

Abstract
Hypercholesterolaemia is considered to be a principle risk factor for cardiovascular disease, having direct negative effects on the myocardium itself, in addition to the development of atherosclerosis. Since hypercholesterolaemia affects the global cardiac gene expression profile, among many other factors, it results in increased myocardial oxidative stress, mitochondrial dysfunction and inflammation triggered apoptosis, all of which may account for myocardial dysfunction and increased susceptibility of the myocardium to infarction. In addition, numerous experimental and clinical studies have revealed that hyperlcholesterolaemia may interfere with the cardioprotective potential of conditioning mechanisms. Although not fully elucidated, the underlying mechanisms for the lost cardioprotection in hypercholesterolaemic animals have been reported to involve dysregulation of the endothelial NOS‐cGMP, reperfusion injury salvage kinase, peroxynitrite‐MMP2 signalling pathways, modulation of ATP‐sensitive potassium channels and apoptotic pathways. In this review article, we summarize the current knowledge on the effect of hypercholesterolaemia on the non‐ischaemic and ischaemic heart as well as on the cardioprotection induced by drugs or ischaemic preconditioning, postconditioning and remote conditioning. Future perspectives concerning the mechanisms and the design of preclinical and clinical trials are highlighted.
Linked Articles
This article is part of a themed section on Redox Biology and Oxidative Stress in Health and Disease. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.12/issuetoc
Abbreviations
- AMI
acute myocardial infarction
- BH4
tetrahydrobiopterin
- CAD
coronary artery disease
- eNOS
endothelial NOS
- iNOS
inducible NOS
- LV
left ventricular
- MPT
mitochondrial permeability transition
- mPTP
mitochondrial permeability transition pore
- PC
ischaemic preconditioning
- PostC
postconditioning
- RAS
renin‐angiotensin system
- RIPC
remote preconditioning
- RISK
reperfusion injury salvage kinase
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Enzymes f |
| Bax | Akt |
| Bcl‐2 | ERK1 |
| GPCRs b | ERK2 |
| AT1 receptor | GSK3β |
| Voltage‐gated ion channels c | MMP2 |
| KATP (Kir6.x) channels | NOS |
| Other ion channels d | PCSK9 |
| Connexin 43 (Cx43) | PI3K |
| Nuclear hormone receptors e | PKG |
| PPARα |
| LIGANDS | |
|---|---|
| ADMA | NO |
| cGMP | Sevoflurane |
| Cyclosporine A (CsA) | VCAM‐1 |
| Fasudil |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,d,e,fAlexander et al., 2015a,b,c,d,e,f).
Introduction
Although cholesterol is one of the main constituents of cellular membranes and plays an important role in hormone and bile acid synthesis, increased circulating levels, especially if oxidized, are detrimental resulting in atherosclerosis and thus in the development and progression of coronary, carotid and peripheral vessels' disease (Félix‐Redondo et al., 2013). Several years ago, clinical studies clearly showed a linear relation between the regression of cholesterol levels and the consequent improvement of clinical outcome in patients suffering from hypercholesterolaemia (Castelli et al., 1989; Rubin et al., 1990). Hypercholesterolaemia is widely accepted as a principal risk factor for coronary artery disease (CAD) (Tiwari and Khokhar 2014), and patients with extremely increased levels of cholesterol have an elevated risk of ischaemic events regardless of their genotype (Sniderman, et al., 2014). Moreover, analysis of the Kaiser Permanente Heart Study and Framingham Heart Study cohorts showed significant associations between cholesterol levels and the risk for cardiovascular mortality in individuals with and without a history of CAD (Castelli et al., 1989; Rubin et al., 1990; Wong et al., 1991).
Although, the observations from clinical trials and experimental studies suggest an effect of cholesterol on myocardial function (Huang et al., 2004), little is known about its effects on cardiac function apart from CAD. Hypercholesterolaemia has been proposed to have direct negative effects on the myocardium itself, in addition to the development of atherosclerosis, with several studies demonstrating increased myocardial injury in hypercholesterolaemic animals (Hoshida et al., 1996a; Ferdinandy et al., 1998a; Scalia et al., 2001; see for review by Ferdinandy, 2003; Osipov et al., 2009). Hypercholesterolaemia alone increased myocardial necrosis by 45% over what was observed in normal fed animals (Osipov et al., 2009), and impaired diastolic function in vitro as well as in vivo (Onody et al., 2003; Huang et al., 2004; Varga et al., 2013). Intracellular lipid accumulation in cardiomyocytes and several alterations in the structural and functional properties of the myocardium have been observed in response to a high cholesterol diet in rodents (Onody et al., 2003; Puskas et al., 2004). Thus, it seems that hypercholesterolaemia is not only detrimental for the vasculature but is also a risk factor for increased cariomyocyte death and poor left ventricular (LV) systolic function in patients following acute myocardial infarction (AMI) (Corti et al., 2001). However, the molecular mechanisms by which chronically elevated cholesterol can detrimentally affect the cardiomyocyte are poorly understood.
In this review article, we summarize the current knowledge on the effect of hypercholesterolaemia on the non‐ischaemic and ischaemic heart, as well as on cardioprotection induced by ischaemic preconditioning (PC), postconditioning (PostC) and remote conditioning. Additionally, we summarize the effects of hypercholesterolaemia on drug‐induced cardioprotection as well as the effect of antihyperlipidaemic drugs on cardioprotection. Future perspectives concerning the mechanisms and the design of preclinical and clinical trials are highlighted.
Effect of hypercholesterolaemia on the myocardium
Hypercholesterolaemia causes endothelial dysfunction
Numerous animal and clinical models have reported impaired endothelium‐dependent and independent relaxation in the presence of hypercholesterolaemia (Kawashima and Yokoyama, 2004). Both acute and chronic elevations in blood cholesterol induce nitro‐oxidative stress in microvascular endothelium that results from an increased generation of ROS and a decreased bioavailability of NO (Davidson, 2010), up‐regulated inflammation (Liu et al., 2009), inhibition of NOS (Prasan et al., 2007), and increased cardiomyocyte apoptosis (Wang et al., 2002).
ROS can be produced by a variety of cells that have been implicated in the inflammatory responses to hypercholesterolaemia, such as neutrophils, monocytes, B‐lymphocytes, platelets, mast cells, endothelial cells and vascular smooth muscle cells (Stokes et al., 2002). Potential sources of ROS in endothelial cells that have been identified so far, include NADPH oxidase (Konior et al., 2014), xanthine oxidase, enzymes involved in the metabolism of arachidonic acid (lipoxygenase and cyclooxygenase) and NO synthases (NOS) (reviewed in Stokes et al., 2002). One of the most important oxygen free radicals that is produced during hypercholesterolaemia is superoxide anion (Landmesser et al., 2000; Napoli and Lerman, 2001). The premise that this enhanced superoxide anion production is due to the elevated blood cholesterol level is further supported by the observation that dietary correction of the hypercholesterolaemia restores superoxide production to normal levels in isolated arterial vessels (Ohara et al., 1995).
Both the expression and activity of NADPH oxidase is responsible, at least in part, for the increased superoxide anion production in cholesterol‐fed apolipoprotein B100 transgenic mice (Csont et al., 2007), and in postcapillary venules in skeletal muscle of hypercholesterolaemic wildtype or p47phox−/− mice (Stokes et al., 2001). The enhanced superoxide anion production in arterial vessels from hypercholesterolaemic rabbits was blunted by treatment with either allopurinol (a xanthine oxidase inhibitor) or heparin, which competes with xanthine oxidase for binding to sulfated glycosaminoglycans on endothelial cells (Landmesser et al., 2000; Napoli and Lerman, 2001). Supporting the role of xanthine oxidase in ROS formation in hypercholesterolaemia, superoxide generation by endothelial NOS (eNOS) occurs as a result of uncoupling of L‐arginine metabolism from NO production and a reduction in the eNOS cofactor tetrahydrobiopterin (BH4) (Cosentino and Katusic, 1995; Vergnani et al., 2000). Furthermore, an increased formation of peroxynitrite, a toxic reaction product of superoxide and NO, has been observed in hyperlcholesterolaemic rat myocardium, and it is accompanied by a decrease in the bioavailability of NO (Onody et al., 2003). Peroxynitrite induces DNA damage, increases lipid peroxidation, and causes post‐translational modification of proteins (e.g. nitration and oxidation of thiol groups) (Pacher et al., 2005), contributing to the development of cardiac dysfunction observed in hyperlcholesterolaemic rats.
The above observations in experimental studies have been confirmed in humans; an increased NADPH oxidase‐mediated superoxide anion generation was observed in vessels of hypercholesterolaemic patients (Assmann et al., 1996; Guzik et al., 2000; Stokes et al., 2001; Itoh et al., 2002).
One additional potential mechanism by which hypercholesterolaemia‐derived oxidative stress could induce cardiac myocyte dysfunction and death is through disruption of mitochondrial function. Increased oxidative stress during hypercholesterolaemia enhances the mitochondrial permeability transition (MPT) response. MPT dissipates the proton electrochemical gradient (ΔΨm), leading to ATP depletion, further ROS production, swelling and rupture of the mitochondria, thereby releasing pro‐apoptotic and pro‐death proteins into the cytosol (Halestrap, 2009). Hypercholesterolaemia increases mitochondrial oxidative stress and enhances the MPT response in the porcine myocardium (McCommis et al., 2011).
In conclusion, when blood cholesterol concentrations are elevated, a low‐grade systemic inflammatory response is elicited in multiple vascular beds and may create an environment in the extracellular compartment, possibly through the generation of cytokines, oxidized molecules, for example, oxidized LDL, and other inflammatory mediators, that predisposes the endothelial cells of large arteries to an inflammatory phenotype. Inflammation is associated with increased ROS production that may overcome cellular defence mechanisms leading to atherogenesis, protein damage and enzyme inactivation, and eventually to loss of contractile function and vascular dysfunction (Misra et al., 2009), which may also account for the increased susceptibility of the myocardium to ischaemia–reperfusion injury and infarction.
Hypercholesterolaemia affects cardiac gene expression profile
Hypercholesterolaemia has been shown to affect the global cardiac gene expression profile at the mRNA level in several studies. In an early study, Puskas et al. reported that in the hearts of rats on a cholesterol‐enriched diet the expression of numerous genes were modulated, including those involved in energy metabolism, heat shock proteins, ion channels and structural proteins (Puskas et al., 2004). Similar results were found in Zucker Diabetic Fatty (ZDF) animals that also show a hypercholesterolaemic profile (Sárközy et al., 2013). Hypercholesterolaemia has also been shown to affect cardiac microRNA profile leading to increased NOX‐4 expression and nitro‐oxidative stress (Varga et al., 2013). These results indicate that hypercholesterolaemia dramatically changes cardiac transcriptomics affecting several known and yet unknown cellular signalling pathways that may impact cardiac function per se and the susceptibility of the heart to ischaemia/reperfusion injury (see for a recent review: Varga et al. 2015). Systematic analysis of the above mentioned large scale transcriptomic data and more ‘omics’ data generated in future studies will be necessary to explore alterations in the cellular signalling network of the hypercholesterolaemic heart in comparison with those of the normal heart (Varga et al. 2015; Perrino et al., 2017).
Effect of hypercholesterolaemia on myocardial ischaemia/reperfusion injury and cardioportection
Effect of hypercholesterolaemia on myocardial ischaemia/reperfusion injury
Larger myocardial infarct sizes have an ominous long‐term prognosis compared to the smaller ones with increased morbidity and mortality. Restriction of the final infarct size is therefore mandatory for the health status and the future of the patients suffering from AMI. How the presence of hypercholesterolaemia affects infarct size is not clear, as the results that exist in the literature in different species and models of hypercholesterolaemia are contradictory. In this regard, larger infarctions have been described within the first hours after acute ischaemia and reperfusion in cholesterol‐fed pigs (Osipov et al., 2009) and rabbits (Golino et al., 1987). In rat isolated hearts exposed to hypercholesterolaemia, induced by a cholesterol enriched diet, the final infarct size and the release of myocardial biomarkers of necrosis and apoptosis were significantly increased, while the recovery of LV function was not affected (Wu et al., 2015b). In contrast, the infarct size was similar in hypercholesterolaemic and normocholesterolaemic rats, but the LV remodelling and risk of developing heart failure were worse in the animals fed the hypercholesterolaemic diet (Maczewski and Maczewska, 2006). New Zealand White rabbits, fed for 4 weeks with a hypercholesterolaemic diet and subjected to 30 min myocardial ischaemia and 2 h reperfusion, exhibited a significantly increased infarct size compared with animals fed a normal diet (Hoshida et al., 1996b; Jung et al., 2004). Additionally, greater cardiac damage, as compared with normal‐fed rabbits, was also observed in cholesterol‐fed rabbits that were subjected to 60 min of myocardial ischaemia followed by 60 min of reperfusion (Ma et al., 1996).
In contrast, using a model of 30 min ischaemia and 3 h reperfusion a similar degree of myocardial infarction was observed in cholesterol‐fed and normal fed rabbits (Andreadou et al., 2006; 2012; 2016; Iliodromitis et al., 2006; 2010). Many other studies have also shown similar infarct sizes in normal and cholesterol‐fed rats with no presence of significant atherosclerosis (Giricz et al., 2009; Görbe et al., 2011; Csont et al., 2013; Csonka et al., 2014). These divergent results are possibly related to different species, diet and experimental protocols. However, it can be concluded that the majority of the studies show that hypercholesterolaemia increases infarct size to some extent in animal models.
The mechanism of the effect of hypercholesterolaemia on myocardial ischaemia–reperfusion injury is still not well understood. It has been demonstrated that decreased cardiac NO content, increased oxidative/nitrosative stress, enhanced apoptotic cell death and dramatic changes in the cardiac gene expression profile, as a consequence of hypercholesterolaemia (see for earlier review: Ferdinandy, 2003; Ferdinandy et al., 2007), may play an essential role in the manipulation of myocardial ischaemia/reperfusion injury in the presence of hypercholesterolaemia. Hypercholesterolaemia decreases the bioavailability of NO with a down‐regulation of eNOS, in association with increased production of oxygen‐derived free radicals that may inactivate NO. More specifically, increased plasma LDL inhibits the active transport of L‐arginine by endothelial cells, uncoupling the eNOS pathway, hence limiting NO synthesis and leading to superoxide anion production (Pritchard et al., 1995; Wilson et al., 2001). The contribution of increased nitrotyrosine formation to the development of atherosclerosis and thus to CAD has been demonstrated in patients with hypercholesterolaemia in combination with CAD (Shishehbor et al., 2003). Experimental hypercholesterolaemia is associated with increased myocardial oxidative stress and inflammation, attenuation of cell survival pathways and the induction of apoptosis (Wang et al., 2002; Osipov et al., 2009).
Additionally, it should be noted that the extent of myocardial injury after ischaemia/reperfusion is determined not only by the tolerance of cardiomyocytes to ischaemia/reperfusion injury but also by the coronary collateral blood flow and rate‐pressure product at the time of myocardial ischaemia (Reimer et al., 1985). LDL up‐regulates the renin‐angiotensin system (RAS), leading to increased generation of angiotensin II, which in turn, binds to the type 1 angiotensin II receptor (AT1 receptor) and activates a signalling cascade that results in the enhanced accumulation of the cholesteryl ester (Rafatian et al., 2013). In particular, hypercholesterolaemia seems to promote the up‐regulation of AT1 receptor genes followed by the structural overexpression of vascular AT1 receptors for angiotensin II (Borghi et al., 2016). Studies in rabbits showed that hypercholesterolaemia increased cardiac diastolic pressure after 1 month of cholesterol treatment, and this was correlated with increased levels of superoxide in the aortas, and to a higher expression of NADPH subunits, associated with altered vasorelaxation (Collin et al., 2007). Clinical studies have also suggested a role of some lipoprotein subfractions as risk factors for the development of hypertension (see for a recent review: Borghi et al., 2016). The development of vascular damage in patients with hypercholesterolaemia could also involve the activation of the RAS, and although the mechanisms of interaction between hypercholesterolaemia and hypertension have not been completely elucidated, there is growing evidence that the involvement of RAS can be considered as a common link between hypertension and hypercholesterolaemia.
Effect of hypercholesterolaemia on cardioprotection induced by ischaemic preconditioning
Initially, hypercholesterolaemia was shown to alter responses to ischaemic preconditioning (PC); pacing‐induced PC was blocked in hypercholesterolaemic rabbits and rats (Szilvassy et al., 1995). This loss of pacing‐induced PC was further noted in another experimental model, in rats administered a cholesterol‐enriched diet for 24 weeks (Ferdinandy et al., 1997). Furthermore, isolated papillary muscle from rats fed a high‐fat diet was more susceptible to the effects of ischaemia and less protected by the effects of PC compared with controls (Kocić et al., 1999). Although studies performed in mice and rats showed consistent results, divergent results have been observed in anaesthetised rabbits. The infarct size‐limiting effect of one cycle PC (5 min occlusion and 10 min reperfusion) was shown to be blunted in the hypercholesterolaemic (16 weeks) rabbit heart subjected to ischaemia/reperfusion (Ueda et al., 1999), whereas hypercholesterolaemia (8 weeks) did not attenuate the reduction in myocardial infarction in rabbits subjected to one cycle of PC, comprising 5 min of regional ischaemia plus 10 min reperfusion (Kremastinos et al., 2000). The difference between the above protocols was the duration of cholesterol feeding, that is, 16 versus 8 weeks. Similar results were obtained in a later study, using two cycles of 5 min ischaemia followed by 10 min reperfusion before sustained ischaemia as a PC stimulus, corroborating the finding that PC limits the infarct size in hypercholesterolaemic animals (Iliodromitis et al., 2006). Consistent with the latter results, Jung et al. showed that hypercholesterolaemia did not affect the beneficial influence of PC on infarct mass (Jung et al., 2000).
In patients with hypercholesterolaemia, an early cardiomyopathy characterized by systolic and diastolic dysfunction has been observed, producing a substratum for an ‘impaired preconditioning’ (Talini et al., 2008). In this respect, two clinical studies investigated the effect of conditioning interventions in the context of hypercholesterolaemia. Both studies examined the effects of repeated balloon inflations at the time of angioplasty in patients with CAD and demonstrated that hypercholesterolaemia attenuated the anti‐ischaemic effect of preconditioning during coronary angioplasty (Kyriakides et al., 2002; Ungi et al., 2005). Moreover, hypercholesterolaemia accelerated the development of intracoronary ST‐segment elevation in humans (Ungi et al., 2005).
In summary, divergent results exist in the literature concerning cardioprotection by PC in the presence of hypercholesterolaemia. While shorter durations of hypercholesterolaemia may not affect the cardioprotective signalling of PC, longer durations may disrupt this cardioprotective effect. This was observed in animal studies and has been confirmed in clinical trials.
Mechanisms of the interaction between ischaemic preconditioning and hypercholesterolaemia
Several potential mechanisms have been proposed to explain the lack of a PC effect in hypercholesterolaemia. Since hypercholesterolaemia is linked to oxidative/nitrosative stress in the vasculature and in the myocardium (Szilvassy et al., 2001; Giricz et al., 2006) and to a decreased NO bioavailability, many studies have focused on the role of NO as a potential mechanism of PC's lost effect. A low concentration of NO is associated with a high concentration of asymmetric dimethylarginine (ADMA), and it has been shown that during hypercholesterolaemia the level of ADMA is elevated and the cardioprotective effects of PC are eliminated in rats (Landim et al., 2013). The beneficial effects of late PC were shown to be abolished in an in vivo rabbit model of hypercholesterolaemia, due to an impaired up‐regulation of BH4, which is essential for inducible NOS (iNOS) (Tang et al., 2005).
With regard to the involvement of apoptosis signalling in the effects of PC, it has been shown that hypercholesterolaemia prevents the effects of sevoflurane‐induced PC by altering the upstream signalling of glycogen synthase kinase 3 β (GSK3β) indicating that acute GSK inhibition may provide a novel therapeutic strategy to protect hypercholesterolaemic hearts against ischaemia/reperfusion injury (Ma et al., 2013). In isolated hearts from cholesterol‐fed rats, the protective effect of PC mediated by moderate inhibition of MMP2 was blocked, whereas a reduction in infarct size could be produced using an MMP inhibitor in non‐preconditioned hearts (Giricz et al., 2006; Bencsik et al., 2014). Hypercholesterolaemia also causes an alteration in one of the main signal transduction elements of the conditioning mechanism, connexin, producing a redistribution of the intracellular localization of connexin 43 in the cardiomyocytes, and this might be a potential explanation for the loss of the myocardial infarction‐limiting effect of PC in the presence of hypercholesterolaemia (Görbe et al., 2011).
The effects of PC in ischaemia/reperfusion injury during hypercholesterolaemia and the proposed mechanisms are summarized in Table 1.
Table 1.
Effect of hypercholesterolaemia (HC) on cardioprotection induced by ischaemic and pharmacological PC
| Experimental model | Effect on PC | Proposed mechanism(s) | Reference |
|---|---|---|---|
| Isolated rat hearts subjected to PC | Elimination of infarct size reduction by PC | HC abolished PC‐induced inhibition of myocardial MMP2 activation and release | Giricz et al., 2009 |
| Isolated rat hearts subjected to PC | Elimination of infarct size reduction by PC | Loss of cardioprotection by PC in HC is associated with a redistribution of both sarcolemmal and mitochondrial connexin 43 | Görbe et al., 2011 |
| Rats exposed to pacing‐induced PC | Elimination of pacing‐induced cardioprotection by PC | HC induced deterioration of cardiac NO metabolism | Ferdinandy et al., 1997 |
| Rats subjected to three cycles of PC | Elimination of infarct size reduction by PC | HC elevated ADMA and eliminated the cardioprotective effects of PC | Landim et al., 2013 |
| Rabbits exposed to pacing‐induced PC | Elimination of pacing‐induced cardioprotection by PC | HC impaired cardiac NO synthesis | Szilvassy et al., 1995; Ferdinandy et al., 1998a,b |
| Rabbits subjected to one cycle PC | Elimination of the infarct size‐limiting effect of PC | HC prevented ecto‐5′‐nucleotidase activation by PC | Ueda et al., 1999 |
| Rabbits subjected to one cycle of PC | Myocardial infarction reduction by PC was not attenuated by HC | Observational study | Kremastinos et al., 2000 |
| Rabbits subjected to two cycles of PC | Myocardial infarction reduction by PC was not attenuated by HC | Observational study | Iliodromitis et al., 2006 |
| Rabbits subjected to one cycle of PC | Myocardial infarction reduction by PC was not attenuated by HC | Reduced calcium‐ionophore stimulated endothelial NO‐release were found in isolated aortic rings of hypercholesterolemic animals suggesting that NO produced by the endothelium is not a prime factor in the cardioprotective mechanism of PC | Jung et al., 2000 |
| Rabbits subjected to late PC | Elimination of infarct size reduction by late PC | Impaired up‐regulation of BH4, which is essential for inducible nitric oxide (NO) synthase | Tang et al., 2005 |
| Sevoflurane‐induced PC in rats | Elimination of sevoflurane‐induced cardioprotection | HC alterated the upstream signalling of GSK3β | Ma et al., 2013 |
| Sevoflurane‐induced PC in rats | Elimination of sevoflurane‐induced cardioprotection | Interference with the iNOS/mitochondrial ATP‐dependent K+ channel pathway | Zhang et al., 2012 |
| Fasudil induced pharmacological PC in rats | Low‐dose fasudil‐induced PC is abolished by HC, but only high‐dose restored the cardioprotection | Fasudil up‐regulated the PI3K/Akt/eNOS pathway and induced the opening of the mito‐KATP channel | Wu et al., 2014b |
| NO donors induced late PC in rabbits | Hypercholesterolaemia blunted NO donor (diethylenetriamine/NO) ‐induced late PC | Disruption of biochemical pathways distal to the generation of NO | Tang et al., 2004 |
Effect of hypercholesterolaemia on cardioprotection induced by ischaemic postconditioning
PostC triggered by two different algorithms (six cycles of 10 s ischaemia separated by 10 s reperfusion, and four cycles of 30 s ischaemia separated by 30 s reperfusion immediately after the end of the index ischaemia) has been found to be ineffective at limiting the infarct size in anaesthetised rabbits with hypercholesterolaemia and atherosclerosis (Iliodromitis et al., 2006). This initial observation was further confirmed in mini swines (Zhao et al., 2007) and in hypercholesterolaemic rats (Kupai et al., 2009). In contrast to the above findings, in rabbit isolated crystalloid‐perfused hearts, PostC reduced the infarct size in hypercholesterolaemic animals (Donato et al., 2007) and the same result was observed in hypercholesterolaemic rats (Zhao et al., 2009). These results may show that some components of the hypercholesterolaemic blood contributes to the attenuation of the effectiveness of PostC in hypercholesterolaemia.
In summary, although studies are not consistent, most of the robust studies showed that cardioprotection by PostC is abolished in the presence of hypercholesterolaemia, indicating that hypercholesterolaemia interferes with the molecular signalling of PostC.
Mechanisms of the interaction between ischaemic postconditioning and hypercholesterolaemia
NO pathway and nitro‐oxidative stress
The myocardial NO‐cGMP pathway seems to be impaired in hypercholesterolaemia. In hypercholesterolaemic rats, phosphorylation of eNOS and Akt was decreased compared with controls, which may result in a decrease in NO production, and the loss of effect of cardioprotective interventions (Penumathsa et al., 2007). Similar results and a decreased phosphorylation of eNOS were found in hypercholesterolaemic rabbits (Andreadou et al., 2012). Similar to the findings mentioned above for PC, a low concentration of NO is associated with a high concentration of ADMA, hypercholesterolaemia elevated ADMA and eliminated the cardioprotective effects of PostC in rats. It has also been shown that although hypercholesterolaemia did not modulate the basal expression of PKG, its oxidized dimeric form was more abundant in hearts of hypercholesterolaemic animals possibly due to increased oxidative stress (Giricz et al., 2009). It is interesting to note that PostC increased cardiac 3‐nitrotyrosine concentrations in the normally fed rats but not in the cholesterol‐fed group, when measured at the fifth minute of reperfusion, indicating that an early increase in peroxynitrite after PostC plays a role in cardioprotection (Kupai et al., 2009). In contrast, in rabbit myocardium, PostC reduced nitrotyrosine concentrations in the normal fed group but not in the cholesterol‐fed group at the 10th minute of reperfusion, indicating that inhibition of nitrosative stress plays a role in cardioprotection (Andreadou et al., 2012). These controversial results concerning the mechanisms of cardioprotection vary according to quality, composition and time of administration of the high‐cholesterol diet, as well as the species used in each experiment.
Apoptosis and reperfusion injury salvage kinase signalling
Other factors that have been proposed to account for the larger myocardial infarction observed in hypercholesterolaemia include heat shock protein‐70, which is down‐regulated in hypercholesterolaemia (Csont et al., 2002) and caspase‐3, the activation of which is increased in hypercholesterolaemic ischaemic rabbit myocardium (Wang et al., 2002). Hypercholesterolaemia prevents the sevoflurane‐induced cardioprotection against ischaemia/reperfusion injury by alterating the upstream signalling of GSK3β (Xu et al., 2013). In another study that investigated the cardioprotection of PostC in hypercholesterolaemic rat isolated hearts, it was observed that infarct size and cardiomyocyte apoptosis were completely abolished by hypercholesterolaemia due to the impairment of phosphorylation of GSK3β and attenuation of mitochondrial permeability transition pore (mPTP) opening (Wu et al., 2014a).
The roles of reperfusion injury salvage kinases (RISK) and apoptosis‐related pathways in the attenuation of cardioprotection of PostC were recently investigated in rat isolated hearts. The results showed that PostC significantly decreased the infarct size and apoptosis, and improved the functional recovery of ischaemic myocardium, but these beneficial effects were reversed by a high‐cholesterol diet. Moreover, hypercholesterolaemia inhibited the phosphorylation of Akt and ERK1/2, which were activated by PostC in normal hearts, and induced excessive apoptosis by down‐regulating B‐cell lymphoma 2 (Bcl‐2) and up‐regulating bcl‐2‐like protein 4, cytochrome c, caspase 9 and caspase 3. These results indicate that the hypercholesterolaemia‐induced loss of cardioprotection conferred by PostC is associated with inactivation of the RISK signalling pathway and dysregulation of the downstream apoptosis‐related pathway (Wu et al., 2015a).
Furthermore, it has been shown that PostC reduces the myocardial injury in hypercholesterolaemic rats probably by the up‐regulation of hypoxia‐inducible factor 1‐α (HIF‐1α), which may be involved in the PostC‐mediated cardioprotective mechanisms (Zhao et al., 2009). This was further confirmed by another study, which showed that when dimethyloxalylglycine was given before PostC to up‐regulate HIF‐1α protein level, the degree of ischaemia/reperfusion injury was attenuated in hypercholesterolaemic rats suggesting that an up‐regulation of HIF‐1α may be one of the cardioprotective mechanisms of PostC against ischaemia/reperfusion injury in hypercholesterolaemia (Li et al., 2014).
ATP‐sensitive potassium channels
Divergent results exist on the role of both mitochondrial and sarcolemal ATP‐sensitive potassium channels (KATP channels also known as Kir6.2 channels) in hypercholesterolaemia. In rabbit isolated hearts, PostC reduced the infarct size in hypercholesterolaemic animals through the activation of adenosine A1 receptors and KATP channels (Donato et al., 2007). In contrast, the infarct‐size reducing effect of either the nonselective KATP activator cromakalim or the selective mitoKATP activator diazoxide was lost in hearts of hyperlcholesterolaemic rats, showing that hypercholesterolaemia may influence KATP channel function in the heart. Although the mechanism by which hypercholesterolaemia inhibits the cardioprotective effect of KATP modulators is not known, altered energy metabolism as well as increased oxidative stress, but not changes in the expression levels of functional KATP protein expression in the heart, due to the cholesterol diet have been shown to be involved (Csonka et al., 2014).
Mitochonrial permeability transition pore
The blocking of the mPTP with cyclosporine A (CsA) was investigated in order to determine whether it can restore the cardioprotection of PostC in hypercholesterolaemic rat hearts. It was concluded that the effect of PostC blocked by hypercholesterolaemia may be due to the excessive opening of the mPTP, and thus, inhibiting the mPTP with CsA is able to reverse this loss of cardioprotection observed during hypercholesterolaemia (Wu et al., 2015c).
The effects of PostC in ischaemia/reperfusion injury in the presence of hypercholesterolaemia and the proposed mechanisms are summarized in Table 2.
Table 2.
Effect of HC on cardioprotection induced by PostC
| Experimental model | Effect of PostC | Proposed mechanism(s) | Reference |
|---|---|---|---|
| Isolated rat hearts subjected to PostC | Elimination of infarct size reduction and cardiomyocyte apoptosis | Impairment of phosphorylation of GSK3β and attenuation of mPTP opening | Wu et al., 2014a |
| Isolated rat hearts subjected to PostC | Elimination of infarct size reduction and apoptosis | Inactivation of RISK signal pathway and dysregulation of downstream apoptosis‐related pathway | Wu et al., 2015a |
| Rats subjected to PostC | Elimination of infarct size limiting effects of PostC | ΗC blocked the cardioprotective effect of PostC at least in part via deterioration of the PostC‐induced early increase in peroxynitrite formation | Kupai et al., 2009 |
| Rats subjected to PostC | Myocardial infarction reduction was not attenuated by hypercholesterolemia | HIF‐1α up‐regulation | Zhao et al. 2009; Li et al., 2014 |
| Rats subjected to PostC | Elimination of PostC cardioprotection | Elevation of ADMA | Landim et al., 2013 |
| Isolated rabbit hearts subjected to PostC | Myocardial infarction reduction was not attenuated by hypercholesterolaemia | Activation of A1 receptors and KATP channels | Donato et al., 2007 |
| Rabbits subjected to PostC triggered by two different algorithms (six cycles of 10 s ischaemia separated by 10 s reperfusion and four cycles of 30 s ischaemia separated by 30 s reperfusion immediately after the end of the index ischaemia) | Elimination of infarct size limiting effects of PostC | Observational study | Iliodromitis et al., 2006 |
| Rabbits subjected to PostC | Elimination of PostC cardioprotection | Increased oxidative and nitrosative stress | Iliodromitis et al., 2010 |
| Rabbits subjected to PostC | Elimination of PostC cardioprotection | Decreased phosphorylation of eNOS | Andreadou et al., 2012 |
| Mini swines subjected to PostC | Elimination of the reduction of the no‐reflow and necrosis areas | Ηypercholesterolaemia increased the area of no‐reflow | Zhao et al., 2007 |
| Sevoflurane‐induced PostC in rats | Elimination of sevoflurane‐induced cardioprotection | Alteration of upstream signalling of GSK3β | Xu et al., 2013 |
Effect of hypercholesterolaemia on cardioprotection induced by remote conditioning
To the best of our knowledge, there is only one very recent study investigating whether remote preconditioning (RIPC)‐induced cardioprotection is intact in hypercholesterolaemia. In this study, RIPC failed to reduce myocardial necrosis and apoptosis in hypercholesterolaemic myocardium in rats. Importantly, the authors found that inhibition of GSK3β reduced myocardial infarct size in hypercholesterolaemic hearts, but no additional cardioprotective effect was achieved when combined with RIPC, suggesting that acute GSK3β inhibition may provide a novel therapeutic strategy for hypercholesterolaemic patients during AMI, whereas RIPC is less effective due to signalling events that adversely affect GSK3β (Ma et al., 2016).
Effect of hypercholesterolaemia on drug‐induced cardioprotection
Although a number of drugs have been shown to be effective in preventing myocardial ischaemic‐reperfusion injury, few are capable of preserving cardioprotection in the presence of hypercholesterolaemia (Balakumar and Babbar 2012). In addition to studies that refer to the application of PC as a manoeuvre, some studies have also examined the role of pharmacologically‐ induced PC during hypercholesterolaemia. It is well established that volatile anaesthetic‐induced PC confers myocardial protection against ischaemia–reperfusion; however, hypercholesterolaemia abolished the sevoflurane‐induced cardioprotection in rats (Ma et al., 2013). Although sevoflurane‐induced PC exerts delayed cardioprotection in normocholesterolaemic rats, this beneficial effect was blocked by hypercholesterolaemia probably by an effect on the iNOS/mitochondrial ATP‐dependent K+ channel pathway (Zhang et al., 2012).
Fasudil, a Rho‐kinase inhibitor, has been shown to induce pharmacological PC in rats. However, low‐dose fasudil‐induced PC is abolished by hypercholesterolaemia and only a high‐dose restored the cardioprotection (Wu et al., 2014b). Conversely, fasudil was effective at restoring the cardioprotection of PostC in the hypercholesterolaemic rat heart. This effect was mediated by the activation of the PI3K/Akt/eNOS signalling pathway and an increase in the myocardial NO content (Wu et al., 2014c).
Another example of the effect of hypercholesterolaemia on drug‐induced PC are NO donors, which have been shown to confer late PC against myocardial ischaemia/reperfusion in healthy rabbits. Hypercholesterolaemia blunted the late PC mediated by the NO donor (diethylenetriamine), indicating that the inhibitory effects of hypercholesterolaemia on NO donor‐induced late PC in conscious rabbits are caused by the disruption of biochemical pathways distal to the generation of NO that triggers these adaptations (Tang et al., 2004). In hyperlcholesterolaemic rat hearts, the NO donor S‐nitroso‐N‐acetyl‐D,L‐penicillamine (SNAP), brain natriuretic peptide (BNP‐32) and exogenous cGMP failed to induce cardioprotection, suggesting that the defect in cytoprotective signalling in the hypercholesterolaemic myocardium may reside downstream of cGMP elevation probably at the level of PKG (Giricz et al., 2009).
The loss of pacing‐induced PC could be recaptured by the key polyprenyl product farnesol in hypercholesterolaemia; however, farnesol‐treatment did not influence cardiac NO content in the cholesterol‐fed rats or in the normal fed rats (Ferdinandy et al., 1998b). Furthermore, the infarct‐size limiting effect of cromakalim (a nonselective KATP channel activator) or diazoxide (a selective mitoKATP channel activator) was lost in hypercholesterolaemic rats (Csonka et al., 2014).
It is well established that hypercholesterolaemia is accompanied by a decrease in cardiac NO content and increased nitro‐oxidative stress; however, the role of NO donors in ischaemia/reperfusion injury with or without hypercholesterolaemia is not well established. Although NO treatment prior to or during the early reperfusion period can limit infarct size in preclinical studies, the excessive production of NO at the beginning of reperfusion reacts with ROS and forms peroxynitrite (see for reviews: Andreadou et al., 2015; Bice et al., 2016). The three clinical studies with NO donors that have been performed so far have revealed no evidence of infarct size reduction in patients treated with NO donors immediately prior to reperfusion. Additionally, high concentrations of NO can promote cellular injury, a situation that is possible in patients being treated with several co‐medications including nitrates. Hence, cardioprotection by NO donors should be demonstrated in experimental models with co‐morbidities and relevant co‐medications prior to clinical translation (Ferdinandy et al., 2014; Andreadou et al., 2015; Bell et al., 2016; Bice et al., 2016).
Effect of antihyperlipidaemic drugs on the ischaemic heart and cardioprotection
Statins
Elevated cholesterol levels can be decreased by diet, exercise and appropriate medical therapy. Among the various hypolipidaemic agents, statins confer the main benefit in treated patients by substantially decreasing cardiovascular morbidity and mortality. Apart from the significant decrease in cholesterol levels, statins also have pleiotropic effects, which, albeit exaggerated, may provide additional benefits. In fact, experimental and clinical studies have shown that statins are involved in a reduction in reperfusion injury and inflammatory reactions and in the improvement in the microcirculation. Statins are implicated in the generation of intracellular mediators, which may prevent mPTP opening and therefore preserve mitochondrial integrity and the survival of cells (Ludman et al., 2009; Antoniades and Channon, 2014; Mihos et al., 2014). However, a growing body of evidence suggests that the cardioprotective potential of statins, associated with their pleiotropic and anti‐inflammatory effects, is mediated by the up‐regulation and activation of PPARα (Balakumar and Mahadevan 2012; Ravingerova et al., 2015). Many experimental studies have shown that statins reduce infarct size in hypercholesterolaemic animals (Penumathsa et al., 2007); therefore, in the present review, we will focus on the role of statin administration on the cardioprotective mechanisms (PC and PostC).
There are biological differences between lipophilic and hydrophilic statins. Lovastatin prevents the cardioprotective effect of PC when applied acutely but not when given chronically. The cardioprotective effect of PostC was attenuated when chronic lovastatin treatment was applied, whereas acute lovastatin treatment had no effect (Kocsis et al., 2008). Furthermore, acute and chronic lovastatin treatment show differential effects on the p42/p44 MAPK pathway; only acute lovastatin treatment significantly increased p42/p44 MAPK phosphorylation. These effects of lovastatin might play a role in its differential action on cardioprotective mechanisms (Kocsis et al., 2008) The most hydrophilic statin, pravastatin, at a dose in which serum cholesterol was not normalized, restored the infarct size‐limiting effect of PC in hypercholesterolaemic rabbits, although it did not reduce the infarct size when it was administered without PC (Ueda et al., 1999). The activation of ecto‐5′‐nucleotidase was suggested as a possible mechanism for the hypercholesterolaemia‐induced retardation and pravastatin‐mediated restoration of the cardioprotective effect of PC (Ueda et al., 1999).
The loss of PostC benefits could be reversed by a 3 week simvastatin treatment, which limits the infarct size both in normo‐ and in hypercholesterolaemic rabbits subjected to ischaemia–reperfusion irrespective of the presence of PostC, while PostC is effective only in normocholesterolaemic animals. One should deduce that simvastatin also reduced total cholesterol and LDL plasma levels and attenuated the oxidative and nitrosative stress in the ischaemic myocardium (Iliodromitis et al., 2010). However, the infarct size limitation by simvastatin was lost in hyperlcholesterolaemic animals, when simvastatin was administered for a short time period and did not possess hypolipidaemic activities (Andreadou et al., 2012). In contrast, short‐term administration of pravastatin at the same dose as simvastatin reduced infarction in cholesterol‐fed rabbits independently of any lipid lowering effect, potentially through eNOS activation and the attenuation of nitro‐oxidative stress. The open lactone ring chemical structure of pravastatin prevents its plasma protein binding by 100 times compared to simvastatin. This may partly be related to the high 45% unbound fraction of pravastatin sodium in plasma, which may interact actively with the endothelium and activate the eNOS/Akt signalling cascade (Andreadou et al., 2012).
Fibrates
Fibrates, such as fenofibrate, bezafibrate, ciprofibrate and clofibrate, are PPARα agonists widely used clinically for treating dyslipidaemias (Staels et al., 1998). Fibrates have been suggested to improve the prognosis for ischaemic heart disease due to other non‐lipid effects that are directly associated with the activation of PPARα, resulting in numerous changes in gene transcription including the genes regulating lipid metabolism (Schoonjans et al., 1996; Ravingerova et al., 2015). Although accumulating data have demonstrated the role of PPAR activation in mediating cardioprotection in the setting of ischaemia/reperfusion in various experimental animal models (Ravingerová et al., 2012; Barlaka et al. 2013; Barlaka et al., 2016), evidence for the effectiveness of fibrates in restoring the lost cardioprotection of PC or PostC in hyperlcholesterolaemic animals is scarce. Treatment with fenofibrate markedly restored the cardioprotective and infarct size limiting properties of PC in hypercholesterolaemic rat hearts, whereas it did not affect the cardioprotection by PC in normal rat hearts (Singh et al., 2014). Although this effect has not yet been corroborated in subsequent studies and the underlying mechanism is unclear, it is pausible that activation of PPARα, which is markedly down‐regulated in hypercholesterolaemic rat heart after ischaemia/reperfusion, and subsequent activation of the PI3K/Akt/eNOS pathway may restore the lost cardioprotection in hypercholesterolaemic hearts. In this context, PPARα up‐regulation confers preconditioning‐like protection against ischaemia/reperfusion via metabolic effects whereas PI3K/Akt activation may also be involved in the downstream mechanisms (Ravingerová et al., 2012; 2015).
Niacin
Another hypolipidaemic drug with pleiotropic properties is nicotinic acid (niacin), which inhibits platelet activation, and reduces the expression of pro‐inflammatory vascular cell adhesion molecule‐1 (VCAM‐1; Stach et al., 2012) and oxidative stress (Gouni‐Berthold and Berthold 2013), in addition to modulating the lipid profile. To the best of our knowledge, there are no data associating niacin with the cardioprotective effect of PC or PostC or its loss in the hypercholesterolaemic heart.
PCSK9 inhibitors
Inhibition of the proprotein convertase subtilisin‐kexin type 9 (PCSK9) leads to an increased density of cell surface LDL receptors and therefore a reduction in serum LDL. Several monoclonal antibodies targeting PCSK9 have been developed recently and used as anti‐hypercholesterolaemic drugs (Cohen et al., 2006). Nevertheless, there are no data in the literature regarding the possible influence of PCSK9 inhibition on the cardioprotective effect of conditioning. Therefore, it would be of great importance to evaluate if these new anti‐hypercholesterolaemic drugs can affect the efficacy or safety of cardioprotection elicited by conditioning strategies.
Conclusions and perspectives
Hypercholesterolaemia changes cardiac transcriptomics, cellular signalling and metabolism leading to mild diastolic dysfunction and endothelial dysfunction. Moreover, hypercholesterolaemia worsens the outcome of ischaemia/reperfusion injury and attenuates the cardioprotective effect of preconditioning, PostC, remote conditioning, as well as pharmacological cardioprotection by interfereing with cardioprotective signalling pathways (Figure 1). Our review highlights the relative lack of experimental and especially clinical data looking at cardioprotection in treated or untreated hypercholesterolaemic animal models and patients, as well as the lack of knowledge on the effect of antihyperlipidaemic drugs on the ischaemic heart and cardioprotective signalling. Therefore, the establishment of more clinically relevant preclinical models of hypercholesterolaemia, together with the identification of novel targets are needed for the development of new cardioprotective drugs that will be able to reverse the increased susceptibility of hypercholesterolaemic hearts to ischaemia/reperfusion injury and to provide cardioprotection.
Figure 1.
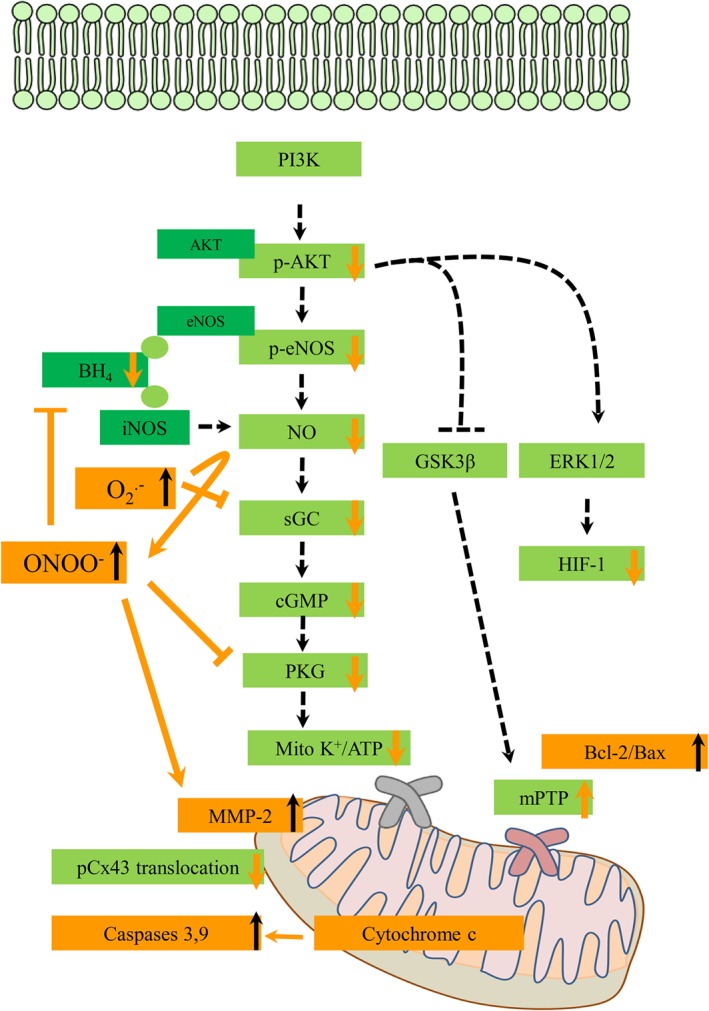
Effect of hypercholesterolaemia on major known cardioprotective cellular mechanisms induced by conditioning interventions: Hypercholesterolaemia inhibits the phosphorylation of Akt and impairs the myocardial NO‐cGMP pathway leading to inhibition of mitochondrial KATP channel opening. Additionally, impairment of the inhibition of GSK3β may cause excessive opening of the mPTP leading to mitochondrial swelling and cell death. Hypercholesterolaemia also inhibits the phosphorylation of extracellular‐ERK1/2 and induces down‐regulation of HIF‐1α, which is one of the cardioprotective mechanisms of PostC. Hypercholesterolaemia produces excessive apoptosis by down‐regulating Bcl‐2 and up‐regulating Bcl‐2‐like protein 4 (Bax), cytochrome c, caspase 9 and caspase 3. Hypercholesterolaemia inhibits mitochondrial translocation of connexin 43 (Cx43). Hypercholesterolaemia produces an increased generation of superoxide anion and a decreased bioavailability of NO through, for example, eNOS and iNOS uncoupling by a reduction in the NOS cofactor BH4. Therefore, during hypercholesterolaemia, an increased formation of peroxynitrite, a toxic reaction product of superoxide and NO, is observed that further depletes bioavailability of NO in the heart. Moreover, the inhibition of oxidative activation of MMP2 by conditioning is blocked in hypercholesterolaemia due to peroxynitrite‐induced activation of MMP2. Green boxes and dashed arrows denote major cardioprotective pathways that are affected by hypercholesterolaemia. Orange boxes and arrows indicate major influence of hypercholesterolaemia on cardioprotective cellular pathways.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
All the authors were members of the European Cooperation in Science and Technology program (COST EU‐ROS). This work was funded by the European Foundation for the Study of Diabetes (EFSD) New Horizons Collaborative Research Initiative from the European Association for the Study of Diabetes (EASD) to RS of PF; the Hungarian Scientific Research Fund (OTKA K 109737 and ANN 107803) to PF; Hungarian National Research, Development, and Innovation Office (NVKP 16‐1‐2016‐0017 – National Heart Program) to AG, ZG and PF, and GINOP‐2.3.2‐15, Myoteam to AG and PF.
Andreadou, I. , Iliodromitis, E. K. , Lazou, A. , Görbe, A. , Giricz, Z. , Schulz, R. , and Ferdinandy, P. (2017) Effect of hypercholesterolaemia on myocardial function, ischaemia–reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. British Journal of Pharmacology, 174: 1555–1569. doi: 10.1111/bph.13704.
Contributor Information
Ioanna Andreadou, Email: jandread@pharm.uoa.gr.
Péter Ferdinandy, Email: peter.ferdinandy@pharmahungary.com.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Other ion channels. Br J Pharmacol 172: 5942–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015e). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015f). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadou I, Iliodromitis EK, Mikros E, Constantinou M, Agalias A, Magiatis P et al. (2006). The olive constituent oleuropein exhibits anti‐ischemic, anti‐oxidative and hypolipidemic effects in anesthetized rabbits. J Nutr 136: 2213–2219. [DOI] [PubMed] [Google Scholar]
- Andreadou I, Farmakis D, Prokovas E, Sigala F, Zoga A, Spyridaki K et al. (2012). Short‐term statin administration in hypercholesterolaemic rabbits resistant to postconditioning: effects on infarct size, endothelial nitric oxide synthase, and nitro‐oxidative stress. Cardiovasc Res 94: 501–509. [DOI] [PubMed] [Google Scholar]
- Andreadou I, Iliodromitis EK, Rassaf T, Schulz R, Papapetropoulos A, Ferdinandy P (2015). The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br J Pharmacol 172: 1587–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadou I, Mitakou S, Paraschos S, Efentakis P, Magiatis P, Kaklamanis L et al. (2016). “Pistacia lentiscus L.” reduces the infarct size in normal fed anesthetized rabbits and possess antiatheromatic and hypolipidemic activity in cholesterol fed rabbits. Phytomedicine 23: 1220–1226. [DOI] [PubMed] [Google Scholar]
- Antoniades C, Channon KM (2014). Statins: pleiotropic regulators of cardiovascular redox state. Antioxid Redox Signal 20: 1195–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmann G, Schulte H, von Eckardstein A (1996). Hypertriglyceridemia and elevated lipoprotein(a) are risk factors for major coronary events in middle‐aged men. Am J Cardiol 77: 1179–1184. [DOI] [PubMed] [Google Scholar]
- Balakumar P, Babbar L (2012). Preconditioning the hyperlipidemic myocardium: fact or fantasy? Cell Signal 24: 589–595. [DOI] [PubMed] [Google Scholar]
- Balakumar P, Mahadevan N (2012). Interplay between statins and PPARs in improving cardiovascular outcomes: a double‐edged sword? Br J Pharmacol 165: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlaka E, Ledvényiová V, Galatou E, Ferko M, Čarnická S, Ravingerová T et al. (2013). Delayed cardioprotective effects of WY‐14643 are associated with inhibition of MMP‐2 and modulation of Bcl‐2 family proteins through PPAR‐α activation in rat hearts subjected to global ischaemia‐reperfusion. Can J Physiol Pharmacol 91: 608–616. [DOI] [PubMed] [Google Scholar]
- Barlaka E, Galatou E, Mellidis K, Ravingerova T, Lazou A (2016). Role of pleiotropic properties of peroxisome proliferator‐activated receptors in the heart: focus on the nonmetabolic effects in cardiac protection. Cardiovasc Ther 34: 37–48. [DOI] [PubMed] [Google Scholar]
- Bell RM, Bøtker HE, Carr RD, Davidson SM, Downey JM, Dutka DP et al. (2016). 19th Hatter Biannual Meeting: position document on ischaemia/ reperfusion injury, conditioning and the ten commandments of cardioprotection. Basic Res Cardiol 111: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencsik P, Pálóczi J, Kocsis GF, Pipis J, Belecz I, Varga ZV et al. (2014). Moderate inhibition of myocardial matrix metalloproteinase‐2 by ilomastat is cardioprotective. Pharmacol Res 80: 36–42. [DOI] [PubMed] [Google Scholar]
- Bice JS, Jones BR, Chamberlain GR, Baxter GF (2016). Nitric oxide treatments as adjuncts to reperfusion in acute myocardial infarction: a systematic review of experimental and clinical studies. Basic Res Cardiol 111: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghi C, Urso R, Cicero AF (2016). Renin‐angiotensin system at the crossroad of hypertension and hypercholesterolemia. Nutr Metab Cardiovasc Dis . doi:10.1016/j.numecd.2016.07.013. [DOI] [PubMed] [Google Scholar]
- Castelli WP, Wilson PW, Levy D, Anderson K (1989). Cardiovascular risk factors in the elderly. Am J Cardiol 63: 12H–19H. [DOI] [PubMed] [Google Scholar]
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH (2006). Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 354: 1264–1272. [DOI] [PubMed] [Google Scholar]
- Collin B, Busseuil D, Zeller M, Perrin C, Barthez O, Duvillard L et al. (2007). Increased superoxide anion production is associated with early atherosclerosis and cardiovascular dysfunctions in a rabbit model. Mol Cell Biochem 294: 225–235. [DOI] [PubMed] [Google Scholar]
- Corti R, Fuster V, Badimon JJ, Hutter R, Fayad ZA (2001). New understanding of atherosclerosis (clinically and experimentally) with evolving MRI technology in vivo. Ann N Y Acad Sci 947: 181–195. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Katusic ZS (1995). Tetrahydrobiopterin and dysfunction of endothelial nitric oxide synthase in coronary arteries. Circulation 91: 139–144. [DOI] [PubMed] [Google Scholar]
- Csonka C, Kupai K, Bencsik P, Görbe A, Pálóczi J, Zvara A et al. (2014). Cholesterol‐enriched diet inhibits cardioprotection by ATP‐sensitive K+ channel activators cromakalim and diazoxide. Am J Physiol Heart Circ Physiol 306: H405–H413. [DOI] [PubMed] [Google Scholar]
- Csont T, Balogh G, Csonka C, Boros I, Horváth I, Vigh L et al. (2002). Hyperlipidemia induced by high cholesterol diet inhibits heat shock response in rat hearts. Biochem Biophys Res Commun 290: 1535–1538. [DOI] [PubMed] [Google Scholar]
- Csont T, Bereczki E, Bencsik P, Fodor G, Görbe A, Zvara A et al. (2007). Hypercholesterolemia increases myocardial oxidative and nitrosative stress thereby leading to cardiac dysfunction in apoB‐100 transgenic mice. Cardiovasc Res 76: 100–109. [DOI] [PubMed] [Google Scholar]
- Csont T, Sárközy M, Szűcs G, Szűcs C, Bárkányi J, Bencsik P et al. (2013). Effect of a multivitamin preparation supplemented with phytosterol on serum lipids and infarct size in rats fed with normal and high cholesterol diet. Lipids Health Dis 12: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM (2010). Endothelial mitochondria and heart disease. Cardiovasc Res 88: 58–66. [DOI] [PubMed] [Google Scholar]
- Donato M, D'Annunzio V, Berg G, Gonzalez G, Schreier L, Morales C et al. (2007). Ischemic postconditioning reduces infarct size by activation of A1 receptors and K+(ATP) channels in both normal and hypercholesterolemic rabbits. J Cardiovasc Pharmacol 49: 287–292. [DOI] [PubMed] [Google Scholar]
- Félix‐Redondo FJ, Grau M, Fernández‐Bergés D (2013). Cholesterol and cardiovascular disease in the elderly. Facts and gaps. Aging Dis 4: 154–169. [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Szilvassy Z, Horvath LI, Csont T, Csonka C, Nagy E et al. (1997). Loss of pacing‐induced preconditioning in rat hearts: role of nitric oxide and cholesterol‐enriched diet. J Mol Cell Cardiol 29: 3321–3233. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Szilvassy Z, Baxter GF (1998a). Adaptation to myocardial stress in disease states: is preconditioning a healthy heart phenomenon? Trends Pharmacol Sci 19: 223–229. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Csonka C, Csont T, Szilvássy Z, Dux L (1998b). Rapid pacing‐induced preconditioning is recaptured by farnesol treatment in hearts of cholesterol‐fed rats: role of polyprenyl derivatives and nitric oxide. Mol Cell Biochem 186: 27–34. [PubMed] [Google Scholar]
- Ferdinandy P (2003). Myocardial ischaemia/reperfusion injury and preconditioning: effects of hypercholesterolemia/hyperlipidemia. Br J Pharmacol 138: 283–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R, Baxter GF (2007). Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev 59: 418–458. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R (2014). Interaction of cardiovascular risk factors, comorbidities and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning: an update. Pharmacol Rev 66: 1142–1174. [DOI] [PubMed] [Google Scholar]
- Giricz Z, Lalu MM, Csonka C, Bencsik P, Schulz R, Ferdinandy P (2006). Hyperlipidemia attenuates the infarct size‐limiting effect of ischemic preconditioning: role of matrix metalloproteinase‐2 inhibition. J Pharmacol Exp Ther 316: 154–161. [DOI] [PubMed] [Google Scholar]
- Giricz Z, Görbe A, Pipis J, Burley DS, Ferdinandy P, Baxter GF (2009). Hyperlipidaemia induced by a high‐cholesterol diet leads to the deterioration of guanosine‐3′,5′‐cyclic monophosphate/protein kinase G‐dependent cardioprotection in rats. Br J Pharmacol 158: 1495–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golino P, Maroko PR, Carew TE (1987). The effect of acute hypercholesterolemia on myocardial infarct size and the no‐reflow phenomenon during coronary occlusion‐reperfusion. Circulation 75: 292–298. [DOI] [PubMed] [Google Scholar]
- Görbe A, Varga ZV, Kupai K, Bencsik P, Kocsis GF, Csont T et al. (2011). Cholesterol diet leads to attenuation of ischemic preconditioning‐induced cardiac protection: the role of connexin 43. Am J Physiol Heart Circ Physiol 300: H1907–H1913. [DOI] [PubMed] [Google Scholar]
- Gouni‐Berthold I, Berthold HK (2013). The role of niacin in lipid‐lowering treatment: are we aiming too high? Curr Pharm Des 19: 3094–3106. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R et al. (2000). Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res 86: E85–E90. [DOI] [PubMed] [Google Scholar]
- Halestrap AP (2009). What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 46: 821–831. [DOI] [PubMed] [Google Scholar]
- Hoshida S, Nishida M, Yamashita N, Igarashi J, Hori M, Kamada T et al. (1996a). Amelioration of severity of myocardial injury by a nitric oxide donor in rabbits fed a cholesterol‐rich diet. J Am Coll Cardiol 27: 902–909. [DOI] [PubMed] [Google Scholar]
- Hoshida S, Yamashita N, Igarashi J, Nishida M, Hori M, Kuzuya T et al. (1996b). A nitric oxide donor reverses myocardial injury in rabbits with acute hypercholesterolemia. J Pharmacol Exp Ther 278: 741–746. [PubMed] [Google Scholar]
- Huang Y, Walker KE, Hanley F, Narula J, Houser SR, Tulenko TN (2004). Cardiac systolic and diastolic dysfunction after a cholesterol‐rich diet. Circulation 109: 97–102. [DOI] [PubMed] [Google Scholar]
- Iliodromitis EK, Zoga A, Vrettou A, Andreadou I, Paraskevaidis IA, Kaklamanis L et al. (2006). The effectiveness of postconditioning and preconditioning on infarct size in hypercholesterolemic and normal anesthetized rabbits. Atherosclerosis 188: 356–362. [DOI] [PubMed] [Google Scholar]
- Iliodromitis EK, Andreadou I, Prokovas E, Zoga A, Farmakis D, Fotopoulou T et al. (2010). Simvastatin in contrast to postconditioning reduces infarct size in hyperlipidemic rabbits: possible role of oxidative/nitrosative stress attenuation. Basic Res Cardiol 105: 193–203. [DOI] [PubMed] [Google Scholar]
- Itoh S, Umemoto S, Hiromoto M, Toma Y, Tomochika Y, Aoyagi S et al. (2002). Importance of NAD(P)H oxidase‐mediated oxidative stress and contractile type smooth muscle myosin heavy chain SM2 at the early stage of atherosclerosis. Circulation 105: 2288–2295. [DOI] [PubMed] [Google Scholar]
- Jung O, Jung W, Malinski T, Wiemer G, Schoelkens BA, Linz W (2000). Ischemic preconditioning and infarct mass: the effect of hypercholesterolemia and endothelial dysfunction. Clin Exp Hypertens 22: 165–179. [DOI] [PubMed] [Google Scholar]
- Jung O, Albus U, Lang HJ, Busch AE, Linz W (2004). Effects of acute and chronic treatment with the sodium hydrogen exchanger 1 (NHE‐1) inhibitor cariporide on myocardial infarct mass in rabbits with hypercholesterolaemia. Basic Clin Pharmacol Toxicol 95: 24–30. [DOI] [PubMed] [Google Scholar]
- Kawashima S, Yokoyama M (2004). Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler Thromb Vasc Biol 24: 998–1005. [DOI] [PubMed] [Google Scholar]
- Kocić I, Konstański Z, Kaminski M, Dworakowska D, Dworakowski R (1999). Experimental hyperlipidemia prevents the protective effect of ischemic preconditioning on the contractility and responsiveness to phenylephrine of rat‐isolated stunned papillary muscle. Gen Pharmacol 33: 213–219. [DOI] [PubMed] [Google Scholar]
- Kocsis GF, Pipis J, Fekete V, Kovács‐Simon A, Odendaal L, Molnár E et al. (2008). Lovastatin interferes with the infarct size‐limiting effect of ischemic preconditioning and postconditioning in rat hearts. Am J Physiol Heart Circ Physiol 294: H2406–H2409. [DOI] [PubMed] [Google Scholar]
- Konior A, Schramm A, Czesnikiewicz‐Guzik M, Guzik TJ (2014). NADPH oxidases in vascular pathology. Antioxid Redox Signal 20: 2794–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremastinos DT, Bofilis E, Karavolias GK, Papalois A, Kaklamanis L, Iliodromitis EK (2000). Preconditioning limits myocardial infarct size in hypercholesterolemic rabbits. Atherosclerosis 150: 81–89. [DOI] [PubMed] [Google Scholar]
- Kupai K, Csonka C, Fekete V, Odendaal L, van Rooyen J, Marais de W et al. (2009). Cholesterol diet‐induced hyperlipidemia impairs the cardioprotective effect of postconditioning: role of peroxynitrite. Am J Physiol Heart Circ Physiol 297: H1729–H1735. [DOI] [PubMed] [Google Scholar]
- Kyriakides ZS, Psychari S, Iliodromitis EK, Kolettis TM, Sbarouni E, Kremastinos DT (2002). Hyperlipidemia prevents the expected reduction of myocardial ischemia on repeated balloon inflations during angioplasty. Chest 121: 1211–1215. [DOI] [PubMed] [Google Scholar]
- Landim MB, Dourado PM, Casella‐Filho A, Chagas AC, da‐Luz PL (2013). High plasma concentrations of asymmetric dimethylarginine inhibit ischemic cardioprotection in hypercholesterolemic rats. Braz J Med Biol Res 46: 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmesser U, Hornig B, Drexler H (2000). Endothelial dysfunction in hypercholesterolemia: mechanisms, pathophysiological importance and therapeutic interventions. Semin Thromb Hemost 26: 529–537. [DOI] [PubMed] [Google Scholar]
- Li X, Zhao H, Wu Y, Zhang S, Zhao X, Zhang Y et al. (2014). Up‐regulation of hypoxia‐inducible factor‐1α enhanced the cardioprotective effects of ischemic postconditioning in hyperlipidemic rats. Acta Biochim Biophys Sin 46: 112–118. [DOI] [PubMed] [Google Scholar]
- Liu HR, Tao L, Gao E, Qu Y, Lau WB, Lopez BL et al. (2009). Rosiglitazone inhibits hypercholesterolaemia‐induced myeloperoxidase upregulation‐‐a novel mechanism for the cardioprotective effects of PPAR agonists. Cardiovasc Res 81: 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludman A, Venugopal V, Yellon DM, Hausenloy DJ (2009). Statins and cardioprotection—More than just lipid lowering? Pharmacol Ther 122: 30–43. [DOI] [PubMed] [Google Scholar]
- Ma XL, Yue TL, Lopez BL, Barone FC, Christopher TA, Ruffolo RR Jr et al. (1996). Carvedilol, a new beta adrenoreceptor blocker and free radical scavenger, attenuates myocardial ischemia–reperfusion injury in hypercholesterolemic rabbits. J Pharmacol Exp Ther 277: 128–136. [PubMed] [Google Scholar]
- Ma LL, Zhang FJ, Qian LB, Kong FJ, Sun JF, Zhou C et al. (2013). Hypercholesterolemia blocked sevoflurane‐induced cardioprotection against ischemia–reperfusion injury by alteration of the MG53/RISK/GSK3β signaling. Int J Cardiol 168: 3671–3678. [DOI] [PubMed] [Google Scholar]
- Ma LL, Kong FJ, Guo JJ, Zhu JB, Shi HT, Li Y et al. (2016). Hypercholesterolemia abrogates remote ischemic preconditioning‐induced cardioprotection: role of reperfusion injury salvage kinase signals. Shock . doi:10.1097/SHK.0000000000000737. [DOI] [PubMed] [Google Scholar]
- Maczewski M, Maczewska J (2006). Hypercholesterolemia exacerbates ventricular remodeling in the rat model of myocardial infarction. J Card Fail 12: 399–405. [DOI] [PubMed] [Google Scholar]
- McCommis KS, McGee AM, Laughlin MH, Bowles DK, Baines CP (2011). Hypercholesterolemia increases mitochondrial oxidative stress and enhances the MPT response in the porcine myocardium: beneficial effects of chronic exercise. Am J Physiol Regul Integr Comp Physiol 301: R1250–R1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihos CG, Pineda AM, Santana O (2014). Cardiovascular effects of statins, beyond lipid‐lowering properties. Pharmacol Res 88: 12–19. [DOI] [PubMed] [Google Scholar]
- Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N (2009). Oxidative stress and ischemic myocardial syndromes. Med Sci Monit 15: RA209–RA219. [PubMed] [Google Scholar]
- Napoli C, Lerman LO (2001). Involvement of oxidation‐sensitive mechanisms in the cardiovascular effects of hypercholesterolemia. Mayo Clin Proc 76: 619–631. [DOI] [PubMed] [Google Scholar]
- Ohara Y, Peterson TE, Sayegh HS, Subramanian RR, Wilcox JN, Harrison DG (1995). Dietary correction of hypercholesterolemia in the rabbit normalizes endothelial superoxide anion production. Circulation 92: 898–903. [DOI] [PubMed] [Google Scholar]
- Onody A, Csonka C, Giricz Z, Ferdinandy P (2003). Hyperlipidemia induced by a cholesterol‐rich diet leads to enhanced peroxynitrite formation in rat hearts. Cardiovasc Res 58: 663–670. [DOI] [PubMed] [Google Scholar]
- Osipov RM, Bianchi C, Feng J, Clements RT, Liu Y, Robich MP et al. (2009). Effect of hypercholesterolemia on myocardial necrosis and apoptosis in the setting of ischemia–reperfusion. Circulation 120: S22–S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Schulz R, Liaudet L, Szabo C (2005). Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol Sci 26: 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penumathsa SV, Thirunavukkarasu M, Koneru S, Juhasz B, Zhan L, Pant R et al. (2007). Statin and resveratrol in combination induces cardioprotection against myocardial infarction in hypercholesterolemic rat. J Mol Cell Cardiol 42: 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrino C, Barabási AL, Condorelli G, Davidson SM, De Windt L, Dimmeler S et al. (2017). ESC working group on cellular biology of the heart – position paper on epigenomic and transcriptomic approaches in cardiac diseases in the post‐genomic era: path to novel targets for diagnosis and therapy of ischemic heart disease? Cardiovasc Res (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasan AM, McCarron HC, Zhang Y, Jeremy RW (2007). Myocardial release of nitric oxide during ischaemia and reperfusion: effects of L‐arginine and hypercholesterolaemia. Heart Lung Circ 16: 274–281. [DOI] [PubMed] [Google Scholar]
- Pritchard KA, Grozek L, Smalley DM, Sessa WC, Wu M, Villalon P et al. (1995). Native low density lipoprotein increases endothelial cell nitric oxide synthase generation of superoxide anion. Circ Res 77: 510–518. [DOI] [PubMed] [Google Scholar]
- Puskas LG, Nagy ZB, Giricz Z, Onody A, Csonka C, Kitajka K et al. (2004). Cholesterol diet‐induced hyperlipidemia influences gene expression pattern of rat hearts: a DNA microarray study. FEBS Lett 562: 99–104. [DOI] [PubMed] [Google Scholar]
- Rafatian N, Milne RW, Leenen FH, Whitman SC (2013). Role of renin‐angiotensin system in activation of macrophages by modified lipoproteins. Am J Physiol Heart Circ Physiol 305: H1309–H1320. [DOI] [PubMed] [Google Scholar]
- Ravingerová T, Čarnická S, Nemčeková M, Ledvényiová V, Adameová A, Kelly T et al. (2012). PPAR‐alpha activation as a preconditioning‐like intervention in rats in vivo confers myocardial protection against acute ischaemia–reperfusion injury: involvement of PI3K–Akt. Can J Physiol Pharmacol 90: 1135–1144. [DOI] [PubMed] [Google Scholar]
- Ravingerova T, Ledvenyiova‐Farkašova V, Ferko M, Bartekova M, Bernatova I, Pechanova O et al. (2015). Pleiotropic preconditioning‐like cardioprotective effects of hypolipidemic drugs in acute ischemia–reperfusion in normal and hypertensive rats. Can J Physiol Pharmacol 93: 495–503. [DOI] [PubMed] [Google Scholar]
- Reimer KA, Jennings RB, Cobb FR, Murdock RH, Greenfield JC Jr, Becker LC et al. (1985). Animal models for protecting ischemic myocardium: results of the NHLBI cooperative study. Comparison of unconscious and conscious dog models. Circ Res 56: 651–665. [DOI] [PubMed] [Google Scholar]
- Rubin SM, Sidney S, Black DM, Browner WS, Hulley SB, Cummings SR (1990). High blood cholesterol in elderly men and the excess risk for coronary heart disease. Ann Intern Med 113: 916–920. [DOI] [PubMed] [Google Scholar]
- Sárközy M, Zvara A, Gyémánt N, Fekete V, Kocsis GF, Pipis J et al. (2013). Metabolic syndrome influences cardiac gene expression pattern at the transcript level in male ZDF rats. Cardiovasc Diabetol 12: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalia R, Gooszen ME, Jones SP, Hoffmeyer M, Rimmer DM, Trocha SD et al. (2001). Simvastatin exerts both anti‐inflammatory and cardioprotective effects in apolipoprotein E‐deficient mice. Circulation 103: 2598–2603. [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Staels B, Auwerx J (1996). Role of the peroxisome proliferator‐activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J Lipid Res 37: 907–925. [PubMed] [Google Scholar]
- Shishehbor MH, Aviles RJ, Brennan ML, Fu X, Goormastic M, Pearce GL et al. (2003). Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 289: 1675–1680. [DOI] [PubMed] [Google Scholar]
- Singh G, Mu K, Khana R (2014). Probable role of peroxisome proliferator activated receptor‐alpha in attenuated cardioprotective effect of ischemic preconditioning in hyperlipidemic rat hearts. Asian J Pharm Clin Res 7: 67–74. [Google Scholar]
- Sniderman AD, Tsimikas S, Fazio S (2014). The severe hypercholesterolemia phenotype: clinical diagnosis, management, and emerging therapies. J Am Coll Cardiol 63: 1935–1947. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44 (D1): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staels S, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart J‐C (1998). Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 98: 2088–2093. [DOI] [PubMed] [Google Scholar]
- Stach K, Zaddach F, Nguyen XD, Elmas E, Kralev S, Weiss C et al. (2012). Effects of nicotinic acid on endothelial cells and platelets. Cardiovasc Pathol 21: 89–95. [DOI] [PubMed] [Google Scholar]
- Stokes KY, Clanton EC, Russell JM, Ross CR, Granger D (2001). NAD(P)H oxidase‐derived superoxide mediates hypercho‐ lesterolemia‐induced leukocyte‐endothelial cell adhesion. Circ Res 88: 499–505. [DOI] [PubMed] [Google Scholar]
- Stokes KY, Cooper D, Tailor A, Granger DN (2002). Hypercholesterolemia promotes inflammation and microvascular dysfunction: role of nitric oxide and superoxide. Free Radic Biol Med 33: 1026–1036. [DOI] [PubMed] [Google Scholar]
- Szilvassy Z, Ferdinandy P, Szilvassy J, Nagy I, Karcsu S, Lonovics J et al. (1995). The loss of pacing‐induced preconditioning in atherosclerotic rabbits: role of hypercholesterolaemia. J Mol Cell Cardiol 27: 2559–2569. [DOI] [PubMed] [Google Scholar]
- Szilvassy Z, Csont T, Pali T, Droy‐Lefaix MT, Ferdinandy P (2001). Nitric oxide, peroxynitrite and cGMP in atherosclerosis‐induced hypertension in rabbits: beneficial effects of cicletanine. J Vasc Res 38: 39–46. [DOI] [PubMed] [Google Scholar]
- Talini E, Di Bello V, Bianchi C, Palagi C, Delle Donne MG et al. (2008). Early impairment of left ventricular function in hypercholesterolemia and its reversibility after short term treatment with rosuvastatin A preliminary echocardiographic study. Atherosclerosis 197: 346–354. [DOI] [PubMed] [Google Scholar]
- Tang XL, Stein AB, Shirk G, Bolli R (2004). Hypercholesterolemia blunts NO donor‐induced late preconditioning against myocardial infarction in conscious rabbits. Basic Res Cardiol 99: 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XL, Takano H, Xuan YT, Sato H, Kodani E, Dawn B et al. (2005). Hypercholesterolemia abrogates late preconditioning via a tetrahydrobiopterin‐dependent mechanism in conscious rabbits. Circulation 112: 2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V, Khokhar M (2014). Mechanism of action of anti‐hypercholesterolemia drugs and their resistance. Eur J Pharmacol 741: 156–170. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Kitakaze M, Komamura K, Minamino T, Asanuma H, Sato H et al. (1999). Pravastatin restored the infarct size‐limiting effect of ischemic preconditioning blunted by hypercholesterolemia in the rabbit model of myocardial infarction. J Am Coll Cardiol 34: 2120–2125. [DOI] [PubMed] [Google Scholar]
- Ungi T, Ruzsa Z, Nagy E, Zimmermann Z, Csont T, Ferdinandy P (2005). Hypercholesterolemia attenuates the anti‐ischemic effect of preconditioning during coronary angioplasty. Chest 128: 1623–1628. [DOI] [PubMed] [Google Scholar]
- Varga ZV, Kupai K, Szűcs G, Gáspár R, Pálóczi J, Faragó N et al. (2013). MicroRNA‐25‐dependent up‐regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia‐induced oxidative/nitrative stress and subsequent dysfunction in the heart. J Mol Cell Cardiol 62: 111–121. [DOI] [PubMed] [Google Scholar]
- Varga ZV, Giricz Z, Bencsik P, Madonna R, Gyongyosi M, Schulz R et al. (2015). Functional genomics of cardioprotection by ischemic conditioning and the influence of comorbid conditions: implications in target identification. Curr Drug Targets 16: 904–911. [DOI] [PubMed] [Google Scholar]
- Vergnani L, Hatrik S, Ricci F, Passaro A, Manzoli N, Zuliani G et al. (2000). Effect of native and oxidized low‐density lipoprotein on endothelial nitric oxide and superoxide production: key role of L‐arginine availability. Circulation 101: 1261–1266. [DOI] [PubMed] [Google Scholar]
- Wang TD, Chen WJ, Su SS, Lo SC, Lin WW, Lee YT (2002). Increased cardiomyocyte apoptosis following ischemia and reperfusion in diet‐induced hypercholesterolemia: relation to Bcl‐2 and Bax proteins and caspase‐3 activity. Lipids 37: 385–394. [DOI] [PubMed] [Google Scholar]
- Wilson SH, Simari RD, Best PJM, Peterson TE, Lerman LO, Aviram M et al. (2001). Simvastatin preserves coronary endothelial function in hypercholesterolemia in the absence of lipid lowering. Arterioscler Thromb Vasc Biol 21: 122–128. [DOI] [PubMed] [Google Scholar]
- Wong ND, Wilson PW, Kannel WB (1991). High blood cholesterol in elderly men and the excess risk for coronary heart disease. Ann Intern Med 113: 916–920. [DOI] [PubMed] [Google Scholar]
- Wu N, Zhang X, Guan Y, Shu W, Jia P, Jia D (2014a). Hypercholesterolemia abrogates the cardioprotection of ischemic postconditioning in isolated rat heart: roles of glycogen synthase kinase‐3β and the mitochondrial permeability transition pore. Cell Biochem Biophys 69: 123–130. [DOI] [PubMed] [Google Scholar]
- Wu N, Li W, Shu W, Lv Y, Jia D (2014b). Inhibition of Rho‐kinase by fasudil restores the cardioprotection of ischemic postconditioninng in hypercholesterolemic rat heart. Mol Med Rep 10: 2517–2524. [DOI] [PubMed] [Google Scholar]
- Wu N, Zhang X, Jia D (2014c). High‐dose fasudil preconditioning and postconditioning attenuate myocardial ischemia–reperfusion injury in hypercholesterolemic rats. Mol Med Rep 9: 560–566. [DOI] [PubMed] [Google Scholar]
- Wu N, Zhang X, Jia P, Jia D (2015a). Hypercholesterolemia aggravates myocardial ischemia reperfusion injury via activating endoplasmic reticulum stress‐mediated apoptosis. Exp Mol Pathol 99: 449–454. [DOI] [PubMed] [Google Scholar]
- Wu N, Zhang X, Jia P, Jia D (2015c). Hypercholesterolemia abrogates the protective effect of ischemic postconditioning by induction of apoptosis and impairment of activation of reperfusion injury salvage kinase pathway. Biochem Biophys Res Commun 458: 148–153. [DOI] [PubMed] [Google Scholar]
- Wu N, Li WN, Shu WQ, Lv Y, Jia DL (2015b). Blocking the mitochondrial permeability transition pore with cyclosporine‐A can restore cardioprotection of ischemic postconditioning in hypercholesterolemic rat heart. Eur Rev Med Pharmacol Sci 19: 446–454. [PubMed] [Google Scholar]
- Xu Y, Ma LL, Zhou C, Zhang FJ, Kong FJ, Wang WN et al. (2013). Hypercholesterolemic myocardium is vulnerable to ischemia–reperfusion injury and refractory to sevoflurane‐induced protection. PLoS One 8: e76652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang FJ, Ma LL, Wang WN, Qian LB, Yang MJ, Yu J et al. (2012). Hypercholesterolemia abrogates sevoflurane‐induced delayed preconditioning against myocardial infarct in rats by alteration of nitric oxide synthase signaling. Shock 37: 485–491. [DOI] [PubMed] [Google Scholar]
- Zhao H, Wang Y, Wu Y, Li X, Yang G, Ma X et al. (2009). Hyperlipidemia does not prevent the cardioprotection by postconditioning against myocardial ischemia/reperfusion injury and the involvement of hypoxia inducible factor‐1alpha upregulation. Acta Biochim Biophys Sin 41: 745–753. [DOI] [PubMed] [Google Scholar]
- Zhao JL, Yang YJ, You SJ, Cui CJ, Gao RL (2007). Different effects of postconditioning on myocardial no‐reflow in the normal and hypercholesterolemic mini‐swines. Microvasc Res 73: 137–142. [DOI] [PubMed] [Google Scholar]