levels, even when serum was removed to maximally activate CMA30 (Fig. 3A and B). In contrast, treatment with the proteasome inhibitor MG132 induced significant (P ≤ 0.01) accumulation of MYC protein regardless of serum presence or absence (Fig. 3A and B). Positive controls of the efficiency of these inhibitors are shown in Fig. S3A, B and C: TP53 (p53/TP53/TRP53 (please note that the mouse [MGI] nomenclature is TRP53, but we use TP53 [HGNC/HUGO nomenclature] hereafter to refer to both the human and mouse genes/proteins for simplicity), and ubiquitin K48 [UB (K48)] linkage (proteasome substrates, Fig. S3A and B) and MAP1LC3B-II/LC3B-II (autophagosome-associated form of LC3B-I and a lysosomal substrate, Fig. S3A and C).
Figure 3.

MYC is a proteasome substrate and does not undergo degradation by CMA. (A) Immunoblot for the indicated proteins in total cell extracts of NIH-3T3 cells at time zero (Time 0) and 12 h after incubation without additions (None) or in the presence of lysosomal proteolysis inhibitors (ammonium chloride and leupeptin, N/L) or a proteasome inhibitor (MG132) in media supplemented (Serum +) or not (Serum -) with serum. (B) Densitometry analysis of MYC levels in blots as the representative one shown in A. Values are expressed relative to untreated (None) serum + conditions and normalized to Ponceau S staining (N = 6). (C) Immunoblot for the indicated proteins in total cell extracts from MYC-transduced cells, control (ctrl) or LAMP2A knockdown (LAMP2A [-]) treated for 12 h with N/L and MG132 in serum-supplemented media. (D) Densitometry analysis of MYC in blots as the representative one shown in (C). Values are expressed relative to untreated (None) ctrl MYC-transduced cells and normalized to Ponceau S staining (N = 4). (E) Immunoblot for MYC and LAMP1 of homogenates (HOM.) and lysosome-enriched fractions (Lys enr.) isolated from control and LAMP2A (-) cells, transduced with MYC. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) is shown as an example of a well-characterized CMA substrate. Right: Densitometry analysis of MYC levels in blots as the one shown here. Values are expressed relative to control homogenates after normalization by Ponceau S staining (N = 2). All values are presented as mean ± SEM. Two-way ANOVA and the Bonferroni post-hoc test were used and differences were considered significant for *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. ns represents nonsignificant changes.
We next assessed the possible role of CMA on the degradation of exogenous MYC. For this purpose, CMA-competent and CMA-deficient cells transduced with MYC were treated with inhibitors of lysosomal proteolysis or of the proteasome (Fig. 3C and D). Similar to endogenous MYC, exogenously expressed MYC failed to accumulate upon inhibition of lysosomal degradation with ammonium chloride and leupeptin (Fig. 3C and D), even after doubling the duration of this treatment (Fig. S4A, B; note that as levels of proteins degraded in lysosomes increase upon ammonium chloride and leupeptin treatment, the relative contribution of nonlysosomal substrates, such as MYC, to the total cellular protein content is less resulting in its lower levels in the immunoblot). Lastly, we found negligible levels of MYC in lysosome-enriched fractions isolated from control and LAMP2A (-) cells (Fig. 3E; compared with the bona fide CMA substrate GAPDH), further supporting that CMA does not directly contribute to MYC degradation in these cells under these conditions.
However, inhibition of the proteasome stimulated significant (P ≤ 0.01) accumulation of MYC, but only in CMA-competent (ctrl) cells (Fig. 3C and D). The higher MYC protein levels observed in LAMP2A (-) upon MYC transduction were not significantly altered by treatment with MG132 (Fig. 3D), suggesting that MYC protein expressed in CMA deficient cells is less susceptible to proteasomal degradation. Analysis of the efficiency of the different protease inhibitors (Fig. S3D and E) revealed that failure to degrade MYC in CMA-incompetent cells was not due to overall defect of the proteasome system since degradation of other proteasome substrates such as UB (K48) proteins or TP53 was comparable between ctrl and LAMP2A (-) cells (Fig. S3E). Functionality of other autophagy pathways such as macroautophagy was also preserved in these cells as demonstrated by an intact autophagic flux (measured as the rates of lysosomal degradation of LC3) (Fig. S3F). Lastly, we confirmed that the decrease in MYC proteasomal degradation upon CMA blockage was not limited to the transduced protein, since a similar decrease in proteasomal degradation was observed for endogenous MYC in LAMP2A (-) cells when compared with control cells (Fig. S3G).
Overall, these results support that neither endogenous nor exogenously expressed MYC proteins are targeted for CMA degradation in fibroblasts and that instead, MYC proteasomal degradation is the one compromised under CMA-deficient conditions.
CMA control of MYC accumulation is dependent on MYC phosphorylation on Ser62
MYC phosphorylation status determines its stability or degradation by the ubiquitin-proteasome system.28,31,32 Phosphorylation at the MYC Ser62 residue has been shown sufficient to prevent MYC ubiquitination and targeting for proteasomal degradation.28 To investigate whether reduced degradation of MYC through the proteasome upon CMA blockage associates with changes in phosphorylated MYC levels, we used an antibody specific against phosphorylated MYC on Ser62 (Fig. 4A). We found significantly (P ≤ 0.01) higher levels of MYC but also phospho-MYC (Ser62) in LAMP2A (-) cells than in ctrl ones (Fig. 4A to C). These results support that a large fraction of the MYC that accumulates upon CMA inhibition is phosphorylated in Ser62.
Figure 4.
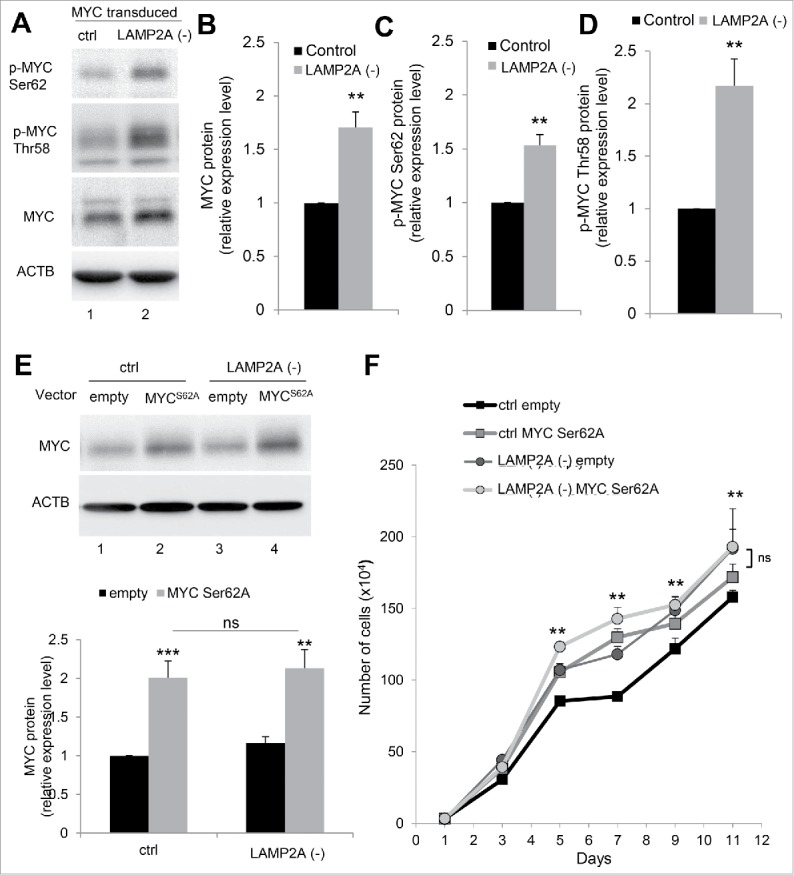
MYC phosphorylation is required for its cellular accumulation upon CMA blockage. (A) Immunoblot for the indicated proteins of NIH-3T3 MYC-transduced cells, control (ctrl) or LAMP2A knockdown (LAMP2A [-]). Densitometry quantification of total MYC (B), phospho-MYC on Ser62 (C) and Thr58 (D) in blots as the one shown in (A). Values are expressed relative to ctrl MYC-transduced cells and normalized to Ponceau S staining (N = 4). (E and F) NIH-3T3 cells, ctrl or LAMP2A (-) were transduced with a MYC mutated at the Ser62 phosphorylation site (MYCS62A). (E) Immunoblot for MYC and ACTB and quantification of total MYC in relation to ctrl empty vector cells, normalized to Ponceau S staining (N = 6). (F) Growth curve of these cells (N = 6). In all graphs values are presented as mean ± SEM. Using the Student t test (B, C, and D), as well as 2-way ANOVA and the Bonferroni post-hoc test ((E)and F), differences were calculated and considered significant for *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. ns represents nonsignificant changes
Dephosphorylation of Ser62 in MYC and its subsequent proteasomal degradation is triggered by MYC phosphorylation in residue Thr58. To determine if the reduced proteasomal degradation of MYC in CMA-deficient cells was the consequence of failure in Thr58 phosphorylation, we compared levels of phospho-MYC (Thr58) in control and LAMP2A (-) cells. CMA-defective cells displayed significantly higher levels of phospho-MYC (Thr58) than control cells (Fig. 4A and D) suggesting that MYC was properly primed for proteasomal degradation in these cells and that the defect was at a later step, likely the removal of the Ser62 phosphorylation. As for total MYC, we confirmed that the increase in levels of MYC phosphorylated in Ser62 and Thr58 in CMA-incompetent cells was not a reflection of their direct degradation in lysosomes since inhibition of lysosomal degradation in control cells with ammonium chloride and leupeptin did not increase levels of these 2 phosphorylated forms of MYC (Fig. S4D).
The relevance of Ser62 phosphorylation on MYC accumulation in CMA-incompetent cells was confirmed by analyzing the effect of a mutation at this specific phosphorylation site (S62A) on MYC levels (Fig. 4E) and on the proliferative advantage (Fig. 4F) of LAMP2A (-) cells. Immunoblot of ctrl and LAMP2A (-) cells transduced with a vector expressing MYC in which serine 62 was replaced by alanine (MYCS62A) demonstrated that CMA blockage was not able to induce accumulation of mutant MYC (Fig. 4E) leading to comparable MYC expression levels to those observed in ctrl cells. Moreover, differences in MYC driven proliferation rates between ctrl and LAMP2A (-) cells were no longer detected when the nonphosphorylatable mutant MYC was expressed.
Altogether, our results reveal the central role of the Ser62 phosphorylation on MYC protein accumulation and enhanced proliferation observed upon CMA inhibition.
Degradation of CIP2A by CMA regulates MYC levels
When Ser62 is phosphorylated, MYC cannot be delivered to the proteasome,33 making MYC dephosphorylation by PPP2/PP2A (protein phosphatase 2) a requirement for its proteasomal degradation. We found that the higher levels of phosphorylated MYC (Ser62) in CMA-defective cells were due, at least in part, to reduced PPP2-mediated dephosphorylation because treatment of LAMP2A (-) cells with the PPP2 activator FTY720 (that we confirmed was effective in enhancing dephosphorylation of phospho-MYC [Ser62] Fig. S5A) was able to reduce MYC levels in a proteasome-dependent manner (Fig. 5A, B and S5A,B; note that, as expected, the protein that accumulates upon proteasome inhibition was no longer phosphorylated in Ser62, in further support that the protein undergoing degradation was dephosphorylated MYC). Treatment of LAMP2A (-) cells with the PPP2 activator also abrogated the hyperproliferative phenotype of these cells (Fig. 5C). Defective PPP2-mediated dephosphorylation of MYC may account for most of MYC accumulation upon CMA blockage, because treatment with okadaic acid to inhibit PPP2 activity increased MYC levels in control cells but did not have an additive effect on the already high MYC levels in LAMP2A (-) (Fig. 5D).
Figure 5.
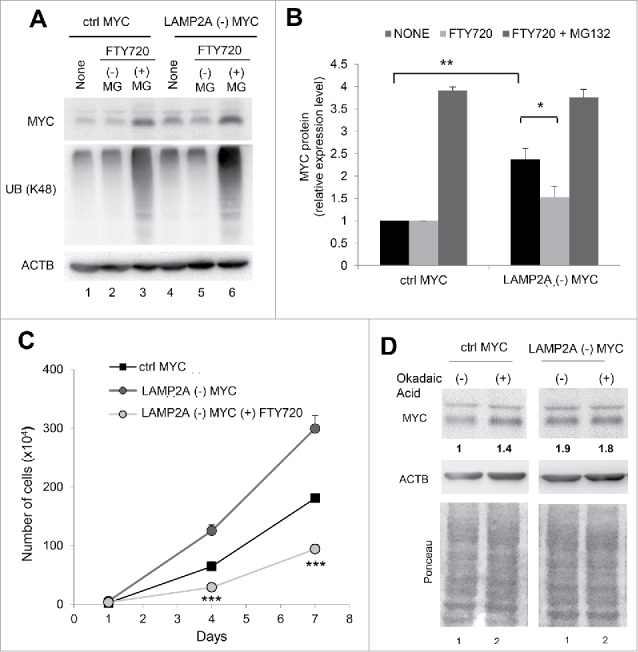
Reduced PPP2 activity upon CMA blockage is responsible for elevated MYC levels. (A, B and C) NIH-3T3 cells, ctrl or LAMP2A (-), transduced with MYC were treated for 12 h with 5 µM of FTY720 (PPP2 activator) in the presence or not of a proteasome inhibitor (MG132). (A) Representative immunoblot for the indicated proteins in cells after the indicated treatments. (B) Densitometry quantification of total MYC in blots as the one shown in (A). Values are expressed relative to untreated (None) ctrl MYC-transduced cells and normalized to Ponceau S staining (N = 4). (C) Growth curve of cells under the indicated treatments (N = 6). (D) Immunoblot for the indicated proteins and Ponceau S staining of NIH-3T3 cells, ctrl or LAMP2A (-), transduced with MYC and treated with 2 nM okadaic acid (PPP2 inhibitor) for 6 h. The densitometric values of MYC are indicated below the immunoblot. In all graphs, values are presented as mean ± SEM. Using 2-way ANOVA and the Bonferroni post-hoc test differences were calculated and considered significant for *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
The phosphorylated MYC (Ser62) is positively regulated by the recently described CIP2A that has been proposed as a critical guardian of this oncoprotein. 33–35 CIP2A stabilizes MYC through its inhibitory effect on PPP2. Immunoblot for CIP2A in ctrl and LAMP2A (-) cells with or without transduction with wild-type MYC (Fig. 6A and B) revealed that blockage of CMA alone was already enough for inducing significant increase (P ≤ 0.01) of CIP2A levels, further increased (P ≤ 0.001) in LAMP2A (-) cells transduced with a plasmid encoding MYC (Fig. 6B).
Figure 6.

CIP2A is a CMA substrate. (A) Immunoblot for CIP2A and ACTB protein in total protein lysates of NIH-3T3 cells, control (ctrl) or LAMP2A knocked down (LAMP2A [-]), transduced with a plasmid encoding MYC or the empty vector. (B) Densitometric quantification for CIP2A in blots as the one shown in (A). Values are relative to ctrl empty vector cells and normalized to Ponceau S staining (N = 6). ((C)and D) NIH-3T3 MYC-transduced cells, ctrl or LAMP2A (-) 12 h after incubation without additions (None) or in the presence of lysosomal proteolysis inhibitors (ammonium chloride and leupeptin, N/L) or a proteasome inhibitor (MG132). (C) Immunoblot for the indicated proteins. (D) Densitometry analysis of CIP2A in blots as the one shown in C. Values are relative to None condition and normalized to Ponceau S staining (N = 6). In both graphs values are presented as mean ± SEM. Two-way ANOVA and the Bonferroni post-hoc test were used and differences were considered significant for *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. ns represents nonsignificant changes.
This accumulation of CIP2A in LAMP2A (-) cells led us to investigate the possible involvement of CMA on its degradation. Inhibition of lysosomal and proteasomal degradation in MYC-transduced ctrl cells revealed a significant (P ≤ 0.01) increase of CIP2A levels upon treatment with ammonium chloride and leupeptin or MG132 (Fig. 6C and D), suggesting degradation of CIP2A by both proteasome and lysosomes. While proteasomal degradation of CIP2A was still preserved in MYC-transduced LAMP2A (-) cells, degradation of CIP2A in lysosomes was completely blocked in these cells, supporting that functional CMA is required for lysosomal CIP2A degradation (Fig. 6C and D).
In summary, these results suggest that failure to undergo lysosomal degradation by CMA is responsible for CIP2A accumulation in CMA-deficient cells.
MYC accumulation driven by CMA deficiency requires CIP2A expression
To confirm whether CIP2A was required for CMA-mediated accumulation of MYC, CIP2A was knocked down in ctrl and LAMP2A (-) cells, with or without transduction with MYC (Fig. 7). CIP2A negative cells (both ctrl and LAMP2A [-]) presented significantly (P ≤ 0.05) decreased levels of phosphorylated MYC (Ser62) (Fig. 7A and B). Total MYC levels in ctrl cells were not markedly affected by Cip2a silencing, suggesting that Ser62-phophorylated MYC represented only a small fraction of MYC in those cells (likely because of rapid dephosphorylation by PPP2). In contrast, total levels of MYC in CMA deficient cells were significantly (P ≤ 0.05) reversed by CIP2A blockage (Fig. 7C), in support of a larger fraction of MYC remaining phosphorylated in those cells because of the higher CIP2A levels. In fact, without the inhibitory effect of CIP2A, CMA-incompetent cells recovered their sensitivity to chemical inhibition of PPP2 with okadaic acid. Treatment with this agent in CIP2A knockdown cells increased levels of MYC and phosphorylated MYC (Ser62) both in control but also in LAMP2A (-) cells (Fig. S5C, D). Furthermore, we confirmed that the proliferation enhancement detected in MYC-transduced LAMP2A (-) cells when compared with ctrl was also eliminated by CIP2A knockdown (Fig. 7D).
Figure 7.
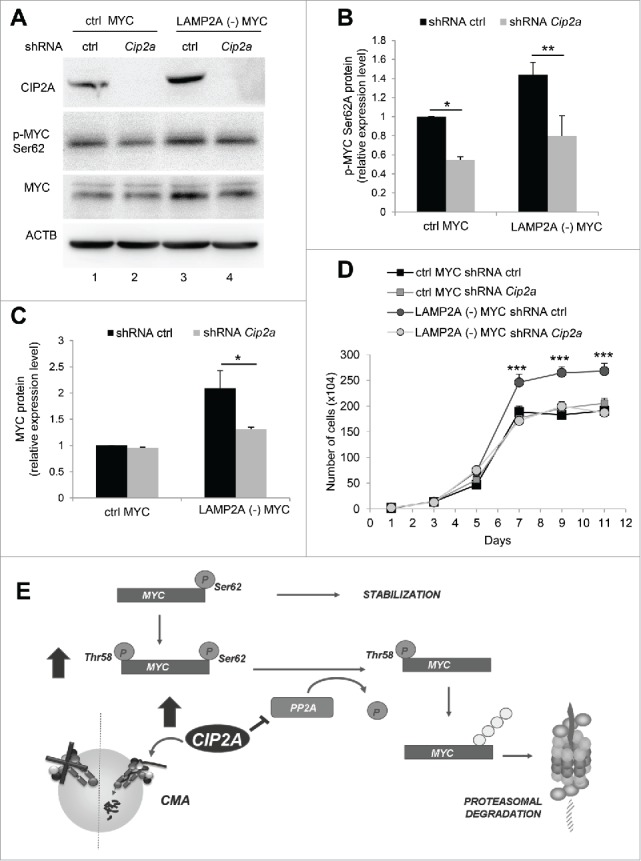
Increased CIP2A expression is required for the regulatory effect of CMA on MYC levels. NIH-3T3 cells overexpressing MYC, control (ctrl) or LAMP2A knockdown (LAMP2A [-]) were transduced with lentivirus carrying shRNA against Cip2a. (A) Total extracts from these cells were used for immunoblotting for the indicated proteins. Densitometric quantification of immunoblots as the one in A for phospho-MYC (Ser62) (B) and total MYC (C). Values are relative to those in ctrl cells and normalized to Ponceau S staining (N = 4). (D) Growth curve of these cells (N = 6). (E) Schema of the proposed regulatory effect of CMA on MYC levels: CMA favors activation of the phosphatase PPP2 by directly degrading its endogenous inhibitor CIP2A in lysosomes. Higher PPP2 activity results in dephosphorylation of MYC on Ser62 promoting its subsequent degradation by the proteasome. CMA inhibition leads to CIP2A accumulation and, consequently, decreased PPP2 activity and MYC stabilization by preventing its proteasomal degradation. In the graphs, values are presented as mean ± SEM. Two-way ANOVA and the Bonferroni post-hoc test were used and differences were considered significant for *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Overall, our results support a novel role for CMA in the control of intracellular MYC levels that is exerted through changes in its proteasomal susceptibility. The regulated selective degradation of CIP2A via CMA modulates levels of MYC phosphorylation on Ser62 and its subsequent delivery to the proteasome (Fig. 7E). Thus, this study unveils a protective role for CMA against malignant cellular transformation by regulating levels of oncogenic proteins such as MYC.
Discussion
Early studies from our group and others described constitutive upregulation of CMA in most cancer cells and demonstrated the need for functional CMA in lung, mammary and skin cancer cells to sustain tumor growth and metastasis.19,20 However, like other autophagic pathways such as macroautophagy,6,36,37 the contribution of CMA to cancer cell proliferation and tumor growth seems to be context dependent. Thus, studies in other types of cancer cells have proposed a negative effect of CMA on cancer cell biology through degradation of oncogenic proteins such as TP53 protein38,39 or HK2 (hexokinase 2).40–42 In this work, we have instead focused in the early events that lead to malignant transformation of noncancerous fibroblasts and have identified an antioncogenic tumor suppressive role for CMA, mediated in this case by negative control of MYC cellular levels.
A protective role for CMA against malignant transformation was suspected based on the physiological functions of this autophagic pathway. Added to the well-established contribution of CMA to protein quality control, recent studies demonstrate that CMA also contributes to maintain genome stability in response to DNA damage through regulated degradation of CHEK1 (checkpoint kinase 1).43 Furthermore, mouse models incompetent for CMA in liver display higher incidence of spontaneous hepatic tumors.18 We initially proposed that the poor protein quality control, increased oxidative stress and severe lipotoxicity observed in CMA-defective hepatocytes provided conditions favorable for oncongenic transformation in these animals. However, in this study we have found that the antioncogenic capacity of CMA is not limited to its role in the cellular defense against toxicity (proteotoxicity, genotoxicity or lipotoxicity) and that instead it is exerted, at least in part, by negatively regulating the cellular levels of the prooncogenic protein MYC.
Metabolic changes have been implicated in the CMA-mediated control of tumor growth and survival.7,23,41 Interestingly, the metabolic consequences of CMA blockage seem opposite in transformed and untransformed cells, and our current findings support that this switch occurs during transformation. Thus, CMA blockage in untransformed cells leads to increase glycolysis because of reduced degradation of glycolytic enzymes (well-characterized CMA substrates).17 In contrast, blockage of CMA in different types of cancer cells reduces glycolytic flux, in part, through transcriptional downregulation of glycolytic enzymes 7,23,41 and in part through failure to eliminate acetylated forms of the less glycolytically active M2 isoform of PKM/PKM2 (pyruvate kinase, muscle), the enzyme responsible for the metabolic shift from oxidative phosphorylation to aerobic glycolysis.23 We propose that the burst of glycolytic activity observed as CMA activity declines may provide the energetic boost required for transformation. Once transformed, cells develop yet-to-identify mechanisms to avoid the negative regulatory effect of CMA on glycolytic enzymes. How glycolytic enzymes escape CMA degradation in the transformed cells and whether the increase in MYC levels directly contributes to that effect requires future investigation.
We have found that CMA deficiency led to MYC protein accumulation and identified a regulatory role for CMA in the degradation of MYC. Contrary to our initial prediction that the higher MYC levels in CMA-incompetent cells reflected direct contribution of CMA to MYC degradation, we find instead that active CMA was necessary to secure proteasomal degradation of MYC. Although MYC does not undergo in this context direct degradation via CMA, the fact that the protein bears in its amino acid sequence 2 KFEQR-like motifs (267VEKRQ271 and 361VLERQ365), leaves open the possibility that in other cellular contexts HSPA8 may use these motifs to target MYC directly for degradation by CMA. Changes in MYC conformation, posttranslational modifications or even interaction of MYC with other proteins during cellular transformation could prevent accessibility of HSPA8 to the KFEQR-like motif of MYC and spare it from CMA degradation. During the process of cellular transformation, we found that phosphorylation of MYC is the central event in the CMA-driven effect on MYC degradation. Site-specific phosphorylation of MYC controls its proteolysis through the ubiquitin-proteasome pathway33 and in fact, the N-terminal phosphorylation sites (Thr58 and Ser62) are located within a mutational hotspot found in Burkitt lymphomas.33,44 These phosphorylation sites modulate crucial biological activities of MYC,33,44,45 and, at least for Ser62, phosphorylation in this site is required for the oncogenic activity of MYC, since its substitution severely inhibited transformation.44,45 Control of phosphorylated MYC (Ser62) levels is a new example of crosstalk between autophagy and the ubiquitin-proteasome system.46 Failure to degrade MYC by the proteasome in CMA-incompetent cells was not a consequence of overall proteasome proteolytic failure. In fact, although blockage of CMA in mouse fibroblasts did not lead to increased proteasome activity as reported in mouse liver, we found comparable proteasome-dependent degradation of polyubiquitinated (K48) proteins and other proteasome substrates such as TP53 between control and CMA-defective cells (Fig. S3D and E). The selective effect of CMA on regulating MYC degradation by the proteasome is exerted through changes in total levels of CIP2A, a direct determinant of MYC proteasomal degradation.
In this work, we have identified CIP2A as a new degradation target of CMA. Both LAMP2A knockdown and inhibition of lysosomal proteolysis by ammonium chloride and leupeptine stabilized CIP2A, but only LAMP2A knockdown stabilized MYC levels, supporting that most of the effect of CMA on MYC stability is dependent on the presence of CIP2A in the cytosol. Thus, LAMP2A knockdown inhibits CMA substrate translocation into lysosomes, leading to accumulation of CIP2A in cytosol (increased function). On the other hand, lysosomal proteolysis inhibitors only block CIP2A degradation that occurs after its lysosomal translocation, leading to its accumulation inside lysosomes where is no longer function. Puustinen and collaborators, working in cancer cells (MCF7 cells) have recently identified an inhibitory effect of CIP2A on macroautophagy, by modulating its major negative regulator, the mechanistic target of rapamycin (serine/threonine kinase) complex 1.47,48 Despite the higher levels of CIP2A in the CMA-incompetent cells, we did not find reduced macroautophagy activity in these cells, suggesting that the effect of CIP2A on macroautophagy may be different in cancer cells and in cells undergoing transformation. However, it is interesting that contrary to most untransformed cells where blockage of CMA leads to marked upregulation of macroautophagy, we find only a discrete increase in macroautophagy levels in the CMA-defective cells in presence of MYC (Fig. S3D and F). Our previous studies also support that once transformed, the compensatory upregulation of macroautophagy in response to CMA blockage is no longer activated.7 An attractive possibility, worth exploring in the future, is that the increase in CIP2A observed upon CMA blockage in transforming cells may help explain the lack of compensatory upregulation of macroautophagy in cancer cells. The findings in the same study reporting that CIP2A in MCF7 cells undergoes autophagy-mediated degradation 47,48 do not conflict with our results regarding the CIP2A degradation by CMA. Most of the autophagy stimuli (amino acid deprivation) and inhibitors (lysosomal pH neutralization) used in the study will also modify CMA activity, leaving open the possibility that, at least part of the observed changes in CIP2A levels were happening through CMA. In our study, using genetic blockage of CMA, that we show does not affect macroautophagy, we found that the lysosomal degradation of CIP2A was no longer observed, suggesting that under our experimental conditions most of CIP2A lysosomal degradation occurred through CMA (Fig. 6). In addition, it is well established that the same protein can undergo degradation simultaneously through different pathways (i.e. we found both proteasome and lysosomal degradation of CIP2A in our study). Differentially modified forms of the same protein may be the determinants of the different degradation pathways, and the relative contribution of each pathway may be cell-type dependent, providing thus a possible explanation from findings in different studies.
Overall, our findings reveal a context dependent role for CMA in tumorigenesis, whereby contrary to the pro-oncogenic effect observed for this autophagic pathways in cancer cells, CMA serves as a cellular defense mechanism against malignant transformation driven by the proto-oncogene MYC in normal cells. In addition to the already proposed benefits of activating CMA in the treatment of neurodegeneration, metabolic syndromes, aging and other disorders associated with CMA reduced activity, chemical enhancement of CMA may also prevent cancer initiation.
Materials and methods
Cell culture and treatment
The NIH-3T3 mouse embryonic fibroblast cells (American Type Culture Collection, CRL-1658™) were cultured in a 37°C incubator with 5% CO2 in DMEM medium supplemented with 10% newborn calf serum (NCS). Mouse embryonic fibroblasts (MEFs) were generated from lamp2a null mice and corresponding wild-type littermates as described previously.18 For degradation assays, cells where treated with lysosomal proteolysis inhibitors ammonium chloride (20 mM; Sigma-Aldrich, A9434) and leupeptin (100 µM; Fisher Scientific, BP26621) or the proteasome inhibitor MG132 (10 µM; Sigma-Aldrich, M7449) for 12 h in the presence or absence of serum, as indicated.
Antibodies, plasmids and reagents
The following antibodies were used for immunoblot analysis: human MYC (Santa Cruz Biotechnology, sc-40), ACTB/β-actin (Abcam, ab6276), HSP90 (Cell Signaling Technology, 4877), LMNA/Lamin A/C (Santa Cruz Biotechnology, sc-20681), phospho-MYC (Ser62) (Cell Signaling Technology, 13748), mouse MYC (Cell Signaling Technology, 5605), CIP2A (Santa Cruz Biotechnology, sc-80659), LAMP1 (Hybridoma Bank, 1D4B), LAMP2A (Invitrogen™, 512200), UB (K48) (EMD Millipore, 05–1307), TP53 (Cell Signaling Technology, 2524S), LC3B (Cell Signaling Technology, 2775). The plasmids for overexpression of wild-type (pCDH-puro-cMyc) and mutated (pD40-His/V5-MYCS62A) MYC were deposited by Dr. Jialiang Wang49 (Addgene, plasmid 46970) and Dr. Rosalie Sears50 (Addgene, plasmid 45599), respectively. Upon cell infection with these lentiviruses, they were selected with 5 µg/mL of puromycin (Sigma-Aldrich, P8833) for 7 d. Stable RNA interference was performed by lentiviral delivery of shRNA against the desired targets: for mouse Lamp2a we used the same PGK-driven construct generated in our laboratory as described previously51 that targets the region 5-CTGCAATCTGATTGATTA-3, corresponding to bases 1331 to 1359. For mouse Cip2a we used the shRNA in the pLKO.1-C330027C09Rik from Sigma-Aldrich (SHCLNG-NM_172616). Cultured cells were transduced with 1 × 107 transducing units of concentrated lentivirus at 37°C for 24 h when media was added for a 1:1 dilution. Following this 72 h transduction, cells were washed and transduction efficiency was verified by fluorescent microscopy for GFP expression followed by immunoblot for the targeted proteins.
Transformation assays
For focus formation assay 10 × 104 cells/cm2 were plated in triplicate in 6-well plates and maintained in culture for 3 wk (the medium was replaced every 3 d). For soft agar colony formation, 1.5 mL of 0.7% low-meting-point agarose (Lonza, 50100) in DMEM (supplemented with 10% NCS) were added per well of 6-well plates. Cells (1 × 104) were suspended in 1mL of 0.35% low-meting-point agarose (Lonza, 50100) in the same medium and plated in triplicate on top of the presolidified agarose, that was then layered with 1 mL of 0.7% low-meting-point agarose and 1.5 mL of DMEM 10% NCS. These cells were incubated at 37°C and 5% CO2 for 3 weeks and the medium was replaced every 3 d. After this period the plates for focus formation and soft-agar colony formation assays were stained with crystal violet and scanned. The ImageJ software (National Institutes of Health) was used for quantification of focus and colony formation capacities.
Metabolic measurements
Real-time measurement of pH and oxygen concentration for the determination of ECAR and OCR were performed in XF96 plates with the XF Extracellular Flux Analyzer (Seahorse Bioscience-Agilent (Santa Clara, CA). Key parameters of mitochondrial function were measured by using specific mitochondria respiration inhibitors (Seahorse XF Cell Mito Stress Test Kit, 103015–100, Agilent Technologies) while recording OCR values. The kit contains: oligomycin (ATP synthase/complex V inhibitor), carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (uncoupling agent responsible for disrupting of the mitochondrial membrane potential) and rotenone (complex I inhibitor). Contribution of glycolysis to ECAR was determined by adding 2-DG (glycolysis inhibitor; Sigma Aldrich, D8375). The results obtained were normalized by the number of cells.
Electrophoresis, immunoblot and densitometric quantification
Cells were lysed in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% NaDoc, 0.1% SDS, 50 mM Tris, pH 8). For nuclear and cytoplasmic fractionation we used the NER-PER™ kit (Thermo Fisher Scientific, 78833). Protein concentration was determined by the Lowry method.52 The ImageJ software was used for densitometry quantification of bands obtained in the western-blot assays. The quantification was performed by volume densitometry and normalized to the intensity of the Ponceau S red stained immunoblot membrane (Fig. S6) whereby each protein of interest band was normalized to the total densitometric value of the Ponceau S staining for its corresponding gel line. Levels of oxidized proteins were determined using the OxyBlot Oxidized Protein Detection Kit (Chemicon International, S7150). Briefly, carbonyl groups of oxidized proteins were derivatized to 2,4-dinitrophenylhydrazone by reaction with 2,4-dinitrophenylhydrazine and detected by immunoblot with an antibody specific for the dinitrophenyl (DNP) derivatized groups. Cell viability was determined by using the CellTiter-Blue Cell Viability Assay kit (Promega, G8081).
Statistical analysis
All results are presented as MEAN ± SEM. Statistical significance was determined using the Student t test to compare the means of 2 groups and the 2-way analysis of variance (ANOVA) and Bonferroni post-hoc test for multiple group analysis of means. A P value less than 0.05 was considered to be statistically significant. All the experiments were repeated at least 3 times and duplicates or triplicates in the same experiment were used to control for technical variability.
Supplementary Material
Abbreviations
- ACTB
actin, β
- CHEK1
checkpoint kinase 1
- CMA
chaperone-mediated autophagy
- ECAR
extracellular acidification rate
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HK2
hexokinase 2
- HSP
heat shock protein
- KO
knockout
- LAMP1
lysosomal-associated membrane protein 1
- LAMP2A
lysosomal-associated membrane protein 2A
- LMNA
lamin A
- MEF
mouse embryonic fibroblasts
- MYC/c-Myc
myelocytomatosis oncogene
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- N/L
ammonium chloride and leupeptin
- NCS
newborn calf serum
- OCR
oxygen consumption rate
- p-MYC
phosphorylated form of the MYC protein
- PKM/PKM2
pyruvate kinase, muscle
- PPP2/PP2A
protein phosphatase 2
- shRNA
short hairpin RNA
- TP53
transformation related protein 53
- UB (K48)
ubiquitin (K48-specific)
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Antonio Diaz for his assistance with lentiviral production; the Stable Isotope and Metabolomics Core Facility and Dr. Susmita Kaushik for their support with the Seahorse Bioscience assays. We also thank Dr. Susmita Kaushik for her assistance in the preparation of lysosomal-enriched protein fractions and the Oxyblot™ assays.
Funding
This work was funded by NIH grants AG021904, AG031782, CA013330 and FAPESP (São Paulo, Brazil) grants 2011/50856–3, 2014/15982–6, CAPES and CNPq (Brasilia, Brazil) awards.
References
- [1].Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069–75; PMID:18305538; http://dx.doi.org/ 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24–41; PMID:24366339; http://dx.doi.org/ 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab 2011; 13:495–504; PMID:21531332; http://dx.doi.org/ 10.1016/j.cmet.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 2012; 22:407–17; PMID:22748206; http://dx.doi.org/ 10.1016/j.tcb.2012.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta 2003; 1603:113–28; PMID:12618311 [DOI] [PubMed] [Google Scholar]
- [6].White E. The role for autophagy in cancer. J Clin Invest 2015; 125:42–6; PMID:25654549; http://dx.doi.org/ 10.1172/JCI73941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med 2011; 3:109ra17; ; http://dx.doi.org/ 10.1126/scitranslmed.3003182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bejarano E, Cuervo AM. Chaperone-mediated autophagy. Proc Am Thorac Soc 2010; 7:29–39; PMID:20160146; http://dx.doi.org/ 10.1513/pats.200909-102JS [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kgdalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989; 246:382–5; PMID:2799391; http://dx.doi.org/ 10.1126/science.2799391 [DOI] [PubMed] [Google Scholar]
- [10].Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996; 273:501–3; PMID:8662539; http://dx.doi.org/ 10.1126/science.273.5274.501 [DOI] [PubMed] [Google Scholar]
- [11].Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem 2000; 275:27447–56; PMID:10862611 [DOI] [PubMed] [Google Scholar]
- [12].Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci 2001; 114:2491–9; PMID:11559757 [DOI] [PubMed] [Google Scholar]
- [13].Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 2008; 28:5747–63; PMID:18644871; http://dx.doi.org/ 10.1128/MCB.02070-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM. Identification of regulators of chaperone-mediated autophagy. Mol Cell 2010; 39:535–47; PMID:20797626; http://dx.doi.org/ 10.1016/j.molcel.2010.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res 2014; 24:92–104; PMID:24281265; http://dx.doi.org/ 10.1038/cr.2013.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schneider JL, Cuervo AM. Autophagy and human disease: emerging themes. Curr Opin Genet Dev 2014; 26C:16–23; ; http://dx.doi.org/ 10.1016/j.gde.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 2014; 20:417–32; PMID:25043815; http://dx.doi.org/ 10.1016/j.cmet.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schneider JL, Villarroya J, Diaz-Carretero A, Patel B, Urbanska AM, Thi MM, Villarroya F, Santambrogio L, Cuervo AM. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 2015; 14:249–64; PMID:25620427; http://dx.doi.org/ 10.1111/acel.12310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Saha T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 2012; 8:1643–56; PMID:22874552; http://dx.doi.org/ 10.4161/auto.21654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhou J, Yang J, Fan X, Hu S, Zhou F, Dong J, Zhang S, Shang Y, Jiang X, Guo H, et al. . Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 2016:12(3): 515-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Quintavalle C, Di Costanzo S, Zanca C, Tasset I, Fraldi A, Incoronato M, Mirabelli P, Monti M, Ballabio A, Pucci P, et al. . Phosphorylation-regulated degradation of the tumor-suppressor form of PED by chaperone-mediated autophagy in lung cancer cells. J Cell Physiol 2014; 229:1359–68; PMID:24477641; http://dx.doi.org/ 10.1002/jcp.24569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lu W, Zhang Y, McDonald DO, Jing H, Carroll B, Robertson N, Zhang Q, Griffin H, Sanderson S, Lakey JH, et al. . Dual proteolytic pathways govern glycolysis and immune competence. Cell 2014; 159:1578–90; PMID:25525876; http://dx.doi.org/ 10.1016/j.cell.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, et al. . Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 2011; 42:719–30; PMID:21700219; http://dx.doi.org/ 10.1016/j.molcel.2011.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vennstrom B, Sheiness D, Zabielski J, Bishop JM. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J Virol 1982; 42:773–9; PMID:6284994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 2005; 6:635–45; PMID:16064138; http://dx.doi.org/ 10.1038/nrm1703 [DOI] [PubMed] [Google Scholar]
- [26].Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res 2012; 18:5546–53; PMID:23071356; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-0977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 1999; 19:1–11; PMID:9858526; http://dx.doi.org/ 10.1128/MCB.19.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 2004; 23:2116–25; PMID:15103331; http://dx.doi.org/ 10.1038/sj.emboj.7600217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol 2015; 17:759–70; PMID:25961502; http://dx.doi.org/ 10.1038/ncb3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Autophagy Mizushima N.: process and function. Genes Dev 2007; 21:2861–73; PMID:18006683; http://dx.doi.org/ 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- [31].Noguchi K, Kitanaka C, Yamana H, Kokubu A, Mochizuki T, Kuchino Y. Regulation of c-Myc through phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J Biol Chem 1999; 274:32580–7; PMID:10551811; http://dx.doi.org/ 10.1074/jbc.274.46.32580 [DOI] [PubMed] [Google Scholar]
- [32].Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 2000; 14:2501–14; PMID:11018017; http://dx.doi.org/ 10.1101/gad.836800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sears RC. The life cycle of C-myc: from synthesis to degradation. Cell Cycle 2004; 3:1133–7; PMID:15467447; http://dx.doi.org/ 10.4161/cc.3.9.1145 [DOI] [PubMed] [Google Scholar]
- [34].Junttila MR, Puustinen P, Niemelä M, Ahola R, Arnold H, Böttzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, et al. . CIP2A inhibits PP2A in human malignancies. Cell 2007; 130:51–62; PMID:17632056; http://dx.doi.org/ 10.1016/j.cell.2007.04.044 [DOI] [PubMed] [Google Scholar]
- [35].Jung HM, Patel RS, Phillips BL, Wang H, Cohen DM, Reinhold WC, Chang LJ, Yang LJ, Chan EK. Tumor suppressor miR-375 regulates MYC expression via repression of CIP2A coding sequence through multiple miRNA-mRNA interactions. Mol Biol Cell 2013; 24:1638–48, S1–7; ; http://dx.doi.org/ 10.1091/mbc.E12-12-0891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401–10; PMID:22534666; http://dx.doi.org/ 10.1038/nrc3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell 2013; 155:1216–9; PMID:24315093; http://dx.doi.org/ 10.1016/j.cell.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, Pan L, Ince TA, Kroemer G, Brugge JS, et al. . Chaperone-mediated autophagy degrades mutant p53. Genes Dev 2013; 27:1718–30; PMID:23913924; http://dx.doi.org/ 10.1101/gad.220897.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vakifahmetoglu-Norberg H, Yuan J. A degradative detour for mutant TP53. Autophagy 2013; 9:2158–60; PMID:24145670; http://dx.doi.org/ 10.4161/auto.26338 [DOI] [PubMed] [Google Scholar]
- [40].Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, et al. . Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013; 24:213–28; PMID:23911236; http://dx.doi.org/ 10.1016/j.ccr.2013.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xia HG, Najafov A, Geng J, Galan-Acosta L, Han X, Guo Y, Shan B, Zhang Y, Norberg E, Zhang T, et al. . Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol 2015; 210:705–16; PMID:26323688; http://dx.doi.org/ 10.1083/jcb.201503044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Galan-Acosta L, Xia H, Yuan J, Vakifahmetoglu-Norberg H. Activation of chaperone-mediated autophagy as a potential anticancer therapy. Autophagy 2015; 11:2370–1; PMID:26577179; http://dx.doi.org/ 10.1080/15548627.2015.1106666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun 2015; 6:6823; PMID:25880015; http://dx.doi.org/ 10.1038/ncomms7823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hann SR. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin Cancer Biol 2006; 16:288–302; PMID:16938463; http://dx.doi.org/ 10.1016/j.semcancer.2006.08.004 [DOI] [PubMed] [Google Scholar]
- [45].Pulverer BJ, Fisher C, Vousden K, Littlewood T, Evan G, Woodgett JR. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene 1994; 9:59–70; PMID:8302604 [PubMed] [Google Scholar]
- [46].Wong E, Cuervo AM. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol 2010; 2:a006734; PMID:21068151; http://dx.doi.org/ 10.1101/cshperspect.a006734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Puustinen P, Rytter A, Mortensen M, Kohonen P, Moreira JM, Jäättelä M. CIP2A oncoprotein controls cell growth and autophagy through mTORC1 activation. J Cell Biol 2014; 204:713–27; PMID:24590173; http://dx.doi.org/ 10.1083/jcb.201304012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Puustinen P, Jäättelä M. KIAA1524/CIP2A promotes cancer growth by coordinating the activities of MTORC1 and MYC. Autophagy 2014; 10:1352–4; PMID:24905455; http://dx.doi.org/ 10.4161/auto.29076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, Thompson RC, Muller S, Knapp S, Wang J. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin Cancer Res 2013; 19:1748–59; PMID:23403638; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. . A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 2004; 6:308–18; PMID:15048125; http://dx.doi.org/ 10.1038/ncb1110 [DOI] [PubMed] [Google Scholar]
- [51].Massey AC, Follenzi A, Kiffin R, Zhang C, Cuervo AM. Early cellular changes after blockage of chaperone-mediated autophagy. Autophagy 2008; 4:442–56; PMID:18253088; http://dx.doi.org/ 10.4161/auto.5654 [DOI] [PubMed] [Google Scholar]
- [52].Lowry OH, J. RN, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951; 193:265–75; PMID:14907713 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.