

Abstract
The intestine is composed of many distinct cell types that respond to commensal microbiota or pathogens with immune tolerance and proinflammatory signals respectively. ROS produced by mucosa‐resident cells or by newly recruited innate immune cells are essential for antimicrobial responses and regulation of signalling pathways including processes involved in wound healing. Impaired ROS production due to inactivating patient variants in genes encoding NADPH oxidases as ROS source has been associated with Crohn's disease and pancolitis, whereas overproduction of ROS due to up‐regulation of oxidases or altered mitochondrial function was linked to ileitis and ulcerative colitis. Here, we discuss recent advances in our understanding of how maintaining a redox balance is crucial to preserve gut homeostasis.
Linked Articles
This article is part of a themed section on Redox Biology and Oxidative Stress in Health and Disease. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.12/issuetoc
Abbreviations
- 5‐ASA
5‐aminosalicylic acid
- CD
Crohn's disease
- CGD
chronic granulomatous disease
- DC
dendritic cells
- DSS
dextran sodium sulphate
- FAK
focal adhesion kinase
- GI
gastrointestinal
- IBD
inflammatory bowel disease
- IEC
intestinal epithelial cells
- LP
lamina propria
- mETC
mitochondrial electron transport chain
- mtROS
mitochondrial ROS
- NSAIDs
nonsteroidal anti‐inflammatory drugs
- PHB
prohibitin
- PMN
polymorphonuclear leukocyte
- RNS
reactive nitrogen species
- SI
small intestine
- SOD
superoxide dismutase
- TNBS
2,4,6‐trinitrobenzenesulfonic acid
- UC
ulcerative colitis
- VEOIBD
very early onset IBD
Tables of Links
| TARGETS | |
|---|---|
| Enzymes a | p38 |
| eNOS, NOS3 | RIPK3 |
| iNOS, NOS2 | Catalytic receptors b |
| nNOS, NOS1 | NLRP3 |
| ERK | PRR, pattern recognition receptors |
| Focal adhesion kinase | TLR2 |
| Mitochondrial F1F0‐ATPase | GPCRs c |
| Myeloperoxidase | FPR1, formyl peptide receptor 1 |
| LIGANDS | |
|---|---|
| 5‐ASA, 5‐aminosalicylic acid (mesalazine) | IL‐13 |
| Allopurinol | IL‐17 |
| Epiregulin | Infliximab |
| H2O2 | LPS |
| IFN‐γ | NO |
| IL‐1β | PGI2 |
| IL‐10 | TGF‐β |
| TNF‐α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a, b, cAlexander et al., 2015a, 2015b, 2015c).
Introduction
The production of ROS is a key event in the progression of many inflammatory disorders, including those involving the gastrointestinal (GI) tract. In the gut, disruption of the mucosal barrier will rapidly activate the innate immune system and set off an acute inflammatory response that begins in the lamina propria (LP). Polymorphonuclear leukocytes (PMNs) migrate to the site of infection or injury, engulf invading pathogens and secrete ROS, granular enzymes, and vasoactive and pro‐inflammatory mediators (Mittal et al., 2014). Production of ROS by PMNs can create a hypoxic niche due to oxygen consumption, which may aid in the resolution of inflammation (Campbell et al., 2014). However, uncontrolled and persistent oxidative stress with overproduction of ROS and/or inadequate removal of ROS by antioxidant systems will cause apoptosis and tissue injury (Rezaie et al., 2007). In particular, deregulation of the mitochondrial electron transport chain (mETC), with increased mitochondrial ROS (mtROS) levels, was observed in inflammatory bowel disease (IBD) patients and decreasing mtROS ameliorated colitis (Dashdorj et al., 2013). Tightly controlled ROS generation by enzymes expressed in non‐phagocytic cells, for example enterocytes or fibroblasts, paved the way towards the concept of ROS as regulators of intracellular processes. Indeed, low physiological levels of ROS are indispensable for a plethora of signalling pathways (Holmstrom and Finkel, 2014), including gene transcription, protein kinase activation and phosphatase inhibition, thereby regulating cytokine production and coordinated cell motility (Peterson and Artis, 2014). The importance of maintaining physiological levels of ROS in the gut is highlighted by the chronic intestinal inflammation present in patients with inactivating gene defects in NADPH oxidase. The consequences of altered ROS levels on multifactorial GI inflammation are still not well understood, but the importance of maintaining the redox balance is emerging. This review aims to highlight the most relevant findings in the field of redox regulation of GI inflammation by discussing the intriguing and debated role of ROS at the crossroad between antimicrobial and pro‐inflammatory agents, and our view that a tight regulation of ROS is a prerequisite for a healthy gut.
Mucosal immunity in the GI tract
From the oral cavity to the anus, the GI epithelium functions as a physical barrier confining an extraordinary number of bacteria, collectively known as microbiota, and potentially harmful pathogens or substances, to the lumen of the GI tract. The GI epithelium is composed of a single cell layer that undergoes turnover approximately every 4–5 days and has the task of sensing the phyla and quantity of bacteria and their distance from the cellular surface. Total bacterial numbers increase along the GI tract, ranging from 102–103 per mL in the highly acidic environment of the stomach to 105 per mL in the upper small intestine (SI) and up to 1012 per mL in the colon (Sekirov et al., 2010). The intestinal mucosa responds to infectious pathogens with inflammatory signals and to harmless commensals with immune tolerance signals. General defence mechanisms include the secretion of polysaccharides by goblet cells that form a mucus bilayer towards the lumen, release of antimicrobial peptides by Paneth cells and secretory IgA by plasma cells (McGuckin et al., 2011; Peterson and Artis, 2014). Maintaining the integrity of the barrier is an essential prerequisite for intestinal homeostasis. In the case of barrier damage, the capacity of rapidly resealing the epithelial layer is critical to avoid or minimize the exposure of immune cells to microbiota, which would lead to the initiation of an inflammatory response (Figure 1).
Figure 1.
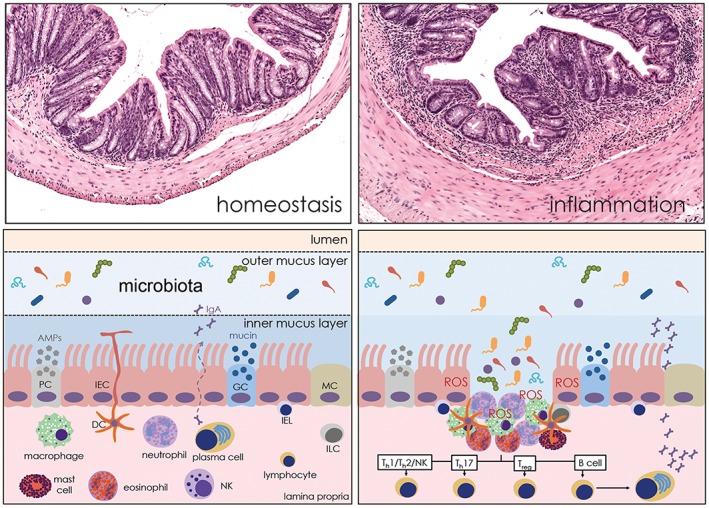
Schematic representation of the GI mucosal immunity. Gut tolerance results from the coordinated action of epithelial, innate and adaptive immune cells. In the event of epithelial barrier failure, the microbiota is detected by innate immune cells of the lamina propria, which then orchestrate appropriate inflammatory and healing responses, in part by releasing ROS. AMP, antimicrobial peptides; GC, goblet cell; IEL, intraepithelial lymphocyte; ILC, innate lymphoid cell; MC, microfold cell; PC, Paneth cell.
During homeostasis, when the epithelial barrier is intact, the sampling of luminal gut antigens that determines the mucosal ‘tolerance’ is mediated by microfold cells in the dome epithelium overlying Peyer's patches, intestinal epithelial cells (IECs), subsets of innate lymphoid cells (Hepworth et al., 2015), γδ T cells (Nanno et al., 2008) and dendritic cells (DCs), the latter directly extending dendrites into the gut lumen (Chieppa et al., 2006). Antigenic signals are transduced into the LP, a loose connective tissue containing lymphocytes, plasma cells, eosinophils, mast cells, neutrophils and a large number of macrophages (Figure 1). Macrophages and DCs are maintained in an anergic, non‐inflammatory state by the predominance of T regulatory cells, which secrete the immunosuppressive cytokines IL‐10, an inhibitor of effector T cell responses, and TGF‐β, an inducer for differentiation of conventional CD4+ T cells into T regulatory cells. The epithelial cell, once thought to be a purely absorptive cell type, is now considered similarly to LP cells a crucial effector of mucosal immunity. IECs express a collection of membrane and cytosolic pattern recognition receptors (PRRs) that recognize distinct microbial ligands. These receptors include toll‐like receptors (TLRs), NOD‐like receptors and RIG‐I‐like receptors (Peterson and Artis, 2014). Seminal studies have defined beneficial roles of PRR signalling by IECs. These include the expression of the EGF receptor ligands, amphyregulin and epiregulin, the production of the cytoprotective trefoil factor‐3, the activation of the NLRP3 inflammasome and NEMO (IκB kinase‐γ)‐dependent NF‐κB signalling (Wullaert et al., 2011; Fukata and Arditi, 2013).
Altered barrier function (or ‘leaky gut’) permits the translocation of bacteria and microbial products into the LP, inducing a series of events ranging from activation of professional phagocytes (PMNs and macrophages) and ROS production for antimicrobial defence to pro‐inflammatory cytokine generation by effector Th1, Th17 and NK cells. At the same time, epithelial cells produce ROS, which participate in regulating intracellular signals for barrier repair and healing (Figure 1).
Dysfunction of biological systems regulating the balance between ‘low, tolerable’ and ‘high, toxic’ levels of ROS can lead to persistent and unresolved mucosal inflammation. Incorrect levels of oxidants, too low or too high, alter the redox balance, thereby triggering pathological processes. Deregulation of the redox balance compromises antimicrobial defence, prolongs immune activation and alters innate and adaptive immune responses such as activation of transcription factors, inflammasome function or autophagy. The ensuing incomplete pathogen clearance and/or oxidative stress not only perpetuates inflammation but may also eventually lead to other chronic complications, such as fibrosis, neoplasia and extra‐intestinal symptoms (Neurath, 2014).
Sources of ROS in the GI tract
ROS are small molecules, including the oxygen radicals [superoxide (O2 •−) and hydroxyl (•OH)] and non‐radicals such as hypochlorous acid (HOCl), singlet oxygen (1O2) and hydrogen peroxide (H2O2). Unstable oxygen‐centred radicals are highly reactive and can lead to deleterious effects in cells and tissues. O2 •−can be converted to more stable and diffusible H2O2 by superoxide dismutase (SOD)‐mediated catalysis (Figure 2). H2O2 is decomposed to water and oxygen or can be converted to HOCl (100 times more toxic than H2O2) by peroxidases, such as phagocytic myeloperoxidase, in the presence of chloride ions (Cl−). H2O2 can also react non‐enzymically with O2 •− to form •OH in the presence of ferrous (Fe2 +) iron in the so‐called Fenton reaction (Winterbourn, 1995). In the presence of NO, a crucial mediator of GI physiology, O2 •− rapidly reacts with NO, thereby forming the highly reactive peroxynitrite (ONOO−) (Figure 2). Additional reactions with peroxynitrite give rise to many other NO‐derived compounds, collectively known as reactive nitrogen species (RNS) (Mittal et al., 2014). Prolonged production of high concentrations of ROS can irreversibly destroy or alter the function of target molecules, causing DNA damage, lipid peroxidation and protein oxidation. Although historically viewed as harmful, low and moderate concentrations of ROS are vital for homeostasis and beneficial in physiological processes, including the regulation of intracellular signalling pathways such as reversible thiol oxidation of reactive cysteine residues within regulatory proteins (Holmstrom and Finkel, 2014). In the GI tract, the main sources of ROS and RNS are NADPH oxidase enzymes [NOX/dual oxidase (DUOX)], the mETC and NOSs.
Figure 2.
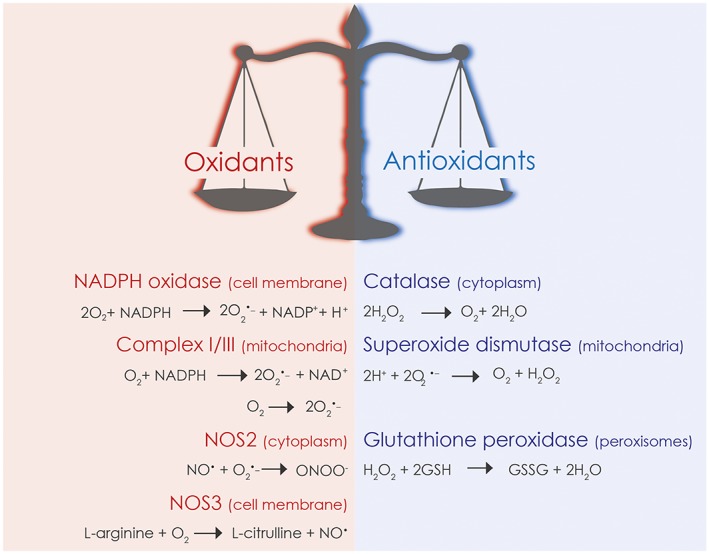
Main sources of ROS/RNS and antioxidant defence systems in the GI tract. Gut homeostasis will be compromised when the capacity of antioxidant enzymes is overcome by enhanced oxidant production.
NADPH oxidases
The membrane‐bound multimeric NOX and DUOX complexes are the only known enzymes generating ROS not as byproduct but rather as their primary function (Lambeth and Neish, 2014). The mammalian oxidase family comprises five NOX members (NOX1–5) and two dual oxidases (DUOX1–2). With the exception of NOX5 that is not expressed in rodents, both humans and mice express all oxidase isoforms. NOX and DUOX homologs differ in their structures, mechanisms of activation and tissue expression patterns. The main oxidases expressed along the GI tract are NOX1 and DUOX2 found in the epithelium (NOX1 in the ileum, cecum and colon; DUOX2 in all segments of the gut), NOX2 expressed by professional phagocytes and DCs, and NOX4 present in the epithelium, fibroblasts and smooth muscle cells (Bedard and Krause, 2007; Mandal et al., 2010) (Figure 3).
Figure 3.
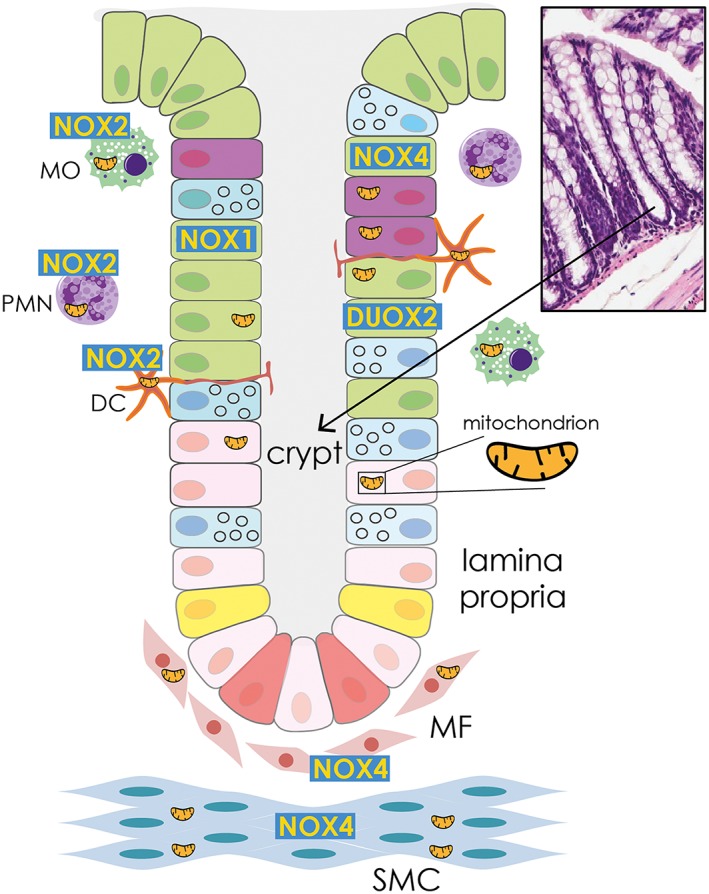
Localization of NADPH oxidase isoforms in the GI mucosa. NOX1 and DUOX2 are present in the epithelium, NOX2 is mainly expressed in professional phagocytes and DCs, while NOX4 is found in fibroblasts, smooth muscle cells and epithelium. MO, macrophage; PMN, polymorphonuclear cell; MF, myofibroblast; SMC, smooth muscle cell. Colour code for crypt cells: green, enterocyte absorptive cell; purple, enteroendocrine cell; pink, progenitor cell; blue, goblet cell; yellow, +4 cell; red, crypt base columnar cells.
Originally discovered in neutrophils in the late 1980s, the O2 •− generating NOX2 oxidase constitutes an essential part of the antimicrobial repertoire of innate immune cells. The NOX2 enzyme is a multi‐subunit complex with a catalytic core termed flavocytochrome b558, composed of the two transmembrane proteins p22phox and gp91phox (renamed NOX2). NOX2 is typically inactive in resting cells, but in the presence of pathogen‐derived stimuli, multiple activation steps take place, initiating post‐translational modifications and translocation of cytosolic subunits (p40phox, p47phox and p67phox) and activated Rac GTPase to the membrane. Association of the NOX2‐p22phox heterodimer, and assembly of a functional complex, permits electron flow from NADPH as electron donor to FAD, both located in the cytosolic region of NOX2, to two low potential haems to convert molecular oxygen (O2) to O2 •−. This so‐called oxidative burst into the phagosome or to the outside environment supports elimination of pathogens. Activated neutrophils can capture and kill pathogens by receptor‐mediated phagocytosis and by releasing complex three‐dimensional structures called neutrophil extracellular traps containing nuclear chromatin, associated mainly with nuclear histones, and several antimicrobial granule proteins, a process that requires NOX2‐derived superoxide (Kirchner et al., 2013). If the ability of phagocytes to generate O2 •− is compromised, the host's antimicrobial defence is weakened leading to an increased risk of developing infections with significant morbidity and/or mortality. This condition is known as chronic granulomatous disease (CGD) (van den Berg et al., 2009; O'Neill et al., 2015) and is an inherited immunodeficiency disorder caused by inactivating mutations in the genes encoding the NOX2 complex (CYBB, CYBA, NCF1, NCF2 and NCF4) and is characterized by recurring and life‐threatening bacterial and fungal infections due to diminished or abolished pathogen killing. CGD consists of a heterogeneous group of disorders ranging from pulmonary to dermatological and GI complications, often accompanied by the formation of granulomas in several organs. Loss‐of‐function variants of X‐linked CYBB occur in about 70% of the reported CGD cases, while variants in CYBA, NCF1 and NCF2, encoding p22phox, p47phox and p67phox, respectively, are less frequent, with NCF1 occurring in 25% of CGD patients. So far, only one NCF4 variant (encoding p40phox) has been described (Matute et al., 2009; O'Neill et al., 2015).
Following the discovery of NOX2, homology screening revealed six additional NADPH oxidase family members in mammals. Here, we will focus primarily on oxidases expressed in the gut. X‐linked NOX1, evolutionarily close to NOX2 (sharing ~60% homology with NOX2), requires heterodimerization with p22phox and assembly with cytosolic subunits and activated Rac GTPase to produce O2 •−. In the case of NOX1, the cytosolic subunits are termed NOXO1 and NOXA1 (for NOX1 organizer and activator respectively) and are homologues of p47phox and p67phox. Although found in LP lymphocytes and other cell types (Szanto et al., 2005), NOX1 is often called ‘colon NADPH oxidase’ due to the high expression in colon epithelium. The enzyme remains inactive until pro‐inflammatory cytokines, such as IFN‐γ (Geiszt et al., 2003a), or bacterial products, such as LPS induce up‐regulation, translocation and activation (Corcionivoschi et al., 2012; O'Leary et al., 2012).
Commonly found in kidney cortex, but also in fibroblasts, smooth muscle cells and the intestinal epithelium, NOX4 shares only 39% identity with NOX2. NOX4 regulation is distinct as it is considered constitutively active and preferentially releases H2O2. NOX4 was shown to be involved in oxygen sensing and cellular senescence (Geiszt et al., 2000; Shiose et al., 2001). Unlike other NOXs, NOX4 does not require regulatory binding partners after the NOX4‐p22phox complex is formed (von Lohneysen et al., 2008). Certain stimuli including TGF‐β or hypoxia induce transcriptional up‐regulation of NOX4 accompanied by continuous H2O2 generation (Hecker et al., 2009; Diebold et al., 2010).
Another important gut NADPH oxidase is DUOX2 (thyroid oxidase), an EF‐ hand motif containing protein that forms a membrane‐bound heterodimer with its maturation factor DUOXA2 and generates H2O2 in a Ca2+‐dependent manner. In Drosophila, the DUOX orthologue dDuox was required for the clearance of bacterial intestinal infections (Ha et al., 2005; Lee et al., 2015). In mammals, the expression of DUOX2/DUOXA2 was detected throughout all of the digestive tract, particularly in caecum and colon epithelium (El Hassani et al., 2005). Microbial stimuli, and in particular microbiota, pathogenic bacteria and viruses induce marked up‐regulation of the DUOX2 enzyme, and thus, DUOX2 is thought to participate in antimicrobial defence in the host mucosa (Geiszt et al., 2003b; Harper et al., 2005; Lipinski et al., 2009; Grasberger et al., 2015; Sommer and Backhed, 2015). Microbial induction of murine Duox2 expression in the ileum required TIR‐domain containing adapter inducing interferon‐beta (TRIF) signalling to NF‐κB p50/p65, whereas in the colon, MyD88 and p38 MAPK pathways were necessary (Sommer and Backhed, 2015). In IECs, Duox2, similarly to Nox1, is upregulated following in vitro stimulation with colitogenic IL‐13 (Mandal et al., 2010). DUOX2 up‐regulation was also observed upon IFN‐γ stimulation (Harper et al., 2005). Albeit mostly expressed in thyroid and lung epithelium, the closely related DUOX1/DUOXA1 enzyme complex has been detected in the stomach lining and selected gastric and colorectal cancer cell lines (El Hassani et al., 2005; Juhasz et al., 2009; Strickertsson et al., 2013).
The mitochondrial electron transport chain (mETC)
Mitochondria are another source of ROS generation in physiological and pathophysiological conditions (Balaban et al., 2005). mtROS are produced by the oxidative phosphorylation pathways involved in energy production. Mitochondria generate high‐energy ATP molecules by an electrochemical proton gradient maintained by the transfer of electrons to protein carriers in the mitochondrial membrane, namely, Complex I–IV. Mitochondrial oxidative phosphorylation is a tightly controlled process; however, 1–2% of oxygen consumed during this process is converted to O2 •− when electrons leak out from the mETC and are aberrantly transferred to molecular O2 (West et al., 2011). Superoxide generated by mETC reacts with NO radicals formed by the interaction of NO with mitochondrial redox‐metal centres. The reactive ONOO‐ product can cause mitochondrial integrity disruption due to irreversible nitration of proteins, inactivation of enzymes and DNA damage. In the mitochondrial matrix, O2 •− can react with SOD2, leading to the formation of diffusible H2O2 that regulates cellular functions by modulating the activity of redox‐sensitive transcription factors (Nrf2, HIF‐1 and NF‐κB), altering kinase activity (MAPK and IκB kinase‐β) and inhibiting phosphatases (PTEN and PTP1B) (Tanner et al., 2011; Marinho et al., 2014). In health and disease, crosstalk between similar or distinct ROS sources can occur. This includes NOX to NOX/DUOX signalling but may also involve mitochondria‐NOX crosstalk in various scenarios, including serum withdrawal, increased glycolysis or hypoxia, leading to mtROS‐mediated up‐regulation and activation of NOX isoforms (Lee et al., 2006; Daiber, 2010; Lu et al., 2012).
Mitochondrial dysfunction and the overproduction of mtROS play an important role in inflammation, degenerative diseases and ageing (Liochev, 2013). However, similarly to NADPH oxidase‐derived ROS, mtROS may predominantly regulate redox signalling, and inducing senescence could be the result of generally compromised signals, rather than being caused by direct damage to sensitive targets (Liochev, 2013). In a high turnover tissue like the digestive tract, factors that regulate enterocyte lifespan are of special interest when considering inflammatory disease and colon cancer (Kajla et al., 2012; Pelicci et al., 2013).
NO synthases (NOS)
Three isoforms of NOSs have been identified: two are constitutively present in either neuronal (NOS1; nNOS) or endothelial (NOS3; eNOS) tissues and are termed constitutive NOS, while a third isoform is expressed by immune, vasculature and neuronal cells after induction by certain cytokines or microbial products and is thus termed inducible NOS (NOS2; iNOS) (Kolios et al., 2004). All these isoforms of NOS catalyse the oxidation of the terminal nitrogen of the amino acid l‐arginine to produce NO and l‐citrulline (Figure 2). The production of NO by constitutive NOS is low (nanomolar concentrations), short‐lasting and highly controlled by Ca2 +‐mobilizing agents, while NOS2 generates high levels of NO (micromolar concentrations) that can persist for days. Transient, low levels of NO are beneficial to control gastric mucosal blood flow, gut motility and barrier integrity, whereas prolonged NO generation is often accompanied with ROS production, leading to the formation of RNS with potentially deleterious effects.
The interaction of NO with O2 or O2 •− gives rise to two types of chemical stress: nitrosative or oxidative stress (Figure 2). Both types of stress are associated with inflammation and de novo expression of iNOS (Kolios et al., 2004; Lanzetti et al., 2012). The interaction of NO with O2 triggers the auto‐oxidation of NO and the formation of nitrogen dioxide and dinitrogen trioxide, potent nitrosating agents with carcinogenic effects, while the interaction of NO with O2 •− produces cytotoxic peroxynitrite (ONOO−) (Kolios et al., 2004). ONOO− can cause lipid peroxidation, or changes in protein structure and folding by oxidation of sulphydryl groups in proteins, and nitration of tyrosine, yielding nitrotyrosine, and eventually leading to loss of protein function and tissue damage (Pereira et al., 2013).
Inflammation and repair in the GI tract
Here, we discuss the role of ROS or RNS during the inflammatory response in three different regions of the digestive tract: stomach, ileum and colon. As prompt resealing of the epithelial surface barrier following injuries is essential to preserve gut homeostasis, the role of ROS and oxidative stress during wound healing will be also discussed.
Gastritis
Acute inflammation of the stomach mucosa is the consequence of a number of triggers including bacterial infections, uremia, ischaemia, nonsteroidal anti‐inflammatory drugs (NSAIDs) and corrosive agents. Infectious gastritis develops when the gastric pH rises above pH 4 and stomach acid is incapable of neutralizing food‐borne pathogens. The most common bacterium leading to severe stomach inflammation is Helicobacter pylori (H. pylori).
H. pylori is a Gram‐negative, helical, microaerophilic bacterium that selectively colonizes the antrum of the human stomach inducing chronic inflammation that eventually evolves into peptic ulcer (10–15% of cases), gastric adenocarcinoma (1–3% of cases) or mucosa‐associated lymphoid tissue lymphomas (<0.1% of cases) (Wroblewski et al., 2010). H. pylori is highly adapted to occupy the special ecological niche of the stomach lining by producing large amounts of urease, thereby creating an alkaline environment. Although the majority of H. pylori reside within the mucus layer, ~20% of the bacteria will bind to gastric epithelial cells (Peek et al., 2010). H. pylori‐induced gastritis is predominantly caused by a Th1/Th17 cell response (Shi et al., 2010) occurring after recruitment of a large number of ROS‐producing neutrophils into the mucosa. Indeed, an excessive production of ROS and RNS was observed in H. pylori‐infected human gastric mucosa, which correlated with mucosal tissue damage and bacterial colonization (Davies et al., 1994). In H. pylori‐infected stomach tissue, possible sources of ROS/RNS include gastric mucosal cells and H. pylori itself, apart from neutrophils (Handa et al., 2010; Naito and Yoshikawa, 2002). While H. pylori infection promoted the recruitment of neutrophils into the mucosa, the bacterium can survive in this hostile environment by manipulating neutrophil defence mechanisms such as phagocytosis and the oxidative burst (Peek et al., 2010). Secretion of the H. pylori virulence factor neutrophil‐activating protein engaged TLR2 resulting in NF‐κB activation and Th1 cytokine up‐regulation. This pro‐inflammatory response was accompanied by recruitment of PMNs and generation of O2 •− via NOX2 activation. Simultaneously, H. pylori can evade neutrophil‐dependent killing by inhibiting opsonisation (Peek et al., 2010). In epithelial cells, H. pylori infection stimulated ROS production via NOX1 activation (Kawahara et al., 2005). H. pylori can also increase the production of mtROS via type IV secretion system‐mediated injection of the virulence factors CagA and VacA, which alter mETC function and elevate oxidative stress (Galmiche and Rassow, 2010; Handa et al., 2010). These studies suggest that the production of pro‐inflammatory ROS as the host's primordial defence mechanism against bacterial invasion can eventually turn deleterious for the host through ROS‐mediated tissue damage and inflammation.
A role for Duox2 in the pathogenesis of H. pylori‐associated gastritis was first suggested in Rhesus macaques (Hornsby et al., 2008). H. pylori infection in mice is usually characterized by limited colonization and variable pathology, while infection with H. felis is more reliable to reproduce human disease. Grasberger et al. (2013) used l‐thyroxine supplemented mice with combined deficiency in Duoxa1 and Duoxa2 to determine the role of Duox in H. felis infection (Grasberger et al., 2013). Mucosal colonization with H. felis was increased in Duoxa −/− mice. The elevated bacterial load caused enhanced shedding of bacterial antigens and virulence factors, leading to severe gastritis in Duoxa −/− mice. These results suggest that the release of H2O2 by Duox at the apical surface of the gastric epithelium controls growth of H. felis in the mucus layer and provides antimicrobial protection (Grasberger et al., 2013).
Ileitis
The intraluminal pH shifts from pH 1.5–3.5 in the stomach to pH 6 in the duodenum and pH 7.4 in the terminal ileum. This alkalization alters the environment and promotes the development of specific microbiota (Hayashi et al., 2005). For example, the SI hosts a considerable number of commensal bacteria (up to 108 per mL in the ileum) and is the site of colonization for a number of enteritis‐causing pathogens, such as Vibrio cholerae, pathogenic forms of Escherichia coli and Salmonella (Garner et al., 2009). Ileitis, an inflammation of the SI, develops often as a consequence of Crohn's Disease (CD); however, a number of other conditions including tuberculosis, spondyloarthropathies, ischaemia or sarcoidosis can be associated with ileitis (Dilauro and Crum‐Cianflone, 2010). Our knowledge regarding redox regulation in ileitis is limited by the paucity of suitable animal models. Transgenic mice with conditional deletion of caspase‐8 (Casp8 ΔIEC) (Gunther et al., 2011) or FADD (FADDIEC‐KO) (Welz et al., 2011) in the intestinal epithelium spontaneously developed inflammatory lesions in the terminal ileum, lacked Paneth cells and exhibited severe colitis in a dextran sodium sulphate (DSS) colitis model. These changes correlated with RIPK3‐dependent IEC necrosis and altered TNF‐α levels. Although RIPK3‐mediated cell death has been associated with oxidative stress, this link was not investigated. Recently, Esworthy et al (2014) reported that transgenic mice with double knockout (dKO) for the antioxidant enzymes GSH peroxidase (GPx)‐1 and GPx‐2 developed spontaneous mild‐to‐moderate ileocolitis. This pathology disappeared in triple knockout (GPx‐dKO/Nox1 −/−) mice, suggesting that Nox1‐generated ROS promotes intestinal inflammation when the antioxidant systems fail. Interestingly, GPx‐dKO/Nox1 −/− mice showed reduced TNF‐α levels in the ileum, indicating crosstalk between TNF‐α and redox signalling in the development of ileitis (Esworthy et al., 2014). Nox1‐generated ROS was also connected to ileal mucositis induced by administration of 5‐fluoruracil. In this mouse model, characterized by an abnormal inflammatory response, intestinal malabsorption and dysfunction, Nox1 deficiency correlated with reduced TNF‐α‐mediated and IL‐1β‐mediated crypt apoptosis (Yasuda et al., 2012).
Similarly to NADPH oxidase‐produced ROS, mtROS were linked to SI inflammation. In a model of NSAID‐induced mucosal injury, increased barrier permeability and inflammation were associated with enhanced mtROS production and changes in mitochondrial morphology towards vacuolation, swelling and loss of cristae (Handa et al., 2014). In enteritis induced by Clostridium difficile infection, the increased fluid and electrolyte secretion, intestinal permeability and loss of electrical resistance were accompanied by increased ROS levels, although the source and mechanism of action were not determined (Qiu et al., 1999). Other studies associated Rac1‐dependent intracellular oxidative stress and necrosis in colonic tissue with the action of C. difficile toxin B (Farrow et al., 2013). Taken together, this evidence suggests a pro‐inflammatory role of Nox1‐derived ROS and mtROS in the development of ileitis.
IBD
Oxidative stress is considered a potential etiological and/or triggering factor for the development of colon inflammation (Biasi et al., 2013). IBD, consisting of CD and ulcerative colitis (UC), are defined as idiopathic, chronic and relapsing intestinal disorders occurring in genetically predisposed individuals exposed to environmental risk factors, such as diet, hygiene, stress and microbiome changes (Kaplan, 2015). Both forms of IBD are associated with significant morbidity and characterized by gut inflammation, diarrhoea, abdominal pain, weight loss and a number of extra‐intestinal manifestations including arthritis, psoriasis, erythema, cholangitis and eye inflammation (Levine and Burakoff, 2011).
CD can affect any part of the GI tract including the terminal ileum with deep and extended ulcers involving all layers of the bowel wall, while UC is limited to the colon and rectum with inflammation occurring in the innermost layer of the intestinal lining. The maladaptive chronic inflammatory responses observed in IBD patients are characterized by alterations of the physiological mechanisms regulating the host's immune tolerance, leading to an abnormal sensing of commensal microorganisms by the LP immune system.
NADPH oxidases and IBD
A chronic inflammatory process affecting both small and large bowels and resembling CD occurs in up to 40% of CGD patients (Marciano et al., 2004; Marks et al., 2009). The GI involvement seems to correlate rather with CYBB deficiency than with autosomal‐recessive forms (CYBA, NCF1, NCF2 and NCF4) or with the remaining ROS levels (Agarwal and Mayer, 2013; Holland, 2013). CGD patients may present with perirectal abscesses, GI tract obstruction and recurrent diarrhoea. Colon endoscopy often reveals colonic narrowing, a thickened bowel wall, pancolitis and pseudopolyps, whereas the histology shows submucosal oedema, crypt abscesses, inflammatory cell infiltration, cryptitis, epithelioid granulomas (usually in the muscularis) and large pigment‐laden histiocytes in the LP (Agarwal and Mayer, 2013; Barbato et al., 2014). Decreased NOX2 activity, above the threshold leading to CGD, has also recently been linked to very early onset (VEO)IBD (defined as IBD in children <6 years of age according to the modified Paris classification) (Eszter Muller et al., 2014). VEOIBD patients present predominantly with pancolitis and occasionally perianal disease and are hyporesponsive to conventional therapy leading to high morbidity and mortality (Uhlig et al., 2014). In contrast to the multifactorial nature of adult IBD, VEOIBD is considered a consequence of monogenic defects (Uhlig et al., 2014). Several functionally altered variants in NOX2 complex components have been identified in VEOIBD (Dhillon et al., 2014a). Certain SNPs in NCF1 and NCF2 are described in up to 10% of VEOIBD patients. Up to now, only one VEOIBD patient was identified with a NCF4 variant, which negatively influenced the binding of this protein to p67phox (O'Neill et al., 2015). This variant has been previously associated with ileal adult CD in genome‐wide association studies (Rioux et al., 2007; Roberts et al., 2008). Recently, we identified two functionally altered NOX1 and DUOX2 variants in VEOIBD patients, both associated with severe pancolitis and reduced ROS production in vitro (Hayes et al., 2015), suggesting an important role of these epithelial oxidases in the pathogenesis of IBD. Interestingly, patients carrying these DUOX2 variants did not present with hypothyroidism at newborn screening.
Human IBD can be mimicked in mice by altering the epithelial barrier integrity with chemicals such as DSS or 2,4,6‐trinitrobenzenesulfonic acid (TNBS). Additionally, transgenic mice with IL‐10 (Berg et al., 1996), Muc2 (Van der Sluis et al., 2006) or NLRP6 (Elinav et al., 2011) deficiency will permit studying the development of spontaneous chronic enterocolitis. However, with respect to ROS generation, current animal models seem often not to reflect human disease. For example, Cybb knockout mice do not develop the spontaneous CD‐like phenotype observed in many CGD patients. Nox2 deficient mice were protected against the mucosal damage induced by DSS (Bao et al., 2011), but succumbed to colitis in the TNBS model (Campbell et al., 2014). In mice harbouring a point mutation in Ncf1, more severe colitis was observed (Rodrigues‐Sousa et al., 2014), while Ncf1 knockout mice showed no colitogenic phenotype (Krieglstein et al., 2001). Mice lacking Ncf4 exhibited exacerbated colitis (Conway et al., 2012) that was absent in Cybb (Nox2)‐deficient mice. As IBD development is not only associated with genetic susceptibility but also with environmental factors, some of the apparent inconsistencies in animal studies are likely due to different housing conditions, nutrition or commensal communities present.
Experimentally, ROS produced by Nox1 during colitis has been studied using Il10 −/− mice as background, as Nox1 deficiency alone did not produce a colitis phenotype in DSS or TNBS models (Leoni et al., 2013; Treton et al., 2014). IL‐10 negatively regulates Nox1‐derived ROS production after barrier insult (Li et al., 2014) as well as following TNF‐α (Kuwano et al., 2008; Kamizato et al., 2009), IFN‐γ (Kuwano et al., 2006; Kamizato et al., 2009) and LPS (O'Leary et al., 2012) stimulation. When Nox1 −/− mice were crossed with Il10 −/− mice, spontaneous colitis developed mimicking clinical and histological features of UC patients (Treton et al., 2014). Consistent with findings that Nox1 regulates the balance between goblet and absorptive cell types in the murine colon (Coant et al., 2010), Nox1 −/−/Il10 −/− mice displayed reduced and aberrant goblet cells and decreased Muc2 and Muc4 mucin expression in ulcerated sites of the intestine (Treton et al., 2014). Others reported that ROS generated by an unspecified Nox isoform was critical for mucin granule accumulation in murine colonic spheroids (Patel et al., 2013).
Interaction between the CD susceptibility genes NOD2, ATG16L1 and ROS sources has been proposed, thus connecting microbial signalling, autophagic clearance and ROS production. Stimulation of NOD2 by its ligand muramyl dipeptide increased ROS generation in Caco‐2 cells and in murine colon biopsies derived from wild type but not Nod2‐ deficient mice (Lipinski et al., 2009). A physical interaction between these two antimicrobial effectors was found in TNF‐α/muramyl dipeptide stimulated Caco‐2 cells (Lipinski et al., 2009) but not in DUOX2/DUOXA2 expressing HT29 cells challenged with enteropathogenic E. coli (Hayes et al., 2015). In autophagy, contrasting views exist in regard to ROS as essential physiological regulator or to ROS/oxidative stress being the result of disturbed autophagic processes. Even the source of ROS is debated with some studies favouring mtROS, while others show the importance of Nox‐ROS signalling. A direct effect of ROS on microtubule‐associated protein 1A/1B‐light chain 3 conjugate recruitment to autophagosomes and co‐localization of Nox with light chain 3 was reported (Huang et al., 2009; Patel et al., 2013), while others implicated mtROS in redox regulation of autophagy and particularly in mitophagy (Frank et al., 2012; Kurihara et al., 2012). Crosstalk between inflammasomes and autophagy is also connected to ROS (Saitoh et al., 2008; Scherz‐Shouval and Elazar, 2011) as NLRP3 and AIM2 inflammasomes required mtROS, but not Nox‐derived ROS, for transcriptional up‐regulation and activation respectively (Zhou et al., 2011; Crane et al., 2014). Taken together, both human and animal data suggest that ROS play an important role in preserving processes essential for intestinal homeostasis and that the impairment or deficiency of phagocytic/epithelial oxidases (ROS low) or mitochondrial dysfunction (ROS high) predispose or exacerbate intestinal pathology. In particular, with respect to patients with defective NADPH oxidase function, these data indicate that reduced innate immune cell or intestinal epithelial ROS generation will increase susceptibility to IBD and colon inflammation.
Conversely, up‐regulation of DUOX2 has been detected in paediatric (Haberman et al., 2014) and adult (Csillag et al., 2007) CD and UC patients. Prominent expression of DUOX2 was also found in all segments of UC patients' large bowel (MacFie et al., 2014), accompanied by an increase of Proteobacteria phylum in the microbiota (Haberman et al., 2014). Interestingly, in vitro stimulation of inflamed UC biopsies with 5‐aminosalicylic acid (5‐ASA) used as first‐choice intervention in the treatment of UC and mild to moderate CD (Dignass et al., 2012), induced up‐regulation of DUOX2 expression (MacFie et al., 2014). The mechanism underlying these ex vivo observations remains unknown, and so far, the use of Duox2 knockout mice is hindered by the severe congenital hypothyroidism present in animals with loss of Duox2 function (Grasberger et al., 2012) and the required continuous l‐thyroxine treatment, both likely altering the microbiome and metabolome.
Little is known regarding the role of NOX4‐derived ROS in the GI tract. Nox4 has been detected in murine IECs upon stimulation with colitogenic IL‐13 (Mandal et al., 2010) and following Listeria infection (Dolowschiak et al., 2010), but its role in IEC physiology or pathophysiology has not been studied. In contrast, an intimate connection exists between TGF‐β signalling, NOX4‐mediated H2O2 production and myofibroblast transformation in lung, kidney and liver fibrosis (Jiang et al., 2014; Paik et al., 2014). TGF‐β1 is a central mediator of fibrosis in CD (Jiang et al., 2014) with up to 30–40% of CD patients developing intestinal fibrosis and ileal strictures within 10 years of disease onset (Lawrance et al., 2015). Recurrent or persistent epithelial injuries, accompanied by local chronic inflammation, are responsible for initiating intestinal fibrogenesis in CD. At which point of this disease process, ROS will trigger development or progression of intestinal fibrosis has not yet been assessed, but NOX4 is a prime candidate for one of the key steps involved.
Mitochondria and IBD
Functionally compromised mitochondria are an important source of ROS in chronic inflammation. mtROS are involved in overproduction of the cytokines IL‐1β and IL‐18 by up‐regulating the nucleotide‐binding domain, leucine rich family, pyrin containing 3 inflammasome (Mittal et al., 2014) and in intestinal tumorigenesis (Bellafante et al., 2014). Marked mitochondrial damage in the colonic mucosa was found in IBD patients, correlating with increased oxidative stress (Biasi et al., 2013). Furthermore, the expression of prohibitin (PHB), an important membrane regulator of mitochondrial respiratory function, was decreased during oxidative stress (Theiss et al., 2007). PHB levels were reduced in mucosal biopsies of IBD patients and in animal models of colitis (Hsieh et al., 2006; Theiss et al., 2007). PHB overexpression in intestinal cells protected against oxidant‐induced permeability changes, suggesting a beneficial role of this protein in preventing mucosal barrier disruption by excessive production of ROS (Theiss et al., 2007). A pro‐inflammatory role of mtROS in intestinal inflammation is supported by analysis of transgenic mice with compromised mitochondrial function due to genetic deficiencies. Ucp2 −/− mice lacking the negative regulation of mtROS production exhibited severe colitis (Zhang et al., 2012). This phenotype correlates with findings in patients, where UCP2 polymorphisms were associated with UC and CD (Yu et al., 2009). mtROS homeostasis in the epithelium is also regulated by the stress‐inducible immediate early response gene IEX‐1 through a mechanism that targets the mitochondrial F1F0‐ATPase inhibitor IF1 for degradation, resulting in elevated F1F0‐ATPase activity, acceleration of ATP hydrolysis and reduced mtROS (Shen et al., 2009). Iex1 −/− mice showed diminished pathology in DSS colitis, possibly by up‐regulating IL‐17 (Ustyugova et al., 2012).
NOS enzymes and IBD
NO directly or indirectly regulates several manifestations of IBD, including mucosal vasodilatation, increased epithelial permeability and altered motility. Cumulative evidence indicates that NO production is enhanced during IBD (Kolios et al., 2004; Winter et al., 2013), and recently two hyperactive NOS2 variants were linked to increased susceptibility to VEOIBD (Dhillon et al., 2014b). The main cellular sources of NO in the inflamed gut tissue are monocytes/macrophages (Bain and Mowat, 2014), IECs (Kolios et al., 2004) and myofibroblasts (Wu et al., 2013). In both animals and humans with colitis, the generation of ONOO− is favoured by the concomitant presence of NO and O2 •− produced via NOS2 and a superoxide source such as NOX1 or NOX2 respectively. This peroxynitrite stress can cause nitrosative DNA damage by forming 8‐nitroguanine (Kawanishi et al., 2006). Several studies have been carried out to uncover the importance of enhanced production of O2 •− and NO and/or ONOO− in the aetiology of IBD; however, so far, this issue remains controversial. For example, one hypothesis is that ONOO− by inducing DNA damage can activate poly(ADP‐ribose) synthetase, which in turn induces increased epithelial permeability as a result of IEC apoptosis (McCafferty, 2000; Pacher et al., 2007). In general, it seems that low and transient levels of NO are an important regulator of intestinal homeostasis as mice deficient in NOS3 (eNos −/−) are highly susceptible to colitis (Sasaki et al., 2003). On the other hand, higher concentrations and prolonged production of NO generated by NOS2 seem to augment the inflammatory response in the colon because the lack of inducible NOS (iNOS −/−) conferred protection in the DSS model (Krieglstein et al., 2001). These data suggest that the protective role of endothelial NO in disease may disappear at some point, possibly overwhelmed by an excess of OONO− supplied by activated immune cells, which ultimately contributes to chronic intestinal inflammation.
Wound healing
Intestinal inflammation severely compromises the epithelial barrier function, resulting in the exposure of underlying tissues to luminal bacteria and their antigenic products. Hence, wound closure of the epithelium must occur efficiently to rapidly restore barrier function and homeostasis. Certain drugs such as NSAIDs can exacerbate pre‐existing IBD, increase flare‐ups and may lead to the development of gastric and intestinal ulcers. Recently, the pivotal role of PGI2 in preventing penetrating ulcers after colonic mucosal injury was reported (Manieri et al., 2015). In general, a deregulated wound healing response often results in fibrotic disease. H2O2 is the most relevant ROS produced during wound healing, and it plays a role in every phase of tissue repair, including haemostasis, inflammation, vascularization and re‐epithelialization (Cordeiro and Jacinto, 2013). Following tissue injury, ROS participate in wound healing and tissue regeneration by conferring resistance to wound infection, altering signalling pathways, modulating leukocyte adhesion molecule expression and inducing fibroblast, smooth muscle and epithelial cell proliferation and migration (Chan et al., 2009). Cooperation between phagocytic and epithelial NADPH oxidases in this process has been visualized in a Xenopus wounding model (Love et al., 2013), while others reported mtROS‐induced redox modifications of the GTPase Rho1 as crucial for promoting skin wound healing responses in Caenorhabditis elegans (Xu and Chisholm, 2014). Although perhaps counterintuitive, the oxidative burst of PMNs accumulating in crypt abscesses during mucosal inflammation is crucial for the resolution of gut inflammation (Campbell et al., 2014). ROS generation by transmigrating PMNs during acute colitis causes an oxygen depleted microenvironment, which transcriptionally up‐regulates hypoxia genes in IECs, resulting in protection of the mucosa (Robinson et al., 2008; Campbell et al., 2014). In general, acute mild to moderate hypoxia supports adaptation and survival, while chronic extreme hypoxia leads to cell death (Sen, 2009).
A growing body of evidence indicates that Nox1‐generated ROS co‐ordinately drive colon epithelial proliferation. Although absent (Kawahara et al., 2004) or barely detectable (Laurent et al., 2008) in the SI mucosa by real‐time PCR, Nox1 is highly expressed in the large bowel (Kikuchi et al., 2000), where it may participate in regulating IEC proliferation and differentiation. Nox1 deficiency in mice promoted the rapid differentiation of intestinal progenitor cells into post‐mitotic goblet cells, suggesting that Nox1 signalling is required within the crypt compartment to maintain the undifferentiated state of crypt progenitors. These data are important in the context of epithelial barrier repair, but they also indicate that Nox1 may have oncogenic potential (Coant et al., 2010). Indeed, Nox1 is overexpressed in human colon cancers harbouring activating mutations of K‐Ras (Laurent et al., 2008). The K‐Ras oncogene‐mediated up‐regulation of Nox1 was required for transformation and resulted in premature senescence (Mitsushita et al., 2004; Kodama et al., 2013). In contrast, oxidative damage of mtDNA by mtROS with an increased rate of mutations was observed in UC patients developing colorectal cancers (Nishikawa et al., 2005).
The microbiota is an important player in gut barrier repair. Signals delivered by pathogens and commensals up‐regulate Nox1 and/or Duox2 and stimulate ROS generation (Corcionivoschi et al., 2012; Sommer and Backhed, 2015). Lactobacilli spp., particularly Lactobacillus rhamnosus, stimulated Nox1‐dependent ROS generation in murine enterocytes, driving cell proliferation (Jones et al., 2013). The importance of Nox1‐derived ROS and mucosal wound repair was also evident in ligand‐mediated stimulation of the formyl peptide receptor FPR1 present on IECs (Leoni et al., 2013). This GPCR, mainly characterized on neutrophils, responds to microbial‐derived formylated peptides and gives rise to Nox‐dependent ROS production. Specific members of the commensal flora can stimulate FPR1 signalling leading to the generation of ROS via enterocyte Nox1 and to phosphorylation of focal adhesion kinase and Erk1,2 MAPK, which ultimately enhances proliferation and migration of enterocytes towards the site of injury (Alam et al., 2014).
Current IBD treatments
The eradication of inflammation results in decreased surgery and hospitalization rates in both UC and CD patients (Costa et al., 2013); therefore, one of the goals of IBD therapy is to reduce intestinal inflammation and immune system hyper‐responsiveness. Conventional therapy involves aminosalicylates, corticosteroids, thiopurines, antimetabolites, calcineurin inhibitors and biological agents. Although debated, the use of 5‐ASA as the first‐choice drug in the treatment of UC is positively influenced by tolerability, dose schedule and cost (Mowat et al., 2011). Different mechanisms of action have been attributed to 5‐ASA (Desreumaux and Ghosh, 2006) including the potential to scavenge ROS/RNS (Couto et al., 2010; Managlia et al., 2013) and to potently inhibit ROS‐dependent neutrophil extracellular trap formation in activated neutrophils (Kirchner et al., 2013). Although the notion that balanced ROS levels are essential for gut health is gaining traction, in general oxidative stress with increased ROS levels has been considered a culprit in the GI inflammatory response. Whether antioxidant therapy will be a valid means in arresting inflammation in patients with gastric or intestinal inflammation remains largely unresolved. For example, the beneficial effect of SOD‐based treatments in experimental models of colitis (Keshavarzian et al., 1990) was not replicated by SOD therapy in human IBD (Emerit et al., 1991). Although 5‐ASA is considered an antioxidant (Managlia et al., 2013), two clinical studies indicated that 5‐ASA is more effective in the treatment of UC when combined with other antioxidants, such as N‐acetyl cysteine (Guijarro et al., 2008), or allopurinol, a xanthine oxidase inhibitor (Jarnerot et al., 2000), suggesting that prevention of oxidative damage may be beneficial when treating IBD. The therapeutic value of 5‐ASA most likely relies, not on its putative antioxidant activity, but on a broad anti‐inflammatory mechanism as patients with reduced ROS generation due to NADPH oxidase loss‐of‐function variants respond well to 5‐ASA treatments. Antioxidant and vitamin dietary supplements are still considered a therapeutic option due to their low risk of side effects. Polyphenols were found clinically beneficial in both UC (Dryden et al., 2013) and CD patients (Kolacek et al., 2013), whereas vitamin supplementation particularly vitamin D, whose deficiency is associated with IBD incidence/activity (Torki et al., 2015), reduced the risk of relapse in a cohort of patients with CD (Jorgensen et al., 2010).
Managing IBD in CGD patients
Special therapeutic considerations are vital for managing IBD in CGD patients. These patients are severely immunocompromised due to impaired host defence by ROS‐deficient innate immune cells and tend to present with hyperinflammation as regulation of cytokine signalling is dependent on NOX2‐derived ROS. Hyperinflammation is frequently observed in CGD patients, predominantly due to deregulation of cytokine signalling pathways and the inability of macrophages to efficiently perform efferocytosis (uptake of apoptotic cells) (O'Neill et al., 2015). The average age of onset of GI symptoms in susceptible CGD patients is approximately 5 years, but some intestinal manifestations may even precede the CGD diagnosis in 5% of cases (Huang et al., 2006; Towbin and Chaves, 2010). When GI symptoms occur, a misdiagnosis of IBD remains a possibility, thereby compromising the therapeutic management of CGD. Although the optimal treatment of CGD colitis is not established yet, the first recommended approach is similar to IBD, often involving the administration of 5‐ASA.
Corticosteroids and other immunomodulatory agents such as methotrexate, azathioprine and its metabolite 6‐mercaptopurine are indicated when patients show a diminished response (or are completely refractory) to 5‐ASA. On the other hand, the use of the monoclonal antibody Infliximab, a selective TNF‐α blocker, is considered a major advance in IBD therapy, although its use is strongly discouraged in CGD patients, even when combined with antibiotic prophylaxis, due to the high risk of developing severe infections (Uzel et al., 2010). Therefore, more effective management of intestinal inflammation in CGD patients is needed. Recently, a pro‐oxidant therapy has been suggested. Pro‐oxidants are molecules capable of inducing mild oxidative stress and therefore could partially restore ROS levels in CGD patients. For instance, some vitamins such as vitamin C (ascorbate) can induce the generation of free radicals (•OH) by reducing Fe2 + to Fe3 + via the Fenton reaction, although the efficacy of this treatment for colitis remains to be established (Aghdassi et al., 2003). NOX2 agonists as ROS‐inducing drugs have been identified by large‐scale screening, but their pharmacological use, therapeutic index and side effects have not yet been determined (Hultqvist et al., 2015).
To date, the only curative treatment for CGD and accompanying GI symptoms is haematopoietic stem cell transplantation from human leukocyte antigen‐matched siblings or unrelated donors, correcting the immune deficiency with engrafted donor cells. In a recent multi‐centre study, haematopoietic stem cell transplantation cured colitis in 22 out of 24 patients with CGD (Gungor et al., 2014). Current clinical trials, using gene therapy with improved vectors or gene editing approaches in CGD iPS cells, may point to a valid alternative for effective treatment of CGD patients when a match is not available or the patient is too ill for a transplant (Flynn et al., 2015; Grez et al., 2011; Kang and Malech, 2012).
Conclusions and future remarks
Over the last few years, the pivotal contributions of the redox balance to GI physiology and gut health have become apparent, significantly changing our understanding of how oxidants maintain the intestinal barrier and control complex host–pathogen interactions in the stomach and intestine. It is now clear that ROS are not only harmful byproducts of cell metabolism but also crucial homeostatic regulators in the gut. Physiological levels of ROS are likely to act as signals governing the interaction of the mucosa with the microbiome, potentially even shaping commensal communities. Likewise, mtROS exert essential immunoregulatory functions, but their link to endoplasmatic reticulum stress, the unfolded protein response and apoptosis/necrosis may be essential in triggering intestinal inflammation and tissue injury via ROS overproduction. Until now, antioxidants have failed in the treatment of IBD in clinical trials, possibly by not reaching therapeutic concentrations or due to their broad inhibitory spectrum. One may argue that more specificity in terms of targeting the deregulated ROS source is required or that even ROS‐enhancing options will be beneficial for particular patient cohorts. We believe that future therapeutic approaches will need to consider the dual role of ROS in health and disease.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors apologize to those researchers whose work could not be included in this review due to space limitation. We thank S. O'Neill for critical reading of the manuscript. Funding was provided by the National Childrens' Research Centre (UGK, GA), Science Foundation Ireland (UGK), and the European Crohn's and Colitis Organization (GA). The present work was supported by the European Cooperation in Science and Technology (COST Action BM1203/EU‐ROS).
Aviello, G. , and Knaus, U. G. (2017) ROS in gastrointestinal inflammation: Rescue Or Sabotage?. British Journal of Pharmacology, 174: 1704–1718. doi: 10.1111/bph.13428.
References
- Agarwal S, Mayer L (2013). Diagnosis and treatment of gastrointestinal disorders in patients with primary immunodeficiency. Clin Gastroenterol Hepatol 11: 1050–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghdassi E, Wendland BE, Steinhart AH, Wolman SL, Jeejeebhoy K, Allard JP (2003). Antioxidant vitamin supplementation in Crohn's disease decreases oxidative stress: a randomized controlled trial. Am J Gastroenterol 98: 348–353. [DOI] [PubMed] [Google Scholar]
- Alam A, Leoni G, Wentworth CC, Kwal JM, Wu H, Ardita CS, et al. (2014). Redox signaling regulates commensal‐mediated mucosal homeostasis and restitution and requires formyl peptide receptor 1. Mucosal Immunol 7: 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain CC, Mowat AM (2014). The monocyte–macrophage axis in the intestine. Cell Immunol 291: 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T (2005). Mitochondria, oxidants, and aging. Cell 120: 483–495. [DOI] [PubMed] [Google Scholar]
- Bao S, Carr ED, Xu YH, Hunt NH (2011). Gp91(phox) contributes to the development of experimental inflammatory bowel disease. Immunol Cell Biol 89: 853–860. [DOI] [PubMed] [Google Scholar]
- Barbato M, Ragusa G, Civitelli F, Marcheggiano A, Di Nardo G, Iacobini M, et al. (2014). Chronic granulomatous disease mimicking early‐onset Crohn's disease with cutaneous manifestations. BMC Pediatr 14: 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH (2007). The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313. [DOI] [PubMed] [Google Scholar]
- Bellafante E, Morgano A, Salvatore L, Murzilli S, Di Tullio G, D'Orazio A, et al. (2014). PGC‐1beta promotes enterocyte lifespan and tumorigenesis in the intestine. Proc Natl Acad Sci U S A 111: E4523–E4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, et al. (1996). Enterocolitis and colon cancer in interleukin‐10‐deficient mice are associated with aberrant cytokine production and CD4(+) TH1‐like responses. J Clin Invest 98: 1010–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasi F, Leonarduzzi G, Oteiza PI, Poli G (2013). Inflammatory bowel disease: mechanisms, redox considerations, and therapeutic targets. Antioxid Redox Signal 19: 1711–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes RP, Weissmann N, Schroder K (2014). Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med 76: 208–226. [DOI] [PubMed] [Google Scholar]
- Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, et al. (2014). Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 40: 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EC, Jiang F, Peshavariya HM, Dusting GJ (2009). Regulation of cell proliferation by NADPH oxidase‐mediated signaling: potential roles in tissue repair, regenerative medicine and tissue engineering. Pharmacol Ther 122: 97–108. [DOI] [PubMed] [Google Scholar]
- Chieppa M, Rescigno M, Huang AY, Germain RN (2006). Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med 203: 2841–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coant N, Ben Mkaddem S, Pedruzzi E, Guichard C, Treton X, Ducroc R, et al. (2010). NADPH oxidase 1 modulates WNT and NOTCH1 signaling to control the fate of proliferative progenitor cells in the colon. Mol Cell Biol 30: 2636–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KL, Goel G, Sokol H, Manocha M, Mizoguchi E, Terhorst C, et al. (2012). p40phox expression regulates neutrophil recruitment and function during the resolution phase of intestinal inflammation. J Immunol 189: 3631–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcionivoschi N, Alvarez LA, Sharp TH, Strengert M, Alemka A, Mantell J, et al. (2012). Mucosal reactive oxygen species decrease virulence by disrupting Campylobacter jejuni phosphotyrosine signaling. Cell Host Microbe 12: 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro JV, Jacinto A (2013). The role of transcription‐independent damage signals in the initiation of epithelial wound healing. Nat Rev Mol Cell Biol 14: 249–262. [DOI] [PubMed] [Google Scholar]
- Costa J, Magro F, Caldeira D, Alarcao J, Sousa R, Vaz‐Carneiro A (2013). Infliximab reduces hospitalizations and surgery interventions in patients with inflammatory bowel disease: a systematic review and meta‐analysis. Inflamm Bowel Dis 19: 2098–2110. [DOI] [PubMed] [Google Scholar]
- Couto D, Ribeiro D, Freitas M, Gomes A, Lima JL, Fernandes E (2010). Scavenging of reactive oxygen and nitrogen species by the prodrug sulfasalazine and its metabolites 5‐aminosalicylic acid and sulfapyridine. Redox Rep 15: 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane DD, Bauler TJ, Wehrly TD, Bosio CM (2014). Mitochondrial ROS potentiates indirect activation of the AIM2 inflammasome. Front Microbiol 5: 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csillag C, Nielsen OH, Vainer B, Olsen J, Dieckgraefe BK, Hendel J, et al. (2007). Expression of the genes dual oxidase 2, lipocalin 2 and regenerating islet‐derived 1 alpha in Crohn's disease . Scand J Gastroenterol 42: 454–463. [DOI] [PubMed] [Google Scholar]
- Daiber A (2010). Redox signaling (cross‐talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta 1797: 897–906. [DOI] [PubMed] [Google Scholar]
- Dashdorj A, Jyothi KR, Lim S, Jo A, Nguyen MN, Ha J, et al. (2013). Mitochondria‐targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome‐mediated inflammatory cytokines. BMC Med 11: 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies GR, Simmonds NJ, Stevens TR, Sheaff MT, Banatvala N, Laurenson IF, et al. (1994). Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut 35: 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desreumaux P, Ghosh S (2006). Review article: mode of action and delivery of 5‐aminosalicylic acid – new evidence. Aliment Pharmacol Ther 24 (Suppl 1): 2–9. [DOI] [PubMed] [Google Scholar]
- Dhillon SS, Fattouh R, Elkadri A, Xu W, Murchie R, Walters T, et al. (2014a). Variants in nicotinamide adenine dinucleotide phosphate oxidase complex components determine susceptibility to very early onset inflammatory bowel disease. Gastroenterology 147: 680–689. [DOI] [PubMed] [Google Scholar]
- Dhillon SS, Mastropaolo LA, Murchie R, Griffiths C, Thoni C, Elkadri A, et al. (2014b). Higher activity of the inducible nitric oxide synthase contributes to very early onset inflammatory bowel disease. Clin Transl Gastroenterol 5: e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold I, Petry A, Hess J, Gorlach A (2010). The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia‐inducible factor‐1. Mol Biol Cell 21: 2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignass A, Lindsay JO, Sturm A, Windsor A, Colombel JF, Allez M, et al. (2012). Second European evidence‐based consensus on the diagnosis and management of ulcerative colitis part 2: current management. J Crohns Colitis 6: 991–1030. [DOI] [PubMed] [Google Scholar]
- Dilauro S, Crum‐Cianflone NF (2010). Ileitis: when it is not Crohn's disease . Curr Gastroenterol Rep 12: 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolowschiak T, Chassin C, Ben Mkaddem S, Fuchs TM, Weiss S, Vandewalle A, et al. (2010). Potentiation of epithelial innate host responses by intercellular communication. PLoS Pathog 6: e1001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryden GW, Lam A, Beatty K, Qazzaz HH, McClain CJ (2013). A pilot study to evaluate the safety and efficacy of an oral dose of (−)‐epigallocatechin‐3‐gallate‐rich polyphenon E in patients with mild to moderate ulcerative colitis. Inflamm Bowel Dis 19: 1904–1912. [DOI] [PubMed] [Google Scholar]
- El Hassani RA, Benfares N, Caillou B, Talbot M, Sabourin JC, Belotte V, et al. (2005). Dual oxidase2 is expressed all along the digestive tract. Am J Physiol Gastrointest Liver Physiol 288: G933–G942. [DOI] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao‐Mejia J, Thaiss CA, Booth CJ, et al. (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145: 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerit J, Pelletier S, Likforman J, Pasquier C, Thuillier A (1991). Phase II trial of copper zinc superoxide dismutase (CuZn SOD) in the treatment of Crohn's disease . Free Radic Res Commun 12‐13 (Pt 2): 563–569. [DOI] [PubMed] [Google Scholar]
- Esworthy RS, Kim BW, Chow J, Shen B, Doroshow JH, Chu FF (2014). Nox1 causes ileocolitis in mice deficient in glutathione peroxidase‐1 and −2. Free Radic Biol Med 68: 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eszter Muller K, Laszlo Lakatos P, Papp M, Veres G (2014). Incidence and paris classification of pediatric inflammatory bowel disease. Gastroenterol Res Pract 2014: 904307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow MA, Chumbler NM, Lapierre LA, Franklin JL, Rutherford SA, Goldenring JR, et al. (2013). Clostridium difficile toxin B‐induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc Natl Acad Sci U S A 110: 18674–18679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn R, Grundmann A, Renz P, Haenseler W, James WS, Cowley SA, et al. (2015). CRISPR‐mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp Hematol 43: 838–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M, Duvezin‐Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, et al. (2012). Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 1823: 2297–2310. [DOI] [PubMed] [Google Scholar]
- Fukata M, Arditi M (2013). The role of pattern recognition receptors in intestinal inflammation. Mucosal Immunol 6: 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galmiche A, Rassow J (2010). Targeting of Helicobacter pylori VacA to mitochondria. Gut Microbes 1: 392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner CD, Antonopoulos DA, Wagner B, Duhamel GE, Keresztes I, Ross DA, et al. (2009). Perturbation of the small intestine microbial ecology by streptomycin alters pathology in a Salmonella enterica serovar typhimurium murine model of infection. Infect Immun 77: 2691–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiszt M, Kopp JB, Varnai P, Leto TL (2000). Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci U S A 97: 8010–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiszt M, Lekstrom K, Brenner S, Hewitt SM, Dana R, Malech HL, et al. (2003a). NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91phox in the regulated production of superoxide by phagocytes. J Immunol 171: 299–306. [DOI] [PubMed] [Google Scholar]
- Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL (2003b). Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J 17: 1502–1504. [DOI] [PubMed] [Google Scholar]
- Grasberger H, De Deken X, Mayo OB, Raad H, Weiss M, Liao XH, et al. (2012). Mice deficient in dual oxidase maturation factors are severely hypothyroid. Mol Endocrinol 26: 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasberger H, El‐Zaatari M, Dang DT, Merchant JL (2013). Dual oxidases control release of hydrogen peroxide by the gastric epithelium to prevent Helicobacter felis infection and inflammation in mice. Gastroenterology 145: 1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasberger H, Gao J, Nagao‐Kitamoto H, Kitamoto S, Zhang M, Kamada N, et al. (2015). Increased expression of DUOX2 is an epithelial response to mucosal dysbiosis required for immune homeostasis in mouse intestine. Gastroenterology 149: 1849–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grez M, Reichenbach J, Schwable J, Seger R, Dinauer MC, Thrasher AJ (2011). Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol Ther 19: 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guijarro LG, Mate J, Gisbert JP, Perez‐Calle JL, Marin‐Jimenez I, Arriaza E, et al. (2008). N‐acetyl‐l‐cysteine combined with mesalamine in the treatment of ulcerative colitis: randomized, placebo‐controlled pilot study. World J Gastroenterol 14: 2851–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gungor T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. (2014). Reduced‐intensity conditioning and HLA‐matched haemopoietic stem‐cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet 383: 436–448. [DOI] [PubMed] [Google Scholar]
- Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, et al. (2011). Caspase‐8 regulates TNF‐alpha‐induced epithelial necroptosis and terminal ileitis. Nature 477: 335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha EM, Oh CT, Bae YS, Lee WJ (2005). A direct role for dual oxidase in Drosophila gut immunity. Science 310: 847–850. [DOI] [PubMed] [Google Scholar]
- Haberman Y, Tickle TL, Dexheimer PJ, Kim MO, Tang D, Karns R, et al. (2014). Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest 124: 3617–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa O, Majima A, Onozawa Y, Horie H, Uehara Y, Fukui A, et al. (2014). The role of mitochondria‐derived reactive oxygen species in the pathogenesis of non‐steroidal anti‐inflammatory drug‐induced small intestinal injury. Free Radic Res 48: 1095–1099. [DOI] [PubMed] [Google Scholar]
- Handa O, Naito Y, Yoshikawa T (2010). Helicobacter pylori: a ROS‐inducing bacterial species in the stomach. Inflamm Res 59: 997–1003. [DOI] [PubMed] [Google Scholar]
- Harper RW, Xu C, Eiserich JP, Chen Y, Kao CY, Thai P, et al. (2005). Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett 579: 4911–4917. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y (2005). Molecular analysis of jejunal, ileal, caecal and recto‐sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol 54 (Pt 11): 1093–1101. [DOI] [PubMed] [Google Scholar]
- Hayes P, Dhillon S, O'Neill K, Thoeni C, Hui KY, Elkadri A, et al. (2015). Defects in NADPH Oxidase Genes NOX1 and DUOX2 in Very Early Onset Inflammatory Bowel Disease. Cell Mol Gastroenterol Hepatol 1: 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, et al. (2009). NADPH oxidase‐4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 15: 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. (2015). Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria‐specific CD4(+) T cells. Science 348: 1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland SM (2013). Chronic granulomatous disease. Hematol Oncol Clin North Am 27: 89–99, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom KM, Finkel T (2014). Cellular mechanisms and physiological consequences of redox‐dependent signalling. Nat Rev Mol Cell Biol 15: 411–421. [DOI] [PubMed] [Google Scholar]
- Hornsby MJ, Huff JL, Kays RJ, Canfield DR, Bevins CL, Solnick JV (2008). Helicobacter pylori induces an antimicrobial response in rhesus macaques in a cag pathogenicity island‐dependent manner. Gastroenterology 134: 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh SY, Shih TC, Yeh CY, Lin CJ, Chou YY, Lee YS (2006). Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics 6: 5322–5331. [DOI] [PubMed] [Google Scholar]
- Huang A, Abbasakoor F, Vaizey CJ (2006). Gastrointestinal manifestations of chronic granulomatous disease. Colorectal Dis 8: 637–644. [DOI] [PubMed] [Google Scholar]
- Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, et al. (2009). Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A 106: 6226–6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultqvist M, Olofsson P, Wallner FK, Holmdahl R (2015). Pharmacological potential of Nox2 agonists in inflammatory conditions. Antioxid Redox Signal 23: 446–459. [DOI] [PubMed] [Google Scholar]
- Jarnerot G, Strom M, Danielsson A, Kilander A, Loof L, Hultcrantz R, et al. (2000). Allopurinol in addition to 5‐aminosalicylic acid based drugs for the maintenance treatment of ulcerative colitis. Aliment Pharmacol Ther 14: 1159–1162. [DOI] [PubMed] [Google Scholar]
- Jiang F, Liu GS, Dusting GJ, Chan EC (2014). NADPH oxidase‐dependent redox signaling in TGF‐beta‐mediated fibrotic responses. Redox Biol 2: 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RM, Luo L, Ardita CS, Richardson AN, Kwon YM, Mercante JW, et al. (2013). Symbiotic lactobacilli stimulate gut epithelial proliferation via Nox‐mediated generation of reactive oxygen species. EMBO J 32: 3017–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen SP, Agnholt J, Glerup H, Lyhne S, Villadsen GE, Hvas CL, et al. (2010). Clinical trial: vitamin D3 treatment in Crohn's disease ‐ a randomized double‐blind placebo‐controlled study. Aliment Pharmacol Ther 32: 377–383. [DOI] [PubMed] [Google Scholar]
- Juhasz A, Ge Y, Markel S, Chiu A, Matsumoto L, van Balgooy J, et al. (2009). Expression of NADPH oxidase homologues and accessory genes in human cancer cell lines, tumours and adjacent normal tissues. Free Radic Res 43: 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajla S, Mondol AS, Nagasawa A, Zhang Y, Kato M, Matsuno K, et al. (2012). A crucial role for Nox 1 in redox‐dependent regulation of Wnt‐beta‐catenin signaling. FASEB J 26: 2049–2059. [DOI] [PubMed] [Google Scholar]
- Kamizato M, Nishida K, Masuda K, Takeo K, Yamamoto Y, Kawai T, et al. (2009). Interleukin 10 inhibits interferon gamma‐ and tumor necrosis factor alpha‐stimulated activation of NADPH oxidase 1 in human colonic epithelial cells and the mouse colon. J Gastroenterol 44: 1172–1184. [DOI] [PubMed] [Google Scholar]
- Kang EM, Malech HL (2012). Gene therapy for chronic granulomatous disease. Methods Enzymol 507: 125–154. [DOI] [PubMed] [Google Scholar]
- Kaplan GG (2015). The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol 12: 720–727. [DOI] [PubMed] [Google Scholar]
- Kawahara T, Kohjima M, Kuwano Y, Mino H, Teshima‐Kondo S, Takeya R, et al. (2005). Helicobacter pylori lipopolysaccharide activates Rac1 and transcription of NADPH oxidase Nox1 and its organizer NOXO1 in guinea pig gastric mucosal cells. Am J Physiol Cell Physiol 288: C450–C457. [DOI] [PubMed] [Google Scholar]
- Kawahara T, Kuwano Y, Teshima‐Kondo S, Takeya R, Sumimoto H, Kishi K, et al. (2004). Role of nicotinamide adenine dinucleotide phosphate oxidase 1 in oxidative burst response to toll‐like receptor 5 signaling in large intestinal epithelial cells. J Immunol 172: 3051–3058. [DOI] [PubMed] [Google Scholar]
- Kawanishi S, Hiraku Y, Pinlaor S, Ma N (2006). Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation‐related carcinogenesis. Biol Chem 387: 365–372. [DOI] [PubMed] [Google Scholar]
- Keshavarzian A, Morgan G, Sedghi S, Gordon JH, Doria M (1990). Role of reactive oxygen metabolites in experimental colitis. Gut 31: 786–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi H, Hikage M, Miyashita H, Fukumoto M (2000). NADPH oxidase subunit, gp91(phox) homologue, preferentially expressed in human colon epithelial cells. Gene 254: 237–243. [DOI] [PubMed] [Google Scholar]
- Kirchner T, Hermann E, Moller S, Klinger M, Solbach W, Laskay T, et al. (2013). Flavonoids and 5‐aminosalicylic acid inhibit the formation of neutrophil extracellular traps. Mediators Inflamm 2013: 710239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama R, Kato M, Furuta S, Ueno S, Zhang Y, Matsuno K, et al. (2013). ROS‐generating oxidases Nox1 and Nox4 contribute to oncogenic Ras‐induced premature senescence. Genes Cells 18: 32–41. [DOI] [PubMed] [Google Scholar]
- Kolacek M, Muchova J, Dvorakova M, Paduchova Z, Zitnanova I, Cierna I, et al. (2013). Effect of natural polyphenols (Pycnogenol) on oxidative stress markers in children suffering from Crohn's disease–a pilot study. Free Radic Res 47: 624–634. [DOI] [PubMed] [Google Scholar]
- Kolios G, Valatas V, Ward SG (2004). Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology 113: 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieglstein CF, Cerwinka WH, Laroux FS, Salter JW, Russell JM, Schuermann G, et al. (2001). Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: divergent roles of superoxide and nitric oxide. J Exp Med 194: 1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T, et al. (2012). Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J Biol Chem 287: 3265–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwano Y, Kawahara T, Yamamoto H, Teshima‐Kondo S, Tominaga K, Masuda K, et al. (2006). Interferon‐gamma activates transcription of NADPH oxidase 1 gene and upregulates production of superoxide anion by human large intestinal epithelial cells. Am J Physiol Cell Physiol 290: C433–C443. [DOI] [PubMed] [Google Scholar]
- Kuwano Y, Tominaga K, Kawahara T, Sasaki H, Takeo K, Nishida K, et al. (2008). Tumor necrosis factor alpha activates transcription of the NADPH oxidase organizer 1 (NOXO1) gene and upregulates superoxide production in colon epithelial cells. Free Radic Biol Med 45: 1642–1652. [DOI] [PubMed] [Google Scholar]
- Lambeth JD, Neish AS (2014). Nox enzymes and new thinking on reactive oxygen: a double‐edged sword revisited. Annu Rev Pathol 9: 119–145. [DOI] [PubMed] [Google Scholar]
- Lanzetti M, da Costa CA, Nesi RT, Barroso MV, Martins V, Victoni T, et al. (2012). Oxidative stress and nitrosative stress are involved in different stages of proteolytic pulmonary emphysema. Free Radic Biol Med 53: 1993–2001. [DOI] [PubMed] [Google Scholar]
- Laurent E, McCoy JW 3rd, Macina RA, Liu W, Cheng G, Robine S, et al. (2008). Nox1 is over‐expressed in human colon cancers and correlates with activating mutations in K‐Ras. Int J Cancer 123: 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrance IC, Rogler G, Bamias G, Breynaert C, Florholmen J, Pellino G, et al. (2015). Cellular and molecular mediators of intestinal fibrosis. J Crohns Colitis pii: j.crohns.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KA, Kim B, Bhin J, do Kim H, You H, Ek K, et al. (2015). Bacterial uracil modulates Drosophila DUOX‐dependent gut immunity via Hedgehog‐induced signaling endosomes. Cell Host Microbe 17: 191–204. [DOI] [PubMed] [Google Scholar]
- Lee SB, Bae IH, Bae YS, Um HD (2006). Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem 281: 36228–36235. [DOI] [PubMed] [Google Scholar]
- Leoni G, Alam A, Neumann PA, Lambeth JD, Cheng G, McCoy J, et al. (2013). Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. J Clin Invest 123: 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JS, Burakoff R (2011). Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol Hepatol (N Y) 7: 235–241. [PMC free article] [PubMed] [Google Scholar]
- Li B, Alli R, Vogel P, Geiger TL (2014). IL‐10 modulates DSS‐induced colitis through a macrophage‐ROS‐NO axis. Mucosal Immunol 7: 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liochev SI (2013). Reactive oxygen species and the free radical theory of aging. Free Radic Biol Med 60: 1–4. [DOI] [PubMed] [Google Scholar]
- Lipinski S, Till A, Sina C, Arlt A, Grasberger H, Schreiber S, et al. (2009). DUOX2‐derived reactive oxygen species are effectors of NOD2‐mediated antibacterial responses. J Cell Sci 122 (Pt 19): 3522–3530. [DOI] [PubMed] [Google Scholar]
- Love NR, Chen Y, Ishibashi S, Kritsiligkou P, Lea R, Koh Y, et al. (2013). Amputation‐induced reactive oxygen species are required for successful Xenopus tadpole tail regeneration. Nat Cell Biol 15: 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Hu Y, Chen G, Chen Z, Zhang H, Wang F, et al. (2012). Novel role of NOX in supporting aerobic glycolysis in cancer cells with mitochondrial dysfunction and as a potential target for cancer therapy. PLoS Biol 10: e1001326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFie TS, Poulsom R, Parker A, Warnes G, Boitsova T, Nijhuis A, et al. (2014). DUOX2 and DUOXA2 form the predominant enzyme system capable of producing the reactive oxygen species H2O2 in active ulcerative colitis and are modulated by 5‐aminosalicylic acid. Inflamm Bowel Dis 20: 514–524. [DOI] [PubMed] [Google Scholar]
- Managlia E, Katzman RB, Brown JB, Barrett TA (2013). Antioxidant properties of mesalamine in colitis inhibit phosphoinositide 3‐kinase signaling in progenitor cells. Inflamm Bowel Dis 19: 2051–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal D, Fu P, Levine AD (2010). REDOX regulation of IL‐13 signaling in intestinal epithelial cells: usage of alternate pathways mediates distinct gene expression patterns. Cell Signal 22: 1485–1494. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Manieri NA, Mack MR, Himmelrich MD, Worthley DL, Hanson EM, Eckmann L, et al. (2015). Mucosally transplanted mesenchymal stem cells stimulate intestinal healing by promoting angiogenesis. J Clin Invest 125: 3606–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya‐O'Brien S, et al. (2004). Gastrointestinal involvement in chronic granulomatous disease. Pediatrics 114: 462–468. [DOI] [PubMed] [Google Scholar]
- Marinho HS, Real C, Cyrne L, Soares H, Antunes F (2014). Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol 2: 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks DJ, Miyagi K, Rahman FZ, Novelli M, Bloom SL, Segal AW (2009). Inflammatory bowel disease in CGD reproduces the clinicopathological features of Crohn's disease . Am J Gastroenterol 104: 117–124. [DOI] [PubMed] [Google Scholar]
- Matute JD, Arias AA, Wright NA, Wrobel I, Waterhouse CC, Li XJ, et al. (2009). A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood 114: 3309–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCafferty DM (2000). Peroxynitrite and inflammatory bowel disease. Gut 46: 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuckin MA, Linden SK, Sutton P, Florin TH (2011). Mucin dynamics and enteric pathogens. Nat Rev Microbiol 9: 265–278. [DOI] [PubMed] [Google Scholar]
- Mitsushita J, Lambeth JD, Kamata T (2004). The superoxide‐generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res 64: 3580–3585. [DOI] [PubMed] [Google Scholar]
- Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB (2014). Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20: 1126–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat C, Cole A, Windsor A, Ahmad T, Arnott I, Driscoll R, et al. (2011). Guidelines for the management of inflammatory bowel disease in adults. Gut 60: 571–607. [DOI] [PubMed] [Google Scholar]
- Naito Y, Yoshikawa T (2002). Molecular and cellular mechanisms involved in Helicobacter pylori‐induced inflammation and oxidative stress. Free Radic Biol Med 33: 323–336. [DOI] [PubMed] [Google Scholar]
- Nanno M, Kanari Y, Naito T, Inoue N, Hisamatsu T, Chinen H, et al. (2008). Exacerbating role of gammadelta T cells in chronic colitis of T‐cell receptor alpha mutant mice. Gastroenterology 134: 481–490. [DOI] [PubMed] [Google Scholar]
- Neurath MF (2014). Cytokines in inflammatory bowel disease. Nat Rev Immunol 14: 329–342. [DOI] [PubMed] [Google Scholar]
- Nishikawa M, Oshitani N, Matsumoto T, Nishigami T, Arakawa T, Inoue M (2005). Accumulation of mitochondrial DNA mutation with colorectal carcinogenesis in ulcerative colitis. Br J Cancer 93: 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary DP, Bhatt L, Woolley JF, Gough DR, Wang JH, Cotter TG, et al. (2012). TLR‐4 signalling accelerates colon cancer cell adhesion via NF‐kappaB mediated transcriptional up‐regulation of Nox‐1. PLoS One 7: e44176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill S, Brault J, Stasia MJ, Knaus UG (2015). Genetic disorders coupled to ROS deficiency. Red Biol 6: 135–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L (2007). Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87: 315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KK, Miyoshi H, Beatty WL, Head RD, Malvin NP, Cadwell K, et al. (2013). Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J 32: 3130–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik YH, Kim J, Aoyama T, De Minicis S, Bataller R, Brenner DA (2014). Role of NADPH oxidases in liver fibrosis. Antioxid Redox Signal 20: 2854–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SPH, Buneman OP, et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek RM Jr, Fiske C, Wilson KT (2010). Role of innate immunity in Helicobacter pylori‐induced gastric malignancy. Physiol Rev 90: 831–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicci PG, Dalton P, Giorgio M (2013). The other face of ROS: a driver of stem cell expansion in colorectal cancer. Cell Stem Cell 12: 635–636. [DOI] [PubMed] [Google Scholar]
- Pereira C, Ferreira NR, Rocha BS, Barbosa RM, Laranjinha J (2013). The redox interplay between nitrite and nitric oxide: from the gut to the brain. Redox Biol 1: 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LW, Artis D (2014). Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14: 141–153. [DOI] [PubMed] [Google Scholar]
- Qiu B, Pothoulakis C, Castagliuolo I, Nikulasson S, LaMont JT (1999). Participation of reactive oxygen metabolites in Clostridium difficile toxin A‐induced enteritis in rats. Am J Physiol 276 (2 Pt 1): G485–G490. [DOI] [PubMed] [Google Scholar]
- Rezaie A, Parker RD, Abdollahi M (2007). Oxidative stress and pathogenesis of inflammatory bowel disease: an epiphenomenon or the cause? Dig Dis Sci 52: 2015–2021. [DOI] [PubMed] [Google Scholar]
- Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. (2007). Genome‐wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 39: 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RL, Hollis‐Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR (2008). Confirmation of association of IRGM and NCF4 with ileal Crohn's disease in a population‐based cohort. Genes Immun 9: 561–565. [DOI] [PubMed] [Google Scholar]
- Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP (2008). Mucosal protection by hypoxia‐inducible factor prolyl hydroxylase inhibition. Gastroenterology 134: 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues‐Sousa T, Ladeirinha AF, Santiago AR, Carvalheiro H, Raposo B, Alarcao A, et al. (2014). Deficient production of reactive oxygen species leads to severe chronic DSS‐induced colitis in Ncf1/p47phox‐mutant mice. PLoS One 9: e97532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1beta production. Nature 456: 264–268. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Bharwani S, Jordan P, Elrod JW, Grisham MB, Jackson TH, et al. (2003). Increased disease activity in eNOS‐deficient mice in experimental colitis. Free Radic Biol Med 35: 1679–1687. [DOI] [PubMed] [Google Scholar]
- Scherz‐Shouval R, Elazar Z (2011). Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 36: 30–38. [DOI] [PubMed] [Google Scholar]
- Sekirov I, Russell SL, Antunes LC, Finlay BB (2010). Gut microbiota in health and disease. Physiol Rev 90: 859–904. [DOI] [PubMed] [Google Scholar]
- Sen CK (2009). Wound healing essentials: let there be oxygen. Wound Repair Regen 17: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Zhi L, Hu W, Wu MX (2009). IEX‐1 targets mitochondrial F1Fo‐ATPase inhibitor for degradation. Cell Death Differ 16: 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Liu XF, Zhuang Y, Zhang JY, Liu T, Yin Z, et al. (2010). Helicobacter pylori‐induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J Immunol 184: 5121–5129. [DOI] [PubMed] [Google Scholar]
- Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, et al. (2001). A novel superoxide‐producing NAD(P)H oxidase in kidney. J Biol Chem 276: 1417–1423. [DOI] [PubMed] [Google Scholar]
- Sommer F, Backhed F (2015). The gut microbiota engages different signaling pathways to induce Duox2 expression in the ileum and colon epithelium. Mucosal Immunol 8: 372–379. [DOI] [PubMed] [Google Scholar]
- Strickertsson JA, Desler C, Martin‐Bertelsen T, Machado AM, Wadstrom T, Winther O, et al. (2013). Enterococcus faecalis infection causes inflammation, intracellular oxphos‐independent ROS production, and DNA damage in human gastric cancer cells. PLoS One 8: e63147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szanto I, Rubbia‐Brandt L, Kiss P, Steger K, Banfi B, Kovari E, et al. (2005). Expression of NOX1, a superoxide‐generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J Pathol 207: 164–176. [DOI] [PubMed] [Google Scholar]
- Tanner JJ, Parsons ZD, Cummings AH, Zhou H, Gates KS (2011). Redox regulation of protein tyrosine phosphatases: structural and chemical aspects. Antioxid Redox Signal 15: 77–97. [DOI] [PubMed] [Google Scholar]
- Theiss AL, Idell RD, Srinivasan S, Klapproth JM, Jones DP, Merlin D, et al. (2007). Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J 21: 197–206. [DOI] [PubMed] [Google Scholar]
- Torki M, Gholamrezaei A, Mirbagher L, Danesh M, Kheiri S, Emami MH (2015). Vitamin D deficiency associated with disease activity in patients with inflammatory bowel diseases. Dig Dis Sci 60: 3085–3091. [DOI] [PubMed] [Google Scholar]
- Towbin AJ, Chaves I (2010). Chronic granulomatous disease. Pediatr Radiol 40: 657–668; quiz 792‐653. [DOI] [PubMed] [Google Scholar]
- Treton X, Pedruzzi E, Guichard C, Ladeiro Y, Sedghi S, Vallee M, et al. (2014). Combined NADPH oxidase 1 and interleukin 10 deficiency induces chronic endoplasmic reticulum stress and causes ulcerative colitis‐like disease in mice. PLoS One 9: e101669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, et al. (2014). The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 147: 990–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustyugova IV, Zhi L, Wu MX (2012). Reciprocal regulation of the survival and apoptosis of Th17 and Th1 cells in the colon. Inflamm Bowel Dis 18: 333–343. [DOI] [PubMed] [Google Scholar]
- Uzel G, Orange JS, Poliak N, Marciano BE, Heller T, Holland SM (2010). Complications of tumor necrosis factor‐alpha blockade in chronic granulomatous disease‐related colitis. Clin Infect Dis 51: 1429–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. (2009). Chronic granulomatous disease: the European experience. PLoS One 4: e5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. (2006). Muc2‐deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131: 117–129. [DOI] [PubMed] [Google Scholar]
- von Lohneysen K, Noack D, Jesaitis AJ, Dinauer MC, Knaus UG (2008). Mutational analysis reveals distinct features of the Nox4‐p22 phox complex. J Biol Chem 283: 35273–35282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez‐Majada V, Ermolaeva M, et al. (2011). FADD prevents RIP3‐mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477: 330–334. [DOI] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S (2011). Mitochondria in innate immune responses. Nat Rev Immunol 11: 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, et al. (2013). Host‐derived nitrate boosts growth of E. coli in the inflamed gut. Science 339: 708–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn CC (1995). Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett 82‐83: 969–974. [DOI] [PubMed] [Google Scholar]
- Wroblewski LE, Peek RM Jr, Wilson KT (2010). Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 23: 713–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Chitapanarux T, Chen Y, Soon RK, Jr, Yee HF, Jr (2013). Intestinal myofibroblasts produce nitric oxide in response to combinatorial cytokine stimulation. J Cell Physiol 228: 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullaert A, Bonnet MC, Pasparakis M (2011). NF‐kappaB in the regulation of epithelial homeostasis and inflammation. Cell Res 21: 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Chisholm AD (2014). C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev Cell 31: 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M, Kato S, Yamanaka N, Iimori M, Utsumi D, Kitahara Y, et al. (2012). Potential role of the NADPH oxidase NOX1 in the pathogenesis of 5‐fluorouracil‐induced intestinal mucositis in mice. Am J Physiol Gastrointest Liver Physiol 302: G1133–G1142. [DOI] [PubMed] [Google Scholar]
- Yu X, Wieczorek S, Franke A, Yin H, Pierer M, Sina C, et al. (2009). Association of UCP2 ‐866 G/A polymorphism with chronic inflammatory diseases. Genes Immun 10: 601–605. [DOI] [PubMed] [Google Scholar]
- Zhang H, Kuai XY, Yu P, Lin L, Shi R (2012). Protective role of uncoupling protein‐2 against dextran sodium sulfate‐induced colitis. J Gastroenterol Hepatol 27: 603–608. [DOI] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225. [DOI] [PubMed] [Google Scholar]