

Abstract
Temozolomide (TMZ) has been widely used in conjunction with radiotherapy for treating various types of cancers. However, tumor cells arrested in senescence due to TMZ administration can sometimes escape and become drug resistant. In the current study, the possible role of survivin in the senescence escape of TMZ-treated glioma cells was comprehensively studied. The levels of survivin and CDK1 expression in a human glioma cell line (U251) were monitored, and cell apoptosis, cell cycle distribution, anchorage-independent growth, and senescence were studied in U251 cells in different degrees of senescence. To further investigate how survivin affects the TMZ-resistance of gliomas, we modulated the levels of survivin and CKD1 expression in TMZ-treated cells and then examined how the treated cells responded. The results showed that knockdown of the survivin gene increased the sensitivity of glioma cells to TMZ treatment by inducing senescent cells to become apoptotic. Moreover, after senescence was induced, expression of the survivin gene became suppressed, but survivin levels returned to normal after the cells had escaped from senescence. While down-regulation of the survivin gene in senescent and senescence-escaping U251 cells had no effect on cell apoptosis, cell cycle distribution, or senescence status, it dramatically reduced the anchorage-independent growth ability of the cells. Additionally, CDK1 was able to not only enhance the anchorage-independent growth ability of the cells, but also contribute to their further senescence escape by modulating the survivin and other pathways. In conclusion, the survivin gene was necessary for glioma cells to escape from and enter into senescence during treatment with TMZ.
Keywords: CDK1, glioma, senescence escape, survivin, temozolomide
Introduction
Gliomas are tumors formed by neoplastic glial cells, and are the most common type of intracranial tumor found in adults. An investigation conducted in 2011 showed that the annual incidence of gliomas was ~0.005% (www.cbtrus.org). In addition to this high incidence, patients with high-grade gliomas (an astrocytoma or glioblastoma multiforme) are often impaired and have a poor prognosis. Due to the high mobility and infiltration capability of glioma cells, radiotherapy followed by surgical removal is central to treating all types of gliomas. Radiotherapy combined with chemotherapy is often used to eliminate any remaining tumor cells; however, most chemotherapeutic agents provide only marginal clinical benefits [1]. Among the different chemotherapeutic agents used to treat gliomas, temozolomide (TMZ) has been proved to be the optimal choice for treating recurrent high-grade gliomas [2-4].
TMZ is a DNA-methylating agent that produces an O6-methylguanine (O6MeG) moiety in DNA [5]. O6MeG mispairs with thymine during the subsequent DNA replication cycle; and thereafter, the TMZ-induced O6MeG/T mismatches are recognized by the cell’s mismatch repair (MMR)system, which triggers futile repair cycles [6,7]. This erroneous repair process induces secondary chemical lesions and leads to DNA double-strand breaks (DSBs) [8,9]. These breaks then stimulate signals for the initiation of apoptosis, autophagy, and the cellular senescence process [3]. Unlike the apoptotic process which is completely destructive to cells, TMZ-induced cellular senescence is increasingly viewed as a survival strategy for tumor cells, as cell viability is maintained and apoptosis is avoided [10,11]. When considering that the entry of cancer cells into apoptosis or senescence following TMZ treatment largely relies on the collective effects of multiple signaling pathways, it is reasonable to investigate the mechanism that drives the transition between apoptosis and senescence.
The term senescence was first introduced in 1961 [12], and applied to a cell phenotype associated with advanced chronological age [11]; namely, cells that were enlarged, flattened, and displayed increased cytoplasmic granularity [11]. Although therapies based on senescence may appear attractive, inducing an irreversible state of cellular arrest does pose potential dangers [3,10,11]. In addition to therapy-induced cellular senescence, the senescence process can also be initiated when cells are challenged with external/internal pressures; and in ideal situations, senescence acts as a self-protection mechanism [11]. It was found that inhibition of p53 expression in senescent fibroblasts dramatically decreased the expression of senescence-associated genes. A recent study by Roberson et al [10] demonstrated that lung carcinoma cells can escape senescence by upregulating their levels of CDC2/CDK1 expression [10]. A further examination of the senescence escape response facilitated by elevation of CDC2/CDK1 levels revealed that survivin might be the downstream effector of this modulation [13]. The possible involvement of survivin in senescence escape also elicits interest because survivin expression improves cellular resistance to paclitaxel, which not only promotes senescence escape, but also post-escape drug resistance [14]. Therefore, the successful application of senescence-induction therapies might critically depend on the prevention of senescence by-pass or senescence re-emergence, either of which will strengthen the drug resistance of emergent tumor cells.
Given the universal use of TMZ for treating gliomas, and the central role of survivin-related signaling in senescence escape, the possible role of survivin in the TMZ-resistance of gliomas was comprehensively examined in the current study. The expression levels of both survivin and CDK1 in the human glioma cell line U251 were modulated, and the responses of TMZ-treated U251 cells to different conditions were investigated. In the current study, we found that knockdown of the survivin gene increased the sensitivity of glioma cells to treatment with TMZ, and activation of survivin signaling promoted senescence escape in glioma cells.
Methods
Cell culture and chemicals
Human glioma cell line U251 was obtained from Bioleaf Corporation (Shanghai, China) and cultured in DMEM supplemented with 15% fetal bovine serum (FBS) and a 1% (v/v) antibiotic mixture in an atmosphere of 95% air and 5% CO2 at 37°C. Antibodies against survivin and CKD1 were purchased from Abcam (Cambridge, UK). The specific siRNA for survivin (5’-CCCAGCCTTCCAGCTCCTTG-3’) and a scrambled version of the siRNA (5’-GGAGCCAGGGGGGAGCAGGG-3’) were synthesized by GenePharma Co. (Shanghai, China) and utilized as described by Wang et al [13]. The whole sequence of CDK1 was obtained from GenBank and ligated into a pcDNA plasmid to create an expression vector for the gene.
Experimental designs
The current study was conducted to investigate the role played by survivin-related signaling in the senescence escape of TMZ-treated glioma cells. The effect of survivin knockdown on the sensitivity of U251 cells to TMZ treatment was first assessed in a TMZ+SC group and a TMZ+siRNA group. In the TMZ+SC group, U251 cells were pretreated with 100 μM TMZ for 24 h and then cultured for 15 days before being transfected with scrambled (SC) siRNA. In the TMZ+siRNA group, U251 cells were pretreated with 100 μM TMZ for 24 h and then cultured for 15 days before being transfected with survivin specific siRNA.
Thereafter, comparisons between senescent U251 cells and senescence-escaping U251 cells regarding their TMZ sensitivity, proliferation, apoptosis levels, and senescence status were performed using the following groups: (1) a senescent U251 group comprised of U251 cells collected from 10 μM TMZ-treated U251 cells that displayed features of senescence; (2) a SE3 U251 group comprised of one subclone of senescence-escaping U251 cells; (3) a SE5 U251 group comprised of one subclone of senescence-escaping U251 cells.
Next the effects survivin knockdown in senescent U251 cells and senescence-escaping U251 cells were examined in four different groups of cells: (1) a senescent U251+SC group comprised of senescent U251 cells transfected with SC siRNA; (2) a senescent U251+siRNA group comprised of senescent U251 cells transfected with survivin-specific siRNA; (3) a SE5 U251+SC group comprised of SE5 U251 cells transfected with SC siRNA; (4) a SE5 U251+siRNA group comprised of SE5 U251 cells transfected with survivin-specific siRNA.
In the last part of this study, we conducted a preliminary investigation of how CDK1 functions in survivin knockdown senescence-escaping U251 cells. For those studies, we used a normal control (NC) group consisting of SE5 U251 cells transfected with NC siRNA, a siRNA group consisting of SE5 U251 cells transfected with survivin-specific siRNA, and a siRNA+CDK1 group consisting of survivin knockdown SE5 U251 cells transfected with CDK1 expression vector pcDNA. Three replicates were created for each group of cells, and all experiments were performed in triplicate.
Transfection and induction of senescent U251 cells and senescence-escaping U251 cells
Transfections of siRNA and plasmids were performed using a lipophilic transfection reagent (Lipofectamine 2000, Invitrogen; Carlsbad, CA, USA) according to the manufacturer’s instructions. To induce senescence, U251 cells were repeatedly treated with 10 µM TMZ until ~80% of the cells displayed a senescence phenotype. Afterwards, several proliferating colonies in these senescent cells were selected and allowed to recover from TMZ treatment until their growth rates resembled those of non-senescent U251 cells, and were then employed as senescence-escaping cells.
Trypanblue exclusion test for cell viability
The viability of cells that had received different treatments was detected using the trypan blue exclusion assay as previously described [15]. The numbers of stained cells were counted with a hemacytometer.
Flow cytometry
The cell cycles of U251 cells that had received different treatments were determined using FACS. Briefly, 300 μL of propidiumiodide (PI)-FITC was added to different aliquots of cells to stain their DNA in the dark. After a 20 min incubation at room temperature, the DNA contents of the cells were analyzed using a FACS flow cytometer (Accuri C6, Becton Dickinson; Franklin Lakes, NJ, USA).
The apoptosis rates in different groups of cells were detected using an Annexin V-FITC Apoptosis Detection Kit (JingMei Biotech; Beijing, China) according to the manufacturer’s instructions; after which, the cells were analyzed by FACScan flow cytometry (Accuri C6, Becton Dickinson, USA). The rates of cellular apoptosis (UR+LR-all apoptotic cell percentage) were calculated as the sum of the late apoptosis rate (UR, upper right quadrant-advanced stage apoptotic cell percentage) and the early apoptosis rate (LR, lower right quadrant-prophase apoptotic cell percentage).
SA-beta-gal assay
To measure the senescence status of cells in different groups, the cells were replated in a 12-well plate at a density of 2×104 cells/well. After seven days of culture, the cells were washed with PBS and fixed with 2% paraformaldehyde for 30 min at room temperature. Next, the cells were incubated with fresh SA-beta-gal staining solution [1 mg/mL X-gal (Sigma; St. Louis, MO, USA), 40 mM citric acid/sodium phosphate (pH 6.0), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, and 2 mM MgCl2] for 12 h at 37°C. Following incubation, the cells were marked with a solution containing 300 nM DAPI and 0.1% Triton X-100 (v/v in PBS) for 30 min at room temperature. Staining was observed under a microscope (×200), and the results were calculated as the ratio of SA-beta-gal-positive cells to total cells.
Hoechst staining
The morphological changes which occurred in the nuclei of apoptotic cells in the different groups were also observed under a fluorescence microscope (×400) after staining was performed with a Hoechst staining kit (Beyotime Biotechnology, China).
Western blot assays
The total proteins were extracted from the different groups of cells using a Total Protein Extraction Kit according to the manufacturer’s instructions (Catalog No. WLA019; Wanleibio, China). β-actin served as an internal reference protein. After the protein concentration in each group was determined using the BCA method, a 40 μg aliquot of total protein from each group was separated by electrophoresis on a 10% sodium dodecylsulfate polyacrylamide gel. After separation, the individual protein bands were transferred onto polyvinylidenedifluoride (PVDF) sheets, which were then washed with TTBS for 5 min prior to incubation for 1 hour with a powdered skim milk solution. The membranes were then incubated overnight at 4°C with primary antibodies against survivin (Abcam; Cambridge, UK), CDK1 (Abcam; Cambridge, UK) or β-actin (1:1000). After incubation, the membranes were washed four times with TTBS, and then incubated with secondary HRP IgG antibodies (BOSTER; Wuhan, China) for 45 min at 37°C. Next, the membranes were washed six times with TTBS; after which, the blots were developed using Beyo ECL Plus reagent, and the results were recorded with a gel imaging system. The relative expression levels of GREM1 and GLI3 in the different samples were calculated with a Gel-Pro-Analyzer (Media Cybernetics; Rockville, MD, USA).
Immunofluorescent assay
The cellular distribution of survivin in different U251 cells was detected with immunofluorescent microscopy. The cells were seeded into 14-well chambers, washed with PBS, and then fixed with 4% paraformaldehyde for 15 min. The cells were then permeabilized with 0.5% Triton X-100 for 30 min. After three cycles of washing with PBS (5 minutes per cycle), the cells were blocked with 10% goat serum for 15 min, and then incubated overnight at 4°C with primary rabbit polyclonal antibodies to survivin (Abcam; Cambridge, UK) in 1% goat serum. Staining was performed by incubating the cells with fluoresce in isothiocyanate secondary antibody (1:200) for 1 h. After incubation with the secondary antibody, the cells were washed and then stained with 4,6-diamino-2-phenyl indole (DAPI) for 5 min at room temperature. After three 5-minute cycles of washing with PBS buffer, the slides were fixed and imaged with a fluorescent microscope at ×400 magnification.
Colony formation assay
The anchorage-independent growth ability of U251 cells was measured with the colony formation assay. Briefly, the cells were suspended in 0.35% agarose (in 10% serum medium), and then inoculated into plates at a concentration of 200 cells per plate. After one week of culture, the cell colonies were stained with Wright-Giemsa stain and the numbers of colonies were recorded. The colony formation rate was calculated as the ratio of the colony number to the inoculated cell number.
Statistical analysis
All data are expressed as the mean ± SD, and each assay was performed using 3 replicate samples (n = 3). After ANOVA, post doc multiple comparisons were performed by Duncan’s method to control for type I errors. Differences between two groups were analyzed using the Student’s t-test, and two-tailed P-values <0.05 were considered statistically significant. All statistical analyses and graph manipulations were performed using SPSS Statistics for Windows, Version 10.0. Armonk, NY: IBM Corp.
Results
Knockdown of the survivin gene increased U251 cell sensitivity to TMZ
As shown in Figure 1A and 1B, TMZ had no effect on survivin expression, and transfection of survivin siRNA effectively suppressed expression of survivin protein in U251 cells. Based on those results, we further explored how survivin might affect the TMZ-sensitivity of U251 cells. U251 cells in the TMZ+SC and TMZ+siRNA groups were cultured for 15 days, and cell viability was measured every day. We found that cells transfected with survivin siRNA were more sensitive to TMZ (Figure 1C). However, the number of surviving cells didn’t change after day 9 of the assay. Therefore, cell cycle distributions and cell apoptosis levels were examined in samples collected on days 0, 4, and 8 of culture, and the samples collected on day 8 were analyzed via Hoechst staining and the SA-beta-gal assay. As shown in Figure 1D, cells in the TMZ+siRNA group were dramatically arrested in their G2/M phase starting on day 4 of culture, indicating the augmented growth inhibitory effect of TMZ on glioma cells after survivin knockdown. In conjunction with G2/M phase arrest in the TMZ+siRNA group, fewer senescent cells also were recorded in that group (Figure 1E and 1F). Given that TMZ exerts its effects by inducing either apoptosis or senescence, we measured the levels of apoptosis in the two groups of cells, and found that although fewer senescent cells were observed in the TMZ+siRNA group, those cells experienced a stronger apoptotic process after knockdown of the survivin gene. Significantly higher rates of apoptosis were detected in the TMZ+siRNA group when compared with the TMZ+SC group (P<0.05), and larger numbers of Hoechst-positive cells (bright blue) were also detected in the TMZ+siRNA group (Figure 2), which confirmed the antagonistic effect of TMZ on glioma cells.
Figure 1.

Knockdown of the survivin gene increased the sensitivity of U251 cells to TMZ. A: Representative images of survivin expression after 0, 2, 4, and 8 days of treatment with 100 μM TMZ. TMZ produced no obvious effect on survivin expression. B: Representative image from western blot validation studies of survivin expression after transfection of survivin-specific siRNA. C: Cell viability as detected by the trypan blue exclusion test. Knockdown of survivin in TMZ-treated U251 cells decreased the relative cell numbers. D: Representative images and a quantitative analysis of results from cell cycle distribution studies. Transfection of survivin siRNA induced G2/M phase arrest in TMZ-treated U251 cells. E and F: Representative images and a quantitative analysis of results from senescence status studies performed using the SA-beta-gal staining method. Transfection of survivin siRNA blocked the entry of TMZ-treated U251 cells into senescence. “*”, significantly different from the TMZ+SC group, P<0.05.
Figure 2.
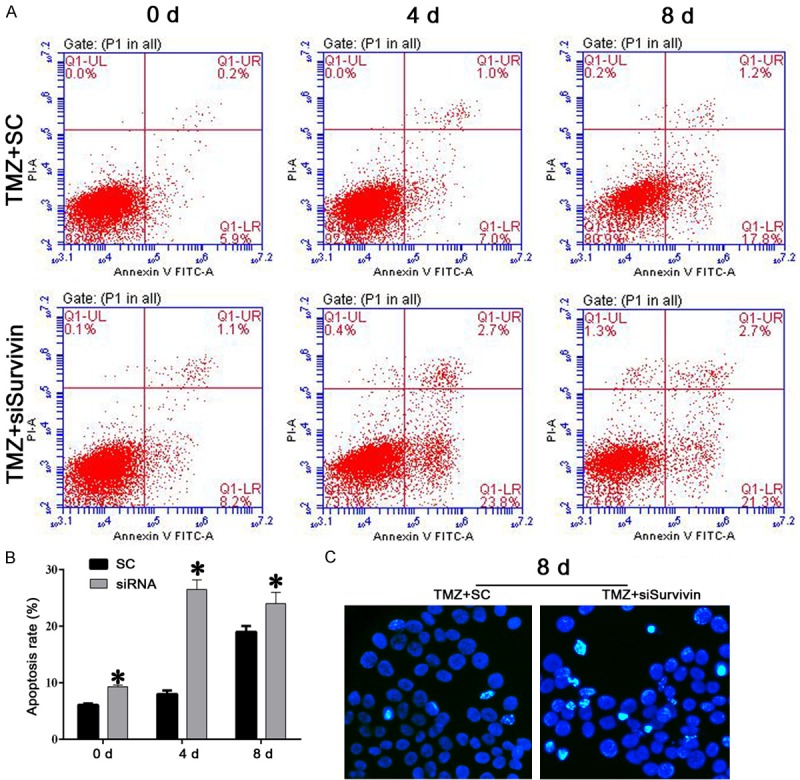
Knockdown of the survivin gene induced apoptosis in TMZ-pretreated U251 cells. A and B: Representative images and results of a quantitative analysis by flow cytometry. Apoptosis was induced beginning on day 4 after survivin knockdown. C: Representative images of Hoechst staining results. Larger numbers of Hoechst-positive cells (bright blue) were recorded in the TMZ+ survivin group. “*”, significantly different from the TMZ+SC group, P<0.05.
Escape from senescence increased the resistance of U251 cells to TMZ
Senescence escape serves as a therapy-resistance mechanism in tumor cells, because if senescent cells are not cleared, any restoration of their proliferative ability increases their resistance to certain therapies [11]. In the current study, the two subclones of senescence-escaping cells showed much greater resistance to TMZ when compared with senescent U251 cells, and especially when they were treated with a high dose of TMZ (Figure 3A).
Figure 3.

Escape from senescence increased the viability and decreased the apoptosis rate of TMZ-treated U251 cells. A: Cell viability as detected by the trypan blue exclusion assay. The relative cell numbers in two subclones of senescence-escaping U251 cells were higher than those in senescent U251 cells. B and C: Representative images and results of a quantitative analysis of cell cycle distribution. Escape from senescence induced a higher proportion of G0/G1 and S phase cells. D: Representative images and results of a quantitative analysis of apoptosis. Apoptosis was suppressed in senescence-escaping cells. E: Representative images of Hoechst staining results. Smaller numbers of Hoechst-positive cells (bright blue) were recorded among senescence-escaping cells. “*”, significant difference between groups, P<0.05.
Moreover, the senescence-escaping cells displayed higher percentages of G0/G1 and S phase cells, confirming the re-entry of those cells into the cell cycle (Figure 3B and 3C). Results of apoptosis assays confirmed the resistance of escaping cells to TMZ treatment: lower apoptotic rates and fewer Hoechst-positive cells were found in the SE3 U251, and SE5 U251 groups (Figure 3D and 3E).
Senescence escape of TMZ-treated glioma cells was associated with activation of CDK1/survivin signaling
The senescence status of cells in the senescent U251, SE3 U251, and SE5 U251 groups was investigated with the SA-beta-gal assay. Representative images and quantitative analyses revealed decreased numbers of senescent cells in the SE3 U251 and SE5 U251 groups, as well as significant difference between the senescent U251 group and the other two groups (P<0.05) (Figure 4A and 4B). To identify the signaling pathway involved in TMZ resistance in senescence-escaping glioma cells, we examined their levels survivin and CDK1 expression. Induction of senescence clearly suppressed both survivin and CDK1 expression (Figure 4C and 4D). In addition, the reduced levels of apoptosis and senescence in SE3 and SE5 cells were associated with an induction of survivin and CDK1 expression. (Figure 4C). The augmented expression and distribution of survivin was further confirmed by immunofluorescent assays (Figure 4D).
Figure 4.
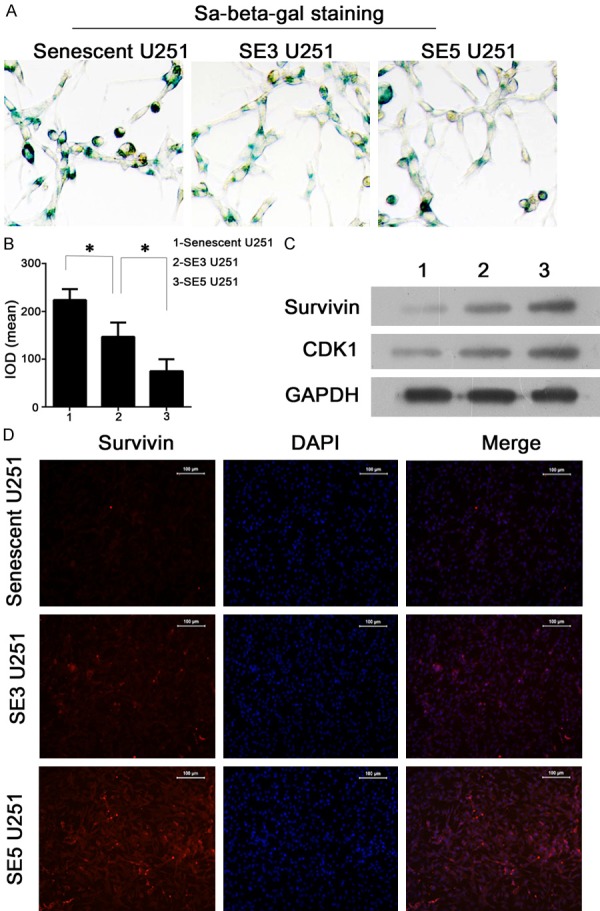
TMZ-treated U251 cells escaping from senescence displayed decreased numbers of senescent cells and activation of CDK1/survivin signaling. A and B: Representative images and results of a quantitative analysis of cell senescence status as detected by SA-beta-gal staining. Groups of cells escaping from senescence contained smaller numbers of senescent cells. C: Representative images from western blot validation studies of CDK1/survivin activity. Senescence was associated with suppressed CDK1 and survivin expression, while cell escaping from senescence displayed an increased expression of both indicators. D: Representative images showing immunofluorescent detection of survivin expression. Cells escaping from senescence displayed increased expression and distribution of survivin. “*”, significant difference between groups, P<0.05.
Knockdown of the survivin gene decreased colony formation, but had no impact on apoptosis or cell cycle distributions in senescent and senescence-escaping U251 cells
The results described above suggest that knockdown of survivin increased the TMZ sensitivity of normal U251 cells, but whether a similar pattern would be observed in senescent and senescence-escaping U251 cells remained unknown. Therefore, we suppressed survivin expression in senescent U251 cells and senescence-escaping cells. Interestingly, knockdown of survivin did not affect apoptosis, the cell cycle distribution or the percent of senescent cells in either type of cells (Figure 5A-D and 5F).
Figure 5.

Knockdown of the survivin gene in senescent and senescence-escaping U251 cells decreased their anchorage-independent growth ability. A and B: Representative images and results of a quantitative analysis of apoptosis. No significant change in apoptosis was detected in either subclone of the TMZ-treated U251 cells after knockdown of the survivin gene. C: Representative images of Hoechst staining results. No significant changes in Hoechst-positive cell numbers (bright blue) were found after knockdown of the survivin gene. D: Representative images and results of a quantitative analysis of cell cycle distribution. No significant changes in cell cycle distribution were detected after knockdown of the survivin gene. E: Representative images and results of a quantitative analysis of colony formation assay data. Knockdown of survivin in TMZ-pretreated U251 cells reduced the colony numbers of both cell lines. F: Representative images of cell senescence status as detected by SA-beta-gal staining. No significant changes in senescent status were detected after knockdown of the survivin gene. “*”, significantly different from the U251+SC group or SE5 U251+SC group, P<0.05.
However, the numbers of colonies formed were significantly decreased by knockdown of survivin (P<0.05) (Figure 5E), which might indicate that survivin affects the anchorage-independent growth ability of senescent and senescence-escaping glioma cells. Moreover, given that apoptosis and the cell cycle distribution were not impacted by survivin knockdown, it remains possible that in TMZ-pretreated U251 cells, those processes are modulated by pathways upstream of survivin signaling.
Overexpression of the CDK1 gene increased the clone formation ability of senescence-escaping U251 cells
The function of CDK1 in senescence-escaping U251 cells was also preliminarily examined in the current study. CDK1 was found to be an up-stream regulator of survivin function. Survivin knockdown SE5 U251 cells showed no obvious change in their cell cycle distribution after being transfected with a CDK1 overexpression vector (Figure 6A, 6B and 6D). However, overexpression of CDK1 increased the colony numbers and attenuated the senescence status of cells in the siRNA+CDK1 group, which to some extent, was indicative of the glioma cells being able to overcome impairments resulting from survivin knockdown (Figure 6C).
Figure 6.

Overexpression of the CDK1 gene in survivin knockdown senescence-escaping U251 cells increased the anchorage-independent growth ability of the cells, and further promoted their senescence escape. A and B: Representative images and results of a quantitative analysis of cell cycle distribution. CDK1 overexpression caused no significant changes in cell cycle distribution when compared with the effects of survivin knockdown. C and D: Representative images and results of a quantitative analysis of colony formation assay data. Overexpression of the CDK1 gene in survivin knockdown U251 cells increased their colony numbers. E: Representative images of cell senescence status as detected by SA-beta-gal staining. Senescent cell numbers in the siRNA+CDK1 group were smaller than those in the siRNA group, indicating the promotion of senescence escape. “*”, significant difference between groups, P<0.05.
Discussion
Cellular senescence, an irreversible arrest of proliferation, is thought to be a universal barrier to tumor progression. Almost all types of cancer cells must overcome this barrier to achieve immortalization [16]. When examining the signaling events which lead to and sustain the senescent phenotype, several events including but not limited to invasion, metastasis, immortalization, proliferation, and immune modulation are can be identified as hallmarks of cellular senescence [17-22], and each of these events is an attractive target therapeutic intervention. Numerous studies have evaluated the therapeutic potential of different drugs or treatment regimens that induce cellular senescence, and considerable progress has been achieved [16]. However, therapy-induced cellular senescence does not always destroy cancer cells. In some circumstances, senescent cancer cells can by-pass replicative senescence and resume their division, leading to tumor progression and therapy-resistance [10,11,23]. Therefore, the successful application of cellular senescence-induction therapies critically depends on gaining an understanding of the mechanism involved in the senescence escape process. In the current study, we demonstrated that a sustained expression of survivin in glioma cells blocked the apoptosis-inducing effect of TMZ. Furthermore, specific inhibition of the survivin gene increased the TMZ-sensitivity of U251 cells, and also suppressed the anchorage-independence of senescent and senescence-escaping cells. Additionally, activation of survivin signaling by overexpression of its upstream regulator, CDK1, contributed to further suppression of the senescence process in senescence-escaping cells, confirming the central role of the survivin gene in tumor senescence.
Survivin belongs to the mammalian inhibitor of apoptosis (IAP) gene family [24]. It is well recognized that overexpression of survivin is associated with inhibition of cell death as initiated via the extrinsic and intrinsic apoptotic pathways [25]. This concept was well validated in the our study, as we found that after knockdown of survivin expression, the apoptotic rates of TMZ-treated U251 cells were dramatically enhanced; but without a normal level of survivin production, the senescence process in TMZ-treated cells was inhibited as well. Furthermore, survivin expression was innately suppressed in senescent cells. Wang et al [13] found that survivin was necessary for cells to escape from senescence and for tumor cells to remain viable after bypassing senescence [13]. Our data showed that survivin was not required for a cell to enter senescence prior to escaping, and once senescence had been induced, the survivin gene became suppressed to avoid the onset of senescence escape.
In addition to its role in initiating senescence escape, we also explored how survivin supports senescent and senescence-escaping cells. In the current study, expression of the survivin gene in both cell lines was suppressed. It was interesting to find that knockdown of survivin had no impact on cell apoptosis, cell cycle distribution or cell senescence in either cell subclone, whereas the colony formation ability of survivin knockdown cells was decreased. The anchorage-independent growth ability of cancer cells is closely related to their oncogenicity [26,27]. Consistent with previous studies, the suppressed anchorage-independent growth found in our study due to down-regulation of survivin represented an antagonizing effect on the oncogenicity of glioma cells. When considering the overall effects of survivin knockdown, it appears that although the roles played by survivin in modulating cell viability, the cell cycle, and cell apoptosis in senescent and senescence-escaping glioma cells were weakened, survivin exerted its function by supporting the oncogenicity of those glioma cells. This function plays an especially important role in senescence-escaping glioma cells, in that it may contribute to regenerated drug-resistance.
As further confirmation of the key role played by survivin signaling in glioma progression, we found that expression of CDK1, the key upstream regulator of survivin [13], was up-regulated. Survivin function is regulated by CDC2/CDK1, which phosphorylates survivin at Thr34. This phosphorylation stabilizes the survivin protein and is necessary for its interaction with mitotic spindles and inhibition of caspase-9 activity [28]. Suppression of survivin phosphorylation with the CDC2/CDK1 kinase inhibitor flavopiridol enhances Adriamycin-induced apoptosis in cervical and breast carcinomas [29]. Regarding the effect on senescence, senescence-escaping cells are always associated with redundant levels of CDC2/CDK1, and are critically reliant on CDC2/CDK1 kinase activity for their viability [13]. In our study, overexpression of CDK1 increased the anchorage-independent growth ability of cells resulting from knockdown of survivin. In contrast to the effect of survivin, up-regulation of CDK1 also promoted senescence escape, as demonstrated by fewer positive cells being found in SA-beta-gal assays. This result indicated that in addition to functioning through survivin, CDK1 can also induce senescence via other pathways.
The major findings outlined in our study suggest that the survivin gene is not only necessary for cells to escape senescence, but is also important for their entry into senescence during chemotherapy. Nevertheless, once senescence is induced, survivin becomes inhibited, which may prevent tumor cells from escaping their senescence. Furthermore, survivin is essential for maintenance of tumor cell oncogenicity. Additionally, an upstream regulator of the survivn gene, CDK1, also contributes to the progression of senescence escape by modulating the survivin and other pathways. Although our study produced some interesting findings regarding how survivin functions in senescence-escaping glioma cells, details of the mechanisms which underlie these functions remain to be discovered. Additional comprehensive studies on the function of survivin-related pathways relevant to cancer senescence need to be conducted to promote the development of therapies based on senescence induction.
Acknowledgements
This research was supported in part by the National Natural Science Foundation of China (NSFC) (81272806 to Y.K.), and the Projects of Public Welfare Research and Capacity Development of Guangdong Province, China (2014A020212171 to M.H.).
Disclosure of conflict of interest
None.
References
- 1.Gunther W, Pawlak E, Damasceno R, Arnold H, Terzis AJ. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br J Cancer. 2003;88:463–469. doi: 10.1038/sj.bjc.6600711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villano JL, Seery TE, Bressler LR. Temozolomide in malignant gliomas: current use and future targets. Cancer Chemoth Pharm. 2009;64:647–655. doi: 10.1007/s00280-009-1050-5. [DOI] [PubMed] [Google Scholar]
- 3.Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski KH, Christmann M, Kaina B. Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS One. 2013;8:e55665. doi: 10.1371/journal.pone.0055665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bower M, Newlands ES, Bleehen NM, Brada M, Begent RJ, Calvert H, Colquhoun I, Lewis P, Brampton MH. Multicentre CRC phase II trial of temozolomide in recurrent or progressive high-grade glioma. Cancer Chemoth Pharm. 1997;40:484–488. doi: 10.1007/s002800050691. [DOI] [PubMed] [Google Scholar]
- 5.Hirose Y, Berger MS, Pieper RO. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001;61:1957–1963. [PubMed] [Google Scholar]
- 6.Caporali S, Falcinelli S, Starace G, Russo MT, Bonmassar E, Jiricny J, D’Atri S. DNA damage induced by temozolomide signals to both ATM and ATR: role of the mismatch repair system. Mol Pharmacol. 2004;66:478–491. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 7.Branch P, Aquilina G, Bignami M, Karran P. Defective mismatch binding and a mutator phenotype in cells tolerant to DNA damage. Nature. 1993;362:652–654. doi: 10.1038/362652a0. [DOI] [PubMed] [Google Scholar]
- 8.Quiros S, Roos WP, Kaina B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle. 2010;9:168–178. doi: 10.4161/cc.9.1.10363. [DOI] [PubMed] [Google Scholar]
- 9.Ochs K, Kaina B. Apoptosis induced by DNA damage O6-methylguanine is Bcl-2 and caspase-9/3 regulated and Fas/caspase-8 independent. Cancer Res. 2000;60:5815–5824. [PubMed] [Google Scholar]
- 10.Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005;65:2795–2803. doi: 10.1158/0008-5472.CAN-04-1270. [DOI] [PubMed] [Google Scholar]
- 11.Gordon RR, Nelson PS. Cellular senescence and cancer chemotherapy resistance. Drug Resist Updat. 2012;15:123–131. doi: 10.1016/j.drup.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q, Wu PC, Roberson RS, Luk BV, Ivanova I, Chu E, Wu DY. Survivin and escaping in therapy-induced cellular senescence. Int J Cancer. 2011;128:1546–1558. doi: 10.1002/ijc.25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puig PE, Guilly MN, Bouchot A, Droin N, Cathelin D, Bouyer F, Favier L, Ghiringhelli F, Kroemer G, Solary E. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol Int. 2008;32:1031–1043. doi: 10.1016/j.cellbi.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 15.Strober W. Trypan blue exclusion test of cell viability. Curr Protoc Immunol. 2015;111:A3.B.1–3. doi: 10.1002/0471142735.ima03bs111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cairney CJ, Bilsland AE, Evans TR, Roffey J, Bennett DC, Narita M, Torrance CJ, Keith WN. Cancer cell senescence: a new frontier in drug development. Drug Discov Today. 2012;17:269–276. doi: 10.1016/j.drudis.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 17.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young AR, Narita M. SASP reflects senescence. EMBO Rep. 2009;10:228–230. doi: 10.1038/embor.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, Valsesia-Wittmann S, Caron de Fromentel C, Puisieux A. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14:79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Feldser DM, Greider CW. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell. 2007;11:461–469. doi: 10.1016/j.ccr.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majumder PK, Grisanzio C, O’Connell F, Barry M, Brito JM, Xu Q, Guney I, Berger R, Herman P, Bikoff R, Fedele G, Baek WK, Wang S, Ellwood-Yen K, Wu H, Sawyers CL, Signoretti S, Hahn WC, Loda M, Sellers WR. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14:146–155. doi: 10.1016/j.ccr.2008.06.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu PC, Wang Q, Grobman L, Chu E, Wu DY. Accelerated cellular senescence in solid tumor therapy. Exp Oncol. 2012;34:298–305. [PubMed] [Google Scholar]
- 24.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Bio. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 25.Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene. 2003;22:8581–8589. doi: 10.1038/sj.onc.1207113. [DOI] [PubMed] [Google Scholar]
- 26.Brockman JA, Gupta RA, Dubois RN. Activation of PPARgamma leads to inhibition of anchorage-independent growth of human colorectal cancer cells. Gastroenterology. 1998;115:1049–1055. doi: 10.1016/s0016-5085(98)70072-1. [DOI] [PubMed] [Google Scholar]
- 27.Fiucci G, Ravid D, Reich R, Liscovitch M. Caveolin-1 inhibits anchorage-independent growth, anoikis and invasiveness in MCF-7 human breast cancer cells. Oncogene. 2002;21:2365–2375. doi: 10.1038/sj.onc.1205300. [DOI] [PubMed] [Google Scholar]
- 28.O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC, Altieri DC. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000;97:13103–13107. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wall NR, O’Connor DS, Plescia J, Pommier Y, Altieri DC. Suppression of survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res. 2003;63:230–235. [PubMed] [Google Scholar]