

Abstract
Microglial over-activation and apoptosis are associated with ischemic brain diseases. These processes may be hindered by oxysophocarpine (OSC) that generates anti-inflammatory and anti-apoptotic activities. However, the precise roles of OSC in microglial inflammation and apoptosis induced by oxygen-glucose deprivation/reoxygenation (OGD/R) remain unclear. In this study, we found that OSC reduced OGD/R-induced inflammation in BV-2 microglia. OSC elevated cell viability and prevented the release of lactate dehydrogenase. OSC downregulated cyclooxygenase 2 and inducible nitric oxide synthase and reduced the levels of inflammatory mediators, including tumor necrosis factor-α, interleukin (IL)-1β, IL-6, monocyte chemoattractant protein-1, prostaglandin E2, and nitric oxide. OSC inhibited the expression of Toll-like receptor 4 (TLR4) and myeloid differentiation protein 88 (MyD88) and blocked the activation of nuclear factor (NF)-κB. In addition, OSC suppressed OGD/R-elicited BV-2 cell apoptosis, as indicated as follows: The restored mitochondrial membrane potential and the reduced caspase-3 activity; the decrease of Bax and cleaved caspase-3 and the increase of Bcl-2; the enhanced phosphorylation of Akt and mTOR. These results implied that OSC impedes OGD/R-induced inflammation and apoptosis of microglial cells. Therefore, OSC may be potentially used for ischemic stroke therapy.
Keywords: Oxysophocarpine, oxygen-glucose deprivation, inflammation, apoptosis, microglia
Introduction
Stroke is the second leading cause of morbidity and mortality worldwide, and ischemic stroke accounts for 80% of total cases [1]. The pathophysiology of ischemic stroke involves a complex series of pathological events, including oxidative stress, inflammation, and apoptosis [2,3]. This disease can be treated by relatively few therapeutic options, such as surgery and anticoagulation. Therefore, discovering novel and effective therapy strategies to hinder ischemic stroke is necessary and urgent.
Microglia, resident macrophage-like cells of the central nervous system, play essential roles in immunological surveillance, foreign invader removal, and brain homeostasis maintenance [4]. Microglia become activated and injured under ischemic and hypoxic conditions [5]. Activated microglia increase the expression of cyclooxygenase 2 (COX2) and inducible nitric oxide synthase (iNOS); these cells also intensively release inflammatory mediators, including tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, IL-6, monocyte chemoattractant protein-1 (MCP-1), prostaglandin E2 (PGE2), and nitric oxide (NO) [5,6]. These uncontrolled inflammatory responses are involved in microglial dysfunction and death in various brain diseases, including ischemic stroke [7,8]. Toll-like receptor 4 (TLR4), primarily expressed in the microglia, evokes inflammatory chain reactions and regulates microglial activation [9]. TLR4/myeloid differentiation protein 88 (MyD88)/nuclear factor (NF)-κB signaling contributes to BV-2 cell injury induced by oxygen and glucose deprivation/reoxygenation (OGD/R) [10-12]. Hypoxia/reperfusion induces microglial apoptosis via a phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR)-regulated autophagy pathway [13]. Thus, preventing microglial over-activation and apoptosis is a potential treatment strategy for ischemic stroke.
Oxysophocarpine (OSC) is an alkaloid extracted from Sophora flavescentis radix and Subprostrate sophora root. OSC has various pharmacological activities, including anti-nociceptive [14], anti-viral [15], anti-inflammatory [16], and anti-apoptotic effects [17]. OSC ameliorates inflammatory pain by reducing the production of TNF-α, IL-1β, IL-6, COX-2, and PGE2 [16]. OSC also elicits significant protective effects on the OGD/R-induced hippocampal neuron injury by inhibiting apoptosis [17]. However, the effects of OSC on OGD/R-induced microglial inflammation and apoptosis remain unclear.
In this study, OSC prevented BV-2 cells from OGD/R-induced injury, as evidenced by the increased cell viability and decreased lactate dehydrogenase (LDH) release. OSC reduced the levels of inflammatory mediators in OGD/R-stimulated BV-2 cells. The anti-inflammatory effects of OSC on OGD/R-exposed BV-2 cells were partly mediated by the TLR4/MyD88/NF-κB pathway. Moreover, OSC increased the phosphorylation of Akt and mTOR in OGD/R-challenged BV-2 cells. Pretreatment with LY294002 or rapamycin attenuated OSC-mediated pro-survival and anti-apoptotic effects, suggesting that PI3K/Akt/mTOR signaling is possibly implicated in OGD/R-induced microglial apoptosis. Thus, OSC may be a potential novel therapeutic agent for ischemic stroke with microglial over-activation.
Materials and methods
Reagents and antibodies
OSC was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China) and dissolved in sterile PBS (pH 7.4) before use. The purity of OSC was above 99% and its chemical structure is shown in Figure 1. All reagents for cell culture were obtained from Gibco BRL (Grand Island, NY, USA). Enzyme-linked immunosorbent assay (ELISA) kits of mouse TNF-α, IL-1β, IL-6, MCP-1, and PGE2 were obtained from R&D Systems (Minneapolis, MN, USA). Rabbit anti-mouse COX-2, iNOS, pro-caspase-3, and cleaved (cl)-caspase-3 polyclonal antibodies (pAbs) were from Santa Cruz Biotechnology (Dallas, TX, USA). Rabbit anti-mouse NF-κB p65, inhibitor of κB (IκB)-α, phosphorylated (p)-IκB-α (Ser32/36) Bax, Bcl-2, Akt, and p-Akt (Ser473) pAbs were supplied by Cell Signaling Technology, Inc. (Danvers, MA, USA). Rabbit anti-mouse TLR4, MyD88, mTOR, and p-mTOR (Ser2448) were purchased from Abcam (Cambridge, UK). Rabbit anti-mouse β-actin and Lamin B monoclonal antibodies (mAbs) were procured from Sigma-Aldrich (St. Louis, MO, USA). Horseradish peroxidase (HRP)-conjugated anti-rabbit IgG was from Chemicon (Temecula, CA, USA). All other chemicals were of analytical grade and obtained from Sigma unless otherwise stated.
Figure 1.
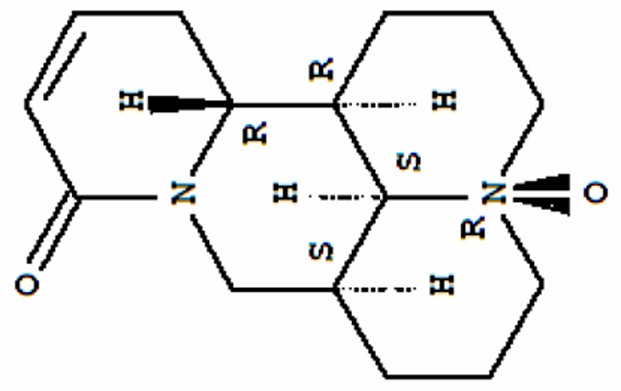
Chemical structure of oxysophocarpine.
Microglial cell culture and OGD/R treatment
An immortalized mouse microglial BV-2 cell line was purchased from American Type Culture Collection (Rockville, MD, USA) and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C and 5% CO2 in a humidified incubator. The OGD/R model was performed as previously described [18]. Briefly, BV-2 cells were digested into a cell suspension, and the cell density was adjusted to 5 × 104 cells/mL. After 80% confluence, the cells were washed thrice with PBS, switched from a normal feeding medium to an oxygen-depleted, glucose-free medium, and incubated in a hypoxic chamber with CO2/N2 (5%/95%) for 3 h. The cultures were then returned to the normal feeding medium and incubated under normal conditions for 0 h, 12 h, 24 h, and 48 h as reperfusion. OSC (1, 3, and 5 μM) was given immediately after the OGD phase, acting through the processes of reoxygenation. The cells with neither OGD/R nor OSC treatment were used as the control group. In blocking experiments, the cells were treated with LY294002 (10 μΜ; PI3K/Akt inhibitor) or rapamycin (30 nM; mTOR inhibitor) for 30 min prior to OGD stimulation.
Cell viability assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed to measure the viability of BV-2 cells. Briefly, after various treatments, the cells were seeded in a 96-well plate at a density of 1 × 104 cells/well. MTT (5 mg/mL) was added into each well and incubated at 37°C for 4 h, followed by 100 μL of dimethyl sulfoxide to dissolve the formazan crystals. The optical density was determined at 570 nm using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA). Cell viability was expressed as a percentage of the control.
LDH assay
Cell damage was detected using a commercial LDH kit in accordance with the manufacturer’s protocol (Nanjing Jianchen Bioengineering Institute, Nanjing, China). The LDH level in the medium was measured to reflect cell membrane integrity. In brief, after the indicated treatments, 20 μL of cell culture medium was added to 300 μL of LDH reagent. Absorbance was recorded at 440 nm using a microplate reader (Bio-Rad).
Quantitative real-time PCR (qPCR)
Total RNA of the BV-2 cells was isolated with a TRIzol commercial kit (Invitrogen, Carlsbad, CA, USA). The cDNA was synthesized using a reverse transcriptase kit (Promega, Madison, WI, USA). Quantitative PCR was performed using SYBR green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). The relative mRNA expressions were determined by 2-ΔΔCt method and calculated after normalization to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primer sequences are listed in Table 1.
Table 1.
Primers used for qPCR
| Gene | Primer sequences (5’ to 3’) | |
|---|---|---|
| TNF-α | Forward | CAAGGGACA AGGCTGCCCCG |
| Reverse | GCAGGGGCTCTTGACGGCAG | |
| IL-1β | Forward | AAGCCTCGTGCTGTCGGACC |
| Reverse | TGAGGCCCAAGGCCACAGGT | |
| IL-6 | Forward | GTACTCCAGAAGACCAGAGG |
| Reverse | TGCTGGTG ACAACCACGGCC | |
| MCP-1 | Forward | CCAGCACCAGCACCAGCCAA |
| Reverse | TGGATGCTCCAGCCGGCAAC | |
| COX2 | Forward | GATGACTGCCCAACTCCC |
| Reverse | AACCCAGGTCCTCGCTTA | |
| iNOS | Forward | CAGCTGGGCTGTACAAACCTT |
| Reverse | CATTGGAAGTGAAGCGTTTCG | |
| GAPDH | Forward | GCCAAGGCTGTGGGCAAGGT |
| Reverse | TCTCCAGGCGGCACGTCAGA | |
ELISA
The levels of inflammatory mediators in the supernatants of BV-2 cells were determined by ELISA kits in accordance with the manufacturer’s instructions. Briefly, the cells were seeded in a 6-well plate at a density of 1 × 106 cells/well. After the indicated treatments, the supernatants were harvested for measuring the levels of TNF-α, IL-1β, IL-6, MCP-1, and PGE2. The absorbance was read at 490 nm on a microplate reader (Bio-Rad).
Nitrite assay
As an indicator of NO production, the nitrite level was assessed using a colorimetric assay (Promega, Madison, WI, USA). In brief, after the various treatments, the supernatants were collected. Equal volumes of supernatant and Griess reagent were mixed and incubated for 10 min at room temperature in the dark. The absorbance at 540 nm was assayed with a microplate reader (Bio-Rad). The concentration of nitrite in samples was determined from a sodium nitrite standard curve.
Western blot analysis
BV-2 cells were lysed using a lysis buffer (Cell Signaling Technology). Cytoplasmic and nuclear proteins were isolated using NE-PERTM Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology, Rockford, IL, USA). Equal amount of protein in each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then electrotransferred onto polyvinylidene fluoride membranes (Millipore, CA, USA). The membrane was blocked with 5% skimmed milk for 2 h at 37°C and incubated at 4°C overnight with the corresponding primary antibodies. Lamin B and β-actin were used as the loading controls. After being washed thrice, the membrane was incubated in the HRP-conjugated secondary antibody for 2 h at 37°C. The reactions were visualized using enhanced chemiluminescence detection reagents (Pierce). The band intensities were quantified using Image J software (NIH, USA).
Flow cytometry analysis
BV-2 cell apoptosis were assessed using Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining. In brief, after the different treatments, the cells were washed twice with cold PBS and resuspended at a concentration of 1 × 106 cells/mL in the binding buffer. Afterward, approximately 10 μL of ready-to-use Annexin V-FITC (R&D Systems) was added to the mixture, incubated at 37°C for 15 min, and counterstained with 5 μL of PI in the dark for 30 min. Cell apoptosis was analyzed on a FACScan flow cytometer with CellQuest software (BD Bioscience, MA, USA).
Mitochondrial membrane potential (MMP) assay
MMP was determined using the lipophilic cationic fluorescence probe, 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1), which exhibits potential-dependent accumulation in the mitochondria. The probe exhibits a red fluorescence when MMP increases and shifts to green fluorescence when the MMP decreases. In brief, after the indicated treatments, the cells were treated with JC-1 (5 μg/mL) for 1.5 h at 37°C in the dark and washed twice with PBS. Fluorescence intensity was measured at 485 nm excitation and 585 and 538 nm emission using a multimode microplate reader (Bio-Rad). The values were expressed as the OD585/OD538 ratio.
Caspase-3 activity assay
Caspase-3 activity was determined using a colorimetric assay kit (Promega, Madison, WI, USA). The activity was assessed by the production of the p-nitroanilide (pNA), which was cleaved from Ac-DEVD-pNA by cleaved caspase-3. Briefly, after the indicated treatments, 1 × 106 cells were harvested, washed with PBS, and lysed in 50 μL lysing buffer. Then, 10 μL of cell lysates were mixed with 10 μL Ac-DEVD-pNA (2 mM) and 80 μL reaction buffer. After incubation for 2 h at 37°C, the release of pNA was measured at 405 nm with a microplate reader (Bio-Rad).
Statistical analysis
The data were expressed as the means ± standard deviation (SD) and analyzed by SPSS 16.0 software (SPSS, Chicago, IL, USA). Statistical analysis was performed using one-way ANOVA followed by Tukey post hoc test. Differences at P < 0.05 were considered statistically significant.
Results
OSC protected BV-2 cells against OGD/R-induced cell injury
We first investigated whether OSC exerts protective effects against OGD/R insult in BV-2 cells. We found that OSC (1, 3, and 5 μM) increased the viability of the OGD/R-treated BV-2 cells partly in a dose- and time-dependent manner (Figure 2A and 2B). Consistently, LDH assays showed that OSC resulted in a marked decrease of LDH release in OGD/R-stimulated BV-2 cells (Figure 2C and 2D). However, no significant differences exist in 3 μM and 5 μM groups and 24 h and 48 h groups, respectively (Figure 2A-D). Thus, 3 μM plus 24 h group was selected for the subsequent experiments. These results indicated that OSC attenuated OGD/R-led BV-2 cell damage.
Figure 2.
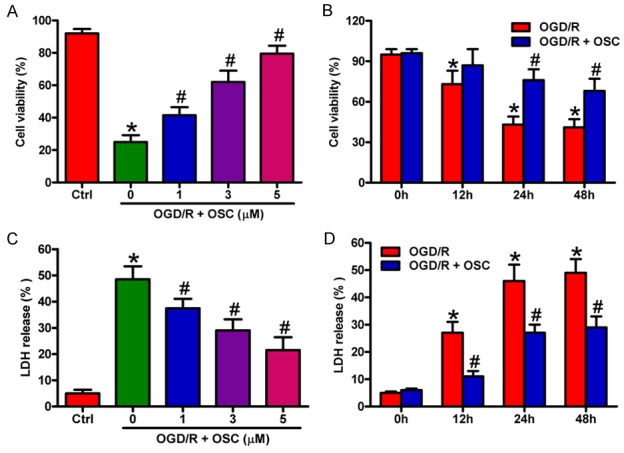
OSC improved the viability and reduced the LDH release in OGD/R-treated BV-2 cells. A and C. BV-2 cells were exposed to OGD for 3 h followed by 24 h of reoxygenation. OSC (1, 3, and 5 μM) was added to the medium immediately after the OGD phase. A. Cell viability was measured by MTT assay. C. LDH release was assessed. B and D. BV-2 cells were exposed to OGD for 3 h followed by 0, 12, 24, and 48 h of reoxygenation. OSC (3 μM) was added to the medium immediately after the OGD phase. B. Cell viability was measured by MTT assay. D. LDH release was assessed. Data are derived from three independent experiments and expressed as means ± SD. *P < 0.05 vs. control (Ctrl) group; #P < 0.05 vs. OGD/R group.
OSC reduced inflammatory mediators in OGD/R-insulted BV-2 cells
OSC inhibits the inflammatory responses in carrageenan-induced pain [16]. Here, we investigated the effect of OSC on OGD/R-induced inflammation in BV-2 cells. qPCR results showed that OGD/R exposure increased the mRNA expression of the pro-inflammatory factors in BV2 cells, including TNF-α (Figure 3A), IL-1β (Figure 3B), IL-6 (Figure 3C), MCP-1 (Figure 3D), COX-2 (Figure 3E), and iNOS (Figure 3F), which was markedly attenuated by OSC. Accordantly, the levels of TNF-α (Figure 3F), IL-1β (Figure 3G), IL-6 (Figure 3H), MCP-1 (Figure 3I), PGE2 (Figure 3J), and NO (Figure 3K) in the supernatants of OGD/R-stimulated cells were remarkably elevated, and these increases were counteracted by OSC. These data suggested that OSC suppressed OGD/R-induced inflammatory responses in BV-2 cells.
Figure 3.

OSC reduced inflammatory mediators in the OGD/R-challenged BV-2 cells. BV-2 cells were exposed to OGD for 3 h followed by 24 h of reoxygenation. OSC (3 μM) was added to the medium immediately after the OGD phase. (A-F) mRNA expression of TNF-α (A), IL-1β (B), IL-6 (C), MCP-1 (D), COX-2 (E), and iNOS (F) was detected by qPCR assay. (G-K) Levels of TNF-α (G), IL-1β (H), IL-6 (I), MCP-1 (J), and PGE2 (K) in the supernatants were measured by ELISA. (L) Nitrite production was measured using Griess reagent. The data are derived from three independent experiments and expressed as means ± SD. *P < 0.05 vs. Ctrl group; #P < 0.05 vs. OGD/R group.
OSC inhibited OGD/R-induced inflammatory response by blocking TLR4 signaling in BV-2 cells
To further investigate the inhibitory effect of OSC on inflammatory response, we detected the protein expression of COX-2 and iNOS. Western blot results showed that COX-2 and iNOS protein levels were significantly decreased by OSC in OGD/R-treated BV-2 cells (Figure 4A and 4B). NF-κB is a critical transcription activator responsible for the expression of inflammatory mediators, including TNF-α, IL-1β, IL-6, COX-2, and iNOS [19]. Here, we detected the activation of NF-κB in OGD/R-exposed BV-2 cells. As shown in Figure 4C-G, NF-κB p65 was markedly accumulated in the nucleus of BV-2 cells after OGD/R exposure, accompanying with the decrease of IκB-α in the cytoplasm. However, OSC inhibited the OGD/R-induced degradation of IκB-α and nuclear translocation of NF-κB p65. TLR4/MyD88/NF-κB signaling is involved in OGD/R-caused BV-2 cell injury [10-12]. In this study, we found that OGD/R markedly upregulated TLR4 and MyD88, which was attenuated by OSC (Figure 4H-J). These results suggested that TLR4/MyD88/NF-κB signaling contributed to the inhibitory effects of OSC on OGD/R-induced inflammation in BV-2 cells.
Figure 4.

OSC inhibited OGD/R-induced inflammatory responses in BV-2 cells by inactivating the TLR4/MyD88/NF-κB pathway. BV-2 cells were exposed to OGD for 3 h followed by 24 h of reoxygenation. OSC (3 μM) was added to the medium immediately after the OGD phase. (A) Representative Western blots showing the expression of COX-2 and iNOS. β-actin was used as the endogenous control. (B) Quantitative analyses of COX-2 and iNOS levels in (A). (C-G) Representative Western blot results of (C) and quantitative data (D-G) on the nuclear level of NF-κB p65 and cytosolic levels of NF-κB p65, p-IκB-α, and IκB-α. β-actin and Lamin B were used as the endogenous controls, respectively. (H-J) Representative Western blot results of (H) and quantitative data (I and J) on the expression of TLR4 and MyD88. β-actin was used as the endogenous control. Data are derived from three independent experiments and expressed as means ± SD. *P < 0.05 vs. Ctrl group; #P < 0.05 vs. OGD/R group.
OSC hindered OGD/R-triggered apoptosis of BV-2 cells
Next, we investigated the protective effects of OSC on BV-2 cell apoptosis. Flow cytometry results showed that the percentage of apoptotic cells increased after OGD/R treatment, which was reduced by OSC (Figure 5A). MMP is known as a critical factor determining mitochondrial integrity, and MMP loss can lead to cell apoptosis. As shown in Figure 5B, OGD/R exposure induced a significant loss of MMP, whereas OSC resulted in a considerable restoration of MMP. In addition, OSC attenuated the increase in caspase-3 activity induced by OGD/R stimulation (Figure 5C). To investigate the underlying mechanisms involved in OSC-reduced apoptosis, we analyzed the expressions of apoptotsis-related molecules, including pro-apoptotic proteins (pro-caspase-3, cl-caspase-3, and Bax), and the anti-apoptotic protein (Bcl-2). OGD/R markedly increased cl-caspase-3 level and Bax/Bcl-2 ratio, which were attenuated by OSC (Figure 5D-F). These results indicated that OSC could protect against OGD/R-induced BV-2 cell apoptosis.
Figure 5.

OSC suppressed OGD/R-elicited BV-2 cell apoptosis. BV-2 cells were exposed to OGD for 3 h followed by 24 h of reoxygenation. OSC (3 μM) was added to the medium immediately after OGD phase. (A) BV-2 cell apoptosis was analyzed by flow cytometry. The percentage of apoptotic cells was calculated. (B) MMP was measured by JC-1 staining. (C) Caspase-3 activity was determined to evaluate cell apoptosis. (D) Western blot analyses of pro-caspase-3, cl-caspase-3, Bax, and Bcl-2 expression. β-actin was used as loading control. (E and F) Quantitative ratios of cl-caspase-3/pro-caspase-3 (E) and Bax/Bcl-2 (F). The data are derived from three independent experiments and expressed as means ± SD. *P < 0.05 vs. Ctrl group; #P < 0.05 vs. OGD/R group.
Pro-survival effects of OSC on OGD/R-treated BV-2 cells were dependent on PI3K/Akt/mTOR signaling
The PI3K/Akt/mTOR pathway plays a key role in microglial cell injury during hypoxia/reperfusion [13]. We investigated whether the PI3K/Akt/mTOR signaling is involved in pro-survival effects of OSC on OGD/R-insulted BV-2 cells. As shown in Figure 6A-C, the phosphorylation of Akt and mTOR significantly increased after OGD/R treatment, which was further enhanced by OSC. By contrast, OSC did not elicit evident effects on the expression of total Akt and mTOR. Moreover, we found that the increased viability of BV-2 cells caused by OSC was abolished by PI3K/Akt inhibitor (LY294002) or mTOR inhibitor (rapamycin) pretreatment (Figure 6D) and the anti-apoptotic effect of OSC on BV-2 cells was weakened by treatment with LY294002 or rapamycin (Figure 6E). These results suggested that PI3K/Akt/mTOR axis partly mediated the pro-survival effects of OSC on OGD/R-exposed BV-2 cells.
Figure 6.

OSC promoted OGD/R-treated BV-2 cell survival by further activating the PI3K/Akt/mTOR pathway. (A-C) BV-2 cells were exposed to OGD for 3 h followed by 24 h of reoxygenation. OSC (3 μM) was added to the medium immediately after OGD phase. (A) Western blot was performed to analyze the expression of Akt, p-Akt, mTOR, and p-mTOR. β-actin was used as the loading control. (B and C) Quantitative ratios of p-Akt/Akt (B) and p-mTOR/mTOR (C). (D and E) BV-2 cells were exposed to OGD for 3 h followed by 24 h of reoxygenation with or without 30 min of pre-treatment of PI3K/Akt inhibitor LY294002 (10 μM) or mTOR inhibitor rapamycin (30 nM). OSC (3 μM) was added to the medium immediately after the OGD phase. (D) MTT assay was adopted to detect cell viability. (E) Cell apoptosis was measured by flow cytometry. Data are derived from three independent experiments and expressed as means ± SD. *P < 0.05 vs. Ctrl group; #P < 0.05 vs. OGD/R group; ★P < 0.05 vs. OGD/R + OSC group.
Discussion
This study illuminated that OSC protected against OGD/R-caused BV-2 microglia injury. The key findings were as follows. First, OSC increased cell viability and reduced LDH release in OGD/R-injured BV-2 cells. Second, OSC decreased inflammatory mediators in OGD/R-exposed BV-2 cells. Third, OSC suppressed the inflammatory responses in OGD/R-challenged BV-2 cells partly by blocking the TLR4/MyD88/NF-κB pathway. Fourth, OSC inhibited OGD/R-induced BV-2 cell apoptosis, as shown by the decrease in MMP loss and caspase-3 activity, the downregulation of pro-apoptotic proteins (cl-caspase-3 and Bax), and the upregulation of anti-apoptotic protein (Bcl-2). Lastly, the anti-apoptotic effects of OSC probably relied on the activation of PI3K/Akt/mTOR signaling. Collectively, OSC attenuated OGD/R-induced BV-2 cell damage by inhibiting inflammation and apoptosis.
Microglia plays a pivotal role in the pathogenesis of ischemic stroke. The excessive activation of microglia contributes to inflammation-mediated neurotoxicity through the release of inflammatory mediators, such as TNF-α, IL-1β, IL-6, PGE2, and NO [20-23]. TLR4 is involved in microglial activation under hypoxic conditions [24]. TLR4 activation triggers the recruitment of MyD88 and sequential activation of IκB kinase (IKK) complex. The activated IKK complex promotes the phosphorylation and degradation of IκB-α and the subsequent release of NF-κB. Then, NF-κB translocates to the nucleus and triggers the transcription of its downstream molecules, such as TNF-α, IL-6, COX-2, and iNOS [25,26]. OGD/R-induced phosphorylation and nuclear NF-κB translocation are mediated by TLR4/MyD88 signaling in BV-2 cells [9-11]. This study demonstrated that OSC reduced the OGD/R-induced upregulation of TLR4 and MyD88 and the activation of NF-κB and the production of its downstream inflammatory mediators in BV-2 cells, suggesting the protection of OSC against OGD/R-induced inflammatory responses.
Apoptosis is a critical event in many neurological disorders, including ischemic stroke. The inflammatory or apoptotic mediators produced by over-activated microglia may cause and exacerbate microglial apoptosis [12]. Lee et al. [27] reported that NO released by microglia may act as an autocrine mediator in the apoptosis of activated microglial cells. The excessive PGE2 can also induce microglial apoptosis [28]. The major components of the apoptotic pathway include caspase and Bcl-2 families [29]. Caspase-3 is a key executor of cell apoptosis. Increased cl-caspase-3 expression is a hallmark of apoptosis in brain diseases [30]. The relative expression of different Bcl-2 family members controls the sensitivity of cells to apoptotic stimuli [31]. Bax overexpression promotes apoptotic cell death, while heterodimer formation between Bax and Bcl-2 inhibits the death-promoting effects of Bax [32]. An increased Bax/Bcl-2 ratio induces cytochrome c release and caspase-3 activation. In this study, OSC reversed the increase of Bax and cl-caspase-3 and the decrease of Bcl-2 in OGD/R-exposed BV-2 cells, suggesting the intensively anti-apoptotic efficacy of OSC. PI3K/Akt signaling has been proposed as an essential strategy to prevent cell death [33]. Endogenous protection of microglia against OGD relies on the pro-survival PI3K/Akt/mTOR pathway because the pharmacological inhibition of mTOR or Akt or Akt silencing significantly exacerbates microglial apoptotic injury under hypoxia/reperfusion [13,34]. In this report, OGD/R insult markedly increased the phosphorylation levels of mTOR and Akt and OSC further enhanced the levels of p-mTOR and p-Akt in BV-2 cells. The pro-survival and anti-apoptotic effects of OSC on OGD/R-stimulated BV-2 cells were counteracted by LY294002 or rapamycin pretreatment. Thus, PI3K/Akt/mTOR signaling contributes to the protective effects of OSC on OGD/R-insulted BV-2 cells.
In summary, this study demonstrated that OSC attenuates OGD/R-induced BV-2 cell damage by its anti-inflammatory and anti-apoptotic properties. OSC reduces the levels of inflammatory mediators partly by inactivating the TLR4/MyD88/NF-κB signaling. The OSC-elicited inhibition of OGD/R-led BV-2 cell apoptosis is probably dependent on PI3K/Akt/mTOR signaling. These findings suggested that OSC may be a potential agent for the treatment of ischemic brain diseases.
Disclosure of conflict of interest
None.
References
- 1.Poisson SN, Schardt TQ, Dingman A, Bernard TJ. Etiology and treatment of arterial ischemic stroke in children and young adults. Curr Treat Options Neurol. 2014;16:315. doi: 10.1007/s11940-014-0315-4. [DOI] [PubMed] [Google Scholar]
- 2.Chamorro A, Hallenbeck J. The harms and benefits of inflammatory and immune responses in vascular disease. Stroke. 2006;37:291–293. doi: 10.1161/01.STR.0000200561.69611.f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nilupul Perera M, Ma HK, Arakawa S, Howells DW, Markus R, Rowe CC, Donnan GA. Inflammation following stroke. J Clin Neurosci. 2006;13:1–8. doi: 10.1016/j.jocn.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Chen J. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 2015;11:56–64. doi: 10.1038/nrneurol.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaur C, Rathnasamy G, Ling EA. Roles of activated microglia in hypoxia induced neuroinflammation in the developing brain and the retina. J Neuroimmune Pharmacol. 2013;8:66–78. doi: 10.1007/s11481-012-9347-2. [DOI] [PubMed] [Google Scholar]
- 6.Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol. 2010;119:89–105. doi: 10.1007/s00401-009-0622-0. [DOI] [PubMed] [Google Scholar]
- 7.Yang Q, Yang ZF, Liu SB, Zhang XN, Hou Y, Li XQ, Wu YM, Wen AD, Zhao MG. Neuroprotective effects of hydroxysafflor yellow A against excitotoxic neuronal death partially through down-regulation of NR2B-containing NMDA receptors. Neurochem Res. 2010;35:1353–1360. doi: 10.1007/s11064-010-0191-6. [DOI] [PubMed] [Google Scholar]
- 8.Kim SJ, Li J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013;4:e716. doi: 10.1038/cddis.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez-Lizarbe S, Pascual M, Guerri C. Critical role of TLR4 response in the activation of microglia induced by ethanol. J Immunol. 2009;183:4733–4744. doi: 10.4049/jimmunol.0803590. [DOI] [PubMed] [Google Scholar]
- 10.Qin X, Sun ZQ, Dai XJ, Mao SS, Zhang JL, Jia MX, Zhang YM. Toll-like receptor 4 signaling is involved in PACAP-induced neuroprotection in BV2 microglial cells under OGD/reoxygenation. Neurol Res. 2012;34:379–389. doi: 10.1179/1743132812Y.0000000028. [DOI] [PubMed] [Google Scholar]
- 11.Qin X, Sun ZQ, Zhang XW, Dai XJ, Mao SS, Zhang YM. TLR4 signaling is involved in the protective effect of propofol in BV2 microglia against OGD/reoxygenation. J Physiol Biochem. 2013;69:707–718. doi: 10.1007/s13105-013-0247-6. [DOI] [PubMed] [Google Scholar]
- 12.Xiang HF, Cao DH, Yang YQ, Wang HQ, Zhu LJ, Ruan BH, Du J, Wang MC. Isoflurane protects against injury caused by deprivation of oxygen and glucose in microglia through regulation of the Toll-like receptor 4 pathway. J Mol Neurosci. 2014;54:664–670. doi: 10.1007/s12031-014-0373-9. [DOI] [PubMed] [Google Scholar]
- 13.Chen CM, Wu CT, Yang TH, Chang YA, Sheu ML, Liu SH. Green Tea Catechin Prevents Hypoxia/Reperfusion-Evoked Oxidative Stress-Regulated Autophagy-Activated Apoptosis and Cell Death in Microglial Cells. J Agric Food Chem. 2016;64:4078–4085. doi: 10.1021/acs.jafc.6b01513. [DOI] [PubMed] [Google Scholar]
- 14.Xu T, Li Y, Wang H, Xu Y, Ma L, Sun T, Ma H, Yu J. Oxysophocarpine induces anti-nociception and increases the expression of GABAAalpha1 receptors in mice. Mol Med Rep. 2013;7:1819–1825. doi: 10.3892/mmr.2013.1414. [DOI] [PubMed] [Google Scholar]
- 15.Ding PL, Liao ZX, Huang H, Zhou P, Chen DF. (+)-12alpha-Hydroxysophocarpine, a new quinolizidine alkaloid and related anti-HBV alkaloids from Sophora flavescens. Bioorg Med Chem Lett. 2006;16:1231–1235. doi: 10.1016/j.bmcl.2005.11.073. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y, Li YX, Wang HL, Jin SJ, Zhou R, Qiao HQ, Du J, Wu J, Zhao CJ, Niu Y, Sun T, Yu JQ. Oxysophocarpine Ameliorates Carrageenan-induced Inflammatory Pain via Inhibiting Expressions of Prostaglandin E2 and Cytokines in Mice. Planta Med. 2015;81:791–797. doi: 10.1055/s-0035-1546153. [DOI] [PubMed] [Google Scholar]
- 17.Zhu QL, Li YX, Zhou R, Ma NT, Chang RY, Wang TF, Zhang Y, Chen XP, Hao YJ, Jin SJ, Ma L, Du J, Sun T, Yu JQ. Neuroprotective effects of oxysophocarpine on neonatal rat primary cultured hippocampal neurons injured by oxygen-glucose deprivation and reperfusion. Pharm Biol. 2014;52:1052–1059. doi: 10.3109/13880209.2013.877039. [DOI] [PubMed] [Google Scholar]
- 18.Zhang F, Wang S, Zhang M, Weng Z, Li P, Gan Y, Zhang L, Cao G, Gao Y, Leak RK, Sporn MB, Chen J. Pharmacological induction of heme oxygenase-1 by a triterpenoid protects neurons against ischemic injury. Stroke. 2012;43:1390–1397. doi: 10.1161/STROKEAHA.111.647420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saliba E, Henrot A. Inflammatory mediators and neonatal brain damage. Biol Neonate. 2001;79:224–227. doi: 10.1159/000047096. [DOI] [PubMed] [Google Scholar]
- 20.Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol. 2013;5:73–90. [PMC free article] [PubMed] [Google Scholar]
- 21.Weinstein JR, Koerner IP, Moller T. Microglia in ischemic brain injury. Future Neurol. 2010;5:227–246. doi: 10.2217/fnl.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abraham H, Losonczy A, Czeh G, Lazar G. Rapid activation of microglial cells by hypoxia, kainic acid, and potassium ions in slice preparations of the rat hippocampus. Brain Res. 2001;906:115–126. doi: 10.1016/s0006-8993(01)02569-0. [DOI] [PubMed] [Google Scholar]
- 23.Tam WY, Ma CH. Bipolar/rod-shaped microglia are proliferating microglia with distinct M1/M2 phenotypes. Sci Rep. 2014;4:7279. doi: 10.1038/srep07279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.You Y, Kaur C. Expression of induced nitric oxide synthase in amoeboid microglia in postnatal rats following an exposure to hypoxia. Neurosci Lett. 2000;279:101–104. doi: 10.1016/s0304-3940(99)00967-2. [DOI] [PubMed] [Google Scholar]
- 25.Jung DY, Lee H, Jung BY, Ock J, Lee MS, Lee WH, Suk K. TLR4, but not TLR2, signals autoregulatory apoptosis of cultured microglia: a critical role of IFN-beta as a decision maker. J Immunol. 2005;174:6467–6476. doi: 10.4049/jimmunol.174.10.6467. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Mediavilla V, Crespo I, Collado PS, Esteller A, Sanchez-Campos S, Tunon MJ, Gonzalez-Gallego J. The anti-inflammatory flavones quercetin and kaempferol cause inhibition of inducible nitric oxide synthase, cyclooxygenase-2 and reactive C-protein, and down-regulation of the nuclear factor kappaB pathway in Chang Liver cells. Eur J Pharmacol. 2007;557:221–229. doi: 10.1016/j.ejphar.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 27.Lee P, Lee J, Kim S, Lee MS, Yagita H, Kim SY, Kim H, Suk K. NO as an autocrine mediator in the apoptosis of activated microglial cells: correlation between activation and apoptosis of microglial cells. Brain Res. 2001;892:380–385. doi: 10.1016/s0006-8993(00)03257-1. [DOI] [PubMed] [Google Scholar]
- 28.Nagano T, Kimura SH, Takemura M. Prostaglandin E2 induces apoptosis in cultured rat microglia. Brain Res. 2014;1568:1–9. doi: 10.1016/j.brainres.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 29.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 30.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korsmeyer SJ. BCL-2 gene family and the regulation of programmed cell death. Cancer Res. 1999;59:1693s–1700s. [PubMed] [Google Scholar]
- 32.Merry DE, Korsmeyer SJ. Bcl-2 gene family in the nervous system. Annu Rev Neurosci. 1997;20:245–267. doi: 10.1146/annurev.neuro.20.1.245. [DOI] [PubMed] [Google Scholar]
- 33.Nakaso K, Ito S, Nakashima K. Caffeine activates the PI3K/Akt pathway and prevents apoptotic cell death in a Parkinson’s disease model of SH-SY5Y cells. Neurosci Lett. 2008;432:146–150. doi: 10.1016/j.neulet.2007.12.034. [DOI] [PubMed] [Google Scholar]
- 34.Chong ZZ, Li F, Maiese K. The pro-survival pathways of mTOR and protein kinase B target glycogen synthase kinase-3beta and nuclear factor-kappaB to foster endogenous microglial cell protection. Int J Mol Med. 2007;19:263–272. [PMC free article] [PubMed] [Google Scholar]