

Abstract
Histone proteins wrap around DNA to form nucleosomes, which further compact into higher order structure of chromatin. In addition to the canonical histones, there are also variant histones that often have pivotal roles in regulating chromatin dynamics and the accessibility of the underlying DNA. H3.3 is the most common non-centromeric variant of histone H3 that differs from the canonical H3 by just 4–5 amino acids. Here we discuss the current knowledge of H3.3 in transcriptional regulation and the recent discoveries and molecular mechanisms of H3.3 mutations in human cancer.
Graphical Abstract
Introduction
In eukaryotes, DNA is wrapped around histones to form nucleosomes, the fundamental unit of chromatin. The nucleosome is composed of two copies each of four core histones, H2A, H2B, H3 and H4, which form a histone octamer that is wrapped nearly twice by an average of 147 bp of DNA [1]. Compelling evidence has demonstrated that histones play a fundamental role in regulating virtually all DNA-templated processes and that many of these processes are modulated through the dynamic post-translational modification (PTM) of histones. Histone PTMs include methylation and acetylation, which occur mainly on the N-terminal unstructured regions of histones called “histone tails” [2]. In addition to the canonical histones H2A, H2B, H3 and H4, metazoans also have histone variants, such as H3.3, H2A.Z and H2A.X that are distinguished from their canonical counterparts by just a few amino acids or a large polypeptide fragment [3]. Increasing numbers of researchers have demonstrated that these histone variants also have pivotal roles in modulating chromatin dynamics and the activities of the underlying DNA [4, 5]. There are three main histone H3 variants in metazoans: H3.1 and H3.2 that are known as the “canonical” histone H3, the replacement variant H3.3, and the centromere-specific variant CENP-A. Other variants include the testis-specific H3 variant H3t, and primate-specific variants H3.X and H3.Y. In this review, we focus on the histone H3.3 variant, especially recent discoveries of H3.3-specific regulators as well as genetic mutations of H3.3 and their correlation with the development of human cancers.
1. Properties of H3.3 genes and proteins
Genes encoding the canonical histone H3 are organized into clusters containing multiple gene copies. In humans, three copies of genes encoding H3.2 are located within a histone cluster on chromosome 1, and 10 copies of genes encoding H3.1 are clustered in the chromosome 6p22 region that also contains 39 clustered genes encoding other core histones. These canonical H3-encoding genes have no introns, and their corresponding transcripts are not polyadenylated [6]. The tandem organization of these genes allows their simultaneous expression, which facilitates production of the massive amount of histone protein required for genome duplication during S phase.
In contrast to the canonical histone H3, histone variant H3.3 genes, namely H3F3A and H3F3B in humans, lie outside the histone gene clusters. They are similar to most other eukaryotic genes in that they contain introns and their mRNAs have poly(A) tails. In general, canonical H3 is expressed and incorporated into chromatin at S phase in a replication-dependent manner [7–10]. In contrast, variant H3.3 is expressed throughout the cell cycle. Consequently, H3.3 can be incorporated into chromatin through either a replication-coupled pathway during S phase or a replication-independent manner outside of S phase [9, 11, 12]. Interestingly, a subset of histone genes are expressed as polyadenylated mRNAs in terminally differentiated tissues, indicating that these genes may also provide replication-independent replacement histones in long-lived non-dividing cells [13]. In yeast, there is only one non-centromeric H3 gene, which encodes an H3.3-like protein, suggesting that H3.3 is more conserved than its canonical counterparts.
At the protein level, H3.3 differs from the canonical histone H3 at only 4 or 5 amino acids, with minor variations among organisms. In humans, the differences between H3.3 and H3 are defined by residues 87, 89, 90 and 96 in the histone fold domain and residue 31 in the N-terminal tail. Although the changes in amino acid sequence are subtle, these specific residues contribute to the distinct properties of variant H3.3. Amino acid residues 87–90 in H3.1 (SAVM) are replaced by AAIG in H3.3. Ala 87 and Gly 90 are important for the recognition of H3.3 by the variant-specific chaperone death domain–associated protein (DAXX) [14, 15], and G90 is also recognized by ubinuclein-1 (UBN1), a subunit of the HIRA histone chaperone complex [16]. At the N-terminus, Ala 31 in H3.1 is replaced by Ser in H3.3, which undergoes phosphorylation in the pericentromeric region during mitosis [17–19].
2. H3.3-specific chaperones
Histone chaperones help assemble histones and DNA into nucleosomes and also disassemble nucleosomes into their subcomponents without being part of the final products. They are essential for the precise control of each step during the assembly and disassembly processes. Without chaperones, simply mixing histones and DNA in vitro at physiological salt concentration can not form nucleosomes, and instead only form aggregates due to the strong, non-specific electrostatic interaction between histones and DNA [20]. Previous studies have identified both canonical histone H3 chaperones, such as anti-silencing factor 1 (ASF1) and nuclear autoantigenic sperm protein (NASP), and variant-specific chaperones, discussed below. These chaperones bind to H3 in the form of H3/H4 dimers. During replication, NASP transfers newly synthesized histone H3 to ASF1, which in turn transfers the histone proteins to their specific chaperones: canonical H3 to chromatin assembly factor 1 (CAF-1) [21, 22] and H3.3 to either histone cell cycle regulation defective homolog A (HIRA) or DAXX. Notably, both HIRA and DAXX deposit H3.3 onto chromatin in a replication independent manner.
The HIRA complex
HIRA was first described as a protein interacting with histones H2B and H4 in a yeast two-hybrid screen and an in vitro GST pull-down assay, respectively [23]. Isolation of the predeposition protein complexes with different H3 variants in cells later revealed that HIRA is an H3.3-specific chaperone [24]. HIRA forms a stable complex with cabin1 and ubinuclein (UBN1/2) proteins. Although the actual functions of cabin1 and UBN1/2 in H3.3 deposition are not clear, the protein-protein interaction between UBN1 and HIRA is required for maintaining complex integrity [25]. The HIRA complex is conserved across species. In S. cerevisiae, HIRA (Hir1p and Hir2p) forms a histone regulatory (HIR) complex with Hir3 and Hpc2, which are orthologs of human cabin1 and UBN, respectively [26]. A similar complex also exists in other species, such as S. pombe [27, 28], Drosophila [29, 30], and Arabidopsis [31], suggesting an evolutionally conserved function of the complex. HIRA mediates the deposition of H3.3 into euchromatic regions. Depletion of HIRA leads to impaired genome-wide enrichment of H3.3 at promoters, the gene bodies of actively transcribed genes, and at specific regulatory elements, thus implying a critical role for HIRA in promoting H3.3 occupancy in these regions [32, 33]. HIRA binds to naked DNA in vitro and to nucleosome-depleted regions in cells, indicating that deposition of H3.3 in transiently accessible, non-nucleosomal DNA (gap filling) is HIRA-dependent [33]. This could be a salvage pathway to maintain chromatin integrity when CAF-1 mediated H3.1 deposition is impaired during replication. In mouse embryonic stem cells (ESCs), Hira directly interacts with the PRC2 complex, facilitating PRC2 recruitment and proper establishment of H3K27me3 at the promoters of developmentally regulated genes [34]. HIRA also deposits newly synthesized H3.3 to damaged DNA sites in response to UV irradiation, suggesting that HIRA-dependent histone deposition serves as a chromatin bookmarking system to facilitate transcriptional recovery after genotoxic stress [35]. HIRA dependent H3.3 deposition is also essential for paternal chromatin assembly in both Drosophila and mouse zygotes [36, 37]. Hira mutant mouse zygotes display a loss of H3.3 incorporation in both oocyte and sperm genomes, as well as impaired DNA replication and rRNA transcription [38, 39]. Specific deletion of Hira in developing mouse oocytes results in altered chromatin homeostasis including decreased DNA methylation, increased DNase I sensitivity and accumulation of DNA damage, along with a severe fertility phenotype [40]. Hira is essential for mouse embryonic development. Hira knockout mice are embryonically lethal and display gastrulation defects and patterning abnormalities [41]. Drosophila HIRA mutants are viable, but all females are infertile [42].
The DAXX-ATRX complex
DAXX was initially identified as a protein associated with the FAS cell surface receptor that enhances FAS dependent cell death [43]. Several subsequent studies showed that DAXX is an H3.3-specific histone chaperone unique to metazoans [32, 44, 45]. DAXX forms an H3.3 preassembly complex with the α-thalassemia/mental retardation syndrome protein (ATRX), a SNF2-like ATP-dependent chromatin remodeling factor. Distinct from HIRA that deposits H3.3 in euchromatin, DAXX deposits H3.3 into telomeres and pericentric heterochromatin [32, 44, 45]. In mouse ESCs, H3K9Ac is enriched in purified HIRA-associated H3.3 complexes, but not DAXX-associated H3.3 complexes [46]. In contrast, the DAXX-ATRX complex, but not the HIRA complex, is required for the deposition of H3.3 at pericentromeric heterochromatin, telomeres and endogenous retroviral elements that are enriched for H3K9me3 [32, 45–50]. The deposition of H3.3 by DAXX is unhindered by the absence of HIRA, and vice versa [46]. Therefore, the HIRA complex and the DAXX-ATRX complex control H3.3 deposition at distinct sites within the genome and the H3.3 each deposits may have distinct PTMs and consequently, unique functions.
DAXX distinguishes between histone H3.3 and H3 via direct interaction with the H3.3-specific amino acid residues 87–90 (AAIG) present in the H3.3 histone fold domain [45]. Crystal structure analysis revealed that DAXX binds to H3.3 via an extensive hydrogen bond network between its histone binding domain (HBD) and the AAIG segment, among which Gly90 and Ala87 are the principal determinants of H3.3 specificity [14, 15].
A large pool of nuclear DAXX does not co-fractionate with ATRX in biochemical fractionations of nuclear extracts, suggesting that DAXX may function independently of ATRX [45]. Indeed in cells, DAXX is essential for H3.3 deposition at both telomeres and in pericentromeric heterochromatin, whereas ATRX is only required for H3.3 enrichment at telomeres [32]. DAXX function is further distinguished from ATRX function as DAXX can also contribute to H3.3 deposition at some transcription factor binding sites without ATRX, and can recruit a pool of non-nucleosomal H3.3 to promyelocytic leukemia (PML) nuclear bodies prior to depositing H3.3 in chromatin independently of ATRX [51]. These findings raise the question of whether another protein or proteins interact with DAXX to deposit H3.3 in genic and intergenic regions. Notably, Daxx is required for mouse viability [52], yet in DAXX-depleted mouse embryonic fibroblasts, a fraction of H3.3 associates with the replication-dependent histone deposition machinery [44], suggesting that cells might adapt to the loss of DAXX by using alternate chaperones to deposit H3.3.
DEK
The proto-oncoprotein DEK interacts with DAXX and the histone deacetylase HDAC2 and is involved in transcriptional repression [53]. In both Drosophila and human cells, DEK is phosphorylated by casein kinase 2 and serves as a histone H3.3 chaperone [54]. DEK regulates both the balance between free and nucleosomal H3.3 and proper distribution of H3.3 in chromatin [55]. As already mentioned, newly synthesized H3.3 is enriched in PML bodies together with DAXX before deposition into pericentromeric heterochromatin [44, 51, 56]. This process requires DEK but is independent of ATRX. Depletion of DEK in ESCs re-routes non-nucleosomal H3.3 to chromatin instead of PML bodies, resulting in not only HIRA-dependent widespread loading of H3.3 onto chromatin, but also DAXX/ATRX-dependent redistribution of H3.3 from telomeres to both chromosome arms and pericentric heterochromatin leading to the formation of fragile telomeres. These results suggest that DEK safeguards chromatin integrity by limiting histone access and specifically controlling H3.3 deposition into specific genomic regions [55].
3. Chromatin remodelers that regulate H3.3
Some chromatin remodeling factors have been shown to play a role in depositing H3.3 into specific genomic regions, although they don’t directly interact with H3.3. Chromodomain helicase DNA-binding domain 1 (CHD1), a SNF2 family member, is a chromatin-remodeling factor that together with the histone chaperone NAP-1, can assemble nucleosome arrays from DNA and histones in vitro [57]. In Drosophila, CHD1 interacts with HIRA, and this interaction is required for incorporation of histones into decondensing sperm chromatin in the male pronucleus as well as during the late stages of embryonic development [58]. Depletion of CHD1 in embryos disrupts HIRA-dependent incorporation of H3.3 into the male pronucleus, and consequently, the paternal genome is unable to participate in zygotic mitosis [58]. CHD2, another SNF2 chromatin remodeling enzyme family member, interacts with the MyoD protein to facilitate deposition of H3.3 to loci containing myogenic gene regulatory sequences prior to cell differentiation [59]. Knockdown of CHD2 specifically decreases H3.3 deposition at differentiation-dependent genes and inhibits the activation of myogenic genes, whereas housekeeping genes are only modestly affected [59]. The sequence-specific DNA-binding protein GAGA factor directs the replacement of histone H3 with H3.3 through association with HIRA, and counteracts the spreading of silent chromatin in Drosophila [60, 61]. GAGA recruits FACT and the Polybromo-associated Brm (PBAP) remodeling complex to chromatin boundaries; both complexes are required for boundary maintenance and H3.3 replacement at the boundaries [62]. In addition, EP400, a SWR1-class ATP-dependent chromatin remodeling protein that deposits H2A.Z variant in the Tip60-P400 complex, can also exchange H3.3 into canonical chromatin both in vitro and in cells, promoting gene activation [63]. Given the close correlation of H2A.Z with H3.3 in the genome, it awaits to see if other H2A.Z remodelers also have a role in regulating H3.3 deposition.
4. H3.3-specific readers
In addition to the H3.3 specific chaperones and remodelers, two recent studies have identified the first H3.3-specific reader, ZMYND11 (Zinc finger MYND-domain containing protein 11, also known as BS69) [64, 65]. ZMYND11 is a candidate tumor suppressor that interacts with adenovirus E1A protein and a number of transcription factors to suppress their transactivating activities [66–68]. ZMYND11 contains tandem “reader” modules of histone modifications. These include an N-terminal plant homeodomain (PHD) zinc finger, bromodomain and PWWP domain, and a C-terminal zinc finger MYND motif that mediates protein-protein interactions with E1A and many other factors [69]. Work from our laboratory and Yang Shi’s group at Harvard revealed that the tandem PHD-bromo-PWWP domains recognize histone H3K36 trimethylation (H3K36me3) and importantly, this recognition is specific to the histone variant H3.3 [64, 70]. Of the three modules, the PWWP domain is predominantly responsible for recognition of the trimethyl moiety. The PWWP domain belongs to the “Royal Family” of protein domains that has been shown to recognize methylation on histone residues H3K36 and H4K20 [71]. Notably, ZMYND11 distinguishes itself from other PWWP proteins in that it is a histone variant H3.3-specific reader of K36me3. A second composite pocket at the junction of the bromodomain, PWWP domain, and an embedded zinc finger motif recognizes the “Serine 31-Threonine 32 (S31-T32)” segment, thus accounting for the H3.3 specificity [70].
H3.3 differs from canonical histone H3 by only 4–5 amino acids, depending on organism. Within the N-terminal unstructured histone tail, Serine 31 is the only residue unique to H3.3. A study from the Allis group showed that H3.3 S31 is phosphorylated in late prometaphase and metaphase during mitosis, and this phosphorylation event specifically occurs in the chromosomal regions immediately adjacent to centromeres [72]. However, neither the role of this modification nor S31 itself in transcriptional regulation is known. Our studies suggest that S31 plays a critical role in substrate recognition by epigenetic readers such as ZMYND11 [64, 70]. Substitution of H3.3 S31 with Alanine (A31), as is present in canonical H3.1, severely diminishes the binding of ZMYND11 to both an H3K36me3 peptide in vitro and histones in cells [70]. Further, phosphorylation of S31 also abolishes the ZMYND11-H3.3 interaction (Guo et al., 2014) underscoring the importance of this residue.
ChIP-seq analyses reveal that ZMYND11 is strongly co-localized with H3.3 in actively transcribed genes, especially in gene bodies that are also enriched with H3K36me3 [64, 70]. The combination of H3K36me3 and H3.3 establishes a unique epigenetic state that defines the genomic distribution of ZMYND11 and provides a means for spatiotemporal control of gene expression in both normal and neoplastic growth. Interestingly, ZMYND11 associates with H3.3K36me3 and is involved in regulation of both transcriptional elongation and mRNA splicing. Knockdown of ZMYND11 in U2OS cells leads to increased RNA Pol II occupancy in gene bodies and consequently, increased transcription, suggesting that ZMYND11, although associated with active genes, exerts a repressive role in controlling transcription elongation [70]. In an independent study, Guo et al. found that knockdown of ZMYND11 in HeLa cells results in reduced intron retention events for hundreds of genes, suggesting that ZMYND11 functions to connect H3.3K36me3 to pre-mRNA splicing (Guo et al., 2014). It is not clear how ZMYND11 regulates these two events, whether they are two independent events, or are somewhat interconnected. Indeed, it has been documented that transcription elongation-associated H3K36me3 positively modulates splicing activities, and similarly, active splicing also promotes transcription elongation [73–75]. As ZMYND11 does not have any intrinsic enzymatic activities, it likely exerts these functions through recruiting different proteins. Biochemical purifications have identified a number of proteins with distinct functions that interact with ZMYND11. For example, chromatin remodelers such as SWI/SNF, histone-modifying enzymes like HDAC1/2, KDM3B, NSD1, and splicing factors such as EFTUD2 [64, 70]. It will be of great interest to determine how ZMYND11 recruits distinct factors in different processes. Furthermore, the H3.3 K36M and G34V/R mutations identified in human cancers (discussed below) abrogate ZMYND11 binding to H3.3K36me3 in vitro [70], suggesting that these cancer related mutations may disrupt the recognition of histone modifications by this H3.3-specific reader. Further study of this potential mechanism in H3.3 mutated tumors is warranted.
5. H3.3 in chromatin dynamics and transcriptional regulation
The incorporation of variant histones into chromatin provides an important means of modulating chromatin dynamics. Because H3.3 differs from canonical H3 at only 4–5 amino acids, it is unlikely that the replacement of H3 by H3.3 changes the overall structure and stability of the nucleosome [76, 77]. Instead, incorporation of H3.3 likely modulates higher-ordered chromatin folding, resulting in an open chromatin conformation [76]. Alteration of inter-fiber interactions by H3.3 is likely through, at least in part, counteracting the incorporation of the linker histone H1. In Drosophila cells, H3.3-enriched regions are generally depleted of H1, and knockdown of H3.3 causes H1 levels to increase at these regions accompanied by a concomitant increase in nucleosome repeat length [78]. H3.3 maintains the balance between open and condensed chromatin states, which is critical for mouse preimplantation development. Loss of H3.3 leads to over-condensation and mis-segregation of chromosomes as early as the two-cell stage, with corresponding high levels of aneuploidy [79]. Although incorporation of H3.3 or the H2A variant H2A.Z into the nucleosome alone does not alter the overall structure of the nucleosome [76, 80], nucleosome core particles containing both H3.3 and H2A.Z seem less stable than nucleosomes containing their canonical counterparts [81]. In vivo, the H3.3/H2A.Z double-variant nucleosomes are found in the ‘nucleosome-free region’ of active chromosomal regions [82]. These nucleosomes are unstable and prone to disassembly. This instability seems to facilitate access of transcription factors or other chromatin-associated factors to these regulatory sites in human cells [82].
H3.3 is enriched at dynamic regions such as promoters, gene bodies and cis-regulatory elements; thus serving as a mark of transcriptionally activated genes. This distribution pattern is conserved from yeast to humans [33, 83–88]. H3.3 was first described as a variant linked to actively transcribed regions in Drosophila by its deposition at active rDNA arrays [89]. Loss of H3.3 genes in Drosophila leads to widespread transcriptional defects, with genes being both up- and down-regulated. The evidence that down-regulated genes were predominantly highly expressed genes supports the idea that H3.3 is required for proper transcription of active genes. Indeed, the turnover rate of H3.3 in promoters and coding regions is correlated with polymerase density and level of transcription [82, 90]. However, loss of H3.3 or its chaperone Hira in mouse ESCs does not affect global transcriptional output, and ESCs lacking H3.3 are still functionally pluripotent [34]. These results argue that H3.3 is not required for maintaining active transcription. Moreover, in Drosophila, the transcriptional defects due to H3.3 depletion can be rescued by the expression of canonical H3, suggesting that expression changes are likely due to nucleosome depletion, not the lack of a particular histone variant [89].
The incorporation of H3.3 at promoter regions is not limited to active genes, it also occurs at inactive genes, possibly due to being in a poised state or the memory of a previous activation [91, 92]. In mouse ESCs, H3.3 is required for proper establishment of H3K27me3 at the promoters of developmentally regulated genes. H3.3 depletion leads to reduced PRC2 occupancy and H3K27me3 levels, whereas the chromatin landscape of actively transcribed regions such as those marked with H3K4me3 are not affected [34]. In chicken erythroid cell lines, H3.3 is enriched in the upstream regulatory region of the folate receptor whether or not the gene is transcriptionally active [93]. In Arabidopsis, promoters with GA motifs are enriched for H3.3 with RNA Pol II occupancies regardless of the transcriptional status of the genes [86]. In Xenopus, as an early and necessary step in the direct reprogramming of somatic cell nuclei by oocytes, HIRA-dependent H3.3 deposition is required for the transcriptional memory of active genes following nuclear transfer [94, 95]. Additionally, overexpression of H3.3 can enhance epigenetic memory in transplanted nuclei [94]. Although little is known about the mechanism of transcriptional memory, it is possible that incorporation of H3.3 may facilitate transcription factor binding to gene promoters. Overall, although H3.3 incorporation in euchromatin is largely associated with actively transcribed genes, the precise role of H3.3 in transcription remains unclear. For example, in addition to its incorporation into regions of active transcription, H3.3 is also deposited in largely transcriptionally inactive regions, including telomeres, pericentric heterochromatin and silent retroviral elements [32, 44, 45]. All of these studies suggest a complex role for H3.3 in the regulation of both activation and repression of gene expression as well as higher order chromatin conformation and integrity, likely in a context-dependent manner.
6. H3.3 mutations in human cancers
In addition to a myriad of known frequent mutations of epigenetic regulators in human cancers, recent exome-sequencing studies revealed that histone H3 is also mutated at high frequency in specific cancer types, including pediatric high-grade glioblastoma (HGG) and certain types of bone tumors. All histone mutations are missense mutations that affect only three amino acids on the N-terminus, K27, G34 and K36. These mutations preferentially occur in H3F3A and H3F3B, which encode H3.3, and to a lesser extent in genes encoding H3.1 [96–98].
In one study that sequenced the exomes of 48 pediatric glioblastoma (GBM) samples, 31% of the tumors were found to have recurrent mutations in the H3F3A gene resulting in K27M or G34R/G34V substitutions in the H3.3 protein [99]. An independent whole genome sequencing study in pediatric diffuse intrinsic pontine glioma (DIPG) patients identified similar mutations in both the H3F3A genes and the related H3.1-encoding HIST1H3B gene. Targeted sequencing of 43 DIPG and 36 non-brainstem pediatric glioblastoma (non-BS-PG) patients further revealed a high frequency of these mutations [100]. Additional sequencing analyses with more patient samples revealed that these two types of mutations have distinct features. K27M mutations occur in younger patients (5–29 years), whereas G34R/V mutations appear in slightly older patients (9–42 years) [101–103]. The majority of tumors associated with H3K27M mutation are restricted to midline locations such as thalamus, pons, brainstem and the spinal cord [102]. Compared to non-BS-PG (<25%), DIPG has a very high incidence (>70%) of K27M mutations [100]. G34R and G34V mutations are primarily localized to the cerebral hemispheres. In contrast, in non-BS-PGs patient samples ~14% had G34R or G34V mutations but the frequency of these mutations was much lower in pediatric GBM patients and non-existent in DIPG patients [99, 100]. Both wild-type and K27M or G34R/V mutant proteins are expressed in heterozygotes, suggesting that the mutant proteins might play a gain-of-function role in tumor cells. Mutations of other genes also co-occurred with H3.3 mutations, including inactivating mutations of the ATRX and DAXX genes [99–101], which is frequently associated with abnormal telomeres in tumors [104, 105], and activating mutations of the activin receptor type 1 (ACVR1) gene, which functions in the BMP signaling pathway [106–109]. Strikingly, >60% of K27M patient samples and nearly 100% of G34V/R samples also display p53 mutations, indicating that the H3.3 mutations alone are not sufficient for neoplastic transformation [99, 101].
Mutations in histone H3.3 have also been found in certain types of bone tumors, including chondroblastoma and giant cell tumors of the bone (GCTB) [110, 111]. In a study of 77 cases of chondroblastoma, 95% of tumors contained the K36M alteration and interestingly, these mutations predominantly occurred in the H3F3B gene, not the H3F3A gene. In contrast, K27M mutations in H3F3A were more common in HGG samples [110]. In a study of 53 GCTB tumors, >90% had G34 mutations in the H3F3A gene. Distinct from the H3.3 mutations in HGGs where G34 is mutated to Arg or Val, mutations in GCTBs tended to be either G34W or G34L alterations [110]. Interestingly, although G34 mutations occur in both HGG and bone tumors, K27M and K36M mutations are restricted to only HGG or chondroblastoma, respectively, suggesting that these mutations may function through distinct mechanisms in different types of tumors.
Several recent exciting studies have begun to uncover the molecular mechanisms by which these H3.3 mutations alter chromatin dynamics and contribute to tumorigenesis. H3K27 is subjected to methylation mediated by the Polycomb repressive complex 2 (PRC2), which is a well-known repressive mark normally associated with gene repression. In HGGs that contain H3K27M mutations, although the mutant proteins comprise <5% of total histone H3 proteins, the global level of H3K27me3 is reduced, suggesting H3K27M acts as a gain-of-function mutant [112, 113]. Elegant biochemical studies revealed that the H3K27M mutant has a higher affinity for EZH2 than its wild-type counterpart, thus the lysine to methionine alteration on H3K27 competes for binding with the wild type histone and when the mutant protein binds, it inhibits the enzymatic activity of EZH2, the enzymatic subunit of the PRC2 complex [112]. Crystal structures of the PRC2-H3K27M complex suggest that the K27M alteration increases the overall strength of the PRC2-nucleosome interaction, thus effectively sequestering PRC2 to prevent it from further propagating the repressive H3K27 methylation mark [114, 115]. Indeed, the “hijacking” of the PRC2 complex by H3K27M results in a global loss of H3K27me3 in K27M HGGs [112, 116], and a reduction in H3K27me3 on large chromatin regions concomitant with DNA hypomethylation [113, 117]. It is worth noting that in K27M cells there are also a number or regions that gain H3K27me3, suggesting that loss of H3K27me3 might not be the only functional consequence of K27M. Nevertheless, as these cells have concomitantly higher levels of H3K27ac, the oncogenic potential of K27M mutation is likely due to overall mismanagement of chromatin.
Interestingly, exogenous expression of mutants containing K-to-M substitutions at other known methylated lysine residues, such as H3K9 and H3K36, are also sufficient to cause a specific reduction in methylation through inhibition of their cognate SET-domain enzymes [112, 116]. Similar phenotypes were also observed in chondroblastoma cells that contain K36M mutations. Similar to K27M that sequesters EZH2, the K36M mutant inhibits H3K36 methyltransferases, including NSD1, NSD2 and SETD2 in vitro [118], and reduces global H3K36 methylation in vivo [119, 120]. Loss of H3K36 methylation leads to increased H3K27me3 in the intergenic regions, redistribution of the PRC1 complex from its target genes, and consequently, upregulation of PRC1-repressed target genes known to block mesenchymal differentiation [119]. All together, these results suggest that lysine-to-methionine mutations share a common mechanism of inhibiting methylation pathways to promote tumorigenesis.
Unlike the K27M and K36M mutations, the mechanism underlying the cellular changes due to H3G34 alterations is not clear. H3G34 itself is not post-translationally modified; however, G34 lies in close proximity to K36, which undergoes methylation during transcriptional elongation [121, 122]. Therefore, it is conceivable that G34 mutations may impact the accessibility of K36 to its methyl-transferases, thus altering H3K36 methylation on the same histone tail. Indeed, G34R and G34V-containing nucleosomes show reduced methylation of H3K36 by SETD2 in vitro [112, 113], suggesting that the G34R/V mutation may inhibit gene expression by attenuating SETD2 function in transcriptional elongation. Surprisingly, in contrast to the in vitro data, G34 mutations lead to increased H3K36me3 levels and RNA Pol II occupancy on several key oncogenes including MYCN, thus resulting in an elevated expression of these potent tumorigenic initiators in the G34R/V HGG cells [123]. It is worth noting that almost all the tumors harboring a H3.3 G34R or G34V mutation in one study, exhibited mutations in ATRX and DAXX as well as alternative lengthening of telomeres [99]. Considering the role of ATRX and DAXX in mediating the deposition of H3.3 in both telomeres and pericentric heterochromatin [32], it is possible that G34R/V mutations promote tumorigenesis by impairing higher order chromatin structure rather than by influencing specific gene expression. More in-depth studies are needed to address the details on their relationships.
Conclusions and future directions
H3.3 is incorporated into regions associated with actively transcribed genes in euchromatin as well as regions that are relatively inactive, such as telomeres and pericentric heterochromatin. These genomic distribution patterns suggest that H3.3 plays a role in both gene activation and silencing in a context dependent manner. Although depletion of H3.3 in some systems (such as mouse ESCs) does not have obvious phonotypes, H3.3 and its chaperones are essential for development, as either H3.3 or ATRX/DAXX knockout mice are embryonic lethal. Recent discoveries of H3.3 mutations in human cancers further reveal the important role of H3.3 in pathological processes. Nevertheless, it is still unclear how H3.3 is precisely regulated during both normal and neoplastic development. As both K27M and K36M directly affect histone PTMs that are associated with gene activity, it is likely these mutations contribute to tumor development through either activation of oncogenes or repression of essential tumor suppressor genes. Given that the heterochromatin-specific H3.3 chaperones ATRX and DAXX are also frequently mutated in human cancers, including the types in which H3.3 is mutated, it is also possible that mutations in H3.3 genes lead to destruction of higher order chromatin conformation and loss of chromosome integrity. Since the first discovery of histone mutations in cancer just a few years ago, much progress has been made in our understanding of the molecular mechanisms of histone mutations in the regulation of chromatin and gene expression. Future studies will soon unravel new insights into this old histone variant in human health and disease.
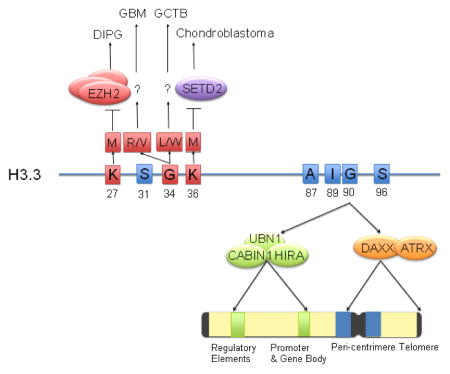
Figure 1. Protein sequence, genomic distributions, and cancer mutations of H3.3.

Amino acids in H3.3 that are distinct from canonical H3 are shown in blue squares and highlighted with residue numbers. G90 is an essential amino acid recognized by UBN1 in the HIRA complex or DAXX in the DAXX/ATRX, the two known variant-specific chaperones that deposit H3.3 into distinct chromosomal regions that is highlighted in different colors in the schematic representation of chromosome. Amino acids that are mutated in human cancers are shown in red squares and highlighted with residue numbers. Mutation of K27M or K36M inhibits the enzymatic activity of EZH2 or SETD2, respectively, leading to global loss of H3K27 methylation in DIPG or H3K36 methylation in chondroblastoma. The mechanism of G34 mutations to various amino acids is current not clear. Abbreviations: A: alanine; G: glycine; I: isoleucine; K: lysine; L: leucine; M: methionine; R: arginine; S: serine; DIPG: diffuse intrinsic pontine glioma; GCTB: giant cell tumors of the bone; GBM: glioblastoma.
Research highlight.
Here we discuss the current knowledge of H3.3 in transcriptional regulation and the recent discoveries and molecular mechanisms of H3.3 mutations in human cancer.
Acknowledgments
We apologize to researchers whose papers are not cited here because of space constraints. We thank Briana Dennehey for critical reading of this paper. This work is supported in part by grants from NIH/NCI (1R01CA204020), Cancer Prevention and Research Institute of Texas (RP160237 and RP140323), American Cancer Society (RSG-13-290-01-TBE), and Welch Foundation (G1719) to X.S. X.S. is a recipient of Leukaemia & Lymphoma Society Career Development Award and a R. Lee Clark Fellow and Faculty Scholar of MD. Anderson Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 angstrom resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 3.Hake SB, Allis CD. Histone H3 variants and their potential role in indexing mammalian genomes: The “H3 barcode hypothesis”. P Natl Acad Sci USA. 2006;103:6428–35. doi: 10.1073/pnas.0600803103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Research. 2011;21:421–34. doi: 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skene PJ, Henikoff S. Histone variants in pluripotency and disease. Development (Cambridge, England) 2013;140:2513–24. doi: 10.1242/dev.091439. [DOI] [PubMed] [Google Scholar]
- 6.Marzluff WF, Gongidi P, Woods KR, Jin J, Maltais LJ. The human and mouse replication-dependent histone genes. Genomics. 2002;80:487–98. [PubMed] [Google Scholar]
- 7.Akhmanova AS, Bindels PC, Xu J, Miedema K, Kremer H, Hennig W. Structure and expression of histone H3.3 genes in Drosophila melanogaster and Drosophila hydei. Genome / National Research Council Canada = Genome / Conseil national de recherches Canada. 1995;38:586–600. doi: 10.1139/g95-075. [DOI] [PubMed] [Google Scholar]
- 8.Frank D, Doenecke D, Albig W. Differential expression of human replacement and cell cycle dependent H3 histone genes. Gene. 2003;312:135–43. doi: 10.1016/s0378-1119(03)00609-7. [DOI] [PubMed] [Google Scholar]
- 9.Krimer DB, Cheng G, Skoultchi AI. Induction of H3.3 replacement histone mRNAs during the precommitment period of murine erythroleukemia cell differentiation. Nucleic acids research. 1993;21:2873–9. doi: 10.1093/nar/21.12.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okada T, Endo M, Singh MB, Bhalla PL. Analysis of the histone H3 gene family in Arabidopsis and identification of the male-gamete-specific variant AtMGH3. The Plant Journal. 2005;44:557–68. doi: 10.1111/j.1365-313X.2005.02554.x. [DOI] [PubMed] [Google Scholar]
- 11.Castiglia D, Cestelli A, Scaturro M, Nastasi T, Di Liegro I. H1(0) and H3.3B mRNA levels in developing rat brain. Neurochemical research. 1994;19:1531–7. doi: 10.1007/BF00969002. [DOI] [PubMed] [Google Scholar]
- 12.Wu RS, Tsai S, Bonner WM. Patterns of histone variant synthesis can distinguish G0 from G1 cells. Cell. 1982;31:367–74. doi: 10.1016/0092-8674(82)90130-1. [DOI] [PubMed] [Google Scholar]
- 13.Lyons SM, Cunningham CH, Welch JD, Groh B, Guo AY, Wei B, et al. A subset of replication-dependent histone mRNAs are expressed as polyadenylated RNAs in terminally differentiated tissues. Nucleic acids research. 2016;44:9190–205. doi: 10.1093/nar/gkw620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu CP, Xiong C, Wang M, Yu Z, Yang N, Chen P, et al. Structure of the variant histone H3.3-H4 heterodimer in complex with its chaperone DAXX. Nature structural & molecular biology. 2012;19:1287–92. doi: 10.1038/nsmb.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elsasser SJ, Huang H, Lewis PW, Chin JW, Allis CD, Patel DJ. DAXX envelops a histone H3.3-H4 dimer for H3.3-specific recognition. Nature. 2012;491:560–5. doi: 10.1038/nature11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricketts MD, Frederick B, Hoff H, Tang Y, Schultz DC, Rai TS, et al. Ubinuclein-1 confers histone H3.3-specific-binding by the HIRA histone chaperone complex. Nat Commun. 2015:6. doi: 10.1038/ncomms8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulmeister A, Schmid M, Thompson EM. Phosphorylation of the histone H3.3 variant in mitosis and meiosis of the urochordate Oikopleura dioica. Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology. 2007;15:189–201. doi: 10.1007/s10577-006-1112-z. [DOI] [PubMed] [Google Scholar]
- 18.Hake SB, Garcia BA, Kauer M, Baker SP, Shabanowitz J, Hunt DF, et al. Serine 31 phosphorylation of histone variant H3.3 is specific to regions bordering centromeres in metaphase chromosomes. Proc Natl Acad Sci U S A. 2005;102:6344–9. doi: 10.1073/pnas.0502413102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang FT, Chan FL, JD RM, Udugama M, Mayne L, Collas P, et al. CHK1-driven histone H3.3 serine 31 phosphorylation is important for chromatin maintenance and cell survival in human ALT cancer cells. Nucleic acids research. 2015;43:2603–14. doi: 10.1093/nar/gkv104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das C, Tyler JK, Churchill MEA. The histone shuffle: histone chaperones in an energetic dance. Trends in Biochemical Sciences. 2010;35:476–89. doi: 10.1016/j.tibs.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaufman PD, Kobayashi R, Kessler N, Stillman B. The p150 and p60 subunits of chromatin assembly factor I: a molecular link between newly synthesized histones and DNA replication. Cell. 1995;81:1105–14. doi: 10.1016/s0092-8674(05)80015-7. [DOI] [PubMed] [Google Scholar]
- 22.Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell. 1996;87:95–104. doi: 10.1016/s0092-8674(00)81326-4. [DOI] [PubMed] [Google Scholar]
- 23.Lorain S, Quivy JP, Monier-Gavelle F, Scamps C, Lecluse Y, Almouzni G, et al. Core histones and HIRIP3, a novel histone-binding protein, directly interact with WD repeat protein HIRA. Molecular and cellular biology. 1998;18:5546–56. doi: 10.1128/mcb.18.9.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- 25.Tang Y, Puri A, Ricketts MD, Rai TS, Hoffmann J, Hoi E, et al. Identification of an ubinuclein 1 region required for stability and function of the human HIRA/UBN1/CABIN1/ASF1a histone H3.3 chaperone complex. Biochemistry. 2012;51:2366–77. doi: 10.1021/bi300050b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green EM, Antczak AJ, Bailey AO, Franco AA, Wu KJ, Yates JR, 3rd, et al. Replication-independent histone deposition by the HIR complex and Asf1. Current biology : CB. 2005;15:2044–9. doi: 10.1016/j.cub.2005.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenall A, Williams ES, Martin KA, Palmer JM, Gray J, Liu C, et al. Hip3 interacts with the HIRA proteins Hip1 and Slm9 and is required for transcriptional silencing and accurate chromosome segregation. The Journal of biological chemistry. 2006;281:8732–9. doi: 10.1074/jbc.M512170200. [DOI] [PubMed] [Google Scholar]
- 28.Anderson HE, Kagansky A, Wardle J, Rappsilber J, Allshire RC, Whitehall SK. Silencing mediated by the Schizosaccharomyces pombe HIRA complex is dependent upon the Hpc2-like protein, Hip4. PloS one. 2010;5:e13488. doi: 10.1371/journal.pone.0013488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirov N, Shtilbans A, Rushlow C. Isolation and characterization of a new gene encoding a member of the HIRA family of proteins from Drosophila melanogaster. Gene. 1998;212:323–32. doi: 10.1016/s0378-1119(98)00143-7. [DOI] [PubMed] [Google Scholar]
- 30.Orsi GA, Algazeery A, Meyer RE, Capri M, Sapey-Triomphe LM, Horard B, et al. Drosophila Yemanuclein and HIRA cooperate for de novo assembly of H3.3-containing nucleosomes in the male pronucleus. PLoS genetics. 2013;9:e1003285. doi: 10.1371/journal.pgen.1003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nie X, Wang H, Li J, Holec S, Berger F. The HIRA complex that deposits the histone H3.3 is conserved in Arabidopsis and facilitates transcriptional dynamics. Biology open. 2014 doi: 10.1242/bio.20148680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–91. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ray-Gallet D, Woolfe A, Vassias I, Pellentz C, Lacoste N, Puri A, et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol Cell. 2011;44:928–41. doi: 10.1016/j.molcel.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 34.Banaszynski LA, Wen D, Dewell S, Whitcomb SJ, Lin M, Diaz N, et al. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell. 2013;155:107–20. doi: 10.1016/j.cell.2013.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adam S, Polo SE, Almouzni G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell. 2013;155:94–106. doi: 10.1016/j.cell.2013.08.029. [DOI] [PubMed] [Google Scholar]
- 36.van der Heijden GW, Dieker JW, Derijck AA, Muller S, Berden JH, Braat DD, et al. Asymmetry in histone H3 variants and lysine methylation between paternal and maternal chromatin of the early mouse zygote. Mechanisms of development. 2005;122:1008–22. doi: 10.1016/j.mod.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 37.Loppin B, Bonnefoy E, Anselme C, Laurencon A, Karr TL, Couble P. The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature. 2005;437:1386–90. doi: 10.1038/nature04059. [DOI] [PubMed] [Google Scholar]
- 38.Lin CJ, Koh FM, Wong P, Conti M, Ramalho-Santos M. Hira-mediated H3.3 incorporation is required for DNA replication and ribosomal RNA transcription in the mouse zygote. Developmental cell. 2014;30:268–79. doi: 10.1016/j.devcel.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inoue A, Zhang Y. Nucleosome assembly is required for nuclear pore complex assembly in mouse zygotes. Nature structural & molecular biology. 2014;21:609–16. doi: 10.1038/nsmb.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nashun B, Hill PWS, Smallwood SA, Dharmalingam G, Amouroux R, Clark SJ, et al. Continuous Histone Replacement by Hira Is Essential for Normal Transcriptional Regulation and De Novo DNA Methylation during Mouse Oogenesis. Molecular Cell. 2015;60:611–25. doi: 10.1016/j.molcel.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts C, Sutherland HF, Farmer H, Kimber W, Halford S, Carey A, et al. Targeted mutagenesis of the Hira gene results in gastrulation defects and patterning abnormalities of mesoendodermal derivatives prior to early embryonic lethality. Molecular and cellular biology. 2002;22:2318–28. doi: 10.1128/MCB.22.7.2318-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonnefoy E, Orsi GA, Couble P, Loppin B. The essential role of Drosophila HIRA for de novo assembly of paternal chromatin at fertilization. PLoS genetics. 2007;3:1991–2006. doi: 10.1371/journal.pgen.0030182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang X, Khosravi-Far R, Chang HY, Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell. 1997;89:1067–76. doi: 10.1016/s0092-8674(00)80294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes & development. 2010;24:1253–65. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A. 2010;107:14075–80. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elsaesser SJ, Allis CD. HIRA and Daxx constitute two independent histone H3.3-containing predeposition complexes. Cold Spring Harbor symposia on quantitative biology. 2010;75:27–34. doi: 10.1101/sqb.2010.75.008. [DOI] [PubMed] [Google Scholar]
- 47.He QY, Kim H, Huang R, Lu WS, Tang MF, Shi FT, et al. The Daxx/Atrx Complex Protects Tandem Repetitive Elements during DNA Hypomethylation by Promoting H3K9 Trimethylation. Cell Stem Cell. 2015;17:273–86. doi: 10.1016/j.stem.2015.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sadic D, Schmidt K, Groh S, Kondofersky I, Ellwart J, Fuchs C, et al. Atrx promotes heterochromatin formation at retrotransposons. Embo Reports. 2015;16:836–50. doi: 10.15252/embr.201439937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Voon HPJ, Hughes JR, Rode C, De La Rosa-Velazquez IA, Jenuwein T, Feil R, et al. ATRX Plays a Key Role in Maintaining Silencing at Interstitial Heterochromatic Loci and Imprinted Genes. Cell Rep. 2015;11:405–18. doi: 10.1016/j.celrep.2015.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elsasser SJ, Noh KM, Diaz N, Allis CD, Banaszynski LA. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature. 2015;522:240–U323. doi: 10.1038/nature14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delbarre E, Ivanauskiene K, Kuntziger T, Collas P. DAXX-dependent supply of soluble (H3.3–H4) dimers to PML bodies pending deposition into chromatin. Genome research. 2013;23:440–51. doi: 10.1101/gr.142703.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michaelson JS, Bader D, Kuo F, Kozak C, Leder P. Loss of Daxx, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes & development. 1999;13:1918–23. doi: 10.1101/gad.13.15.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hollenbach AD, McPherson CJ, Mientjes EJ, Iyengar R, Grosveld G. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. Journal of cell science. 2002;115:3319–30. doi: 10.1242/jcs.115.16.3319. [DOI] [PubMed] [Google Scholar]
- 54.Sawatsubashi S, Murata T, Lim J, Fujiki R, Ito S, Suzuki E, et al. A histone chaperone, DEK, transcriptionally coactivates a nuclear receptor. Genes & development. 2010;24:159–70. doi: 10.1101/gad.1857410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ivanauskiene K, Delbarre E, McGhie JD, Kuntziger T, Wong LH, Collas P. The PML-associated protein DEK regulates the balance of H3.3 loading on chromatin and is important for telomere integrity. Genome research. 2014;24:1584–94. doi: 10.1101/gr.173831.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Corpet A, Olbrich T, Gwerder M, Fink D, Stucki M. Dynamics of histone H3.3 deposition in proliferating and senescent cells reveals a DAXX-dependent targeting to PML-NBs important for pericentromeric heterochromatin organization. Cell cycle (Georgetown, Tex) 2014;13:249–67. doi: 10.4161/cc.26988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lusser A, Urwin DL, Kadonaga JT. Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nature structural & molecular biology. 2005;12:160–6. doi: 10.1038/nsmb884. [DOI] [PubMed] [Google Scholar]
- 58.Konev AY, Tribus M, Park SY, Podhraski V, Lim CY, Emelyanov AV, et al. CHD1 motor protein is required for deposition of histone variant h3.3 into chromatin in vivo. Science. 2007;317:1087–90. doi: 10.1126/science.1145339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harada A, Okada S, Konno D, Odawara J, Yoshimi T, Yoshimura S, et al. Chd2 interacts with H3.3 to determine myogenic cell fate. The EMBO journal. 2012;31:2994–3007. doi: 10.1038/emboj.2012.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shimojima T, Okada M, Nakayama T, Ueda H, Okawa K, Iwamatsu A, et al. Drosophila FACT contributes to Hox gene expression through physical and functional interactions with GAGA factor. Genes & development. 2003;17:1605–16. doi: 10.1101/gad.1086803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakayama T, Nishioka K, Dong YX, Shimojima T, Hirose S. Drosophila GAGA factor directs histone H3.3 replacement that prevents the heterochromatin spreading. Genes & development. 2007;21:552–61. doi: 10.1101/gad.1503407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakayama T, Shimojima T, Hirose S. The PBAP remodeling complex is required for histone H3.3 replacement at chromatin boundaries and for boundary functions. Development (Cambridge, England) 2012;139:4582–90. doi: 10.1242/dev.083246. [DOI] [PubMed] [Google Scholar]
- 63.Pradhan SK, Su T, Yen LD, Jacquet K, Huang CY, Cote J, et al. EP400 Deposits H3.3 into Promoters and Enhancers during Gene Activation. Molecular Cell. 2016;61:27–38. doi: 10.1016/j.molcel.2015.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, et al. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol Cell. 2014;56:298–310. doi: 10.1016/j.molcel.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014;508:263–8. doi: 10.1038/nature13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hateboer G, Gennissen A, Ramos YF, Kerkhoven RM, Sonntag-Buck V, Stunnenberg HG, et al. BS69, a novel adenovirus E1A-associated protein that inhibits E1A transactivation. The EMBO journal. 1995;14:3159–69. doi: 10.1002/j.1460-2075.1995.tb07318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ladendorff NE, Wu S, Lipsick JS. BS69, an adenovirus E1A-associated protein, inhibits the transcriptional activity of c-Myb. Oncogene. 2001;20:125–32. doi: 10.1038/sj.onc.1204048. [DOI] [PubMed] [Google Scholar]
- 68.Masselink H, Bernards R. The adenovirus E1A binding protein BS69 is a corepressor of transcription through recruitment of N-CoR. Oncogene. 2000;19:1538–46. doi: 10.1038/sj.onc.1203421. [DOI] [PubMed] [Google Scholar]
- 69.Velasco G, Grkovic S, Ansieau S. New insights into BS69 functions. The Journal of biological chemistry. 2006;281:16546–50. doi: 10.1074/jbc.M600573200. [DOI] [PubMed] [Google Scholar]
- 70.Wen H, Li YY, Xi YX, Jiang SM, Stratton S, Peng DN, et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014;508:263. doi: 10.1038/nature13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qin S, Min J. Structure and function of the nucleosome-binding PWWP domain. Trends Biochem Sci. 2014;39:536–47. doi: 10.1016/j.tibs.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 72.Hake SB, Garcia BA, Duncan EM, Kauer M, Dellaire G, Shabanowitz J, et al. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. The Journal of biological chemistry. 2006;281:559–68. doi: 10.1074/jbc.M509266200. [DOI] [PubMed] [Google Scholar]
- 73.de Almeida SF, Grosso AR, Koch F, Fenouil R, Carvalho S, Andrade J, et al. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nature structural & molecular biology. 2011;18:977–U1501. doi: 10.1038/nsmb.2123. [DOI] [PubMed] [Google Scholar]
- 74.Kim S, Kim H, Fong N, Erickson B, Bentley DL. Pre-mRNA splicing is a determinant of histone H3K36 methylation. P Natl Acad Sci USA. 2011;108:13564–9. doi: 10.1073/pnas.1109475108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nature Genetics. 2009;41:376–81. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen P, Zhao J, Wang Y, Wang M, Long H, Liang D, et al. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes & development. 2013;27:2109–24. doi: 10.1101/gad.222174.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thakar A, Gupta P, Ishibashi T, Finn R, Silva-Moreno B, Uchiyama S, et al. H2A.Z and H3.3 histone variants affect nucleosome structure: biochemical and biophysical studies. Biochemistry. 2009;48:10852–7. doi: 10.1021/bi901129e. [DOI] [PubMed] [Google Scholar]
- 78.Braunschweig U, Hogan GJ, Pagie L, van Steensel B. Histone H1 binding is inhibited by histone variant H3.3. The EMBO journal. 2009;28:3635–45. doi: 10.1038/emboj.2009.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Torres-Padilla ME, Bannister AJ, Hurd PJ, Kouzarides T, Zernicka-Goetz M. Dynamic distribution of the replacement histone variant H3.3 in the mouse oocyte and preimplantation embryos. The International journal of developmental biology. 2006;50:455–61. doi: 10.1387/ijdb.052073mt. [DOI] [PubMed] [Google Scholar]
- 80.Suto RK, Clarkson MJ, Tremethick DJ, Luger K. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat Struct Biol. 2000;7:1121–4. doi: 10.1038/81971. [DOI] [PubMed] [Google Scholar]
- 81.Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes & development. 2007;21:1519–29. doi: 10.1101/gad.1547707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, et al. H3.3/H2A.Z double variant-containing nucleosomes mark 'nucleosome-free regions' of active promoters and other regulatory regions. Nat Genet. 2009;41:941–5. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schneiderman JI, Orsi GA, Hughes KT, Loppin B, Ahmad K. Nucleosome-depleted chromatin gaps recruit assembly factors for the H3.3 histone variant. Proc Natl Acad Sci U S A. 2012;109:19721–6. doi: 10.1073/pnas.1206629109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wollmann H, Holec S, Alden K, Clarke ND, Jacques PE, Berger F. Dynamic deposition of histone variant H3.3 accompanies developmental remodeling of the Arabidopsis transcriptome. PLoS genetics. 2012;8:e1002658. doi: 10.1371/journal.pgen.1002658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stroud H, Otero S, Desvoyes B, Ramírez-Parra E, Jacobsen SE, Gutierrez C. Genome-wide analysis of histone H3.1 and H3.3 variants in Arabidopsis thaliana. Proceedings of the National Academy of Sciences. 2012;109:5370–5. doi: 10.1073/pnas.1203145109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shu H, Nakamura M, Siretskiy A, Borghi L, Moraes I, Wildhaber T, et al. Arabidopsis replacement histone variant H3.3 occupies promoters of regulated genes. Genome biology. 2014;15:R62. doi: 10.1186/gb-2014-15-4-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ. Dynamics of replication-independent histone turnover in budding yeast. Science. 2007;315:1405–8. doi: 10.1126/science.1134053. [DOI] [PubMed] [Google Scholar]
- 88.Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell. 2007;27:393–405. doi: 10.1016/j.molcel.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 89.Sakai A, Schwartz BE, Goldstein S, Ahmad K. Transcriptional and developmental functions of the H3.3 histone variant in Drosophila. Current biology : CB. 2009;19:1816–20. doi: 10.1016/j.cub.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kraushaar DC, Jin W, Maunakea A, Abraham B, Ha M, Zhao K. Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome biology. 2013;14:R121. doi: 10.1186/gb-2013-14-10-r121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet. 2005;37:1090–7. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- 92.Tamura T, Smith M, Kanno T, Dasenbrock H, Nishiyama A, Ozato K. Inducible deposition of the histone variant H3.3 in interferon-stimulated genes. The Journal of biological chemistry. 2009;284:12217–25. doi: 10.1074/jbc.M805651200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jin C, Felsenfeld G. Distribution of histone H3.3 in hematopoietic cell lineages. Proc Natl Acad Sci U S A. 2006;103:574–9. doi: 10.1073/pnas.0509974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ng RK, Gurdon JB. Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nature cell biology. 2008;10:102–9. doi: 10.1038/ncb1674. [DOI] [PubMed] [Google Scholar]
- 95.Jullien J, Astrand C, Szenker E, Garrett N, Almouzni G, Gurdon JB. HIRA dependent H3.3 deposition is required for transcriptional reprogramming following nuclear transfer to Xenopus oocytes. Epigenetics & chromatin. 2012;5:17. doi: 10.1186/1756-8935-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kallappagoudar S, Yadav RK, Lowe BR, Partridge JF. Histone H3 mutations-a special role for H3. 3 in tumorigenesis? Chromosoma. 2015;124:177–89. doi: 10.1007/s00412-015-0510-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lindroth AM, Plass C. Recurrent H3.3 alterations in childhood tumors. Nature Genetics. 2013;45:1413–4. doi: 10.1038/ng.2832. [DOI] [PubMed] [Google Scholar]
- 98.Yuen BTK, Knoepfler PS. Histone H3.3 Mutations: A Variant Path to Cancer. Cancer Cell. 2013;24:567–74. doi: 10.1016/j.ccr.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 100.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–3. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–47. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–37. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 103.Gessi M, Gielen GH, Hammes J, Dorner E, zur Muhlen A, Waha A, et al. H3. 3 G34R mutations in pediatric primitive neuroectodermal tumors of central nervous system (CNS-PNET) and pediatric glioblastomas: possible diagnostic and therapeutic implications? J Neuro-Oncol. 2013;112:67–72. doi: 10.1007/s11060-012-1040-z. [DOI] [PubMed] [Google Scholar]
- 104.Heaphy CM, de Wilde RF, Jiao YC, Klein AP, Edil BH, Shi CJ, et al. Altered Telomeres in Tumors with ATRX and DAXX Mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lovejoy CA, Li WD, Reisenweber S, Thongthip S, Bruno J, de Lange T, et al. Loss of ATRX, Genome Instability, and an Altered DNA Damage Response Are Hallmarks of the Alternative Lengthening of Telomeres Pathway. PLoS genetics. 2012:8. doi: 10.1371/journal.pgen.1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nature Genetics. 2014;46:451–6. doi: 10.1038/ng.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fisett PO, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nature Genetics. 2014;46:462–6. doi: 10.1038/ng.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Taylor K, Mackay A, Truffaux N, Morozova O, Butterfield Y, Phillipe C, et al. Recurrent Activating Acvr1/Alk2 Mutations in Diffuse Intrinsic Pontine Glioma. Neuro-Oncology. 2013;15:174–5. [Google Scholar]
- 109.Wu G, Diaz AK, Paugh BS, Rankin SL, Ju BS, Li YJ, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nature Genetics. 2014;46:444–50. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–82. doi: 10.1038/ng.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cleven AHG, Hocker S, Briaire-de Bruijn I, Szuhai K, Cleton-Jansen AM, Bovee JVMG. Mutation Analysis of H3F3A and H3F3B as a Diagnostic Tool for Giant Cell Tumor of Bone and Chondroblastoma. Am J Surg Pathol. 2015;39:1576–83. doi: 10.1097/PAS.0000000000000512. [DOI] [PubMed] [Google Scholar]
- 112.Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–61. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes & development. 2013;27:985–90. doi: 10.1101/gad.217778.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jiao LY, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science. 2015:350. doi: 10.1126/science.aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Justin N, Zhang Y, Tarricone C, Martin SR, Chen SY, Underwood E, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 2016:7. doi: 10.1038/ncomms11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Herz HM, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, et al. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science. 2014;345:1065–70. doi: 10.1126/science.1255104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DTW, Kool M, et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell. 2013;24:660–72. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 118.Yang S, Zheng X, Lu C, Li GM, Allis CD, Li H. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes & development. 2016;30:1611–6. doi: 10.1101/gad.284323.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. 2016;352:844–9. doi: 10.1126/science.aac7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fang D, Gan H, Lee J-H, Han J, Wang Z, Riester SM, et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science. 2016 doi: 10.1126/science.aae0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–92. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 122.Li J, Moazed D, Gygi SP. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. The Journal of biological chemistry. 2002;277:49383–8. doi: 10.1074/jbc.M209294200. [DOI] [PubMed] [Google Scholar]
- 123.Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, et al. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer discovery. 2013;3:512–9. doi: 10.1158/2159-8290.CD-12-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]