

Abstract
Colorectal cancer is one of the major health problems, with invade surrounding tissues, and migrate to distant organs being the most critical concern, thus identified metastasis associated hallmarks and more efficacious treatment are urgently needed. It found that forkhead box q1 (FOXQ1) is aberrant expression in variety of human cancers and FOXQ1 is involved in oncogenic pathways. However, the role of FOXQ1 has been unexplored in colorectal cancer metastasis to date. Here, expression of FOXQ1 was higher in colorectal cancer tissue samples and cancer cell lines than in normal colorectal tissue and cell lines. Further research suggested that FOXQ1 positively regulated cell proliferation in colorectal cancer and down-regulation of CDK6, extracellular regulated protein kinases 1/2 (ERK1/2) and mammalian target of rapamycin (mTOR). In corresponding to this result, over-expression of FOXQ1 significantly promoted colorectal cancer growth in vivo. Moreover, down regulation of FOXQ1 expression in colorectal carcinoma cell HCT116 and LOVO strikingly inhibits tumor growth in vivo. Finally, FOXQ1-dependent inhibition of colorectal cancer cell migration and invasion and down-regulation of focal adhesion kinase (FAK), phosphatidyl inositol 3-kinase (PI3K) phosphorylation, AKT (v-akt murine thymoma viral oncogene) phosphorylation and matrix metalloproteinase-2/9 (MMP-2/9) expression. These integrated efforts have identified FOXQ1 as a tumor promoter and might provide promising approaches for colorectal cancer metastasis treatment.
Keywords: FOXQ1, colorectal carcinoma, migration, invasion
Introduction
Colorectal cancer represents one of the most common solid gastrointestinal cancers worldwide [1]. The main curative treatments for colorectal cancer are the surgical removal of lesions, radiotherapy, and chemotherapy. Although outcomes of clinical treatment for colon carcinoma have improved in recent decades, the overall 5-year survival rate for patients is only 50% because new diagnostic and treatment methods are lacking [2]. Therefore, it is of great clinical value to explore the molecular mechanisms underlying the development of colon carcinoma and to identify effective treatment strategies to improve the survival rate of patients with colorectal carcinoma [3]. Accumulating evidence suggests that the poor therapeutic effects and dismal survival rate with colorectal carcinoma are associated with aberrantly activated signaling pathways, including epidermal growth factor receptor (EGFR) signaling [4]. In late-stage colorectal cancer, the most commonly used targeted therapies are the monoclonal antibodies cetuximab and panitumumab, which prevent EGFR activation [5]. EGFR activation has been reported to play key roles in the development and progression of colorectal carcinoma and to contributes to the malignant phenotype of human colon cancer cells. For instance, activated EGFR signaling results in colorectal carcinoma cell resistance to chemotherapy and promotes cell survival, whereas inhibition of EGFR signaling dramatically reduces the proliferation of colorectal carcinoma cells [6]. EGFR signaling is involved in the EGF-induced epithelial-mesenchymal transition (EMT) and is positively associated with lymph node metastasis of colorectal cancer. Metastasis, which precipitates death in the vast majority of colorectal cancer patients, is a multi-step process by which tumor cells disseminate from their primary site and form secondary tumors at a distant site [7]. These studies highlight molecular mechanisms that contribute to colorectal carcinoma metastasis and indicate the great need for new colorectal cancer interventions.
Recent studies have identified alterations in forkhead box q1 (FOXQ1) and other genes as likely mechanisms of secondary resistance to anti-EGFR antibody therapy that participate in colorectal cancer metastasis [8]. As a member of the forkhead box protein family, FOXQ1 is a transcription factor that regulates cell differentiation. FOXQ1 was recently found to be expressed in carcinomas of the breast, ovaries, and endometrium, where activation of the receptor by its ligand, produced either by the tumor cells or by stromal elements, stimulated tumor cell invasion and metastasis [9]. Furthermore, the activated or phosphorylated form of FOXQ1 has been associated with enhanced invasive and metastatic potential in advanced epithelial ovarian carcinoma. Phosphorylation of FOXQ1 on specific tyrosine residues enables it to bind directly to cytoplasmic effector proteins, which in turn relays receptor-induced signals through multiple signal transduction pathways [10]. Phosphorylation of FOXQ1 creates binding sites for a variety of cytoplasmic proteins that activate signal transduction pathways, including that of ERK1/2 and PI3K [11]. Several modes of regulating FOXQ1 expression have been demonstrated in normal and tumor cells, such as microRNA and the Wnt signaling pathway. Based on the hypothesis that blockade of FOXQ1 would be beneficial in cancer treatment, specific FOXQ1 inhibitors were developed to target malignant tumors. Collectively, these results identify FOXQ1 as a promising therapeutic target for solid tumors [12]. However, further studies are needed to fully elucidate how the multifunctional properties of FOXQ1 contribute to colorectal carcinoma invasion and metastasis.
In the present study, we found that FOXQ1 expression was higher in colorectal cancer tissues and cancer cell lines than in normal colorectal tissue and a normal cell line. FOXQ1 positively regulated cell proliferation in colorectal cancer cells lines and promoted metastasis of colorectal cancer cells. Downregulation of FOXQ1 hindered cellular migration and invasion of colorectal cancer cells, whereas silencing endogenous FOXQ1 resulted in the opposite outcome. Our findings suggest that FOXQ1 plays a critical oncogenic role in colorectal cancer progression and highlights its potential as a target for colorectal cancer therapies.
Materials and methods
microRNA database analysis
To examine the FOXQ1 expressed gene, the GENE-E tool (http://www.broadinstitute.org/cancer/software/GENE-E/download.html) was utilized to conduct the expression analysis of the microarray dataset. The dataset (GDS4382) were selected for the heterogeneity analysis of paired colorectal cancer tumors and adjacent non-cancerous tissues [13]. P values lower than 0.05 were considered significantly regulated and the top 10% of genes within the dataset were selected.
Cell culture
The human colorectal cancer cell lines (LOVO, HT-29, HCT116, Caco-2) and non-tumorigenic human colonic epithelial cells FHC used as control were obtained from the Chinese Academy of Sciences Cell Bank of Type Culture Collection (CBTCCCAS, Shanghai, China). The colorectal cancer cell lines were cultured as monolayers in ATCC-formulated DMEM: F12 media supplemented with 10% fetal bovine serum (FBS), penicillin (100 μg/mL), and streptomycin (100 μg/mL) and FHC was cultured in serum-free LHC-8 medium (Invitrogen, Carlsbad, CA) maintained in an incubator with a humidified atmosphere of 5% CO2 at 37°C.
Stable cell lines
A siRNA plasmid targeting human FOXQ1 (sc-60660; Santa Cruz Biotechnology) or a non-silencing control siRNA plasmid (sc-37007; Santa Cruz Biotechnology) was transfected into HCT116 and LOVO, using Lipofectamine® 2000 transfection reagent (Life Technologies). A FOXQ1 expression plasmid was generated by PCR sub-cloning full-length human FOXQ1 coding sequence into the retroviral transfer plasmid pQCXIP (Clontech, CA) to generate plasmid FOXQ1. Retroviral production and infection were performed as manufacturer’s instructions. Stable cell lines expressing FOXQ1 were selected for with 0.5 g/ml puromycin 10 days after infection [14].
Cell proliferation
The cell proliferation was determined by 3-(4, 5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay. Cells (1 × 104) seeded in 96-well tissue culture plates were incubated for 24, 48 and 72 hours, and at the end of the culture 10 μl MTT solution (5 mg/mL; Sigma) was added to each well with 100 μL of medium for 4 hours at 37°C. The medium was replaced with 200 μl imethyl sulfoxide (DMSO), followed by incubation for 10 minutes at room temperature. The absorbance was then measured at a wavelength of 490 nm using SynergyTM HT Multi-Mode Microplate Reader (Bio-Tek, Winooski, VT, USA). The effect of siRNA FOXQ1 or FOXQ1 over-expression on colorectal cancer cell viability was assessed as the percent of cell viability compared with vehicle-treated control cells, which were arbitrarily assigned 100% viability.
Colony formation
LOVO and HCT116 cells (1 × 102) were seeded in 25 mm2 petri dishes and were allowed to grow. Subsequently, growth medium was added every week for 21 days until colonies were easily distinguishable. Then, the cell clones were fixed with methanol and stained with 0.1% crystal violet for number counting [15].
Cell migration
The scratch wound healing assay was adapted to evaluate the ability of colorectal cancer cells migration. Cells were cultured in 6-well plates until reach to 90% confluence. The cell were monolayers scratched manually with a 200 µl pipette tip, washed with PBS for twice and cultured in serum-free medium for 48 hours. Then, cells were fixed with methanol, and the scratch area was photographed [16].
Cell invasion
Cell invasiveness was determined, using a Matrigel (BD Biosciences)-coated Transwell chamber (pore size of 8-um, Corning). Briefly, LOVO or HCT116 cells (5 × 104 cells per well) in 100 µl of serum-free medium were seeded into the upper chamber of the Transwell, whereas 600 μl of medium containing 10% FBS was applied to the lower chamber. After incubation for 24 hours at 37°C in a CO2 incubator, invasive cells were fixed with 4% formaldehyde, stained with 0.1% crystal violet, and counted in five random microscopic fields under a light microscope [17].
Flow cytometry
FHC, LOVO and HCT116 cells were detached by 2 mM PBS/EDTA and fixed with 4% paraformaldehyde, washed twice with PBS and re-suspended for antibody staining at 1 × 106 cells/100 ul in 1% PBS/BSA. Gates (in the live cell population) were drawn in order to exclude > 99% of background staining (based on the isotype-stained samples). For FOXQ1 staining, anti-FOXQ1-APC conjugated and its highly adsorbed isotype matched control (Biolegend) were used [18].
Western blotting
Cell protein extraction were prepared with RIPA lysis buffer containing both protease and phosphatase inhibitors. Equal amounts of cell lysates (30 µg) were loaded on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes. After membranes were blocked, they were incubated with monoclonal antibody against FAK (1:500, Signalway Antibody) and phosphor-FAK Tyr407 (1:10000, Epitomics), AKT (1:5000, Epitomics) and phosphor-AKT Ser473 (1:1000, Cell Signaling Technology), PI3K and phosphor-PI3K Tyr458 (1:1000, Cell Signaling Technology), MMP-2 (1:1000, Cell Signaling Technology), MMP-9 (1:1000, Cell Signaling Technology), CD44 (1:1000, Cell Signaling Technology), PCNA (1:1000, Cell Signaling Technology), mTOR (1:1000, Cell Signaling Technology), ERK1/2 (1:1000, Cell Signaling Technology), FOXQ1 (1:1000, Cell Signaling Technology) and GPADH (1:5000, Bioworld Technology) followed by incubation with horseradish peroxidase-conjugated IgGs (1:10000, Bioworld Biotechnology). Target proteins were detected by the ECL system (Millipore, Braunschweig, Germany) and visualized with the ChemiDoc XRS system (Bio-Rad, Hercules, CA, USA) [19].
Quantitative real-time PCR
Total RNA (0.5 μg) was isolated from cultured cells using TRIzol reagent (Invitrogen, USA). First-strand cDNA was synthesized with 1 μg total RNA using a PrimeScript RT reagent kit (TakaraBio, Japan). Quantitative real-time PCR was performed using iQTM SYBR® Green Supermix and the iQ5 real-time detection system (Bio-Rad Laboratories, CA). The comparative cycle threshold (Ct) method was applied to quantify the expression levels through calculating the 2(-ΔΔCt) method. The primer pairs used for PCR were as follows (sense and antisense, respectively): GAPDH (forward, 5’-GGAGCGAGATCCCTCCAAAAT-3’; reverse, 5’-GGCTGTTGTCATACTTCTCATGG-3’) was used as an internal control; FOXQ1 (forward, 5’-CACGCAGC-AAGCCATATACG-3’; reverse, 5’-CGTTGAGCGAAAGGTTGTGG-3’). cDNAs amplification and relative expression values were obtained from three independent experiments.
Mouse xenograft model
Six-week-old athymic male BALB/c nude mice (Shanghai Slack Laboratory Animal co., Ltd) were acclimatized under controlled standard conditions for a week prior to the experiment according to Guide for the Care and Use of Laboratory Animals of Nanjing University of Chinese Medicine. LOVO or HCT116 (5 × 107) cells re-suspended in 100 μl of PBS and were subcutaneously injected into the back flank area of nude mice. The diameter of the tumor was measured externally with a caliper ruler every 3 days, and the volume of the tumor (mm3) was estimated by the following formula: (short diameter) 2 × (longest diameter) × 0.5 [20]. For immunohistochemical analysis, tumor tissues were removed, formalin-fixed and paraffin-embedded. Serial 4-μm tumor sections were deparaffinized in xylene, rehydrated through descending grades of ethanol, and subjected to immunohistochemical analysis using antibodies against CD44, extracellular regulated protein kinases 1/2 (ERK1/2) and mTOR.
Statistical analysis
The Fisher exact test or the χ2 test was used to evaluate the associations between categorized variables. The numerical data were compared by Student’s t-test or Mann-Whitney U test. Survival rates in patients were calculated by the Kaplan-Meier method and comparison was made by log-rank test. P values less than 0.05 were considered to be statistically significant.
Results
FOXQ1 expression is elevated in colorectal cancer
We tested five representative colorectal cancer cell lines and non-tumorigenic human colonic epithelial cell line FHC for the expression of FOXQ1. Quantitative PCR revealed that all colorectal cancer cell lines expressed detectable amounts of FOXQ1 mRNA (Figure 1A). Western blotting with anti-FOXQ1 antibody confirmed higher detectable amounts of FOXQ1 in all five cell lines (Figure 1B). To determine changes in FOXQ1 in clinical specimens of colorectal cancer tissue, the level of FOXQ1 in colorectal carcinoma tissues was examined using a microarray expression database (DataSet Record GDS4382). Based on gene summary views (neoplastic vs. normal tissue), FOXQ1 was significantly upregulated in colorectal cancer tissue (Figure 1C). The top 10 differentially regulated in the dataset were selected for further analysis. The FOXQ1 gene was identified as significantly upregulated in colorectal cancer tissue based on their presence in non-cancerous tissue (Figure 1C). Fluorescence-activated cell sorting analysis after staining with anti-FOXQ1 antibody revealed the existence of HCT116 and LOVO cell subpopulations that expressed FOXQ1 (range 28.3-35.9%, Figure 1D), whereas no FOXQ1-expressing FHC cells were identified (2.9%).
Figure 1.

FOXQ1 expression is up-regulated in human colorectal cancer. A. qRT-PCR analysis of FOXQ1 mRNA levels in various colorectal cancer cell lines. Histograms reporting the levels of FOXQ1 mRNA (normalized to the GAPDH mRNA) as assessed in five representative colorectal cancer cell lines (LOVO, HT-29, HCT116, Caco2) and non-tumorigenic human colonic epithelial cells FHC. Data were presented as the mean ± SD from three independent measurements. For indicated comparisons, *P < 0.05 and **P < 0.01 versus FOXQ1 mRNA in FHC cells. B. Western blotting with anti-FOXQ1 antibody of cell lysates from colorectal cancer cell lines (LOVO, HT-29, HCT116, Caco2) and non-tumorigenic human colonic epithelial cells FHC. GAPDH was used as a loading control. Expression of FOXQ1 was quantified, normalized to GAPDH in different cells, and shown in the graph to the right. C. Analysis of paired colorectal cancer tumors and adjacent non-cancerous tissues. Blue indicated down-regulation. Red indicated up-regulation. Black indicates no change. D. Cells stained with anti-FOXQ1 antibody or with an isotype matched antibody (as a background control). Representative flow cytometry plots of FOXQ1 surface protein expression by FHC cells and representative tumor cells (n = 3 independent experiments, respectively).
FOXQ1 regulates cell proliferation and colony formation in colorectal cancer cells
To investigate the role of FOXQ1 in colorectal cells proliferation, we transfected HCT116 and LOVO cells with FOXQ1 siRNA or a wild-type FOXQ1 expression plasmid (FOXQ1) to silence or overexpress FOXQ1, respectively. Lower levels of FOXQ1 expression were observed in the cells transfected with FOXQ1 siRNA, and higher expression levels were observed in cells transfected with the FOXQ1 plasmid (Figure 2A). As shown in Figure 2B, downregulation of FOXQ1 with specific siRNA inhibited proliferation of both HCT116 and LOVO cells. A growth curve analysis showed that growth of HCT116 and LOVO cells with downregulated FOXQ1 decreased relative to that of control cells. However, overexpression of FOXQ1 in HCT116 and LOVO cells significantly promoted cell proliferation. The soft agar colony formation assay is a method used to confirm cellular anchorage-independent growth in vitro. As expected, inhibiting FOXQ1 expression in HCT116 and LOVO cells markedly decreased the efficiency of cell colony formation, whereas HCT116 and LOVO cells transfected with FOXQ1 showed an enhanced ability to form colonies (Figure 2C). To study the molecular mechanisms responsible for FOXQ1-regulated colon cancer cellular proliferation, we evaluated, by quantitative PCR, whether the effect of FOXQ1 on HCT116 and LOVO cell proliferation was accompanied by modulation of proteins that are involved in tumor cell growth and survival. This revealed significant downregulation of CD44, extracellular regulated protein kinases 1/2 (ERK1/2), and mammalian target of rapamycin (mTOR) in the FOXQ1 siRNA-treated HCT116 and LOVO cells (Supplementary Figure 1). Additionally, we observed that FOXQ1 transfection increased the levels of CD44, ERK1/2, and mTOR (Supplementary Figure 1). We then assessed the impact of FOXQ1 on the protein levels of these targets by overexpressing FOXQ1 and knocking down FOXQ1 in HCT116 and LOVO cells. Consistently, western blots of these putative targets (Figure 2D) showed that high levels of FOXQ1 expression increased levels of CD44, ERK1/2, and mTOR, and that FOXQ1 knockdown resulted in significantly decreased levels of these proteins (Figure 2D). Overall, these results indicated that FOXQ1 positively regulated cell proliferation and clone formation ability in colorectal cancer cells in vitro.
Figure 2.

FOXQ1 regulates cell proliferation in colorectal cancer cell lines. A. Western blot shown the protein expression levels of FOXQ1 in the human colorectal cancer cell lines HCT116 and LOVO after transfected with FOXQ1 siRNA or FOXQ1. GAPDH was used as a loading control. B. The proliferate abilities of HCT116 and LOVO cells transfected with FOXQ1 siRNA and cells transfected with FOXQ1 were examined based on MTT assay. Each bar represents the mean ± SD of three independent experiments. For indicated comparisons, *P < 0.05 and **P < 0.01. C. The efficiencies of cell colony formation in HCT116 and LOVO cells after transfected with FOXQ1 siRNA and cells transfected with FOXQ1. Mean number of tumor spheres ± SD in FOXQ1-siRNA versus control (left). Mean number of tumor spheres ± SD in FOXQ1 over-expressing colorectal cancer cell versus control (right). The data were presented as mean ± SD. For indicated comparisons, *P < 0.05 and **P < 0.01. D. CD44, ERK1/2 and mTOR protein expression levels were analyzed by western blot in in the indicated cells. GAPDH was used as a loading control.
Downregulation of FOXQ1 blocks cellular migration and invasion of colorectal cancer cells
Increased mobility is a crucial property of invasive cancer cells. To determine the effect of FOXQ1 on cellular migration, HCT116 and LOVO cells were transfected with a FOXQ1 siRNA plasmid and subjected to wound-healing migration and Transwell invasion assays. Non-specific siRNA (control siRNA) was used as a negative control. As shown in Figure 3A, the cell migration rate was significantly decreased in FOXQ1-downregulated cells compared with the rate in control cells. The invasion ability of colorectal cancer cells was examined in HCT116 and LOVO cells transfected with the FOXQ1 siRNA plasmid cultured in a 24-well Transwell plate with an 8-μm pore polyester insert. As shown in Figure 3B, significantly fewer FOXQ1-downregulated HCT116 and LOVO cells showed invasion as compared to control cells. To further investigate the molecular mechanism by which FOXQ1 contributes to cellular migration and invasion in colon carcinoma, the expression of several metastasis-related proteins were determined. An immunoblotting assay was performed to assess changes in the phosphorylation state of important metastasis-associated kinases, including focal adhesion kinase (FAK), protein kinase B (AKT), and phosphatidylinositol 3-kinase (PI3K). Figure 3C shows that FAK, AKT, and PI3K phosphorylation was effectively inhibited in FOXQ1 siRNA-transfected HCT116 and LOVO cells, but not in control cells. Matrix metalloproteinases (MMPs), which are regarded as capable of degrading extracellular matrix, play a critical role in the processes of tumor invasion, migration, and metastasis. To determine whether FOXQ1 downregulation affected MMP-2/9 levels, we applied a western blot to FOXQ1 siRNA-transfected cells to examine the expression of MMP-2/9. As expected, we observed a significant reduction in MMP-2/9 expression after RNAi-mediated FOXQ1 knockdown (Figure 3D). Consistent with this result, the activity of MMP-2/9 in HCT116 and LOVO cells was also markedly decreased by siRNA mediated knockdown of FOXQ1 (Supplementary Figure 2). In summary, these results suggested that FOXQ1-dependent inhibition of colorectal cancer cell migration and invasion was accompanied by reduced the level of metastasis molecules and MMPs.
Figure 3.

Knock-down FOXQ1 hinders cellular migration and invasion in colorectal cancer cells. A. LOVO (left panel) and HCT116 (right panel) cells were transfected with either control siRNA or FOXQ1-targeted siRNAs for 48 h prior to wound healing assays. Bars show means ± SD of three independent experiments. B. LOVO (upper panel) and HCT116 (lower panel) cells were transfected with either control siRNA or FOXQ1-targeted siRNAs for 48 h prior to Transwell invasion assays. After 24 h, the cells invaded through the membrane were stained (left) and the number of cells was quantified in five random microscopic fields (right). The data were shown as mean ± SD. For indicated comparisons, *P < 0.05 and **P < 0.01. C. LOVO (left panel) and HCT116 (right panel) cells were seeded in six well plates and were transfected with control siRNA or FOXQ1 siRNA. After for 48 h post transfections, cells were subjected to immunoblotting assay for measuring protein levels of phosphor-FAK (Ser9), phosphor-AKT (Ser9) and phosphor-PI3K (Ser9), respectively. D. Control siRNA or siRNA against FOXQ1 were transfected into HCT116 (upper panel) and LOVO (lower panel) cells. After 48 h post transfections, western blot assay was performed to evaluate the expression levels of MMP-2 and MMP-9.
Overexpression of FOXQ1 promotes cellular migration and invasion in colorectal cancer cells
To determine the functional role of FOXQ1 in cellular migration and invasion, HCT116 and LOVO cells that stably expressed FOXQ1 were established and subjected to wound-healing migration and Transwell invasion assays. As shown in Figure 4A, ectopic expression of FOXQ1 in colorectal cancer cells markedly increased the cell migration rate compared with the rate of cells transfected with the control vector. In addition, cell invasion potential was examined using Matrigel-coated Transwell plates. As shown in Figure 4B, the invasive abilities of HCT116 and LOVO cells were significantly increased in cells that overexpressed FOXQ1. Furthermore, phosphorylation of numerous well-characterized metastasis-related kinases was shown to be increased in FOXQ1 overexpressing cells (Figure 4C). Additionally, a western blot assay showed that overexpression of FOXQ1 remarkably upregulated MMP-2/9 expression (Figure 4D) and activity (Supplementary Figure 3). Collectively, these results suggested that FOXQ1 upregulation promotes the aggressiveness of colorectal cancer cells in vitro.
Figure 4.

Up-regulation of FOXQ1 accelerates cellular migration and invasion in colorectal cancer cells. A. Based on the scratch migration assay, the migration ability of HCT116 and LOVO cells transfected with FOXQ1 was augmented. B. The invasion ability of HCT116 and LOVO cells transfected with FOXQ1 was increased. LOVO (upper panel) and HCT116 (lower panel) cells were transfected with either control vector or FOXQ1 for 48 h prior to Transwell invasion assay. Bars show means ± SD of three independent experiments. The number of cells was quantified and was shown in a column graph. *P < 0.05 and **P < 0.01, compared between FOXQ1 over-expression cells and control HCT116 cells. C. Control vector or FOXQ1 plasmid were transfected into HCT116 (upper panel) and LOVO (lower panel) cells. After 48 h post transfections, western blot assay shown that the over-expression FOXQ1 remarkably up-regulated phosphor-FAK (Ser9), phosphor-AKT (Ser9) and phosphor-PI3K (Ser9). D. Control vector or FOXQ1 plasmid were transfected into HCT116 (upper panel) and LOVO (lower panel) cells. After 48 h post transfections, western blot assay was carried out to evaluate the expression level of MMP-2 and MMP-9 in indicated cells.
Downregulation of FOXQ1 significantly inhibited cellular growth in vivo
To investigate the effect of FOXQ1 expression on colorectal cancer cell growth in vivo, HCT116 and LOVO cells with downregulated FOXQ1 were subcutaneously injected into the posterior flanks of nude mice. Xenograft tumor volumes were measured every 3 d when they were palpable. On day 21 after implantation, tumors from FOXQ1-knockdown cells were significantly smaller than those from control cells (Figure 5A and 5B). Nude mice were euthanized on day 21 after implantation, and xenografts were collected and weighed (Supplementary Figure 4). When compared with controls, the average tumor weights from the FOXQ1-downregulated cells were found to be reduced by 80%. Consistent with the in vitro results, CD44, PCNA, and mTOR expression was significantly decreased in tumors derived from cells transfected with FOXQ1 siRNA (Figure 5C). The results showed that downregulation of FOXQ1 significantly suppressed tumor growth relative to that of control cells.
Figure 5.
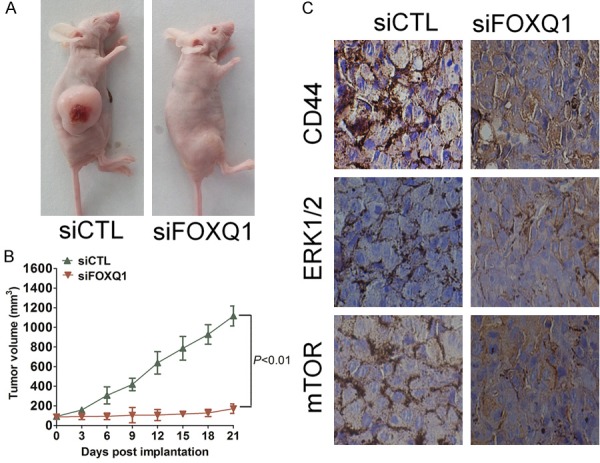
Down-expression of FOXQ1 significantly inhibits colorectal cancer cells growth in vivo. A. Control siRNA or siRNA against FOXQ1 were transfected into HCT116 cells and then cells was subcutaneously injected into back flank of nude mice (n = 6 each). Representative pictures of tumor in control siRNA or FOXQ1 siRNA HCT116 cell-inoculated nude mice. B. Tumor growth kinetics (mean ± SD) of FOXQ1 siRNA cells versus control HCT116 cells in nude mice (n = 6 each). The tumor volumes were measured at the indicated number of days after mice were transplanted with indicated cells. For indicated comparison, **P < 0.01. C. 21 days after xenograft implantation, mice were sacrificed. HCT116 colorectal cancer cells xenograft tumor sections were subjected to immunohistochemical analysis for CD44 (upper panel), ERK1/2 (middle panel) and mTOR (lower panel).
Upregulation of FOXQ1 significantly increased tumor growth in vivo
To further determine the effect of FOXQ1 on colorectal carcinoma cell growth in vivo, we then xenografted the following cell types into nude mice: HCT116 cells transfected with the FOXQ1 or control plasmid. As shown in Figure 6A, tumors derived from cells overexpressing FOXQ1 exhibited a greater size and mass than those derived from control cells. We found that upregulation of FOXQ1 markedly increased tumor volume and weight compared to tumors in counterpart mice injected with control cells (Figure 6B). Nude mice were sacrificed on day 21 after implantation, and xenografts were collected and weighed (Supplementary Figure 5). When compared with control mice, the average tumor weights from the FOXQ1-overexpressing cells were increased by 60%. We observed similar results as those obtained in vitro: immunohistochemistry analysis revealed that CD44, ERK1/2, and mTOR staining was significantly increased in tumors derived from cells that overexpressed FOXQ1 (Figure 6C). These results strongly indicate FOXQ1 overexpression contributed to colon carcinoma growth in vivo.
Figure 6.

Over-expression of FOXQ1 contributes cellular growth in vivo. A. Control vector or FOXQ1 was transfected into HCT116 cells and then cells was subcutaneously injected into back flank of nude mice. Representative images of tumor-bearing mice after inoculated control vector or FOXQ1 transfected HCT116 cells. B. Tumor growth kinetics (mean ± SD) of FOXQ1-over-expressiong versus vector control HCT116 cells in nude mice (n = 6 each). The tumor volumes were measured at the indicated number of days after mice were transplanted with indicated cells. For indicated comparison, **P < 0.01. C. 21 days after xenograft implantation, mice were sacrificed. Representative CD44 (upper panel), ERK1/2 (middle panel) and mTOR (lower panel) immunohistochemistry staining of HCT116 colorectal cancer cells graft grown in nude mice.
Discussion
Colorectal cancer remains a major clinical challenge because of poor prognosis and limited treatment options that prevent recurrence [13]. The oncogenesis of colorectal cancer involves changes in multiple oncogenes and suppressor genes. Therefore, many molecular biomarkers can be utilized for viable approaches to improving cancer prognosis and treatment [14]. Our study showed the role of a specific tumor-promotion protein, FOXQ1, in colorectal cancer. Therefore, integrated studies on the contribution of FOXQ1, with its multifunctional properties, to tumorigenesis will be important. In the current study, we found that FOXQ1 might play an important role in malignant progression of colon cancer and regulation of the FAK/PI3K/AKT signaling pathway. Preparation of a microRNA database and western blot analysis revealed that FOXQ1 was significantly upregulated in colon cancer tissues and cancer cell lines. Overexpression of FOXQ1 augmented the anchorage-independent growth and invasive abilities of human colon cancer cells, promoting their growth in vivo. Downregulation of FOXQ1 hindered cellular migration and invasion of colon cancer cells, as well as tumor growth in vivo. These findings provide novel insights into the potential roles of FOXQ1 dysregulation in promoting carcinogenesis and progression of colorectal cancer.
FOXQ1 expression levels are significantly higher in primary cancers than in matched noncancerous tissues [15], which suggests that downregulation of FOXQ1 in colorectal cancer might regulate the initiation of colorectal cancer [16]. Next, we investigated the role of FOXQ1 in the proliferation of cancer cells. When FOXQ1 was silenced in HCT116 and LOVO cells, the proliferation of cells decreased. In nude mice xenograft experiments, we found that downregulation of FOXQ1 significantly inhibited cellular growth in vivo. CD44, ERK1/2, and mTOR are reported to play fundamental roles in many biological processes, including cell proliferation [17]. Herein, we found that silencing FOXQ1 significantly inhibited the expression of various target genes that specifically regulation proliferation of colorectal cancer cells. Additionally, overexpression of FOXQ1 augmented the anchorage-independent growth and proliferation abilities of colorectal cancer cells in vitro, suggesting a potential role for FOXQ1 dysregulation in carcinogenesis and colon carcinoma progression.
Clinical specimens from ovarian cancer metastases have shown strong FOXQ1 immunostaining, in contrast to noninvasive borderline tumors, which do not express FOXQ1, and benign ovarian tissue, which expresses little to no FOXQ1 [18]. However, blockade of FOXQ1 signaling, which does not affect primary tumor growth, can enhance spontaneous metastasis of breast carcinoma to both the lung and bone. Given the pro-metastasis and anti-metastasis potential of FOXQ1 in cancer progression, an in vitro model that allows the role of FOXQ1 in the migration and invasion of colorectal cancer cells to be characterized may provide insight into the etiologic role of FOXQ1 in colorectal carcinoma metastasis [19]. We found, in wound healing and migration assays, that metastasis of HCT116 and LOVO human colon cancer cells was significantly decreased when the FOXQ1 gene was disrupted by siRNA. In contrast, overexpression of FOXQ1 greatly enhanced migration and invasion of HCT116 and LOVO cells. The FAK/PI3K/AKT pathway is involved in many cellular processes, including proliferation, differentiation, apoptosis, cell cycle progression, cell motility, tumorigenesis, tumor growth, and angiogenesis. PI3K is a member of the intracellular lipid kinase family, which catalyzes the generation of phosphatidylinositol-3,4,5-triphosphate (PIP3) from phosphatidylinositol-4,5-triphosphate (PIP2) [20]. Activated AKT translocates to the nucleus and activates mTOR and downstream targets [21,22]. Phosphorylated PI3K/AKT also acts as a scaffold, primarily regulating focal signaling related to cell adhesion to the extracellular matrix and MMP-mediated matrix degradation [23,24]. Several studies have indicated that the PI3K/AKT signaling pathway is involved in regulation of MMP-2 and MMP-9 activity [25-27]. Our results showed that overexpression of FOXQ1 in HCT116 and LOVO cells upregulates phosphorylation of FAK/PI3K/AKT, and FOXQ1 silencing reduces FAK/PI3K/AKT phosphorylation. Our findings identify FOXQ1 as a potential enhancer of metastatic potential in colorectal cancer.
Collectively, our results show that FOXQ1 was markedly upregulated in colorectal carcinoma cells and clinical colorectal cancer samples, which might be significant in colorectal cancer diagnosis. Moreover, overexpression of FOXQ1 increased colorectal carcinoma aggressiveness, including cell proliferation, migration, and invasion in vitro and growth in vivo, and activated the FAK/PI3K/AKT signaling pathway. Understanding the biological function of FOXQ1 in colorectal carcinoma progression both advances our knowledge of the mechanisms that underlie colorectal carcinoma tumorigenesis and establishes FOXQ1 as a potential therapeutic target for clinical management of and therapies for human colorectal cancers [28].
Acknowledgements
This research were funded by the National science foundation of China (81402523, 81672990 to WXY), Jiangsu Province “Six talents” high peak plan (2015-WSN-052 to WXY), Project of Jiangsu Provincial Bureau of traditional Chinese Medicine (JD201510 to WXY), the six “1” Project of Jiangsu Province (LGY2016012 to WXY).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Hu L, Wang RY, Cai J, Feng D, Yang GZ, Xu QG, Zhai YX, Zhang Y, Zhou WP, Cai QP. Overexpression of CHKA contributes to tumor progression and metastasis and predicts poor prognosis in colorectal carcinoma. Oncotarget. 2016;7:66660–66678. doi: 10.18632/oncotarget.11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JR, Kwon CH, Choi Y, Park HJ, Kim HS, Jo HJ, Oh N, Park do Y. Transcriptome analysis of paired primary colorectal carcinoma and liver metastases reveals fusion transcripts and similar gene expression profiles in primary carcinoma and liver metastases. BMC Cancer. 2016;16:539. doi: 10.1186/s12885-016-2596-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu C, Li M, Hu Y, Shi N, Yu H, Liu H, Lian H. miR-486-5p attenuates tumor growth and lymphangiogenesis by targeting neuropilin-2 in colorectal carcinoma. Onco Targets Ther. 2016;9:2865–2871. doi: 10.2147/OTT.S103460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi YH, Zhao DM, Wang YF, Li X, Ji MR, Jiang DN, Xu BP, Zhou L, Lu CZ, Wang B. The association of three promoter polymorphisms in interleukin-10 gene with the risk for colorectal cancer and hepatocellular carcinoma: a meta-analysis. Sci Rep. 2016;6:30809. doi: 10.1038/srep30809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Chen Y, Dai C, Shang Y, Xie J. Ginsenoside Rh2 alleviates tumor-associated depression in a mouse model of colorectal carcinoma. Am J Transl Res. 2016;8:2189–2195. [PMC free article] [PubMed] [Google Scholar]
- 6.Wu L, Shi B, Huang K, Fan G. MicroRNA-128 suppresses cell growth and metastasis in colorectal carcinoma by targeting IRS1. Oncol Rep. 2015;34:2797–2805. doi: 10.3892/or.2015.4251. [DOI] [PubMed] [Google Scholar]
- 7.Yang B, Tan Z, Song Y. Study on the molecular regulatory mechanism of MicroRNA-195 in the invasion and metastasis of colorectal carcinoma. Int J Clin Exp Med. 2015;8:3793–3800. [PMC free article] [PubMed] [Google Scholar]
- 8.Abba M, Patil N, Rasheed K, Nelson LD, Mudduluru G, Leupold JH, Allgayer H. Unraveling the role of FOXQ1 in colorectal cancer metastasis. Mol Cancer Res. 2013;11:1017–1028. doi: 10.1158/1541-7786.MCR-13-0024. [DOI] [PubMed] [Google Scholar]
- 9.Peng X, Luo Z, Kang Q, Deng D, Wang Q, Peng H, Wang S, Wei Z. FOXQ1 mediates the crosstalk between TGF-beta and Wnt signaling pathways in the progression of colorectal cancer. Cancer Biol Ther. 2015;16:1099–1109. doi: 10.1080/15384047.2015.1047568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Liu Y, Zhang J, Cui X, Li G, Wang J, Ren H, Zhang Y. FOXQ1 promotes gastric cancer metastasis through upregulation of snail. Oncol Rep. 2016;35:3607–3613. doi: 10.3892/or.2016.4736. [DOI] [PubMed] [Google Scholar]
- 11.Fan DM, Feng XS, Qi PW, Chen YW. Forkhead factor FOXQ1 promotes TGF-beta1 expression and induces epithelial-mesenchymal transition. Mol Cell Biochem. 2014;397:179–186. doi: 10.1007/s11010-014-2185-1. [DOI] [PubMed] [Google Scholar]
- 12.Feng J, Zhang X, Zhu H, Wang X, Ni S, Huang J. FoxQ1 overexpression influences poor prognosis in non-small cell lung cancer, associates with the phenomenon of EMT. PLoS One. 2012;7:e39937. doi: 10.1371/journal.pone.0039937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khamas A, Ishikawa T, Shimokawa K, Mogushi K, Iida S, Ishiguro M, Mizushima H, Tanaka H, Uetake H, Sugihara K. Screening for epigenetically masked genes in colorectal cancer Using 5-Aza-2’-deoxycytidine, microarray and gene expression profile. Cancer Genomics Proteomics. 2012;9:67–75. [PubMed] [Google Scholar]
- 14.Zhang J, Zhu L, Fang J, Ge Z, Li X. LRG1 modulates epithelial-mesenchymal transition and angiogenesis in colorectal cancer via HIF-1alpha activation. J Exp Clin Cancer Res. 2016;35:29. doi: 10.1186/s13046-016-0306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan G, Yan S, Xue H, Zhang P, Sun J, Li G. JSI-124 suppresses invasion and angiogenesis of glioblastoma cells in vitro. PLoS One. 2015;10:e0118894. doi: 10.1371/journal.pone.0118894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su X, Wang J, Chen W, Li Z, Fu X, Yang A. Overexpression of TRIM14 promotes tongue squamous cell carcinoma aggressiveness by activating the NF-kappaB signaling pathway. Oncotarget. 2016;7:9939–9950. doi: 10.18632/oncotarget.6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuang J, Li L, Guo L, Su Y, Wang Y, Xu Y, Wang X, Meng S, Lei L, Xu L, Shao G. RNF8 promotes epithelial-mesenchymal transition of breast cancer cells. J Exp Clin Cancer Res. 2016;35:88. doi: 10.1186/s13046-016-0363-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pass HI, Lavilla C, Canino C, Goparaju C, Preiss J, Noreen S, Blandino G, Cioce M. Inhibition of the colony-stimulating-factor-1 receptor affects the resistance of lung cancer cells to cisplatin. Oncotarget. 2016;7:56408–56421. doi: 10.18632/oncotarget.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian X, Yu J, Yin Y, He J, Wang L, Li Q, Zhang LQ, Li CY, Shi ZM, Xu Q, Li W, Lai LH, Liu LZ, Jiang BH. MicroRNA-143 inhibits tumor growth and angiogenesis and sensitizes chemosensitivity to oxaliplatin in colorectal cancers. Cell Cycle. 2013;12:1385–1394. doi: 10.4161/cc.24477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi ZM, Wang J, Yan Z, You YP, Li CY, Qian X, Yin Y, Zhao P, Wang YY, Wang XF, Li MN, Liu LZ, Liu N, Jiang BH. MiR-128 inhibits tumor growth and angiogenesis by targeting p70S6K1. PLoS One. 2012;7:e32709. doi: 10.1371/journal.pone.0032709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarfati D, Shaw C, McLeod M, Blakely T, Bissett I. Screening for colorectal cancer: spoiled for choice? N Z Med J. 2016;129:120–128. [PubMed] [Google Scholar]
- 22.Al-Maghrabi J, Emam E, Gomaa W, Al-Qaydy D, Al-Maghrabi B, Buhmeida A, Abuzenadah A, Al-Qahtani M, Al-Ahwal M. Overexpression of PAK-1 is an independent predictor of disease recurrence in colorectal carcinoma. Int J Clin Exp Pathol. 2015;8:15895–15902. [PMC free article] [PubMed] [Google Scholar]
- 23.Liang SH, Yan XZ, Wang BL, Jin HF, Yao LP, Li YN, Chen M, Nie YZ, Wang X, Guo XG, Wu KC, Ding J, Fan DM. Increased expression of FOXQ1 is a prognostic marker for patients with gastric cancer. Tumour Biol. 2013;34:2605–2609. doi: 10.1007/s13277-013-0808-x. [DOI] [PubMed] [Google Scholar]
- 24.Christensen J, Bentz S, Sengstag T, Shastri VP, Anderle P. FOXQ1, a novel target of the Wnt pathway and a new marker for activation of Wnt signaling in solid tumors. PLoS One. 2013;8:e60051. doi: 10.1371/journal.pone.0060051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang XJ, Jia SS. Fisetin inhibits laryngeal carcinoma through regulation of AKT/NF-kappaB/mTOR and ERK1/2 signaling pathways. Biomed Pharmacother. 2016;83:1164–1174. doi: 10.1016/j.biopha.2016.08.035. [DOI] [PubMed] [Google Scholar]
- 26.Huang W, Chen Z, Shang X, Tian D, Wang D, Wu K, Fan D, Xia L. Sox12, a direct target of FoxQ1, promotes hepatocellular carcinoma metastasis through up-regulating twist1 and FGFBP1. Hepatology. 2015;61:1920–1933. doi: 10.1002/hep.27756. [DOI] [PubMed] [Google Scholar]
- 27.Sun HT, Cheng SX, Tu Y, Li XH, Zhang S. FoxQ1 promotes glioma cells proliferation and migration by regulating NRXN3 expression. PLoS One. 2013;8:e55693. doi: 10.1371/journal.pone.0055693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Hong Z, Zhang RR, Sun XZ, Yuan YF, Hu J, Wang X. Bakkenolide A inhibits leukemia by regulation of HDAC3 and PI3K/Akt-related signaling pathways. Biomed Pharmacother. 2016;83:958–966. doi: 10.1016/j.biopha.2016.07.049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.