

Abstract
Background
Trimethylamine-N-oxide (TMAO) is produced in the liver from trimethylamine, which is exclusively generated by gut bacteria.
Objective
The objective of this article is to investigate the relationship between TMAO and primary sclerosing cholangitis (PSC) and its clinical characteristics.
Methods
Serum TMAO was measured in 305 PSC patients, 90 ulcerative colitis patients and 99 healthy controls.
Results
In PSC patients with normal liver function (n = 197), TMAO was higher in patients reaching liver transplantation or death during follow-up than those who did not, with an optimal TMAO cut-off of 4.1 µM (AUC = 0.64, p < 0.001). PSC patients with high TMAO (>4.1 µM, n = 77) exhibited shorter transplantation-free survival than patients with low TMAO (n = 120, log-rank test: p < 0.0001). High TMAO (>4.1 µM) was associated with reduced transplantation-free survival (HR 1.87, p = 0.011), independently of the Mayo risk score (HR 1.74, p < 0.001). Overall, PSC patients demonstrated reduced TMAO values compared with ulcerative colitis and healthy controls, mainly caused by PSC patients with reduced liver function (INR > 1.2), suggesting impaired oxidation of trimethylamine to TMAO. PSC patients with and without inflammatory bowel disease had similar TMAO levels.
Conclusion
In PSC patients with normal liver function, elevated TMAO was associated with shorter transplantation-free survival, potentially reflecting clinically relevant metabolic changes resulting from dietary interactions with the gut microbiota.
Keywords: Primary sclerosing cholangitis, gastrointestinal microbiome, trimethylamine-N-oxide
Introduction
Primary sclerosing cholangitis (PSC) is a chronic and immune-mediated liver disease of unknown aetiology.1 It is characterised by cholestasis, and inflammation and fibrosis of the intra- and extra-hepatic bile ducts. Up to 80% of patients with PSC are also diagnosed with inflammatory bowel disease (IBD), most often categorised as ulcerative colitis (UC).1 This striking association suggests that alterations in the gut of patients with PSC are important in the disease process.1
PSC is in part heritable,2 and multiple genetic risk factors have been identified in large-scale studies.3 Several genetic risk factors are also shared between PSC and IBD, and some of these implicate a role for the gut microbiota in disease development.3 However, this collection of risk genes collectively explain less than 10% of the estimated overall susceptibility to this disease.3 Little is known about how environmental factors influence the development of PSC, although both smoking and coffee have been associated with protection against the disease.4 Changes in the gut microbial community composition have been demonstrated in several diseases including gut disorders like IBD,5 as well as systemic inflammatory and metabolic diseases.6 Recently, we also showed that there are extensive differences between the faecal microbiota in PSC patients compared with healthy controls (HCs) and IBD patients without liver disease.7
Diet plays a key role in shaping the gut microbiota.8 In turn, the microbiota plays an intrinsic part of human metabolism, e.g. through the production of vitamins, secondary bile acids and short-chain fatty acids.9,10 It is likely that microbial metabolites influence human disease development as well via the gut–liver axis.10 One documented example is trimethylamine-N-oxide (TMAO), a metabolite whose precursor trimethylamine (TMA) is completely microbiota dependent.11–15 TMA is a volatile gas generated by gut bacteria from phosphatidylcholine, l-carnitine or gamma-butyrobetaine.11–14 Phosphatidylcholine and l-carnitine are abundant in e.g. eggs, milk and red meat.12 After absorption in the gut TMA reaches the liver where it is converted to TMAO by flavin-containing monooxygenase enzymes (FMOs).12 TMAO has recently been associated with atherosclerosis and cardiovascular disease,11–13,16 although the mechanism of this relationship is so far not completely understood.16,17 TMAO has further been shown to influence cholesterol metabolism, bile composition and lowering of key bile acid synthesis- and transport-proteins, all with a potential role in regulation of inflammation and hepatic metabolic pathways.11,12,15 Of note, methylamines (e.g. TMA) and TMAO have been shown to cause cholestasis, cholangiocyte proliferation and cholangiofibrosis in rats, pathological changes that are hallmarks of human PSC.1,18
Given the possible role of gut microbiota in PSC and the potential link between TMAO and key metabolic pathways in the liver and inflammation, we aimed to investigate TMAO as a surrogate marker for diet and microbiota, and determine the impact from TMAO on clinical characteristics of a large Norwegian cohort of patients with PSC and relevant controls.
Materials and methods
Patient population and data collection
Diagnosis of PSC was based on typical cholangiographic findings according to acknowledged criteria.19,20 PSC patients were recruited at admission to Oslo University Hospital Rikshospitalet in the period from 1992 to 2012, and blood samples were included in the Norwegian PSC Research Center (NoPSC) Biobank (Oslo, Norway). All available serum samples for the time period were included in the present study. The first pathological cholangiography defined the time of diagnosis of PSC, and the duration of PSC was defined as the time from the date of diagnosis to the date of serum sampling. Cause of death was extracted from the Cause of Death Registry (Oslo, Norway, reference 16-0230). Sera from HCs were included from individuals recruited from the Norwegian Bone Marrow Donor Registry and sera from UC controls from a population-based Norwegian cohort.21 IBD diagnosis was based on histology, findings at endoscopy and accepted criteria.22
Ethics
Written informed consent was obtained from all participants, and the study protocol was in accordance with the 1975 Declaration of Helsinki. The Regional Committee for Medical and Health Research Ethics in South-Eastern Norway approved the study (references 2011/2572, 2012/286 and 2015/2140).
Blood sampling and routine biochemical analyses
Non-fasting blood was collected and serum prepared in a standardised fashion, followed by storage at –70 degrees. Routine biochemical analyses, e.g. liver function tests and measures of prothrombin, were retrieved from hospital laboratory databases, if available within one week of collection of biobank serum. Reduced liver function was defined as international normalised ratio (INR) > 1.2 and/or Normotest < 70.0. Mayo risk score was calculated using the algorithm for the revised Mayo risk score.23 Estimated glomerular filtration rate (eGFR) was calculated according to the Chronic Kidney Disease Epidemiology Collaboration equation.24
Measurement of TMAO
Levels of TMAO were measured as previously described.13 In short, stable isotope dilution liquid chromatography-tandem mass spectrometry (LC/MS/MS) was used for the quantification of TMAO and was monitored in positive liquid chromatography-tandem mass spectrometry (MRM) MS mode, using characteristic precursor–product ion transition: m/z 76→58. The internal standard TMAO-trimethyl-d9 was added to samples before protein precipitation and was monitored in MRM mode at m/z 85→66. Various concentrations of TMAO and a fixed amount of internal standard were spiked into 4% bovine serum albumin to prepare the calibration curves for the quantification of analytes. TMAO measurements below the limit of quantification (0.1 µM, n = 5) were set to zero. All stable isotope-labelled internal standards were purchased from Cambridge Isotope Laboratories Inc (Andover, MA, USA).
Statistical analyses
For continuous variables, t test or Mann-Whitney U was used depending on distribution. Comparison of categorical variables was performed using the Chi-square test or Fisher’s exact test where appropriate. For correlation analyses, Spearman’s rank correlation test was used. Kaplan-Meier plots were used for visualisation of transplantation-free survival, and crude risk was compared by log-rank test. Due to the low number of patients with longer follow-up time, patients were censored at 15 years. Receiver operating characteristics curve (ROC-AUC) analyses and Youden’s index were used to define the optimal TMAO cut-off for discriminating between patients with and without death or liver transplantation.25 Differences between areas under the curve (AUC) were compared according to the method of DeLong.26 Cox proportional hazards regression analyses were used to investigate associations between TMAO levels, clinical and biochemical variables and endpoints (death or liver transplantation), and variables with a right-skewed distribution were transformed by the natural logarithm prior to Cox proportional hazards regression analyses. All calculations were performed in SPSS Statistics for Macintosh, version 22 (IBM, Armonk, NY, USA) except Youden’s index, calculated in MedCalc (MedCalc Software bvba, Ostend, Belgium).
Results
In total, 305 PSC patients were included in the analyses, together with 90 UC patients and 99 HCs (Table 1). IBD was present in 242 (79.3%) of the patients with PSC, 194 (63.6%) with UC, 35 (11.5%) with Crohn’s disease (CD) and 13 (4.3%) with indeterminate colitis. Prothrombin time measurements (INR or Normotest) were available for 250 PSC patients (82.0%), of whom 197 (64.6%) had normal liver function. Of the UC controls, 45 (50.0%) had active disease and 45 (50.0%) were in remission.
Table 1.
Clinical characterisation of patients with PSC, ulcerative colitis and healthy controls included in the study
| PSC |
Healthy controls |
Ulcerative colitis |
PSC vs HC | PSC vs UC | UC vs HC | ||||
|---|---|---|---|---|---|---|---|---|---|
| n = 305 | n = 99 | n = 90 | p value | p value | p value | ||||
| Age, years, mean (95% CI) | 40.6 | (39.1–42.1) | 40.3 | (38.9–41.6) | 39.3 | (36.5–42.1) | 0.80 | 0.41 | 0.52 |
| Sex (male), n (%) | 231 | (75.7) | 59 | (59.6) | 46 | (51.1) | <0.01 | <0.01 | 0.31 |
| Age at diagnosis, mean (95% CI) | 36.3 | (34.8–37.8) | |||||||
|
PSC-specific variables
|
Available for n (%) | ||||||||
| PSC duration, years, median (range) | 1.4 | (–0.6 to 29.0) | 305 | (100) | |||||
| IBD (yes), n (%) | 242 | (79.3) | 305 | (100) | |||||
| UC (yes), n (%) | 194 | (63.6) | |||||||
| Crohn’s disease (yes), n (%) | 35 | (11.5) | |||||||
| Indeterminate colitis (yes), n (%) | 13 | (4.3) | |||||||
| Total bilirubin, mg/dl, median (range) | 1.4 | (0.18–33.2) | 283 | (92.8) | |||||
| Albumin, g/l, median (range) | 39.0 | (15.0–50.1) | 271 | (88.9) | |||||
| ALP, U/l, median (range) | 350.0 | (51.0–3100.0) | 269 | (88.2) | |||||
| AST, U/l, median (range) | 80.0 | (8.0–1219.0) | 283 | (92.8) | |||||
| ALT, U/l, median (range) | 99.0 | (8.0–885.0) | 284 | (93.1) | |||||
| Creatinine, µmol/l, median (range) | 69.0 | (37.0–216.0) | 264 | (86.6) | |||||
| eGFR, ml/min/1.73 m2, median (range) | 110.7 | (27.2–157.5) | 264 | (86.6) | |||||
| Platelet count, 109/l, median (range) | 255 | (10.0–903.0) | 254 | (83.3) | |||||
| INR, median (range) | 1.1 | (0.8–2.5) | 208 | (68.2) | |||||
| Normotest, median (range) | 97 | (30.0-150.0) | 46 | (15.1) | |||||
| Variceal bleeding (yes), n (%) | 9 | (3.0) | 260 | (85.2) | |||||
| p-ANCA, positive, n (%) | 161 | (52.8) | 227 | (74.4) | |||||
| Mayo risk score | 0.7 | (–2.37 to 5.26) | 254 | (83.3) | |||||
ALP: alkaline phosphatase; ALT: alanine transaminase; AST: aspartate transaminase; CI: confidence interval; eGFR: estimated glomerular filtration rate; HC: healthy control; IBD: inflammatory bowel disease; INR: international normalised ratio; p-ANCA: perinuclear anti-neutrophil cytoplasmic antibodies; PSC: primary sclerosing cholangitis; UC: ulcerative colitis.
TMAO is reduced in PSC patients with reduced liver function
TMAO levels were reduced in patients with PSC compared both with UC and HCs (Figure 1(a)). HCs and patients with UC exhibited similar TMAO levels (p = 0.35), as did PSC patients with and without IBD (Figure 1(b)), and UC with active disease and UC in remission (Figure 1(c)), indicating that the presence of bowel inflammation does not have a substantial impact on TMAO levels.
Figure 1.

TMAO levels are affected by disease status, but not by bowel inflammation. (a) Patients with PSC showed reduced TMAO levels compared both with UC and HCs, while HCs and UC patients exhibited similar TMAO levels (p = 0.35). (b) On the other hand PSC with and without IBD did not differ in TMAO levels, (c) and neither did UC patients with active disease compared with those in remission. HC: healthy controls; IBD: inflammatory bowel disease; PSC: primary sclerosing cholangitis; TMAO: trimethylamine-N-oxide; UC: ulcerative colitis. Data shown as median and interquartile range (IQR). Mann-Whitney U was used for comparisons. *p < 0.05, **p < 0.01.
We hypothesised that reduced TMAO levels in PSC in part could be explained by impaired hepatic metabolism of TMA to TMAO. In line with this, no high values of TMAO were observed in PSC patients with reduced liver function (Figure 2(a)), and the TMAO levels in PSC patients with normal liver function (n = 197) did not differ significantly from HCs (Figure 2(b)). Patients with PSC with or without IBD had similar TMAO levels irrespective of liver function (Supplementary Figure 1). The patients with PSC and reduced liver function were slightly older and had lower creatinine, but similar eGFR, compared with those with normal liver function, in addition to the expected differences in clinical and biochemical liver-related markers (Supplementary Table 1).
Figure 2.
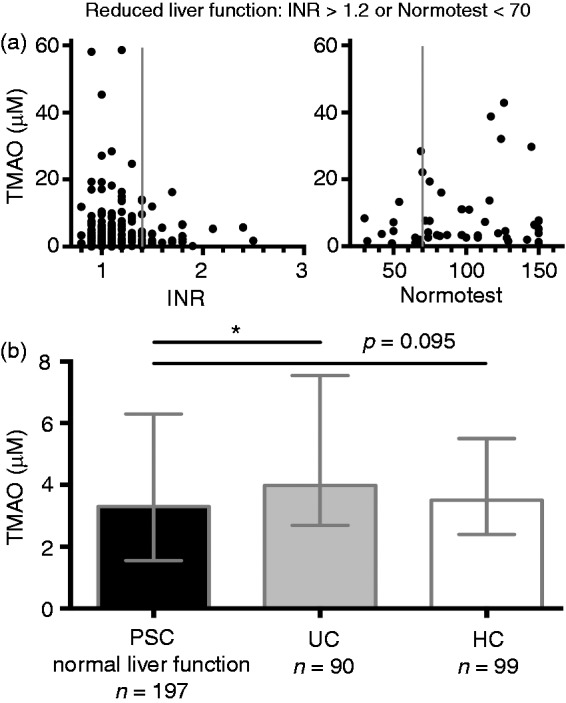
TMAO levels are affected by liver function in patients with PSC. (a) We hypothesised that TMAO values in PSC could be partially explained by reduced liver functioned affecting TMAO production. Reduced liver function was defined by increased prothrombin time (INR > 1.2 or Normotest < 70, cut-offs indicated by solid grey lines). (b) Total prothrombin time was available for 250 patients with PSC (82.0% of PSC patients included in the study). When including only PSC patients with normal liver function in the analyses (n = 197), patients with PSC no longer showed reduced TMAO compared with HCs, but still showed reduced values compared with UC patients. HCs: healthy control; INR: international normalised ratio; PSC: primary sclerosing cholangitis; TMAO: trimethylamine-N-oxide; UC: ulcerative colitis. Data in (b) shown as median and interquartile range (IQR). Mann-Whitney U was used for comparisons. *p < 0.05.
TMAO levels are associated with reduced transplantation-free survival in PSC patients with normal liver function
In the study group as a whole (n = 305) TMAO were compared between PSC patients that reached a clinical endpoint during follow-up (death: n = 44, liver transplantation: n = 118) and those without such endpoints, detecting an elevated level of TMAO in the former (Figure 3(a)). The median follow-up time was 2.7 years (interquartile range 0.81–6.1), and median age at endpoint 45.1 years (interquartile range 38.2–54.2). As our results so far indicated that liver function could influence TMAO levels, similar comparisons were performed in the PSC patients with a measure of prothrombin time available, again showing increased values in patients with a clinical endpoint, but only among those with normal liver function while there was no significant difference in the reduced liver function group (Figure 3(b)). High TMAO levels have been associated with increased risk of cardiovascular death related to atherosclerosis, but importantly, out of the 44 patients with death as primary endpoint only two (4.5%) were identified with a cardiovascular cause of death (Supplementary Table 2). TMAO correlated with age and age at diagnosis (r = 0.13, p < 0.05 and r = 0.11, p < 0.05, respectively), in addition to alkaline phosphatase (ALP) (r = 0.12, p < 0.05) and eGFR (–0.14, p <0.05), but we were unable to identify correlations between TMAO and INR, albumin, Mayo risk score or other biochemical measurements (Supplementary Table 3).
Figure 3.

PSC patients with primary endpoint during follow-up have increased TMAO levels. (a) When considering all patients with PSC included in the study, patients with an endpoint during follow-up (death or liver transplantation) showed higher TMAO values compared with transplantation-free survivors. The median follow-up time was 2.7 years (IQR 0.81–6.1). (b) We then performed similar comparisons in the patients with prothrombin time available, and further in the subgroups with normal and reduced liver function. TMAO was elevated in patients with a clinical endpoint (death or transplantation), but only among those with normal liver function. Reduced liver function was defined as INR > 1.2 and/or Normotest < 70.0. INR: international normalised ratio; IQR: interquartile range; LT: liver transplantation; PSC: primary sclerosing cholangitis; TMAO: trimethylamine-N-oxide. Data shown as median and IQR. Mann-Whitney U was used for comparisons. *p < 0.05, ***p < 0.001.
Given the possible interaction between TMAO and liver function, and a lack of association between TMAO and death or liver transplantation in the group with reduced liver function, the further analyses focused on the PSC patients with normal liver function (n = 197). In this group, TMAO and ALP were correlated (r = 0.16, p < 0.05), as were TMAO and duration of PSC disease (r = 0.15, p < 0.05), while there was a negative correlation with kidney function (eGFR: r = –0.16, p < 0.05). No correlation could be detected between TMAO and age, age at diagnosis of PSC, Mayo risk score or other biochemical measurements (Supplementary Table 3).
To further explore the association between high TMAO values and transplantation-free survival, we performed ROC-AUC analysis that yielded an AUC of 0.64 (95% confidence interval (CI) 0.57–0.70, p < 0.001, Figure 4(a)) to separate patients with and without endpoint. The optimal TMAO cut-off was 4.1 µM. Using this as cut-off in a Kaplan-Meier survival plot, patients with PSC and high TMAO values (n = 77) exhibited a marked decrease in transplantation-free survival, with a mean transplantation-free survival of 6.1 years (95% CI 4.6–7.6), versus 9.9 years (95% CI 8.6–11.1) in patients with low TMAO (n = 120, log-rank test: p < 0.0001, Figure 4(b)), with indications for liver transplantation given in Supplementary Table 4. For comparison, patients with reduced liver function at TMAO measurement had even lower liver transplantation-free survival, but this was unrelated to TMAO level (Supplementary Figure 2).
Figure 4.

A high level of TMAO is associated with worse prognosis in PSC patients with normal liver function. (a) We performed a ROC-AUC analysis, giving an optimal TMAO cut-off of 4.1 µM for discriminating PSC patients with an endpoint (death or liver transplantation) during follow-up and those without. (b) When stratified according to this cut-off in a Kaplan-Meier plot, and censoring data at 15 years due to the low number of patients with longer follow-up time, patients with PSC and high TMAO values (>4.1 µM) showed an unfavourable prognosis, with a marked reduction in transplantation-free survival (mean transplantation-free survival: 6.1 years (95% CI 4.6–7.6), versus 9.9 years (95% CI 8.6–11.1) in PSC patients with low TMAO). The table shows number of patients at risk at 0, 3, 5, 10 and 15 years. AUC: area under the curve; CI: confidence interval; PSC: primary sclerosing cholangitis; ROC: receiver operating characteristic; TMAO: trimethylamine-N-oxide.
In univariate Cox regression analyses high TMAO (>4.1 µM) was associated with reduced transplantation-free survival, as were age, PSC duration, bilirubin, albumin, ALP, eGFR and Mayo risk score (Table 2). In multivariate analyses including all patients with normal liver function and a complete dataset available (n = 172), high TMAO was independently associated with a reduction in transplantation-free survival (hazard ratio (HR) = 1.87, 95% CI 1.15–3.04, p = 0.011, Table 2). Besides high TMAO, Mayo risk score was the only variable that remained associated with transplantation-free survival in the multivariate model. Age, bilirubin and albumin were excluded from the final multivariate model since they are all part of the Mayo risk score.
Table 2.
Univariate and multivariate Cox regression
| Univariate Cox regression |
Multivariate Cox model |
||||||
|---|---|---|---|---|---|---|---|
| HR | 95% CI | p | n | HR | 95% CI | p | |
| High TMAO | 2.48 | (1.62–3.77) | <0.001 | 197 | 1.87 | (1.15–3.04) | 0.011 |
| Age | 1.03 | (1.02–1.05) | <0.001 | 197 | |||
| Sex | 1.07 | (0.64–1.80) | 0.802 | 197 | NS | 0.636 | |
| PSC duration | 1.06 | (1.02–1.09) | 0.001 | 197 | NS | 0.185 | |
| IBD status | 0.94 | (0.56–1.57) | 0.802 | 197 | |||
| Bilirubina | 1.71 | (1.42–2.06) | <0.001 | 194 | |||
| Albumin | 0.91 | (0.88–0.94) | <0.001 | 186 | |||
| ALPa | 1.42 | (1.09–1.84) | 0.009 | 186 | NS | 0.779 | |
| ASTa | 1.24 | (0.98–1.58) | 0.075 | 193 | |||
| ALTa | 0.97 | (0.78–1.21) | 0.789 | 194 | |||
| Creatinine | 0.99 | (0.98–1.01) | 0.513 | 183 | |||
| eGFR | 0.99 | (0.97–1.00) | 0.045 | 183 | NS | 0.526 | |
| Plateletsa | 0.87 | (0.62–1.21) | 0.393 | 179 | |||
| INR | 2.76 | (0.24–31.93) | 0.416 | 166 | |||
| Variceal bleeding | 2.12 | (0.77–5.81) | 0.146 | 167 | |||
| p-ANCA | 1.11 | (0.67–1.87) | 0.681 | 144 | |||
| Mayo risk score | 1.73 | (1.45–2.06) | <0.001 | 177 | 1.74 | (1.40–2.17) | <0.001 |
Right-skewed distribution, transformed by the natural logarithm before analyses.
ALP: alkaline phosphatase; ALT: alanine transaminase; AST: aspartate transaminase; CI: confidence interval; eGFR: estimated glomerular filtration rate; HR: hazard ratio; IBD: inflammatory bowel disease; INR: international normalised ratio; NS: not significant; p-ANCA: perinuclear anti-neutrophil cytoplasmic antibodies; PSC: primary sclerosing cholangitis; TMAO: trimethylamine-N-oxide.
Discussion
In this study, the circulating level of the gut microbiota-dependent metabolite TMAO was lower in patients with PSC than in controls. The reduction was confined to patients with reduced liver function, which potentially is caused by impaired oxidation of TMA to TMAO by hepatic FMOs. When evaluating patients with normal liver function, a high TMAO level was associated with reduced liver transplantation-free survival, independent of other surrogate markers of disease activity including Mayo risk score, suggesting that dietary and microbial factors may influence the disease progression in PSC.
In the case-control assessment, TMAO was reduced in patients with PSC compared with HCs, but when removing patients with reduced liver synthesis function, as defined by prothrombin time outside reference, this difference disappeared. There are few data available on TMAO in cholestatic liver diseases. In a study of patients with liver disease in general, conversions of TMA to TMAO were found to be impaired with reduced excretion of TMAO in urine compared with controls, especially in more severe liver disease with a corresponding increase in TMA.27 The latter has also been reported in other studies,28 and is supported by a more recent report, finding increased TMA (which is a volatile gas) in the breath of patients with cirrhosis compared with HCs.29 In line with this, patients with reduced liver function in our study also showed low TMAO values. Other data on TMAO in human liver disease are scarce,30,31 but overall it is a reasonable assumption that hepatic production of TMAO is similar in PSC and HCs when excluding patients with reduced liver function.
Focusing on patients with normal liver function, the study showed an increased risk of liver transplantation or death in patients with elevated TMAO HR 1.87), independent from Mayo risk score. In contrast, TMAO did not seem to be a marker of poor prognosis in the group with reduced liver function. There is a scarcity of data available for comparison. In several studies TMAO has been associated with atherosclerosis and coronary artery disease.11,15,32 It could therefore be speculated that the association between elevated TMAO and transplantation or death in patients with PSC could be related to cardiovascular disease. However, less than 5% of the patients who died without liver transplantation were classified with a cardiovascular cause of death. This is in line with the observation that PSC patients were relatively young at the time of endpoint (median 45.1 years), and that PSC in general has not been associated with increased risk of ischemic cardiac disease.33 TMAO has been associated not only with the prognosis of ischemic heart disease and heart failure,13,34,35 but also kidney disease including a possible influence on fibrosis,36,37 suggesting that circulating TMAO, or related metabolites, may exert an effect on several organs. Whether the bile ducts are vulnerable and PSC may represent ‘the arteriosclerosis of the bile duct’ can only be speculated upon.38 It is interesting, however, that food with high content of methylamines (e.g. TMA) and TMAO elicits bile duct changes in rats including cholangiocyte proliferation, cholangiofibrosis and cholestasis.18,39 In addition, it has been suggested that dietary amines (e.g. methylamines) could be central to the homing of ‘gut primed’ lymphocytes to the liver seen in PSC.40,41 Thus, the observed association could also be speculated to be related to the production of the precursor to TMAO, and not TMAO itself.
In patients with PSC and normal liver function there were no correlations between TMAO and conventional markers of PSC severity including Mayo risk score, suggesting that TMAO is a marker of disease course, which does not represent disease stage. TMAO levels depend on production of TMA by anaerobic bacteria in the gut.11,12,17,42 Carnitine or phosphatidylcholine are important as choline sources for TMA production, and they are abundant in a variety of food groups, including red meat, dairy products and some types of fish and seafood.43 The gut microbiota is altered in PSC,7 but little is known about how it affects disease activity and prognosis. Data on the effect of diet in PSC are also very scarce. TMAO levels are known to be markedly reduced in individuals exposed to certain antibiotics,15 and it is therefore of interest that antibiotic treatment has been shown both to reduce ALP levels in human PSC,44 and ameliorate PSC-like changes in a murine small intestinal bacterial overgrowth-associated model of cholangitis.45 While the present study cannot prove a direct role of TMAO in the disease pathogenesis, the reduced liver transplantation-free survival associated with elevated TMAO suggests that a combination of dietary factors and microbial composition and metabolites are associated with PSC progression. Furthermore, a recent paper reported that specifically targeting the enzymatic production of TMA from gut bacteria reduced TMAO production and atherosclerosis in mice.46 Similar experiments in animal models of biliary disease could clarify the potential role of TMAO in PSC not only as a marker, but also as a potential mediator. Overall, the present findings provide a strong rationale for deeper investigations of the effect of diet and the gut microbiota in patients with PSC. As shown by the present data, metabolic parameters onto which complex microbial and dietary patterns converge may simplify such assessments.
The strengths of the current study include the large number of patients with long follow-up time as well as the use of an established assay analysing all samples in the same batch. There are also several limitations. Since diet and microbial function are major determinants of TMAO, and there is a lack of a detailed baseline dietary characterisation whether the participants were on antibiotics or not, only limited conclusions can be drawn as to the cause of the association between TMAO and PSC severity. Lack of controls with other liver diseases, including cholestatic liver diseases like primary biliary cholangitis (PBC), also limits our ability to discern whether the association between high TMAO values and shorter time to liver transplantation is specific for PSC or not. As this is a historic cohort and gut microbiota profiling has only recently been in widespread use, the composition of the gut microbiota in the present cohort is unknown. It is therefore not possible to link microbial composition to high TMAO levels or prognosis. There is also a need for prospective studies including other liver diseases to further elucidate these complex associations in the future, and validation panels in external cohorts will be essential. The blood samples were also not collected after a strict fasting protocol. Experimental evidence does, however, suggest that major postprandial TMAO-increases are primarily related to e.g. a phosphatidylcholine challenge, because the majority of choline in ordinary meals will be reabsorbed in the small intestine before reaching the TMA-generating bacteria in the colon, as long as the reabsorption is not overloaded.15,47
In conclusion, the data suggest that TMAO is influenced by reduced liver function and that caution should be made in interpreting TMAO measurements in patients with advanced liver disease. The strong association between elevated TMAO and reduced liver transplantation-free survival in PSC patients with normal liver function at time of sample collection suggests that complex dietary and microbial factors may be relevant for the prognosis of patients with PSC as reflected by specific metabolic alterations. Interventions targeting diet or the gut microbiota could therefore potentially be clinically relevant.
Supplementary Material
Acknowledgements
Hege Dahlen Sollid, Mona Bjørnstad and Liv Wenche Torbjørnsen are acknowledged for help at the Norwegian PSC Research Center, Prof Fredrik Bäckhed for helpful discussions, Prof Erik Schrumpf for critical reading of our manuscript, and Torunn Eide for excellent technical assistance with the TMAO analyses. Prof Benedicte A. Lie and the Norwegian Bone Marrow Donor Registry are acknowledged for providing access to HCs.
Declaration of conflicting interests
All authors have completed the International Committee of Medical Journal Editors (ICMJE) uniform disclosure form: Dr Hov reports grants from Biogen Idec., outside the submitted work; all other authors report no conflict of interest.
Funding
This work was supported by the Western Norway Regional Health Authority (grant number 911802 to THK, 2014) and the Norwegian Research Council (project number 240787/F20 to JRH, 2015).
References
- 1.Hirschfield GM, Karlsen TH, Lindor KD, et al. Primary sclerosing cholangitis. Lancet 2013; 382: 1587–1599. [DOI] [PubMed] [Google Scholar]
- 2.Bergquist A, Montgomery SM, Bahmanyar S, et al. Increased risk of primary sclerosing cholangitis and ulcerative colitis in first-degree relatives of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2008; 6: 939–943. [DOI] [PubMed] [Google Scholar]
- 3.Folseraas T, Liaskou E, Anderson CA, et al. Genetics in PSC: What do the ‘risk genes’ teach us? Clin Rev Allergy Immunol 2015; 48: 154–164. [DOI] [PubMed] [Google Scholar]
- 4.Andersen IM, Tengesdal G, Lie BA, et al. Effects of coffee consumption, smoking, and hormones on risk for primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2014; 12: 1019–1028. [DOI] [PubMed] [Google Scholar]
- 5.Kamada N, Seo SU, Chen GY, et al. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 2013; 13: 321–335. [DOI] [PubMed] [Google Scholar]
- 6.Bäckhed F, Fraser CM, Ringel Y, et al. Defining a healthy human gut microbiome: Current concepts, future directions, and clinical applications. Cell Host Microbe 2012; 12: 611–622. [DOI] [PubMed] [Google Scholar]
- 7.Kummen M, Holm K, Anmarkrud JA, et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut. Epub ahead of print 17 February 2016. DOI: 10.1136/gutjnl-2015-310500. [DOI] [PubMed]
- 8.Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 2011; 9: 279–290. [DOI] [PubMed] [Google Scholar]
- 9.Sharon G, Garg N, Debelius J, et al. Specialized metabolites from the microbiome in health and disease. Cell Metab 2014; 20: 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol 2013; 14: 676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 2013; 19: 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011; 472: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trøseid M, Ueland T, Hov JR, et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med 2015; 277: 717–726. [DOI] [PubMed] [Google Scholar]
- 14.Al-Waiz M, Mikov M, Mitchell SC, et al. The exogenous origin of trimethylamine in the mouse. Metabolism 1992; 41: 135–136. [DOI] [PubMed] [Google Scholar]
- 15.Tang WH, Wang Z, Levison BS, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 2013; 368: 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skagen K, Trøseid M, Ueland T, et al. The carnitine-butyrobetaine-trimethylamine-N-oxide pathway and its association with cardiovascular mortality in patients with carotid atherosclerosis. Atherosclerosis 2016; 247: 64–69. [DOI] [PubMed] [Google Scholar]
- 17.Tremaroli V, Karlsson F, Werling M, et al. Roux-en-Y gastric bypass and vertical banded gastroplasty induce long-term changes on the human gut microbiome contributing to fat mass regulation. Cell Metab 2015; 22: 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin JK, Ho YS. Hepatotoxicity and hepatocarcinogenicity in rats fed squid with or without exogenous nitrite. Food Chem Toxicol 1992; 30: 695–702. [DOI] [PubMed] [Google Scholar]
- 19.Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010; 51: 660–678. [DOI] [PubMed] [Google Scholar]
- 20.European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of cholestatic liver diseases. J Hepatol 2009; 51: 237–267. [DOI] [PubMed] [Google Scholar]
- 21.Solberg IC, Lygren I, Jahnsen J, et al. Clinical course during the first 10 years of ulcerative colitis: results from a population-based inception cohort (IBSEN Study). Scand J Gastroenterol 2009; 44: 431–440. [DOI] [PubMed] [Google Scholar]
- 22.Lennard-Jones JE. Classification of inflammatory bowel disease. Scand J Gastroenterol 1989; 170(Suppl): 2–6. [DOI] [PubMed] [Google Scholar]
- 23.Kim WR, Therneau TM, Wiesner RH, et al. A revised natural history model for primary sclerosing cholangitis. Mayo Clin Proc 2000; 75: 688–694. [DOI] [PubMed] [Google Scholar]
- 24.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009; 150: 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Youden WJ. Index for rating diagnostic tests. Cancer 1950; 3: 32–35. [DOI] [PubMed] [Google Scholar]
- 26.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: A nonparametric approach. Biometrics 1988; 44: 837–845. [PubMed] [Google Scholar]
- 27.Wranne L. Urinary excretion of trimethylamine and trimethylamine oxide following trimethylamine-administration to normals and to patients with liver disease. Acta Med Scand 1956; 153: 433–441. [DOI] [PubMed] [Google Scholar]
- 28.Marks R, Dudley F, Wan A. Trimethylamine metabolism in liver disease. Lancet 1978; 1: 1106–1107. [DOI] [PubMed] [Google Scholar]
- 29.Hanouneh IA, Zein NN, Cikach F, et al. The breathprints in patients with liver disease identify novel breath biomarkers in alcoholic hepatitis. Clin Gastroenterol Hepatol 2014; 12: 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YM, Liu Y, Zhou RF, et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci Rep 2016; 6: 19076–19076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitchell S, Ayesh R, Barrett T, et al. Trimethylamine and foetor hepaticus. Scand J Gastroenterol 1999; 34: 524–528. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, Tang WHW, Buffa JA, et al. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur Heart J 2014; 35: 904–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ludvigsson JF, Bergquist A, Montgomery SM, et al. Risk of diabetes and cardiovascular disease in patients with primary sclerosing cholangitis. J Hepatol 2014; 60: 802–808. [DOI] [PubMed] [Google Scholar]
- 34.Tang WH, Wang Z, Fan Y, et al. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: Refining the gut hypothesis. J Am Coll Cardiol 2014; 64: 1908–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki T, Heaney LM, Bhandari SS, et al. Trimethylamine N-oxide and prognosis in acute heart failure. Heart 2016; 102: 841–848. [DOI] [PubMed] [Google Scholar]
- 36.Tang WH, Wang Z, Kennedy DJ, et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res 2015; 116: 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Missailidis C, Hällqvist J, Qureshi AR, et al. Serum trimethylamine-N-oxide is strongly related to renal function and predicts outcome in chronic kidney disease. PLoS One 2016; 11: e0141738–e0141738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fickert P, Moustafa T, Trauner M. Primary sclerosing cholangitis – the arteriosclerosis of the bile duct? Lipids Health Dis 2007; 6: 3–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin JK, Hurng DC. Potentiation of ferrous sulphate and ascorbate on the microbial transformation of endogenous trimethylamine N-oxide to trimethylamine and dimethylamine in squid extracts. Food Chem Toxicol 1989; 27: 613–618. [DOI] [PubMed] [Google Scholar]
- 40.Quraishi MN, Sergeant M, Kay G, et al. The gut-adherent microbiota of PSC-IBD is distinct to that of IBD. Gut. Epub ahead of print 19 April 2016. DOI: 10.1136/gutjnl-2016-311915. [DOI] [PubMed]
- 41.Trivedi PJ, Adams DH. Mucosal immunity in liver autoimmunity: A comprehensive review. J Autoimmun 2013; 46: 97–111. [DOI] [PubMed] [Google Scholar]
- 42.Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A 2012; 109: 21307–21312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeisel SH, DaCosta KA. Increase in human exposure to methylamine precursors of N-nitrosamines after eating fish. Cancer Res 1986; 46: 6136–6138. [PubMed] [Google Scholar]
- 44.Tabibian JH, Weeding E, Jorgensen RA, et al. Randomised clinical trial: Vancomycin or metronidazole in patients with primary sclerosing cholangitis – a pilot study. Aliment Pharmacol Ther 2013; 37: 604–612. [DOI] [PubMed] [Google Scholar]
- 45.Lichtman SN, Keku J, Clark RL, et al. Biliary tract disease in rats with experimental small bowel bacterial overgrowth. Hepatology 1991; 13: 766–772. [PubMed] [Google Scholar]
- 46.Wang Z, Roberts AB, Buffa JA, et al. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 2015; 163: 1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeisel SH, DaCosta KA, Youssef M, et al. Conversion of dietary choline to trimethylamine and dimethylamine in rats: Dose-response relationship. J Nutr 1989; 119: 800–804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.