

Abstract
Accumulating evidence has shown that long noncoding RNAs (lncRNAs) are significant regulators of multiple cellular processes, including the development of chronic myelocytic leukemia (CML). However, the mechanism of how the lncRNA PLIN2 affects CML development remains unclear. In this study, we aimed to investigate the potential roles of CEBPA-mediated upregulation of PLIN2 in the process of CML development by regulating the GSK3 and Wnt/β-catenin signaling pathways. We found that both CEBPA and PLIN2 were expressed at significantly higher levels in CML. Simultaneously, we found that CEBPA upregulated the expression of PLIN2 and that there was a positive correlation between CEBPA and PLIN2 in CML patients. CEBPA promoted the progression of CML by upregulating PLIN2. We also found that PLIN2 increased the expression levels of AKT, p-AKT, GSK-3β, β-catenin and Axin2/Conductin as well as promoted the progression of CML via the GSK3 and Wnt/β-catenin signaling pathways in vitro. Furthermore, we found that CEBPA-mediated upregulation of PLIN2 expression promotes tumor growth via GSK3 and Wnt/β-catenin signaling in vivo. Therefore, our study provided a new theoretical basis for CML treatment through the CEBPA/PLIN2 axis.
Keywords: CEBPA, lncRNA PLIN2, chronic myelogenous leukemia
Introduction
Chronic myeloid leukemia (CML) is a hematopoietic disorder that leads to the accumulation of immature white blood cells in the bone marrow and peripheral blood [1-6]. The etiology of CML is a reciprocal translocation between chromosomes 9 and 22, which generates the BCR-ABL oncogene that encodes a chimeric oncoprotein with constitutive tyrosine kinase activity. In addition, genetic changes contribute to the malignant transformation driven by Bcr-Abl [7] and cause increases in proliferation ability, changes in cell adhesion, inhibition of apoptosis and strengthening of bone marrow angiogenesis during the progression of the disease [8]. However, effective therapeutic targets have still not been found for CML. At present, some side-effects, such as diarrhea and allergic reactions, are still serious problems in the clinical management of CML patients [9]. Therefore, a better understanding of the molecular mechanism responsible for CML development and progression as well as improved therapeutic methods are extremely urgent.
An increasing number of studies has found that noncoding RNAs (ncRNAs), which include long noncoding RNA (lncRNA), microRNA (miRNA), circular RNA (circRNA), small nucleolar RNA, and ribosomal RNA, play critical roles in tumor progression by regulating gene expression [10,11]. Among them, lncRNAs are a recently discovered class of non-coding RNAs longer than 200 nucleotides [12]. Many studies have indicated that lncRNAs regulate gene expression via transcriptional regulation, post-transcriptional regulation, chromatin modifications and genomic imprinting [13,14]. Furthermore, accumulating evidence has consistently indicated that lncRNAs exert specific effects in a variety of cellular processes and therefore may contribute to the development of cancer and other human diseases [15,16]. However, the role of the lncRNA PLIN2 in CML progression is still unknown.
CCAAT/enhancer-binding protein-α (CEBPA), which is a key regulator of myeloid differentiation, regulates many protein-coding genes [17-19]. Recent research has expanded the analysis of CEBPA transcriptional targets to the newly identified class of long non-coding RNAs and found that CEBPA upregulated the expression of PLIN2 [20]. In the present study, we analyzed the expression levels of PLIN2 in human CML cell lines and blood samples from patients as well as explored the effects of PLIN2 on the progression and prognosis of CML. In addition, we also explored the role of the CEBPA/PLIN2 axis on CML proliferation and apoptosis.
Materials and methods
Clinical specimens
Ninety-six patients with CML and ninety-six healthy donors were included in this study. Patients with CML and healthy volunteers were recruited from 2014 to 2016 at Yantai Yuhuangding Hospital affiliated with Qingdao University Medical College. Informed consent was signed by the participants, and this study was approved by the ethics committee at Yantai Yuhuangding Hospital affiliated with Qingdao University Medical College.
Cell lines
HEK293T, KT-1/A3 and K562 cells were purchased from the Institute of Biochemistry and Cell Biology at the Chinese Academy of Sciences (Shanghai, China). HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA, USA); KT-1/A3 and K562 cells were cultured in RPMI 1640 (Invitrogen, Carlsbad, CA, USA). All media were supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA), penicillin (100 U/ml), and streptomycin (100 μg/ml), and all the cell lines were cultured at 37°C in an appropriate incubator.
Lentiviral vector construction
Human CEBPA, PLIN2, GSK-3β and β-catenin DNA was amplified from K562 cells by RT-PCR. Then, the PCR products were inserted into a lentiviral vector with EGF DNA used as a control. Furthermore, we synthesized DNA fragments for shRNA and cloned the shRNAs into pBluescript SK (+) plasmid containing a human U6 promoter (pU6) after annealing. Then, the U6-shRNA cassettes were subcloned into a lentiviral vector [21,22]. shLuc was used as the control. Lentiviral vectors expressing shRNAs targeting either CEBPA or PLIN2 were generated. Then, the lentiviral vectors were packaged in HEK293T cells by co-transfecting the cells with packaging vectors (pCMV-VSVG, pMDLg/pRRE and pRSV-REV). The produced lentivirus was ultracentrifuged, concentrated, and validated.
Transduction
KT-1/A3 and K562 cells (5 × 104 cells/well) were seeded in 24-well plates and transduced with lentivirus supplemented with 8 μg/mL of polybrene (Sigma-Aldrich Chemie, The Netherlands). KT-1/A3 cells were transduced with either Lenti-GFP or Lenti-CEBPA, and K562 cells were transduced with either shLuc and shCEBPA. Additionally, KT-1/A3 cells were transduced with either Lenti-GFP or Lenti-PLIN2, and K562 cells were transduced with either shLuc or shPLIN2, respectively. G418 (Life Technologies, 0.8 mg/mL) was used to select the cell lines with stable expression.
For RNA interference, siRNAs for GSK-3β and β-catenin as well as negative control siRNAs were purchased from GenePharma (GenePharma Co., Ltd., Shanghai, China). The sequences of selected regions to be targeted by siRNAs for GSK-3β and β-catenin were as follows: GSK-3β-siRNA, 5’-CGA GGG AAC UGG UGG CCA UTT-3’ (sense) and 5’-AUG GCC ACC AGU UCC CUC GTT-3’ (antisense); and β-catenin, 5’-AGC TGA TAT TGA TGG ACA G-3’ (sense) and 5’-CAG TTG TGG TTA AGC TCT T-3’ (antisense). KT-1/A3 and K562 cells (2 × 105 cells/well in a 6-well plate) were cultured and transfected with 50 nM negative control (NC) siRNA, siGSK-3β or siβ-catenin using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Quantitative real-time reverse transcription PCR (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen, CA, USA). A RevertAid First Strand cDNA Synthesis kit (Thermo Fisher) was used to synthesize cDNAs with random primers according to the manufacturer’s protocol. The mRNA expression levels were detected using a SYBR-Green PCR Master Mix kit (Takara) in the PCR reaction. The primer sequences were as follows: GAPDH, 5’-TGT TCG TCA TGG GTG TGA AC-3’ (forward) and 5’-ATG GCA TGG ACT GTG GTC AT-3’ (reverse; internal control); CEBPA, 5’-TGG ATA AGA ACA GCA ACG AG-3’ (forward) and 5’-TCA CTG GTC AAC TCC AGC AC-3’ (reverse); PLIN2, 5’-TGC CTG TAA TCC CAG CTA CT-3’ (forward) and 5’-CCT GCC AGG GGA AAG ATC TA-3’ (reverse); GSK-3β, 5’-CGG GAT ATT AAA CCG CAG AAC CTC-3’ (forward) and 5’-TGG TGC CCT ATA GTA CCG AGA ACA G-3’ (reverse); and β-catenin, 5’-CTT ACA CCC ACC ATC CCA CT-3’ (forward) and 5’-CCT CCA CAA ATT GCT GCT GT-3’ (reverse).
Western blot analysis
Total protein was extracted from the treated KT-1/A3 and K562 cells into a radio immunoprecipitation assay (RIPA) buffer (Thermo Scientific, Rockford, IL, USA) containing a protease inhibitor cocktail (P8340; Sigma-Aldrich, St. Louis, MO, USA). The proteins were separated based on their molecular weight on SDS-PAGE gels and transferred to a PVDF membrane (PerkinElmer, Boston, MA). The PVDF membrane was blocked for 2 hrs in 5% skim milk at room temperature and probed with primary antibodies overnight at 4°C. The next day, the PVDF membrane was incubated with HRP-conjugated secondary antibodies. Signals were detected using an enhanced chemiluminescence (ECL) substrate kit (Amersham Biosciences, Inc.) and an enhanced chemiluminescence Western blotting system (ComWin Biotech, Beijing, China). The following antibodies were used in this study anti-GAPDH (1:2000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-AKT (1:1000, Cell Signaling Technology, Danvers, MA), anti-p-AKT (1:200, Cell Signaling Technology, Danvers, MA), anti-GSK-3β (1:500, Abcam, Cambridge, MA, USA), anti-β-catenin (1:500, BD Transduction Laboratories, San Jose, CA) and anti-Axin2/Conductin (1:100, Santa Cruz Biotechnology, Santa Cruz, CA).
Cell proliferation
Cell proliferation was detected using methylthiazole tetrazolium (MTT). Treated KT-1/A3 and K562 cells (3 × 103 cells/well) were cultured in 96-well plates, and 20 μL of MTT solution (5 mg/ml) was added into each well at the indicated time points. Then, the treated cells were incubated for 4 hrs at 37°C. Dimethyl sulfoxide (DMSO, 200 μL) was added to dissolve the precipitates. The absorbance value was detected at 490 nm with a microplate reader.
Flow cytometry analysis of apoptosis
The transduced KT-1/A3 and K562 cells were digested, dispersed, centrifuged, collected, washed with cold PBS and resuspended in 1 × binding buffer at a concentration of 1 × 106 cells/mL. The apoptotic cell death rate was measured by flow cytometry and double staining using phosphatidylethanolamine (PE) and an annexin-V-FITC staining kit (BD Biosciences) according to the manufacturer’s instructions. The flow cytometry results were obtained by a BD FACS Calibur cytometer (BD Biosciences) and analyzed by using FlowJo software (Tree Star, Ashland, OR). The flow cytometry results were divided into four sections: viable cells, dead cells, early stage apoptotic cells, and late stage apoptotic cells.
In vitro migration and invasion assays
In vitro invasion and migration assays were performed using Transwell chambers (pore size of 8 μm; Costar, Corning, NY). Transduced cells were resuspended in serum-free medium, and 200 μl of the cell suspension (4 × 104 cells) was added to the upper chamber. Complete medium was added to the bottom wells of the chambers. After fixing and staining the cells on the lower membrane with a dye solution containing 0.1% crystal violet and 20% methanol, the cells were counted.
Tumor formation in nude mice
KT-1/A3 cells were transduced with Lenti-GFP, Lenti-CEBPA, or Lenti-CEBPA and siPLIN2, and K562 cells transduced with shLuc, shCEBPA, or shCEBPA and PLIN2. Cells (1 × 107 cells in 100 μl) were injected into 6-week-old BALB/c athymic nude mice, which were purchased from the Shanghai LAC Laboratory Animal Co. Ltd. Tumors were excised and photographed after 40 days. Tumor volume was also measured at 10, 15, 20, 25, 30, 35, and 40 days after injection. The tumors were then homogenized for use in qRT-PCR and Western blotting. This study was approved by the Institutional Committee for Animal Research.
Statistical analysis
Statistical significance was analyzed using GraphPad (GraphPad Prism Software, La Jolla, CA, USA) and SPSS 20.0 software (variance and Student’s t-test). All data are shown as the means ± SD. A P value < 0.05 indicated statistical significance.
Results
CEBPA promotes proliferation and inhibits apoptosis of CML
To determine the effects of CEBPA on the proliferation and apoptosis of CML, qRT-PCR was first used to detect the expression levels of CEBPA in CML patients (n = 96) and healthy subjects (n = 96). The results showed that CEBPA expression was significantly higher in CML patients compared with healthy donors (P < 0.05, Figure 1A). The receiver operating characteristic (ROC) curve was used to predict the prognosis of CEBPA in CML, and the results indicated that the area under the curve (AUC) of CEBPA was 0.739 (P < 0.0001, Figure 1B), suggesting that CEBPA expression could serve as a molecular marker for CML. Second, KT-1/A3 cells were transduced with either Lenti-GFP or Lenti-CEBPA, and K562 cells were transduced with either shLuc and shCEBPA. The results indicated that CEBPA levels were successfully increased in KT-1/A3 cells (P < 0.0001, Figure 1C) and that the expression levels of CEBPA were significantly decreased in K562 cells transduced with shCEBPA than with shLuc (P < 0.0001, Figure 1D). Furthermore, the proliferative abilities of KT-1/A3 and K562 cells were detected by the MTT assay, and the results showed that overexpression of CEBPA promoted the proliferative capacity of KT-1/A3 cells (P < 0.0001, Figure 1E) and that silencing CEBPA by lentivirus inhibited the proliferative ability of K562 cells (P < 0.0001, Figure 1F). The apoptosis ability was then detected by annexin VFITC/PE staining, and we found that overexpression of CEBPA inhibited the apoptosis capacity of KT-1/A3 cells (P < 0.0001, Figure 1G) whereas silencing CEBPA by lentivirus accelerated the apoptotic ability of K562 cells (P < 0.0001, Figure 1H).
Figure 1.

CEBPA promotes the proliferation and inhibits apoptosis of CML. A. The expression levels of CEBPA were detected using qRT-PCR in CML patients (n = 96) and healthy subjects (n = 96) (***P < 0.001). B. The receiver operating characteristic curve (ROC) was used to analyze the cut-off score of CEBPA expression. C. qRT-PCR was used to detect the expression levels of CEBPA in KT-1/A3 cells transduced with either Lenti-GFP or Lenti-CEBPA (**P < 0.01). D. The mRNA expression levels of CEBPA were measured by qRT-PCR in K562 cells transduced with either shLuc or shCEBPA (**P < 0.01). E. An MTT assay was performed to detect the proliferative ability of KT-1/A3 cells transduced with either Lenti-GFP or Lenti-CEBPA (**P < 0.01). F. The proliferative ability of K562 cells transduced with either shLuc or shCEBPA was measured by the MTT assay (**P < 0.01). G. The apoptotic ability was measured by annexin V-FITC/PE staining in KT-1/A3 cells transduced with either Lenti-GFP or Lenti-CEBPA. H. The apoptotic ability was detected by annexin V-FITC/PE staining in K562 cells transduced with either shLuc or shCEBPA.
CEBPA levels do not affect cell invasion and migration in CML cells
To determine if CEBPA affects cell invasion and migration, we performed Transwell assays in KT-1/A3 cells transduced with either Lenti-GFP or Lenti-CEBPA as well as in K562 cells transduced with either shLuc or shCEBPA. The invasion and migration assays indicated that overexpression of CEBPA did not significantly influence KT-1/A3 cell invasion and migration at 48 hrs after transduction compared to the control cells (Figure 2A, 2B). Similar results were found in the K562 cells transduced with shCEBPA (Figure 2C, 2D).
Figure 2.
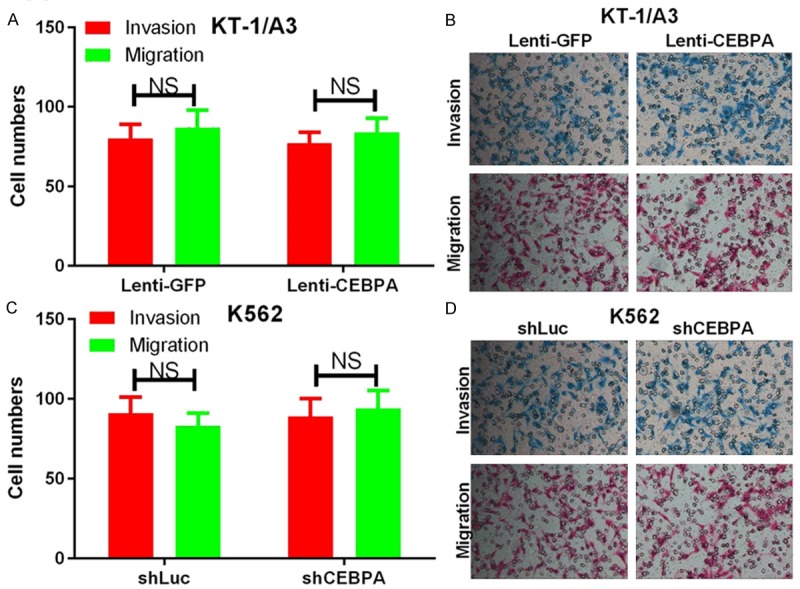
CEBPA does not affect cell invasion and migration in CML cells. A. Transwell invasion and migration assays were carried out using KT-1/A3 cells transduced with either Lenti-GFP or Lenti-CEBPA. The quantification of five randomly selected fields is shown. B. Representative images are shown. C. Transwell invasion and migration assays were carried out using K562 cells transduced with either shLuc or shCEBPA. The quantification of five randomly selected fields is shown. D. Representative images are shown. NS indicates no significance between the two groups.
The lncRNA PLIN2 accelerates proliferation and inhibits apoptosis of CML
At the same time, we explored the effects of PLIN2 on the proliferation, apoptosis, invasion and migration of CML cells. First, we detected the relative expression levels of PLIN2 using qRT-PCR in CML patients and healthy donors and found that PLIN2 was significantly expressed at higher levels in CML patients compared with healthy donors (P < 0.05, Figure 3A). The AUC of PLIN2 was 0.692 (P < 0.0001, Figure 3B), suggesting that PLIN2 expression could act as a molecular marker for CML. Then, KT-1/A3 cells were transduced with either Lenti-GFP or Lenti-PLIN2, and K562 cells were transduced with either shLuc or shPLIN2. The results indicated that PLIN2 levels were successfully increased in KT-1/A3 cells (P < 0.0001, Figure 3C) and that the expression of PLIN2 was significantly decreased in K562 cells transduced with shCEBPA than with shLuc (P < 0.0001, Figure 3D). Furthermore, the proliferative abilities of KT-1/A3 and K562 cells were detected by the MTT assay, and the results revealed that overexpression of PLIN2 promoted the proliferative capacity of KT-1/A3 cells (P < 0.0001, Figure 3E) but that silencing PLIN2 inhibited the proliferative ability of K562 cells (P < 0.0001, Figure 3F). The apoptotic abilities of KT-1/A3 and K562 cells were then detected by annexin V-FITC/PE staining, and we found that overexpression of PLIN2 inhibited the apoptotic capacity of KT-1/A3 cells (P < 0.0001, Figure 3G) but that silencing PLIN2 using lentivirus accelerated K562 cell apoptosis (P < 0.0001, Figure 3H). Simultaneously, we also observed that PLIN2 does not affect cell invasion and migration of CML cells (Figure 4A-D).
Figure 3.

The lncRNA PLIN2 accelerates the proliferation and inhibits apoptosis of CML. A. Relative PLIN2 expression levels were detected by qRT-PCR in CML patients and healthy subjects (***P < 0.001). B. The receiver operating characteristic (ROC) curve predicted the prognosis of PLIN2 in CML, and the area under the ROC curves of PLIN2 was 0.692 (P < 0.0001). C. PLIN2 levels were examined by qRT-PCR in KT-1/A3 cells transduced with either Lenti-GFP or Lenti-PLIN2 (**P < 0.01). D. The expression levels of PLIN2 were measured by qRT-PCR in K562 cells transduced with either shLuc or shPLIN2 (**P < 0.01). E. MTT assays revealed the cell growth curves of the transduced KT-1/A3 cells (***P < 0.001). F. MTT assays revealed the cell growth curves of the transduced K562 cells (***P < 0.001). G. The apoptotic ability was measured by annexin V-FITC/PE staining in transduced KT-1/A3 cells. H. The apoptotic ability was detected by annexin V-FITC/PE staining in transduced K562 cells.
Figure 4.
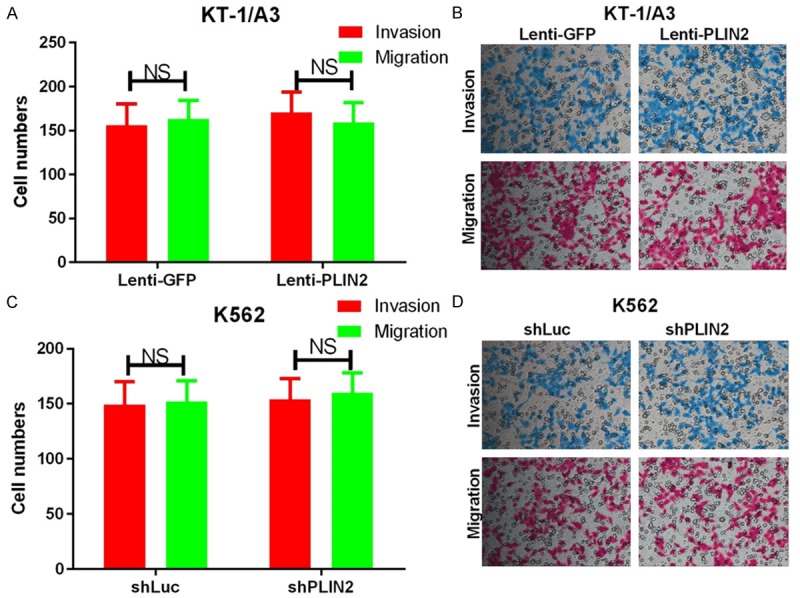
PLIN2 does not affect cell invasion and migration in CML cells. A. Transwell invasion and migration assays were carried out using KT-1/A3 cells transduced with either Lenti-GFP or Lenti-CEBPA. The quantification of five randomly selected fields is shown. B. Representative images are shown. C. Transwell invasion and migration assays were carried out using K562 cells transduced with either shLuc or shCEBPA. The quantification of five randomly selected fields is shown. D. Representative images are shown. NS indicates no significance between the two groups.
CEBPA promotes the progression of CML by upregulating PLIN2
In addition, we analyzed the correlation between CEBPA and PLIN2 using Pearson’s correlation algorithm and found that there was a positive correlation between CEBPA and PLIN2 in CML patients (R2 = 0.372, P < 0.001, Figure 5A). We then detected the influence of CEBPA on the expression of PLIN2 in KT-1/A3 and K562 cells and found that the mRNA expression levels of PLIN2 were significantly increased in KT-1/A3 cells transduced with Lenti-CEBPA compared with those transduced with Lenti-GFP (P < 0.0001, Figure 5B), while the mRNA expression levels of PLIN2 were significantly decreased in K562 cells transduced with shCEBPA compared with those transduced with shLuc (P < 0.0001, Figure 5C). To further study the functions of CEBPA-mediated upregulation of PLIN2 in CML, MTT assays were performed, and we found that CEBPA increased KT-1/A3 cell proliferation and that siPLIN2 could rescue this phenotype (P < 0.001, Figure 5D). Additionally, silencing CEBPA decreased KT-1/A3 cell proliferation, and PLIN2 could rescue this loss of function (P < 0.001, Figure 5E). At the same time, cell apoptosis was determined by flow cytometry analysis with annexin V-FITC and PE staining. The results revealed that siPLIN2 could rescue CEBPA-induced inhibition of apoptosis in KT-1/A3 cells (Figure 5F), while PLIN2 could rescue shCEBPA-induced apoptosis of K562 cells (Figure 5G).
Figure 5.
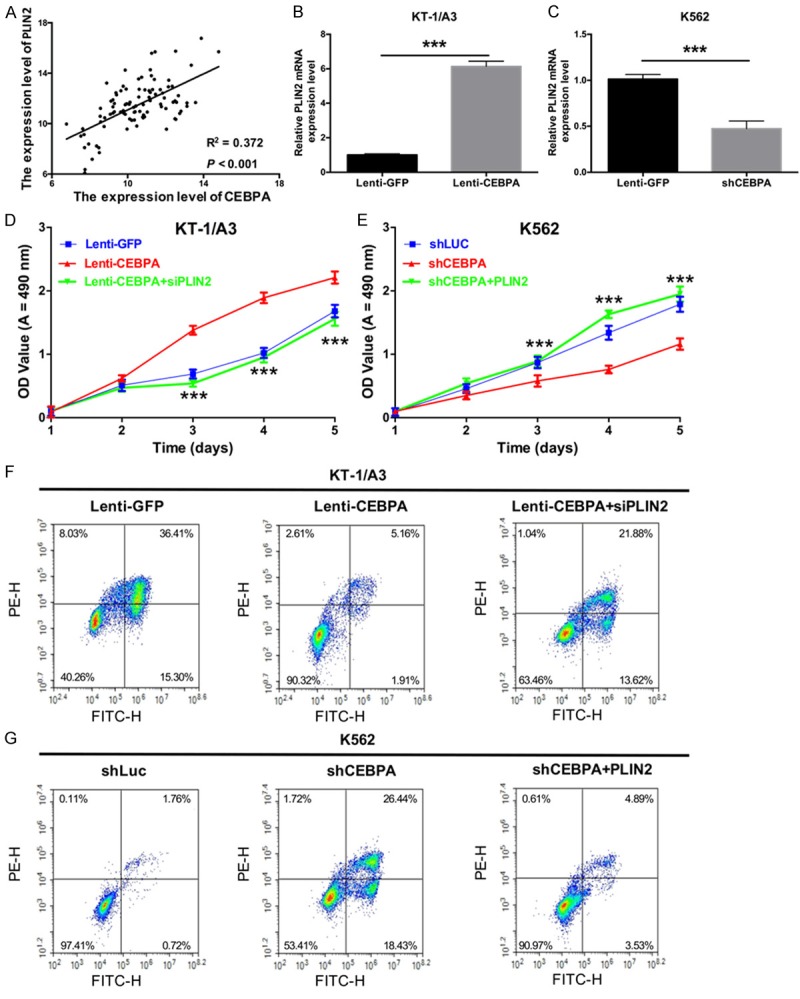
CEBPA promotes the progression of CML by upregulating PLIN2. A. The correlation between CEBPA and PLIN2 expression was in CML patients analyzed by Pearson’s correlation (n = 96, R2 = 0.372, P < 0.001). B. The relative expression levels of PLIN2 were examined by qRT-PCR in KT-1/A3 cells transduced with either Lenti-GFP or Lenti-CEBPA (***P < 0.001). C. The mRNA expression levels of PLIN2 were measured by qRT-PCR in K562 cells transduced with either shLuc or shCEBPA (***P < 0.001). D. The proliferative ability was measured by the MTT assay in KT-1/A3 cells transduced with Lenti-GFP, Lenti-CEBPA, or Lenti-CEBPA and siPLIN2 (***P < 0.001). E. The proliferative ability was measured by the MTT assay in K562 cells transduced with shLuc, shCEBPA, or shCEBPA and PLIN2 (***P < 0.001). F. The apoptotic ability was detected by annexin V-FITC/PE staining in transduced KT-1/A3 cells. G. The apoptotic ability was detected by annexin V-FITC/PE staining in transduced K562 cells.
PLIN2 promotes the progression of CML via GSK3 and Wnt/β-catenin signaling in vitro
Studies have reported that PLIN2 participates in regulating the expression of proteins associated with the Wnt and GSK3 signaling pathways. In our study, we detected the mRNA expression levels of GSK-3β and β-catenin in transduced KT-1/A3 and K562 cells by using qRT-PCR and found that overexpression of PLIN2 increased the mRNA expression levels of GSK-3β and β-catenin while silencing PLIN2 decreased the expression levels of GSK-3β and β-catenin (P < 0.001, Figure 6A). In addition, a Western blot assay was used to analyze the protein expression levels of AKT, p-AKT, GSK-3β, β-catenin and Axin2/Conductin in transduced KT-1/A3 and K562 cells. The results showed that overexpression of PLIN2 upregulated the protein expression levels of AKT, p-AKT, GSK-3β, β-catenin and Axin2/Conductin (Figure 6B), whereas silencing PLIN2 downregulated the protein expression levels of AKT, p-AKT, GSK-3β, β-catenin and Axin2/Conductin (Figure 6C). The MTT results showed that GSK-3β-targeted and β-catenin-targeted siRNAs could rescue PLIN2-mediated proliferation of KT-1/A3 cells (P < 0.001, Figure 6D). Meanwhile, ectopic GSK-3β and β-catenin expression could rescue siPLIN2-mediated inhibition of K562 cell proliferation (P < 0.001, Figure 6E). The flow cytometry results revealed similar trends (Figure 6F, 6G).
Figure 6.
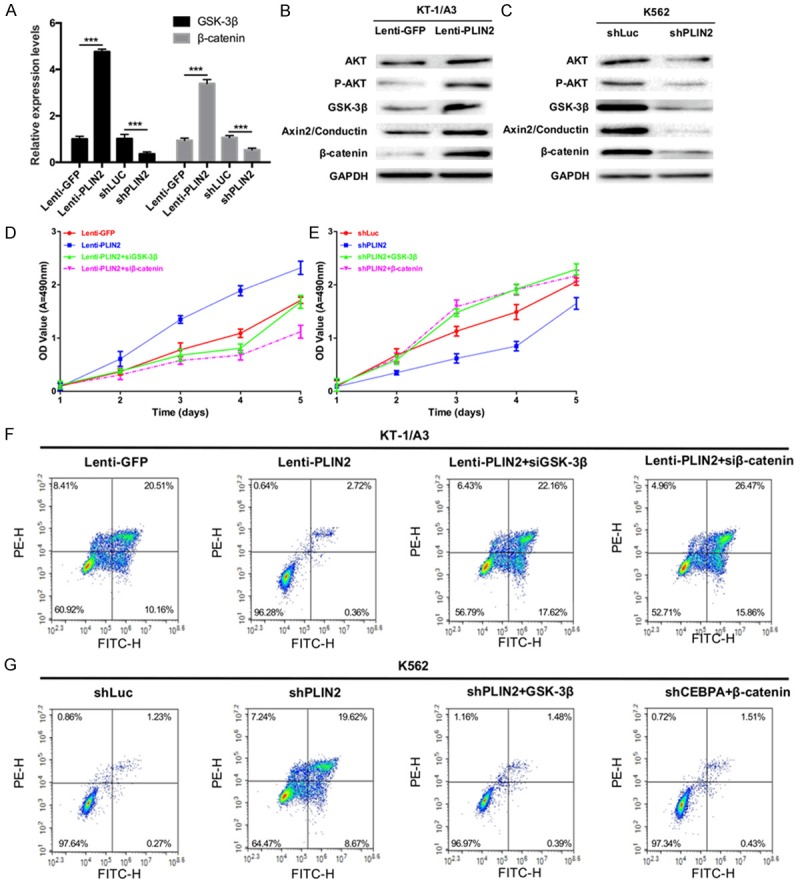
PLIN2 promotes the progression of CML via GSK3 and Wnt/β-catenin signaling in vitro. (A) KT-1/A3 cells were transduced with either Lenti-GFP or Lenti-PLIN2, and K562 cells were transduced with either shLuc or shPLIN2. qRT-PCR was used to detect the expression levels of GSK-3β and β-catenin (***P < 0.001). (B) Western blot analysis of AKT, p-AKT, GSK-3β, β-catenin and Axin2/Conductin protein levels in KT-1/A3 cells transduced with either Lenti-GFP or Lenti-PLIN2; GAPDH was used as a reference protein. (C) Western blot analysis of AKT, p-AKT, GSK-3β, β-catenin and Axin2/Conductin protein levels in K562 cells transduced with either shLuc or shPLIN2; GAPDH was used as a reference protein. (D) The proliferative ability was detected by the MTT assay in KT-1/A3 cells transduced with Lenti-GFP, Lenti-PLIN2, Lenti-PLIN2 and siGSK-3β, or Lenti-PLIN2 and siβ-catenin (P < 0.001). (E) The proliferative ability was measured by the MTT assay in K562 cells transduced with shLuc, shPLIN2, shPLIN2 and GSK-3β, or shPLIN2 and β-catenin (P < 0.001). (F) The apoptotic ability was detected by flow cytometry in KT-1/A3 cells transduced as described in (D). (G) The apoptotic ability was detected by flow cytometry in K562 cells transduced as described in (E).
CEBPA-mediated upregulation of PLIN2 promotes tumor growth via GSK3 and Wnt/β-catenin signaling in vivo
To study the effect of PLIN2 on CML cell proliferation in vivo, KT-1/A3 cells were transduced with Lenti-GFP, Lenti-CEBPA, Lenti-CEBPA or siPLIN2, whereas K562 cells were transduced with shLuc, shCEBPA, shCEBPA or PLIN2. Nude mice were injected with transduced KT-1/A3 or K562 cells, and representative tumors were photographed and excised at 40 days post-injection (Figure 7A). CEBPA expression increased the tumor volume in nude mice injected with transduced KT-1/A3 cells, while co-transfection of CEBPA and PLIN2 siRNAs indicated that siPLIN2 could abolish the effects of CEBPA on CML tumor growth (Figure 7B). Silencing CEBPA decreased the tumor volume in nude mice injected with transduced K562 cells, while co-transfection of shCEBPA and a PLIN2 expression plasmid showed that PLIN2 could abrogate the inhibitory effect mediated by silencing CEBPA (Figure 7C). In addition, we detected the mRNA expression levels of PLIN2, GSK-3β and β-catenin using qRT-PCR assay in nude mouse tumors and found that CEBPA could significantly increase the expression levels of PLIN2, GSK-3β and β-catenin. While siPLIN2 could inhibit the effects of the CEBPA-mediated increase (Figure 7D), Silencing CEBPA could significantly decrease PLIN2, GSK-3β and β-catenin expression levels in K562 cells, while PLIN2 dramatically abolishes the effects mediated by silencing CEBPA (Figure 7E). At the same time, GSK-3β and β-catenin expression levels were analyzed by Western blotting of nude mouse tumors. The results showed that the protein expression levels of GSK-3β and β-catenin agreed with the corresponding mRNA expression levels (Figure 7F, 7G). Therefore, we concluded that within the CEBPA/PLIN2 axis in CML, which is shown in Figure 8, CEBPA could upregulate PLIN2 expression, while PLIN2 could upregulate GSK-3β and β-catenin expression. Thus, the CEBPA/PLIN2 axis accelerates proliferation and inhibits apoptosis through activation of GSK-3β and β-catenin in CML.
Figure 7.
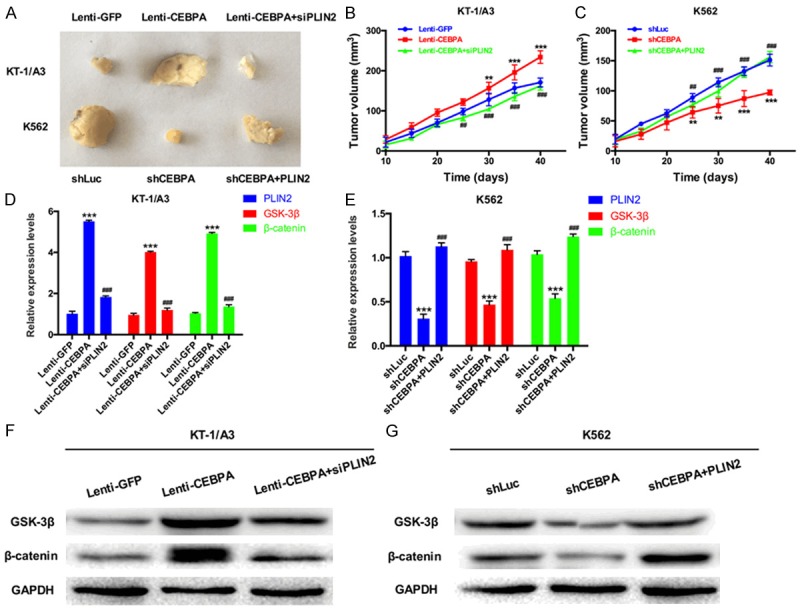
CEBPA-mediated upregulation of PLIN2 promotes tumor growth via GSK3 and Wnt/β-catenin signaling in vivo. A. KT-1/A3 cells were transduced with Lenti-GFP, Lenti-CEBPA, or Lenti-CEBPA and siPLIN2; K562 cells were transduced with shLuc, shCEBPA, or shCEBPA and PLIN2. Nude mice were injected with transduced KT-1/A3 and K562 cells, and then photographs of excised tumors at 40 days post-injection were obtained. B. The mean tumor volume was measured at 10, 15, 20, 25, 30, 35 and 40 days in nude mice injected with transduced KT-1/A3 cells (**P < 0.01 VS. Lenti-GFP group; ***P < 0.001 VS. Lenti-GFP group; ##P < 0.01 VS. Lenti-CEBPA group; ###P < 0.001 VS. Lenti-CEBPA group). C. The mean tumor volume was measured at 10, 15, 20, 25, 30, 35 and 40 days in nude mice injected with transduced K562 cells (**P < 0.01 VS. shLuc group; ***P < 0.001 VS. shLuc group; ##P < 0.01 VS. shCEBPA group; ###P < 0.001 VS. shCEBPA group). D. The mRNA expression levels of PLIN2, GSK-3β and β-catenin were measured by qRT-PCR in nude mice injected with transduced KT-1/A3 cells (***P < 0.001 VS. Lenti-GFP group; ###P < 0.001 VS. Lenti-CEBPA group). E. The mRNA expression levels of PLIN2, GSK-3β and β-catenin were measured by qRT-PCR in nude mice injected with transduced K562 cells (***P < 0.001 VS. shLuc group; ###P < 0.001 VS. shCEBPA group). F. Western blot analysis was used to detect the protein expression levels of GSK-3β and β-catenin in nude mice injected with transduced KT-1/A3 cells. G. GSK-3β and β-catenin expression levels were analyzed by Western blotting in nude mice injected with transduced K562 cells.
Figure 8.
The gene network of CEBPA-mediated upregulation of PLIN2 in CML. CEBPA upregulated PLIN2 expression; PLIN2 upregulated GSK-3β and β-catenin expressions; and CEBPA-mediated upregulation of PLIN2 accelerated proliferation as well as inhibited apoptosis via activation of GSK-3β and β-catenin in CML.
Discussion
LncRNAs are non-protein coding transcripts transcribed by RNA polymerase II [23] that play crucial roles in chromatin structure as well as transcriptional, posttranscriptional, epigenetic, and translational regulation of gene expression [24-26]. Recent research has indicated that lncRNAs are involved in various biological processes such as proliferation [27], carcinogenesis [28], and human diseases [29-31]. Previously, multiple studies have reported that lncRNA expression is a crucial regulator of CML progression and plays an important role in CML [32,33]. In the present study, we found that the novel lncRNA PLIN2 serves as a key regulator of CML and is expressed at significantly high levels in CML as well as promotes proliferation and inhibits apoptosis in CML. In addition, PLIN2 overexpression was attributed to upregulated CEBPA levels in CML. This novel axis may provide new clues for the diagnosis and treatment of CML.
Glycogen synthase kinase-3 (GSK-3) is a highly conserved serine/threonine protein kinase that can generate two related protein homologs (GSK-3α and GSK-3β) [34]. GSK-3 has important functions in many cellular processes via its activity toward glycogen synthase [35]. It is also a major component of the Wnt signaling pathway [35]. One domain on GSK-3 is closely related to the Axin scaffolding protein, which binds adenomatous polyposis coli (APC) and β-catenin during its period of inactivity [36]. GSK-3-mediated phosphorylation of β-catenin targets leads to their ubiquitination and proteosome-mediated degradation [37]. Several target genes of GSK-3, such as Axin2, act as a negative-feedback regulators of Wnt signaling [38,39]. A recent study has confirmed that the growth of both myeloid and lymphoid lineage leukemias is dependent on the Wnt signaling pathway. In our study, we found that PLIN2 increased the mRNA expression levels of GSK-3β and β-catenin and upregulated the protein expression levels of AKT, p-AKT, GSK-3β, β-catenin and Axin2/Conductin; furthermore, PLIN2 promoted the progression of CML via GSK3 and Wnt/β-catenin signaling [40].
In conclusion, we observed these major findings in the present study. 1) We identified that the CEBPA-upregulated lncRNA PLIN2 in the proliferation and apoptosis of CML act by regulating the GSK3 and Wnt/β-catenin signaling pathways. 2) We found that both CEBPA and PLIN2 were upregulated in CML and may serve as a diagnostic marker. 3) We indicated that PLIN2 increased the expression levels of AKT, p-AKT, GSK-3β, β-catenin and Axin2/Conductin. 4) We found that CEBPA-mediated upregulation of PLIN2 promotes tumor growth via GSK3 and Wnt/β-catenin signaling in vivo. These findings suggest that PLIN2 may act as a carcinogenic factor in the progression of CML by regulating the GSK3 and Wnt/β-catenin signaling pathways and may provide a potential therapeutic target for therapy CML in the future.
Acknowledgements
This work was supported the National Fund of Shandong (ZR2015HM073).
Disclosure of conflict of interest
None.
References
- 1.Pane F, Mostarda I, Selleri C, Salzano R, Raiola AM, Luciano L, Saglio G, Rotoli B, Salvatore F. BCR/ABL mRNA and the P210(BCR/ABL) protein are downmodulated by interferon-alpha in chronic myeloid leukemia patients. Blood. 1999;94:2200–2207. [PubMed] [Google Scholar]
- 2.Patnaik MM, Parikh SA, Hanson CA, Tefferi A. Chronic myelomonocytic leukaemia: a concise clinical and pathophysiological review. Br J Haematol. 2014;165:273–286. doi: 10.1111/bjh.12756. [DOI] [PubMed] [Google Scholar]
- 3.Todoric-Zivanovic B, Marisavljevic D, Surace C, Cemerikic V, Markovic O, Krtolica K, Tatomirovic Z, Cikota B, Magic Z, Rocchi M. A Ph-negative chronic myeloid leukemia with a complex BCR/ABL rearrangement and a t(6;9)(p21; q34.1) Cancer Genet Cytogenet. 2006;166:180–185. doi: 10.1016/j.cancergencyto.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 4.Zamecnikova A. Targeting the BCR-ABL tyrosine kinase in chronic myeloid leukemia as a model of rational drug design in cancer. Expert Rev Hematol. 2010;3:45–56. doi: 10.1586/ehm.09.66. [DOI] [PubMed] [Google Scholar]
- 5.Eide CA, O’Hare T. Chronic myeloid leukemia: advances in understanding disease biology and mechanisms of resistance to tyrosine kinase inhibitors. Curr Hematol Malig Rep. 2015;10:158–166. doi: 10.1007/s11899-015-0248-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jabbour E, Fava C, Kantarjian H. Advances in the biology and therapy of patients with chronic myeloid leukaemia. Best Pract Res Clin Haematol. 2009;22:395–407. doi: 10.1016/j.beha.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Ahmed W, Van Etten RA. Alternative approaches to eradicating the malignant clone in chronic myeloid leukemia: tyrosine-kinase inhibitor combinations and beyond. Hematology Am Soc Hematol Educ Program. 2013:189–200. doi: 10.1182/asheducation-2013.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cilloni D, Saglio G. Molecular pathways: BCR-ABL. Clin Cancer Res. 2012;18:930–937. doi: 10.1158/1078-0432.CCR-10-1613. [DOI] [PubMed] [Google Scholar]
- 9.Hehlmann R. How I treat CML blast crisis. Blood. 2012;120:737–747. doi: 10.1182/blood-2012-03-380147. [DOI] [PubMed] [Google Scholar]
- 10.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15:R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 12.Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Morales DR, Thomas K, Presser A, Bernstein BE, van Oudenaarden A. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tano K, Akimitsu N. Long non-coding RNAs in cancer progression. Front Genet. 2012;3:219. doi: 10.3389/fgene.2012.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Medeiros BC. Role of CEBPA in normal karyotype acute myeloid leukemia. J. Clin. Oncol. 2009;27:2105–2105. doi: 10.1200/JCO.2008.21.5418. [DOI] [PubMed] [Google Scholar]
- 18.Dalo F, Di Ruscio A, Guidi F, Fabiam E, Greco M, Rumi C, Hohaus S, Voso MT, Leone G. PU. 1 and CEBPA expression in acute myeloid leukemia. Leuk Res. 2008;32:1448–1453. doi: 10.1016/j.leukres.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Fuchs O, Provamikova D, Novakova L, Benesova K. CEBPA polymorphisms and mutations in patients with acute myeloid leukemia, myelodysplastic syndrome, multiple myeloma and non-Hodgkin’s lymphoma. Blood Cells Mol Dis. 2008;40:401–405. doi: 10.1016/j.bcmd.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 20.Hughes JM, Salvatori B, Giorgi FM, Bozzoni I, Fatica A. CEBPA-regulated lncRNAs, new players in the study of acute myeloid leukemia. J Hematol Oncol. 2014;7:69. doi: 10.1186/s13045-014-0069-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y, Lin MC, Yao H, Wang H, Zhang AQ, Yu J, Hui CK, Lau GK, He ML, Sung J, Kung HF. Lentivirus-mediated RNA interference targeting enhancer of zeste homolog 2 inhibits hepatocellular carcinoma growth through down-regulation of stathmin. Hepatology. 2007;46:200–208. doi: 10.1002/hep.21668. [DOI] [PubMed] [Google Scholar]
- 22.Jiang L, Lai YK, Zhang J, Wang H, Lin MC, He ML, Kung HF. Targeting S100P inhibits colon cancer growth and metastasis by Lentivirus-mediated RNA interference and proteomic analysis. Mol Med. 2011;17:709–716. doi: 10.2119/molmed.2011.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiu MT, Hu JW, Yin R, Xu L. Long noncoding RNA: an emerging paradigm of cancer research. Tumor Biol. 2013;34:613–620. doi: 10.1007/s13277-013-0658-6. [DOI] [PubMed] [Google Scholar]
- 26.Eades G, Zhang YS, Li QL, Xia JX, Yao Y, Zhou Q. Long non-coding RNAs in stem cells and cancer. World J Clin Oncol. 2014;5:134–141. doi: 10.5306/wjco.v5.i2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai M-C, Hung T, Argani P, Rinn JL. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J Pathol. 2010;220:126–139. doi: 10.1002/path.2638. [DOI] [PubMed] [Google Scholar]
- 30.Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gutschner T, Hämmerle M, Eißmann M, Hsu J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Groß M. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180–1189. doi: 10.1158/0008-5472.CAN-12-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blume CJ, Hotz-Wagenblatt A, Hullein J, Sellner L, Jethwa A, Stolz T, Slabicki M, Lee K, Sharathchandra A, Benner A, Dietrich S, Oakes CC, Dreger P, te Raa D, Kater AP, Jauch A, Merkel O, Oren M, Hielscher T, Zenz T. p53-dependent non-coding RNA networks in chronic lymphocytic leukemia. Leukemia. 2015;29:2015–2023. doi: 10.1038/leu.2015.119. [DOI] [PubMed] [Google Scholar]
- 33.Ferreira PG, Jares P, Rico D, Gomez-Lopez G, Martinez-Trillos A, Villamor N, Ecker S, Gonzalez-Perez A, Knowles DG, Monlong J, Johnson R, Quesada V, Djebali S, Papasaikas P, Lopez-Guerra M, Colomer D, Royo C, Cazorla M, Pinyol M, Clot G, Aymerich M, Rozman M, Kulis M, Tamborero D, Gouin A, Blanc J, Gut M, Gut I, Puente XS, Pisano DG, Martin-Subero JI, Lopez-Bigas N, Lopez-Guillermo A, Valencia A, Lopez-Otin C, Campo E, Guigo R. Transcriptome characterization by RNA sequencing identifies a major molecular and clinical subdivision in chronic lymphocytic leukemia. Genome Res. 2014;24:212–226. doi: 10.1101/gr.152132.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexandrova EM, Sokol SY. Xenopus axin-related protein: a link between its centrosomal localization and function in the Wnt/beta-catenin pathway. Dev Dyn. 2010;239:261–270. doi: 10.1002/dvdy.22125. [DOI] [PubMed] [Google Scholar]
- 37.Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, Mann M, Ben-Neriah Y, Alkalay I. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 2002;16:1066–1076. doi: 10.1101/gad.230302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, Behrens J. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22:1184–1193. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, Cho KR, Fearon ER. Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem. 2002;277:21657–21665. doi: 10.1074/jbc.M200139200. [DOI] [PubMed] [Google Scholar]
- 40.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]