

Abstract
The theory that oxidative stress (OS) is at the root of several diseases is extremely popular. However, so far, no antioxidant has been recommended or offered by healthcare systems neither has any been approved as therapy by regulatory agencies that base their decisions on evidence‐based medicine. This is simply because, so far, despite many preclinical and clinical studies indicating a beneficial effect of antioxidants in many disease conditions, randomised clinical trials have failed to provide the evidence of efficacy required for drug approval.
In this review, we discuss the levels of evidence required to claim causality in preclinical research on OS, the weakness of the oversimplification associated with OS theory of disease and the importance of the narrative in its popularity. Finally, from a more translational perspective, we discuss the reasons why antioxidants acting by scavenging ROS might not only prevent their detrimental effects but also interfere with essential signalling roles. We propose that ROS have a complex metabolism and are generated by different enzymes at diverse sites and at different times. Aggregating this plurality of systems into a single theory of disease may not be the best way to develop new drugs, and future research may need to focus on specific oxygen‐toxifying pathways rather than on non‐specific ROS scavengers. Finally, similarly to what is nowadays required for clinical trials, we recommend making unpublished data available in repositories (open data), as this will allow big data approaches or meta‐analyses, without the drawbacks of publication bias.
Linked Articles
This article is part of a themed section on Redox Biology and Oxidative Stress in Health and Disease. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.12/issuetoc
Abbreviations
- EBM
evidence‐based medicine
- MDA
malondialdehyde
- NCI
National Cancer Institute
- NIH
National Institutes of Health
- OS
oxidative stress
- RA
rheumatoid arthritis
- RCT
randomised clinical trial
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| GLP‐1 receptor |
| Enzymes b |
| Monoamine oxidase |
| Xanthine oxidase/dehydrogenase |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
The oxidative stress theory and its translational gap
Background
The theory that oxidative stress (OS) is at the basis of many diseases is widely popular, but so far, antioxidants have not been approved for any indication because they have not met the criteria of efficacy for drug approval. To address this problem from a novel perspective, we will analyse the reasons for the popularity of the OS theory of disease and its scientific basis. In doing so, we need to consider various aspects participating in the development of scientific theories in biology and medicine. First, we will summarize the type of evidence required to approve a drug. Then we will discuss if it is possible to grade the level of evidence in preclinical research to assess the strength of a scientific hypothesis. We will also discuss the concept of causality and the role of mechanisms in scientific hypotheses and their acceptance. Finally, we will discuss the importance of mechanisms in the development of new therapies and analyse the OS theory of disease in the light of some concepts developed in epistemological research.
The classical oxidative stress theory of disease
The concept behind the OS theory of disease is that the metabolism of molecular oxygen (O2) by the cell results in the production of ROS, including hydrogen peroxide (H2O2), the hydroxyl radical (OH•) and the superoxide radical (O2 •‐), all of which can be toxic by reacting with cellular macromolecules. For this reason, organisms have developed antioxidant defence systems to eliminate ROS. These include enzymes, such as SOD and peroxidases, and low molecular weight antioxidants (vitamin E, vitamin C and GSH). Despite the fact that the theory of OS has evolved in recent years (Jones and Sies, 2015), this concept is still popularized as a balance with ROS on one plate and antioxidants on the other (Figure 1). The condition of OS exists when ROS production exceeds the capacity of the antioxidant systems (either because of an increased ROS generation or a decrease in antioxidants).
Figure 1.
The classical schematic representation of the OS theory.
In 1956, Harman, postulated the ‘free radical theory of ageing’ hypothesizing that the degenerative process of ageing has a free radical mechanism in common with cancer and radiation toxicity (Harman, 1956). Harman immediately saw the possible implications of this postulate in writing that ‘This theory is suggestive of chemical means of prolonging effective life’ (Harman, 1956). The early 1970s also saw the publication of Linus Pauling's book ‘vitamin C and the common cold’ [(Pauling, 1970) p.36]. However, the term OS did not appear in the scientific literature until 1970, in a study on erythrocyte damage induced by H2O2 (Paniker et al., 1970). Since then, many papers have been published, suggesting, often with very convincing data, that OS is associated with many diseases and that antioxidants would have a beneficial effect. Today, it is a challenge to find a disease for which a role of OS has not been postulated. Searching the phrase ‘caused by oxidative stress’ in Google in October 2015 gave 220 000 hits. Most websites and review articles on this subject will have a scheme similar to that in Figure 2. The main purpose of this paper is to discuss the exact meaning of those arrows and their directions.
Figure 2.
A typical scheme generalising the OS theory of disease.
The lack of translational success of the classical OS concept
The appeal of the OS theory of disease resides in its translational implication that antioxidants, by scavenging ROS, could be beneficial in all the diseases in Figure 2 and additionally would ensure healthy ageing; too good to be true? So far, no antioxidants have been approved for any indication by regulatory agencies such as the Food and Drug Administration (FDA), or the European Medicines Agency. Despite their low cost, no antioxidant supplements are reimbursed or offered by public health insurance systems of most countries. Only in Japan, a free radical scavenger, edaravone, is approved for stroke and amyotrophic lateral sclerosis (Abe et al., 2014; Wada et al., 2014). Probably, the most famous failure was the free radical‐trapping agent NXY‐059 that, although showing strong preclinical data in stroke, failed to show efficacy in a large clinical trial, casting doubt on the significance of basic research in this field (Feuerstein et al., 2008). Several studies have shown that, in many diseases, antioxidant supplements are ineffective or even harmful (see a meta‐analysis of 78 studies by Bjelakovic et al., 2012). While this could be due to problems of the drugs tested or the trial design, there may also be weaknesses in the OS theory of disease, for instance, because ROS have beneficial physiological roles (discussed later under ‘redox regulation’).
Despite this lack of evidence, the OS theory of disease is so popular that antioxidants, including vitamins C and E, GSH, some metals (e.g. selenium and zinc) or extracts from a wide range of fruits, vegetables or fish, constitute the lion's share of the nutritional supplements used by 150 million Americans (Starr, 2015). These supplements are used as they supposedly strengthen immune defences, prevent cancer or pathological ageing. ‘Antioxidants are good and free radicals are bad’ became one of the ‘science myths that will not die’ (Scudellari, 2015).
Level of evidence required in translational medicine
Antioxidants, like any other drug, need evidence to be approved as treatment for any indication. For a new drug to be approved for a specific indication, strong evidence of efficacy in a large population and on disease‐relevant endpoints must be provided (Katz, 2004; Downing et al., 2014). These criteria are based on a hierarchy of evidence, with expert opinion and small case–control studies at the bottom, and randomised clinical trials (RCTs) and meta‐analyses of RCTs at the top (Levels of Evidence, 2009) (Evidence‐Based Medicine Working Group, 1992; Howick, 2011; Jeremy et al., 2011). On the other hand, the US FDA criteria for making a health claim regarding a nutritional supplement are less stringent. Two types of health claims are allowed: ‘health claims’ and ‘qualified health claims’ (Ellwood et al., 2010). Health claims require ‘significant scientific agreement’ on the relationship between the supplement and a disease, meaning that ‘the validity of the relationship is not likely to be reversed by new and evolving science, although the exact nature of the relationship may need to be refined’ (Ellwood et al., 2010). On the other hand, a ‘qualified health claim’ can be made when ‘the evidence for a substance‐disease relationship is credible but does not meet the significant scientific agreement standard’ (Ellwood et al., 2010), and in this case, the claim should be moderated with an appropriate disclaimer.
Levels of evidence in basic research
Experimental strategies
Basic research lacks commonly accepted criteria to assess the level of experimental evidence required to conclude, for instance, that ‘OS is implicated in disease x’ or ‘antioxidants could be useful in disease y’. The strength of the evidence may, or may not, be questioned during the peer review process, but it is unlikely that the reviewers will question the OS theory of disease. If we analyse the morphology of the literature on the OS theory of disease, we can come up with a few basic types of experimental evidence, described in Table 1.
Table 1.
Main experimental approaches in the study of the OS theory of disease
| Studies based on testing the effect of ROS or antioxidants on a disease‐related process |
|---|
| 1. Studies investigating the effect of ROS on disease‐related pathway. For instance, H2O2 induces aggregation of α‐synuclein, a protein important in Alzheimer's disease (Hashimoto et al., 1999); adding H2O2 to immune cells causes the production of inflammatory mediators (DeForge et al., 1993); and injecting a H2O2‐generating enzyme in the mouse knee joint causes inflammation (Schalkwijk et al., 1986). |
| 2. Studies investigating the effect of an antioxidant on a disease‐related pathway. For instance, polyphenols present in red wine inhibit formation of amyloid fibrils that are important in Alzheimer's disease (Ono et al., 2003); antioxidants inhibit TNF production in vitro and in vivo in mice (Chaudhri and Clark, 1989); antioxidants from red wine are protective in animal models of stroke (Yu et al., 2016); and clinical trials with antioxidants. |
| 3. Study of the effect of genetic modification (knock out and overexpression) or modulation of antioxidant enzymes or ROS‐generating enzymes, on a disease‐related pathway or an animal model of disease (Sorce et al., 2014). |
| Studies based on measuring ROS or antioxidants in a disease or its model |
|---|
| 1. Studies showing that ROS are produced in a disease or a disease model. Thus, ROS are generated during the aggregation of amyloid peptides implicated in Alzheimer's disease (Tabner et al., 2005); ischaemia causes activation of a ROS‐producing enzyme, xanthine oxidase (Granger, 1988); and an indicator of ROS production, MDA, is elevated in blood from patients with systemic lupus erythematosus (Wang et al., 2010). |
| 2. Studies showing that endogenous antioxidants are decreased in a disease or disease models in vitro or in vivo, or in patients. For instance, levels of plasma antioxidants are lower in osteoporotic patients (Maggio et al., 2003). |
| 3. Genome‐wide association studies where a mutation in a gene for a ROS‐generating enzyme or an antioxidant protein is associated with a disease. |
Different levels of experimental systems can be used: (i) cell‐free; (ii) in vitro (cell culture); and (iii) in vivo (animal models or patients). These experimental interventions, however, often suffer from lack of specificity; while testing the effect of ROS, one can for instance add H2O2 or a ROS‐generating system to cell culture, but adding an antioxidant is not the same as ‘removing ROS’. This may be true when adding SOD or catalase, as they specifically remove superoxide and H2O2 respectively, but it is certainly not the case with low molecular weight antioxidants, which usually have other chemical properties and biological activities than removing ROS (Ohlow and Moosmann, 2011; Forman et al., 2014). For instance, exogenously administered antioxidants can down‐regulate endogenous antioxidant enzymes (Gomez‐Cabrera et al., 2008; Ristow et al., 2009).
The difficulty is to rank the level of evidence obtained in different experimental models. Common sense suggests that in vivo models rank higher than in vitro systems or that extrapolations from in vitro experiments with primary cells are more trustworthy than those obtained with immortalised cell lines (Baetu, 2015). While this may be true in some cases, this may not apply to studies on OS. Indeed, it is difficult to appreciate whether the decreased production of an inflammatory mediator observed in vitro following addition of an antioxidant to purified blood cells from rheumatoid arthritis (RA) patients represents a higher level of evidence than the effect of the administration of the same antioxidant on joint swelling in a chronic mouse model of RA in vivo.
In the next sections, we try to apply the knowledge from current epistemological studies to the problem of the OS theory of disease, to identify potential weaknesses and priorities for future research.
Causality and the problem of confounding
In biomedical research, we often search for the specific association between two parameters or events. If we found a statistically significant association between OS and RA (for instance, the levels of oxidized proteins in blood are higher in RA), we could hypothesize a causal link between the two variables, that is, (i) OS causes RA or (ii) RA causes OS. The direction of the arrow is influenced by common wisdom, dominant or popular theories in the field, or other factors; most will probably favour the first hypothesis, with the arrow pointing from OS to RA.
However, the simple association of ROS production and a disease does not mean that ROS cause the disease: an association does not always mean causation. The possible interpretation of an association between two variables is described in Figure 3A and B (p.79, Illari et al., 2011). The first example (a) is an association with a simple causal link (C causes E). The ‘sign’ of the causal link may be positive or negative (for instance, respectively, ‘autoimmunity causes diabetes’ or ‘low insulin causes diabetes’). The second example (b) is a situation where the association between C and E is not due to a causal link but to a third variable F that acts as a confounder. A typical example is the possible causal link between yellow fingers (nicotine–stained) and lung cancer; both are caused by cigarette smoking, the confounder, and, when this is taken into account, the causal link between C and E disappears (Smith and Phillips, 1992).
Figure 3.
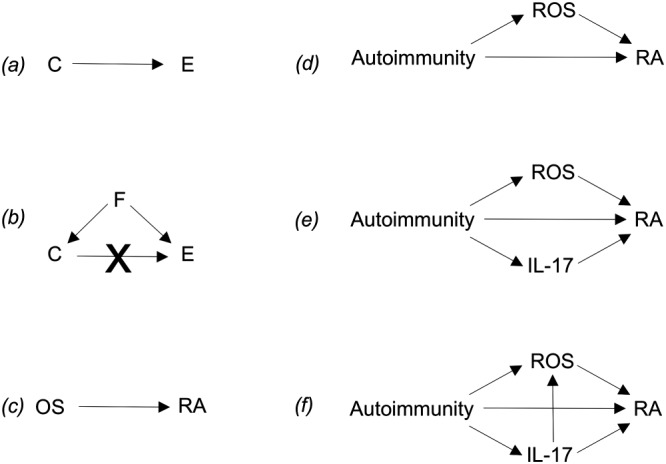
Schematic representations of causal mechanisms.
We can try to apply the scheme of a causal mechanism to the example of RA. Measurements of indicators (biomarkers) of OS and animal experiments suggest a link with OS (Hitchon and El‐Gabalawy, 2004), with papers stating that ‘rheumatologists…may also consider incorporating antioxidant therapeutics in their prescriptions’ (Taysi et al., 2002). If we analyse the causal mechanism described above should we conclude that, because markers of OS are elevated, ‘OS causes RA’ (Figure 3C)? If we consider the scheme (b), could there be a third variable that causes both OS and RA? For example, RA is an autoimmune disease where activated CD4+ T lymphocytes produce mediators such as IL‐17 that, in turn, activate inflammation in the joint, which causes the symptoms of RA (swelling, pain and tissue damage) (Firestein, 2003; van den Berg and Miossec, 2009). Because inflammation can cause production of ROS by inflammatory cells (Smith, 1994), inflammation could well be the confounder and thus the common cause of both RA and OS. In this case, the two variables, RA and OS, are not causally linked. That would make a big difference in translational medicine because only if there was a causal link, ‘OS causes RA’, one could hypothesize that eliminating ROS with antioxidants might improve the disease.
The first two schemes provided in Figure 3, mainly derived from epidemiological research, are sketchy, and causal mechanisms can be more complicated. By adapting schemes developed for experimental studies of mediation in the social sciences to the OS/RA case, (Imai et al., 2011), we could have different mechanisms interacting with each other (Figure 3D–F). Autoimmunity could induce RA directly, for instance, with autoreactive T cells attacking the joints, or indirectly via ROS as mediators (d). Scheme (e) shows a more complex mechanism where two measured mediators are involved, ROS and (for example) the T cell‐derived cytokine IL‐17 (van den Berg and Miossec, 2009). In this scheme, the two mediators are independent, but there are other possibilities, where the two mediators are not independent (f). There are many more possibilities with other mediators and moderators (Edwards and Lambert, 2007; Imai et al., 2011), often with unobserved or unknown mediators involved, and more complex causal schemes can be drawn (Robins, 2001; Hernan et al., 2002).
Causality and the sufficient‐component cause model
The other problem is that most diseases are multifactorial. A conceptual model, known as the ‘sufficient‐component cause model’, originally described by Rothman (1976), states that a disease may be caused by several factors (component causes) that act together to cause the disease. Importantly, a disease may have more than one sufficient cause, each of them made of several component causes. An example describing various component causes of tuberculosis is given in Figure 4.
Figure 4.
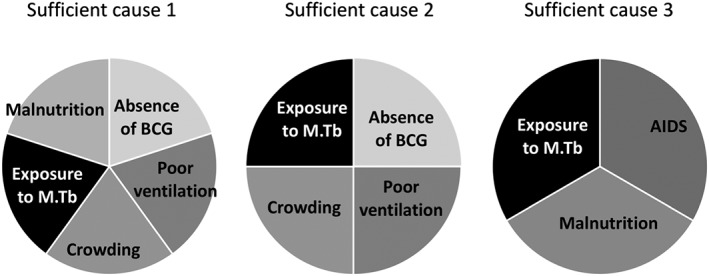
Rothman's sufficient component model of tuberculosis causation. Modified from Prof. Wayne LaMorte, Boston University School of Public Health (personal communication).
In this example, three different situations (sufficient causes) can cause tuberculosis. Each of these sufficient causes is made of more than one ‘component cause’, not necessarily the same ones. In this case, however, one component cause is necessary—exposure to Mycobacterium tuberculosis.
Taking the Rothman model into consideration, we should not just ask if a disease is caused by OS, but whether there are diseases where OS is a component cause, and whether OS is necessary or not. The Rothman model has important implications in translational medicine because a biochemical process (like OS) may not have the same role in all patients with the same disease. In a recent review on biomarkers of OS, we discussed their potential usefulness in patient stratification in clinical trials to identify those who could benefit from therapies that target OS (Frijhoff et al., 2015).
External validity: extrapolation and reductionism
The problem of external validity is how an observation or a theory is generalizable (Johnson, 1997), and if it can be translated to the clinic. This is inextricably linked to the experimental models used. In the laboratory, we use extremely simplified models, and experiments are performed in a controlled environment to avoid exactly those confounders (other biochemical pathways and component causes, factors for which we often have only a partial knowledge) that are present in real life.
This is what philosophers call reductionism. Upon extrapolation from in vitro or animal models to a clinical setting, reductionism hits back with what Steel called the ‘extrapolator's circle’, that is, the challenge ‘to explain how the suitability of the model as a basis for extrapolation can be established given only limited information about the target’ (Steel, 2007) [p.4]. The reductionist approach makes it difficult to design experiments aimed at falsifying the hypothesis, considered by Popper as the key of the scientific method (Popper, 2005). As noted by Bechtel, ‘Confirmation is challenging because there are always alternative possible laws from which one might make the same prediction… Falsification is challenging because a false prediction might be due to an error either in the proposed law or in one of the auxiliary hypotheses that figured in deriving the prediction.’ (p. 61, Bechtel, 2006).
For instance, if we want to test the hypothesis that hydrogen peroxide causes bone damage in RA, we may administer a thiol antioxidant to an animal model where the disease is induced by promoting an autoimmune response to collagen. If the antioxidant did not work, that does not negate the hypothesis as the antioxidant might not reach the site of inflammation at the right concentration, have non‐specific effects or decrease ROS at other sites and should not be considered a clear‐cut tool.
Reductionism in the representation of scientific theories
Reductionism plays an important role in the way we represent mechanistic theories and causal links. Scientists ‘rarely depict all the particular details when describing a mechanism; representations are usually schematic, often depicted in diagrams.’ (Darden, 2005). Mayer pointed out the downside of the overuse of diagrams in modern science: ‘…the typical “cartoon” of signalling pathways, with their reassuring arrows and limited number of states could be the real villain. Instead of simplifying an inherently complex system so that the key points can be grasped, we would argue that such diagrams actively mislead, implying a specificity and homogeneity that does not at all reflect the messy reality of actual signalling complexes.’ (Mayer et al., 2009). In fact, if we think of the representations of the Krebs cycle or the glycolytic pathway, they look like complete representations of the metabolic pathway. However, if we were to draw a metabolic table describing all the protein thiol–disulfide oxidoreductases (thioredoxin, glutaredoxin, peroxiredoxins, sulfiredoxin, etc.) it would be very difficult to give a comprehensive description in one figure to include, for instance, the over 100 protein substrates of thioredoxin and the various sources of the different ROS. When we try to zoom out from a specific biochemical pathway to describe our hypothesis about a role of OS in disease, the focus of the diagram is completely lost. It often results in diagrams, such as that in Figure 5, where we mix different levels: molecules (H2O2, NH4), subcellular organelles, cells, organs and diseases; the resulting diagram will clearly have many implicit black boxes. Of note, there is nothing wrong in these graphs, as long as we are aware of the limitations due to our incomplete knowledge of the system.
Figure 5.
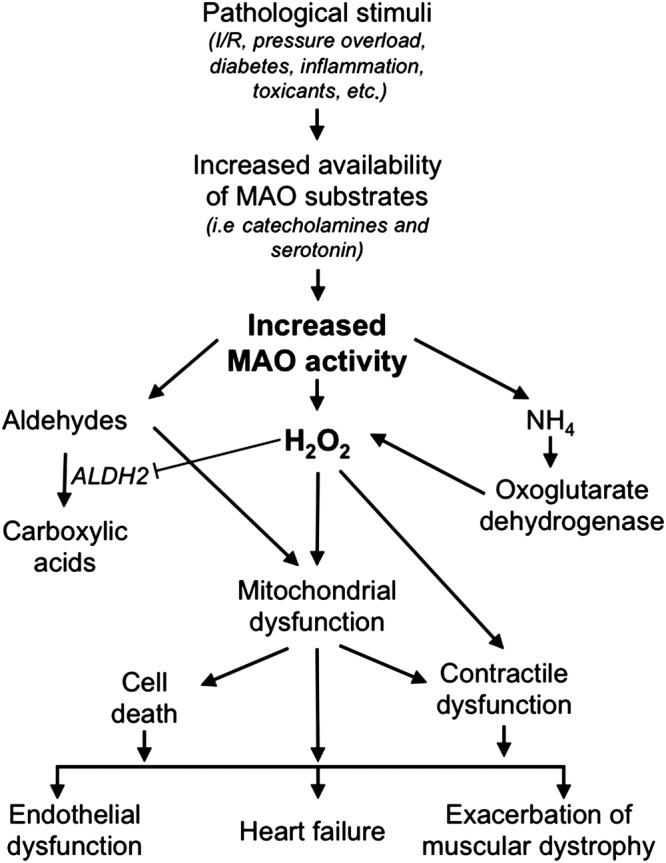
Diagram of a pathogenic mechanism involving ROS. Reproduced with permission from Casas et al. (2015).
Reductionism in the use of proxy biomarkers
For the germ theory of disease, demonstration needs to fit Koch's postulates: (i) detection of the germ in every patient; (ii) the germ is not present in other diseases; and (iii) the isolated germ can induce the disease if re‐injected. Are there any such criteria we could use for the OS theory of disease? Of course, the first two postulates cannot be met in the case of a component cause, and because OS could mediate other diseases. However, the real problem is measuring ROS in disease. ROS are unstable and their half‐lives range from 10−9 s for the hydroxyl radical (OH•) to 10−3 s for H2O2 (D'Autreaux and Toledano, 2007), making them nearly impossible to measure in patients. Therefore, ROS are measured using biomarkers as proxies, which are often insufficiently validated. These are, in most cases, reaction products, resulting from the reaction of ROS with cellular molecules (see Frijhoff et al., 2015), and are seldom specific, as they can be a result of interaction of cellular molecules with more than one ROS, or can be generated by other metabolic pathways. For instance, the most used OS biomarker, malondialdehyde (MDA) (Janero, 1990), (40 000 publications) has been measured by several techniques, often non‐specific and hard to compare. MDA is one of the terminal products of the peroxidation of several polyunsaturated fatty acids. Although this reaction can be initiated by ROS, it has many biases: we do not know exactly from which lipid it is derived, which ROS contributes to its production, and from which tissue it originates, unless it is measured properly by chromatographic methods (Frijhoff et al., 2015); furthermore, MDA has been widely used as a measure of a different pathway, prostaglandin synthesis (Smith et al., 1976).
Biomarkers of OS are probably not the best proxies when compared with other biomarkers (haematocrit for anaemia; glucose or insulin levels for diabetes; C‐reactive protein for inflammation; and fever for infection). These biomarkers only provide very indirect evidence of OS, and even more indirect evidence of ROS production. As of today, no biomarker of OS provides information on the cellular source, tissue or organ involved (when measured in circulation). The technical difficulties of measuring ROS, together with the lack of specificity of the most used biomarkers of OS, are probably the weakest link in the OS theory of disease.
Causality and mechanism. Disease mechanisms versus mechanisms of actions of drugs
Illari and Williamson stated: ‘A mechanism for a phenomenon consists of entities and activities organised in such a way that they are responsible for the phenomenon’. (Illari and Williamson, 2012). Mechanisms include several entities and their causal interactions (Hernandez‐Lemus and Siqueiros‐Garcia, 2015). We are interested in identifying the pathogenesis of diseases, not least because the elucidation of mechanisms can lead to the development of new drugs. For instance, the identification of the cascade of inflammatory cytokines as the (complex) mechanism by which autoimmunity causes inflammation (Feldmann et al., 1996) led to development of the inhibitors of IL‐6, IL‐17 and TNF currently used in the therapy of RA.
However, do we really need a mechanism to develop new drugs? Russo and Williamson maintain that, historically, knowledge of a mechanism is important in accepting a causal claim (Russo and Williamson, 2007). They give the example of Semmelweis' finding that puerperal fever and deaths were associated with cadaveric contamination and could be prevented by washing hands with chlorine; this was only accepted by the scientific community 20 years later, when the development of the germ theory of disease by Pasteur provided a mechanism to this finding (Russo and Williamson, 2007). On the other hand, Howick lists some treatments, in the first place, that were widely accepted before any hint of a mechanism was known (p.131–132, Howick, 2011). A more recent example is the screening programme of the US National Cancer Institute (NCI) in the 1980s, involving empirical screening of chemicals and natural products for their cytotoxicity against a panel of tumour cell lines. This led to the identification of several drugs now used in oncology, before the NCI adopted mechanism‐based screening strategies (Cragg, 1998; Balis, 2002).
Common sense suggests that a drug developed based on a known mechanism of action and a hypothesis for the pathogenesis of a disease should have better probability of showing efficacy in an RCT than a drug tested at random, a view probably shared by many scientists. The importance of the mechanism of action in predicting the efficacy of a drug is a matter of debate, but not from the regulatory point of view. As Katz summarized, the FDA has an empirical approach. Information about the mechanism of action is important but ‘entirely subsidiary to the fundamental questions that must be answered in the course of drug approval; namely, is a drug effective, and is it safe in use’. (Katz, 2004). These questions, Katz writes, cannot be answered by the understanding of the mechanism of action, mainly because this understanding will always be incomplete. The FDA limits the usefulness of animal and in vitro studies to the generation of hypotheses or exploration of mechanisms but states that ‘these studies do not provide information from which scientific conclusions can be drawn regarding a relationship between the substance and disease in humans’ (http://www.fda.gov/food/guidanceregulation/guidancedocumentsregulatoryinformation/ucm073332.htm).
The empiricist position adopted by the FDA does not mean that we should not develop drugs targeting specific mechanisms. It only means that this should not give a drug an advantage over another for which the mechanism is unknown. It is fair to note that some think that an unsuccessful trial does not necessarily falsify the hypothesis on disease mechanisms or on the mechanisms of action of a drug, and might be due to off‐target activities (Mann and Mochly‐Rosen, 2013).
We need to clarify the relationship between the ‘mechanism of action of a drug’ and ‘pathogenic mechanism of a disease’. In many cases, the mechanism of action of a drug is very well characterized, for instance, receptor antagonists or antibodies to an inflammatory cytokine or a cancer antigen. If there are already drugs on the market that have a specific mechanism of action, new drugs with the same mechanism of action on a ‘validated target’ have a better approval rate than the industry average (Kola and Landis, 2004; Falconi et al., 2014). The fact that there are no antioxidants approved means that ROS are not (yet) ‘validated targets’. Furthermore, we have only limited knowledge of the diseases for which OS is a clear cause, at least if we adopt stringent criteria.
Howick stated that one of the reasons why knowledge of the mechanism of action of a drug does not automatically predict its effectiveness is because this knowledge is incomplete, making the example of the mechanism of action of antiarrhythmic drugs, describing this graphically as a black box (Figure 6). The mechanistic reasoning (left) predicted that these drugs would have improved survival in patients but they actually increased mortality (Howick et al., 2013).
Figure 6.
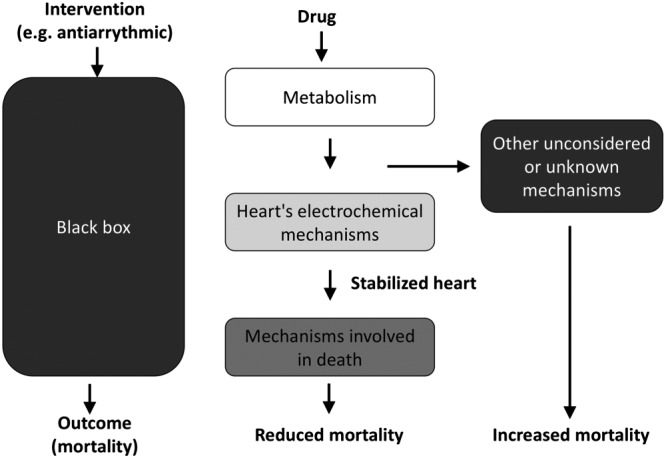
Black boxes in the mechanism of drug action: the example of antiarrhythmic drugs. Modified from Howick et al. (2013).
How much we know of the mechanisms underlying OS, the role of OS in a specific disease, and the mechanism of action of antioxidants and their specificity is important in determining how black is the black box. The fact that the list of diseases found in reviews on OS is so long to include practically all of them is not particularly helpful. It is therefore a priority to identify a hierarchy of levels of evidence for the OS theory of disease, but this will be extremely difficult. When taking the theory of OS from the bench to the bedside using antioxidants as drugs, we are adding the problem that most of them, more than most drugs, will have off‐target effects or that ROS may have physiological functions beyond their toxic effects. The following section describes other aspects that may help explain the popularity of the OS theory of disease and of the use of antioxidants despite evidence‐based medicine (EBM)‐level evidence.
The importance of narrative: teleological perspectives
St. Thomas Aquinas thought that natural objects and organisms have a function, and understanding the natural world is to see how objects fit into a teleological hierarchy (p. 152, Perlman, 2009). Teleology (from the Greek thelos, end, goal) or ‘goal‐directedness’ is deeply rooted in the descriptive language of biology (Toepfer, 2012). Salmon, in the Introduction to the Philosophy of Science (p.14, Salmon, 1999) asks whether explanation involves reduction to the familiar and notes that the type of explanation we are best acquainted with is an anthropomorphic one (Mayr, 1998), in which actions are explained in terms of conscious purposes (p.14, Salmon, 1999). In fact, we often use expressions like ‘the purpose of polymorphonuclear neutrophils is to kill bacteria’ or describe the heart as a machine whose purpose is to pump blood.
However, the term ‘teleological’ is not precise, and scholars distinguish between ‘real’ teleological systems (end‐seeking), teleonomic systems (end‐resulting, with a purpose due to the operation of a programme) and teleomatic systems (end‐resulting but not goal seeking, and having no purpose) (Mayr, 1974; Mayr, 1998). The example of a teleomatic process is any process governed by a natural law: the decay of an isotope or the evaporation of water; there is an end, but this is not the result of a programme or the accomplishment of a purpose.
One pillar of the OS theory of disease is that there are a number of antioxidant systems whose function is to protect the organism from ROS generated by cellular metabolism. If we agree that SOD and catalase evolved to eliminate superoxide (reaction (1)) and hydrogen peroxide (reaction (2)), we could probably refer to them as teleonomic systems—with the purpose of catalysing specific reactions.
| (1) |
| (2) |
Let us now consider other processes associated with OS, such as protein glutathionylation, a process by which a free cysteine in a protein (Prot‐SH), and the cysteine of GSH react to form a mixed disulfide (termed glutathionylated protein, Prot‐SSG). We often read that protein glutathionylation has a purpose, for example, to protect protein thiols from irreversible oxidation, or as a regulatory mechanism (Dalle‐Donne et al., 2007; Mieyal and Chock, 2012); is this what we actually mean? Because GSH is in equilibrium with its oxidized form (its disulfide, GSSG), it will inevitably undergo thiol–disulfide exchange with Prot‐SH (reaction (3)), provided some conditions of accessibility and pK of the Prot‐SH are met. In the same way, Prot‐SH will inevitably react, to an extent determined by the equilibrium, with GSH (reaction (4)).
| (3) |
| (4) |
If we have Prot‐SH, GSH and GSSG, as we have them in every cell, the above processes are inevitable, and we should regard them as teleomatic—without a purpose. These reactions may have results and consequences, but not a purpose. Attributing a purpose to teleomatic reactions oversimplifies complex networks and, by tagging a chemical reaction as ‘good’ or ‘bad’ may create false pharmacological targets. The same line of reasoning could apply to irreversible forms of cysteine oxidation. For instance, oxidation of specific cysteine in a peroxiredoxin can switch its activity from a peroxidase to a chaperone (Lim et al., 2008); thus, even irreversible oxidation should not necessarily be interpreted as a form of protein damage.
The importance of the narrative in the success of a theory and a medical practice: ROS as the axis of evil and antioxidants as the Holy Grail
If we consider the association mentioned above (OS and RA) and ask a medical scientist to draw a causal link for the association between ROS production and RA, it is likely that he or she would draw an arrow from ROS/OS to RA and not vice versa (although that could very well be the case!). This makes a ‘better story’ and fits well in the existing theories. Stories are not just for the layperson; the idea that successful papers should ‘tell a good story’ is hardcoded into the minds of researchers (Shermer, 2007). Even when assessing the validity of a scientific model, some will say ‘a model is valid to the extent that the story being told using that model holds’ (Illari, 2012). However, a good mechanism and a good story are not always predictive of the efficacy of a treatment; bloodletting, used widely in medical practice until a century ago, was popular because it fits with the Greek theory of the four humours (p.136–137, Howick, 2011).
The OS theory of disease is based on an even more attractive narrative, the fight between good and evil, antioxidants preventing ageing like the Holy Grail or acting like shields against ROS, an imagery and wording often used even by scientists in their review articles or presentations. As added value, main sources of antioxidants are fruit and vegetables (Ramos, 2008; Moon and Shibamoto, 2009); ROS are the germs and antioxidants the antibiotics. This has probably a key role in the popularity of the OS theory of disease and of its translational shortcut, the use of antioxidant supplements.
Un‐shading the black box; adding complexity
From the concept of OS to that of redox regulation
Oxidation of protein cysteines has been regarded as one of the many forms of oxidative damage (Stadtman, 1992). The concept of redox regulation (Ray et al., 2012) is based on the fact that the activity of several proteins is regulated by reversible oxidation of protein cysteines (Figure 7). This scheme is very similar to the prototypic mechanism of regulation of protein activity by phosphorylation, where a phosphate group from ATP is reversibly attached to a protein. Examples of redox‐regulated proteins are the transcription factor NF‐kB, which is activated by ROS and inhibited by thiol antioxidants (Schreck et al., 1991), and the transcriptional regulators Keap1‐Nrf2 (Wakabayashi et al., 2004). In the scheme in Figure 7, thiol antioxidants act not only by scavenging ROS (both physiologically produced or pathologically over‐produced) but can also directly reduce protein disulfides, independently of an effect on ROS.
Figure 7.
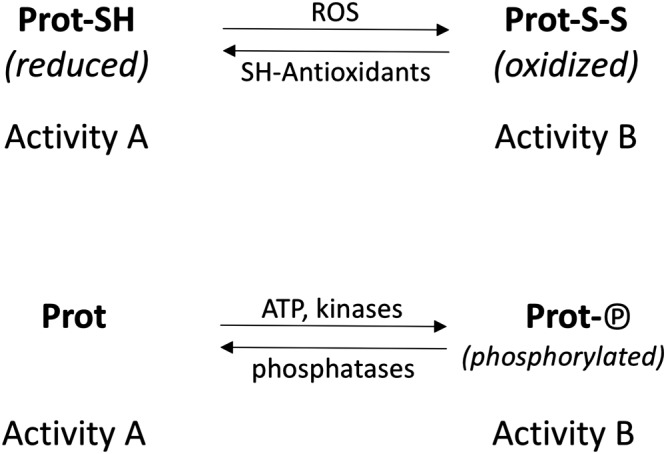
Regulation of protein function by reversible oxidation (top) and by phosphorylation (bottom).
Recent evidence shows that ROS have signalling activity in vivo and that their elimination may have negative effects as this would remove one component of the pathway (Ristow et al., 2009). Furthermore, decreasing ROS may increase the levels of other reactive species, such as nitric oxide (Gryglewski et al., 1986).
One way we attempt to reconcile the two actions of ROS (physiological in redox regulation and pathological in the OS theory of disease) is by assuming that low amounts of ROS are necessary due to their participation in signalling, but excessive production is damaging. This critically affects the therapeutic strategies using antioxidants acting as ROS scavengers to ‘mop up’ ROS. It would be extremely difficult to titrate the dose of antioxidant to inhibit only the excess ROS present in some diseases without decreasing the ‘physiological’ levels.
Obtaining the whole picture; adding complexity rather than reducing
From the pharmacological point of view, one way forward is to look for specific ROS sources as drug targets instead of scavenging ROS in general, as discussed elsewhere (Casas et al., 2015). If an enzyme Z is responsible for overproduction of ROS in a specific tissue and disease, it may be more effective to inhibit its activity rather than ‘mopping up’ all the ROS produced anywhere and by any source. To come back to the analogy of protein phosphorylation, a successful way to target an oncogene has been to inhibit a specific kinase with imatinib rather than developing compounds that would non‐specifically scavenge ATP (Capdeville et al., 2002).
From an epistemological perspective, the complexity of the place of ROS in cellular metabolism may be one of the reasons why it is difficult to answer the research question of the relevance of OS in many diseases. We could take a pragmatic approach, ex juvantibus; if an antioxidant will prove effective in disease X at the highest level of evidence required for drug approval, then we should conclude that OS is a cause of that disease. However, the molecules commonly used as antioxidants are not specific. For instance, all thiol‐based antioxidants, such as GSH, are also reducing agents and will not only scavenge free radicals but will also reduce labile disulfides (Kim et al., 2001; Laragione et al., 2003). Also, the multiple sclerosis drug, dimethyl fumarate, often considered to act by inducing antioxidant systems via Nrf2, has additional activities as it has therapeutic efficacy, regulating immunity, in a model of multiple sclerosis in Nrf2‐deficient mice (Schulze‐Topphoff et al., 2016). Therefore, a proof of efficacy would lead to the approval of the antioxidant tested for that specific indication but might not, alone, be a demonstration of the role of OS in that disease.
Another possibility would be to better integrate and interpret existing knowledge. With hundreds of thousands of scientific publications on this topic, the demand for simplicity is strong. However, given the incredibly complex biology of ROS, we need to open up our small schemes and diagrams to integrate them with other metabolic pathways. There are of course ways of analysing or visualising complex information, aggregating or integrating several experiments. For instance, meta‐analysis can be used to combine the results of many clinical trials, often giving different results, to increase power by aggregating results. While this is typically used to analyse results of interventional trials, the approach has been used to combine studies on the association between biomarkers and disease (Flatow et al., 2013). However, a meta‐analysis can only respond to a simple question, such as the association of one variable with another (typically an outcome) but will not help integrating data from different experiments. Other ‘big data’ approaches can give a better insight into common mechanisms of diseases. Barabasi has built diseasome network maps where diseases are associated with genes (Goh et al., 2007) or protein–protein interactions (Menche et al., 2015). However, it is difficult, at this stage, to integrate experimental evidence obtained in many different models and with very different methodologies.
Open data: a proposal
The real difficulty in using a big data approach is the well‐known publication bias by which only so‐called positive results are published. Anyone who has experience in the field knows that when we encounter two studies reporting that antioxidant X inhibits the production of mediator Z, it is possible that others have performed similar experiments and found no effect, or even the opposite effect but, because this was not the expected result, these ‘negative data’ were not published. This is not specific for studies on OS, and studies have shown that a very large proportion of scientific studies are not reproducible (Ioannidis, 2014). This does not necessarily imply scientific misconduct (such as not publishing negative data to please the funder). Often the experimental design is so circumscribed (see discussion of external validity) that it is only valid in that specific cell line, at that concentration and time point and researchers had to perform several experiments until they found the experimental conditions where ‘it works’. When a specific result is not reproducible in face of variations in the experimental conditions, it lacks robustness (Casadevall and Fang, 2010). Conversely, non‐reproducible results could be due to poorly circumscribed experimental set‐ups (Baetu, 2013): cell lines used at different passages or different density/confluence, in different culture media, time of incubation not properly followed to accommodate working hours, animals of different age and weight, etc.
Publishing negative results, i.e., those where the oxidant or antioxidant X ‘did not work’, would allow us, using proper statistical analysis, to compare different experiments, such as in meta‐analysis. In clinical research, there is now a strong drive to make raw data of all clinical trials public. This philosophy should expand to basic research and repositories of open data. Making laboratory notebooks open and adopting well‐described standard protocols will eventually allow a quantification of the consistency of a given finding. One example is the application of the ARRIVE guidelines for reporting animal research (Kilkenny et al., 2010).
One could think, for instance, of having a network of scientists committed to open their lab notebooks and publish online all their results after a safe period, for instance, 1 year. Funding agencies such as the National Institutes of Health (NIH) or the European Union will be key to these initiatives. An example is the NIH Data Sharing initiative (https://www.nlm.nih.gov/NIHbmic/nih_data_sharing_repositories.html), but one could think of specific initiatives on redox research aimed, for instance, at experiments using antioxidant or pro‐oxidant interventions in vivo or in vitro, or measuring specific biomarkers. Funding agencies could also provide free open‐access hosting, similar to the repositories used for microarray data for specific fields. The availability of open data will eventually allow the set‐up of networks similar to the Cochrane Collaboration. This will clearly need changing the mainstream culture around Impact Factor and the market‐oriented publication business. Only then would it be possible to think of extending the rigour of EBM to the formation of scientific theories.
Conclusions
Viewing the OS theory of disease from different theoretical perspectives developed in epistemological research, the theory can be seen to be significantly weaker than many other pathogenic theories. This might explain why the translational shortcut of this theory, antioxidant therapy, has failed so often and has never shown enough evidence to be incorporated in guidelines, approved by regulatory agencies and recommended and reimbursed by health insurance systems basing their decisions on EBM criteria.
We have seen that this is, in part, due to the technical difficulties of measuring ROS, an essential step to make a claim of a ROS‐mediated disease, which is complicated by the large number of analytical techniques and analytes that have been used to make such claims. Trying to aggregate a plurality of diseases where different ROS are generated in different tissues by different sources may have aggravated the problem.
The fact that antioxidants are not approved therapies for any disease (with the exception indicated above) does not of course rule out that OS may be a causal component of disease. Clinical evidence in favour of an association (but not a causative role) of OS with some diseases are strong. This has been often shown in clinical trials, such as the EPIC trial reporting the benefit of consumption of fruits rich in vitamin C (Khaw et al., 2001). There may be technical reasons why this has not been convincing enough to recommend antioxidants as therapeutic agents. It may be that if the same antioxidants had been given at a different dose, for a longer period of time or to a more selected cohort of the population, the results would have shown an efficacy at a sufficient level to allow their approval for that specific indication.
In the present paper, we tried to use a different, less explored perspective to identify the weakness in the OS theory of disease that we may try to address in the future to improve the translational effect of studies in this field. Some of the issues (measuring ROS and their consequences with sensitivity and specificity) are inherently difficult to solve (Frijhoff et al., 2015).
With the diversity of ROS‐generating systems and the wide variety of researchers (chemists, biochemists, pharmacologists, clinicians) studying ROS and their pathogenic role, if any, we suggest the following aspects are prioritized:
(i) develop networks of international collaborations with shared results (open data, open lab notebooks, big data analysis, a Cochrane‐style collaboration for preclinical studies, www.pre‐clinicaltrials.org), (ii) address the key question of how to demonstrate that OS is a causal component in disease X, (iii) sum up the level of evidence available rather than test another new antioxidant extracted from a different fruit, where a plethora of antioxidants from other sources have already been studied.
Funding agencies need to encourage scientists to take the hardest route rather than go for the easy publication and the conventional wisdom of the dominant group. Finally, with the public's growing expectations and understanding of science thanks to access to the information released by universities, companies and media, it is also the responsibility of scientists to assess more critically the claims of their scientific findings. This includes limitations in terms of component causes, levels of evidence for making a causal claim and the validity of the results outside their experimental system. Only critical, honest and out‐of‐the‐box analysis of redox research will lead to the identification of redox pathways involved in pathologies and only then will innovative therapeutics emerge.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the RM Phillips Trust (to PG) and by the European Cooperation in Science and Research, COST Action BM1203/EU‐ROS (to PG, VJ, HHHWS). VJ is supported by the European Community's Framework Programme (FP7/2007–2013) under Grant 278611 (Neurinox). We thank Ben Alberts, Brighton & Sussex Medical School, for proofreading and editing English.
Ghezzi, P. , Jaquet, V. , Marcucci, F. , and Schmidt, H. H. H. W. (2017) The oxidative stress theory of disease: levels of evidence and epistemological aspects. British Journal of Pharmacology, 174: 1784–1796. doi: 10.1111/bph.13544.
References
- Abe K, Itoyama Y, Sobue G, Tsuji S, Aoki M, Doyu M et al. (2014). Confirmatory double‐blind, parallel‐group, placebo‐controlled study of efficacy and safety of edaravone (MCI‐186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener 15: 610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetu TM (2013). Chance, experimental reproducibility, and mechanistic regularity. Int Stud Philos Sci 27: 253–271. [Google Scholar]
- Baetu TM (2015). The ‘big picture’: the problem of extrapolation in basic research. Br J Philos Sci. doi:10.1093/bjps/axv018. [Google Scholar]
- Balis FM (2002). Evolution of anticancer drug discovery and the role of cell‐based screening. J Natl Cancer Inst 94: 78–79. [DOI] [PubMed] [Google Scholar]
- Bechtel W (2006). Discovering cell mechanisms: The creation of modern cell biology. Cambridge University Press: Cambridge. [Google Scholar]
- Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C (2012). Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev 3 .CD007176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdeville R, Buchdunger E, Zimmermann J, Matter A (2002). Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov 1: 493–502. [DOI] [PubMed] [Google Scholar]
- Casadevall A, Fang FC (2010). Reproducible science. Infect Immun 78: 4972–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas AI, Dao VT, Daiber A, Maghzal GJ, Di Lisa F, Kaludercic N et al. (2015). Reactive oxygen‐related diseases: therapeutic targets and emerging clinical indications. Antioxid Redox Signal 23: 1171–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhri G, Clark IA (1989). Reactive oxygen species facilitate the in vitro and in vivo lipopolysaccharide‐induced release of tumor necrosis factor. J Immunol 143: 1290–1294. [PubMed] [Google Scholar]
- Cragg GM (1998). Paclitaxel (Taxol): a success story with valuable lessons for natural product drug discovery and development. Med Res Rev 18: 315–331. [DOI] [PubMed] [Google Scholar]
- D'Autreaux B, Toledano MB (2007). ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8: 813–824. [DOI] [PubMed] [Google Scholar]
- Dalle‐Donne I, Rossi R, Giustarini D, Colombo R, Milzani A (2007). S‐glutathionylation in protein redox regulation. Free Radic Biol Med 43: 883–898. [DOI] [PubMed] [Google Scholar]
- Darden L (2005). Relations among fields: Mendelian, cytological and molecular mechanisms. Stud Hist Philos Biol Biomed Sci 36: 349–371. [DOI] [PubMed] [Google Scholar]
- DeForge LE, Preston AM, Takeuchi E, Kenney J, Boxer LA, Remick DG (1993). Regulation of interleukin 8 gene expression by oxidant stress. J Biol Chem 268: 25568–25576. [PubMed] [Google Scholar]
- Downing NS, Aminawung JA, Shah ND, Krumholz HM, Ross JS (2014). Clinical trial evidence supporting FDA approval of novel therapeutic agents, 2005‐2012. JAMA 311: 368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JR, Lambert LS (2007). Methods for integrating moderation and mediation: a general analytical framework using moderated path analysis. Psychol Methods 12: 1–22. [DOI] [PubMed] [Google Scholar]
- Ellwood KC, Trumbo PR, Kavanaugh CJ (2010). How the US Food and Drug Administration evaluates the scientific evidence for health claims. Nutr Rev 68: 114–121. [DOI] [PubMed] [Google Scholar]
- Evidence‐Based Medicine Working Group (1992). Evidence‐based medicine. A new approach to teaching the practice of medicine. JAMA 268: 2420–2425. [DOI] [PubMed] [Google Scholar]
- Falconi A, Lopes G, Parker JL (2014). Biomarkers and receptor targeted therapies reduce clinical trial risk in non‐small‐cell lung cancer. J Thorac Oncol 9: 163–169. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Brennan FM, Maini RN (1996). Role of cytokines in rheumatoid arthritis. Annu Rev Immunol 14: 397–440. [DOI] [PubMed] [Google Scholar]
- Feuerstein GZ, Zaleska MM, Krams M, Wang X, Day M, Rutkowski JL et al. (2008). Missing steps in the STAIR case: a translational medicine perspective on the development of NXY‐059 for treatment of acute ischemic stroke. J Cereb Blood Flow Metab 28: 217–219. [DOI] [PubMed] [Google Scholar]
- Firestein GS (2003). Evolving concepts of rheumatoid arthritis. Nature 423: 356–361. [DOI] [PubMed] [Google Scholar]
- Flatow J, Buckley P, Miller BJ (2013). Meta‐analysis of oxidative stress in schizophrenia. Biol Psychiatry 74: 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman HJ, Ursini F, Maiorino M (2014). An overview of mechanisms of redox signaling. J Mol Cell Cardiol 73: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijhoff J, Winyard PG, Zarkovic N, Davies SS, Stocker R, Cheng D et al. (2015). Clinical relevance of biomarkers of oxidative stress. Antioxid Redox Signal 23: 1144–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL (2007). The human disease network. Proc Natl Acad Sci U S A 104: 8685–8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV et al. (2008). Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training‐induced adaptations in endurance performance. Am J Clin Nutr 87: 142–149. [DOI] [PubMed] [Google Scholar]
- Granger DN (1988). Role of xanthine oxidase and granulocytes in ischemia–reperfusion injury. Am J Physiol 255: H1269–H1275. [DOI] [PubMed] [Google Scholar]
- Gryglewski RJ, Palmer RM, Moncada S (1986). Superoxide anion is involved in the breakdown of endothelium‐derived vascular relaxing factor. Nature 320: 454–456. [DOI] [PubMed] [Google Scholar]
- Harman D (1956). Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M et al. (1999). Oxidative stress induces amyloid‐like aggregate formation of NACP/alpha‐synuclein in vitro. Neuroreport 10: 717–721. [DOI] [PubMed] [Google Scholar]
- Hernan MA, Hernandez‐Diaz S, Werler MM, Mitchell AA (2002). Causal knowledge as a prerequisite for confounding evaluation: an application to birth defects epidemiology. Am J Epidemiol 155: 176–184. [DOI] [PubMed] [Google Scholar]
- Hernandez‐Lemus E, Siqueiros‐Garcia JM (2015). Mechanistic‐enriched models: integrating transcription factor networks and metabolic deregulation in cancer. Theor Biol Med Model 12: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchon CA, El‐Gabalawy HS (2004). Oxidation in rheumatoid arthritis. Arthritis Res Ther 6: 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howick J, Glasziou P, Aronson JK (2013). Problems with using mechanisms to solve the problem of extrapolation. Theor Med Bioeth 34: 275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howick JH (2011). The Philosophy Of Evidence‐based Medicine. John Wiley & Sons: Oxford. [Google Scholar]
- Illari P (2012). The Philosophy of Information–A Simple Introduction. Society for the Philosophy of Information: Oxford. [Google Scholar]
- Illari P, Russo F, Williamson J (2011). Causality in the Sciences. OUP: Oxford. [Google Scholar]
- Illari PM, Williamson J (2012). What is a mechanism? Thinking about mechanisms across the sciences. Eur J Philos Sci 2: 119–135. [Google Scholar]
- Imai K, Keele L, Tingley D, Yamamoto T (2011). Unpacking the black box of causality: learning about causal mechanisms from experimental and observational studies. Am Polit Sci Rev 105: 765–789. [Google Scholar]
- Ioannidis JP (2014). How to make more published research true. PLoS Med 11: e1001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janero DR (1990). Malondialdehyde and thiobarbituric acid‐reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radic Biol Med 9: 515–540. [DOI] [PubMed] [Google Scholar]
- Jeremy H, Iain C, Paul G, Trish G, Carl H, Alessandro L et al. (2011). The 2011 Oxford CEBM evidence levels of evidence (Introductory Document) Oxford centre for evidence‐based medicine.
- Johnson RB (1997). Examining the validity structure of qualitative research. Education 118: 282. [Google Scholar]
- Jones DP, Sies H (2015). The redox code. Antioxid Redox Signal 23: 734–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz R (2004). FDA: evidentiary standards for drug development and approval. NeuroRx 1: 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaw KT, Bingham S, Welch A, Luben R, Wareham N, Oakes S et al. (2001). Relation between plasma ascorbic acid and mortality in men and women in EPIC‐Norfolk prospective study: a prospective population study. European prospective investigation into cancer and nutrition. Lancet 357: 657–663. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KY, Rhim T, Choi I, Kim SS (2001). N‐acetylcysteine induces cell cycle arrest in hepatic stellate cells through its reducing activity. J Biol Chem 276: 40591–40598. [DOI] [PubMed] [Google Scholar]
- Kola I, Landis J (2004). Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3: 711–715. [DOI] [PubMed] [Google Scholar]
- Laragione T, Bonetto V, Casoni F, Massignan T, Bianchi G, Gianazza E et al. (2003). Redox regulation of surface protein thiols: identification of integrin alpha‐4 as a molecular target by using redox proteomics. Proc Natl Acad Sci U S A 100: 14737–14741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levels of Evidence . [Online] Available at http://www.cebm.net/oxford‐centre‐evidence‐based‐medicine‐levels‐evidence‐march‐2009/ (accessed 24/6/2016).
- Lim JC, Choi HI, Park YS, Nam HW, Woo HA, Kwon KS et al. (2008). Irreversible oxidation of the active‐site cysteine of peroxiredoxin to cysteine sulfonic acid for enhanced molecular chaperone activity. J Biol Chem 283: 28873–28880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio D, Barabani M, Pierandrei M, Polidori MC, Catani M, Mecocci P et al. (2003). Marked decrease in plasma antioxidants in aged osteoporotic women: results of a cross‐sectional study. J Clin Endocrinol Metab 88: 1523–1527. [DOI] [PubMed] [Google Scholar]
- Mann DL, Mochly‐Rosen D (2013). Translational medicine: proceed at your own risk. Nat Rev Drug Discov 12: 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer BJ, Blinov ML, Loew LM (2009). Molecular machines or pleiomorphic ensembles: signaling complexes revisited. J Biol 8: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr E (1974). Teleological and teleonomic, a new analysis In: Cohen R, Wartofsky M. (eds). Methodological and Historical Essays in the Natural and Social Sciences. Reidl Publishing Company: Dordrecht, Netherlands, pp. 91–117. [Google Scholar]
- Mayr E (1998). The multiple meanings of'teleological’. Hist Philos Life Sci 20: 35–40. [Google Scholar]
- Menche J, Sharma A, Kitsak M, Ghiassian SD, Vidal M, Loscalzo J et al. (2015). Disease networks. Uncovering disease‐disease relationships through the incomplete interactome. Science 347 .1257601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieyal JJ, Chock PB (2012). Posttranslational modification of cysteine in redox signaling and oxidative stress: focus on s‐glutathionylation. Antioxid Redox Signal 16: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JK, Shibamoto T (2009). Antioxidant assays for plant and food components. J Agric Food Chem 57: 1655–1666. [DOI] [PubMed] [Google Scholar]
- Ohlow MJ, Moosmann B (2011). Phenothiazine: the seven lives of pharmacology's first lead structure. Drug Discov Today 16: 119–131. [DOI] [PubMed] [Google Scholar]
- Ono K, Yoshiike Y, Takashima A, Hasegawa K, Naiki H, Yamada M (2003). Potent anti‐amyloidogenic and fibril‐destabilizing effects of polyphenols in vitro: implications for the prevention and therapeutics of Alzheimer's disease. J Neurochem 87: 172–181. [DOI] [PubMed] [Google Scholar]
- Paniker NV, Srivastava SK, Beutler E (1970). Glutathione metabolism of the red cells. Effect of glutathione reductase deficiency on the stimulation of hexose monophosphate shunt under oxidative stress. Biochim Biophys Acta 215: 456–460. [DOI] [PubMed] [Google Scholar]
- Pauling L (1970). Voitamin C and the Common Cold. W.H.Freeman & Co Ltd: New York. [Google Scholar]
- Perlman M (2009). The Modern Philosophical Resurrection of Teleology In: Rosenberg A, Arp R. (eds). Philosophy of Biology: An Anthology. John Wiley & Sons: Chichester, pp. 149–163. [Google Scholar]
- Popper K (2005). The Logic of Scientific Discovery. Routledge: New York. [Google Scholar]
- Ramos S (2008). Cancer chemoprevention and chemotherapy: dietary polyphenols and signalling pathways. Mol Nutr Food Res 52: 507–526. [DOI] [PubMed] [Google Scholar]
- Ray PD, Huang BW, Tsuji Y (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24: 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M et al. (2009). Antioxidants prevent health‐promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A 106: 8665–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins JM (2001). Data, design, and background knowledge in etiologic inference. Epidemiology 12: 313–320. [DOI] [PubMed] [Google Scholar]
- Rothman KJ (1976). Causes. Am J Epidemiol 104: 587–592. [DOI] [PubMed] [Google Scholar]
- Russo F, Williamson J (2007). Interpreting causality in the health sciences. Int Stud Philos Sci 21: 157–170. [Google Scholar]
- Salmon MH (1999). Introduction to the Philosophy of Science. Hackett Publishing: Indianapolis. [Google Scholar]
- Schalkwijk J, van den Berg WB, van de Putte LB, Joosten LA (1986). An experimental model for hydrogen peroxide‐induced tissue damage. Effects of a single inflammatory mediator on (peri)articular tissues. Arthritis Rheum 29: 532–538. [DOI] [PubMed] [Google Scholar]
- Schreck R, Rieber P, Baeuerle PA (1991). Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF‐kappa B transcription factor and HIV‐1. EMBO J 10: 2247–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze‐Topphoff U, Varrin‐Doyer M, Pekarek K, Spencer CM, Shetty A, Sagan SA et al. (2016). Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc Natl Acad Sci U S A 113: 4777–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudellari M (2015). The science myths that will not die. Nature 528: 322–325. [DOI] [PubMed] [Google Scholar]
- Shermer M (2007). The really hard science. Sci Am 297: 44 , 46. [DOI] [PubMed] [Google Scholar]
- Smith GD, Phillips AN (1992). Confounding in epidemiological studies: why “independent” effects may not be all they seem. BMJ 305: 757–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA (1994). Neutrophils, host defense, and inflammation: a double‐edged sword. J Leukoc Biol 56: 672–686. [DOI] [PubMed] [Google Scholar]
- Smith JB, Ingerman CM, Silver MJ (1976). Malondialdehyde formation as an indicator of prostaglandin production by human platelets. J Lab Clin Med 88: 167–172. [PubMed] [Google Scholar]
- Sorce S, Nuvolone M, Keller A, Falsig J, Varol A, Schwarz P et al. (2014). The role of the NADPH oxidase NOX2 in prion pathogenesis. PLoS Pathog 10: e1004531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman ER (1992). Protein oxidation and aging. Science 257: 1220–1224. [DOI] [PubMed] [Google Scholar]
- Starr RR (2015). Too little, too late: ineffective regulation of dietary supplements in the United States. Am J Public Health 105: 478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel D (2007). Across the Boundaries: Extrapolation in Biology and Social Science. Oxford University Press: Oxford. [Google Scholar]
- Tabner BJ, El‐Agnaf OM, Turnbull S, German MJ, Paleologou KE, Hayashi Y et al. (2005). Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J Biol Chem 280: 35789–35792. [DOI] [PubMed] [Google Scholar]
- Taysi S, Polat F, Gul M, Sari RA, Bakan E (2002). Lipid peroxidation, some extracellular antioxidants, and antioxidant enzymes in serum of patients with rheumatoid arthritis. Rheumatol Int 21: 200–204. [DOI] [PubMed] [Google Scholar]
- Toepfer G (2012). Teleology and its constitutive role for biology as the science of organized systems in nature. Stud Hist Philos Biol Biomed Sci 43: 113–119. [DOI] [PubMed] [Google Scholar]
- van den Berg WB, Miossec P (2009). IL‐17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol 5: 549–553. [DOI] [PubMed] [Google Scholar]
- Wada T, Yasunaga H, Inokuchi R, Horiguchi H, Fushimi K, Matsubara T et al. (2014). Effects of edaravone on early outcomes in acute ischemic stroke patients treated with recombinant tissue plasminogen activator. J Neurol Sci 345: 106–111. [DOI] [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova‐Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M et al. (2004). Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A 101: 2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Pierangeli SS, Papalardo E, Ansari GA, Khan MF (2010). Markers of oxidative and nitrosative stress in systemic lupus erythematosus: correlation with disease activity. Arthritis Rheum 62: 2064–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Wang L, Tang F, Zeng L, Zhou L, Song X et al. (2016). Resveratrol pretreatment decreases ischemic injury and improves neurological function via sonic hedgehog signaling after stroke in rats. Mol Neurobiol. doi:10.1007/s12035-015-9639-7. [DOI] [PubMed]