

Abstract
Cardiovascular diseases are associated with and/or caused by oxidative stress. This concept has been proven by using the approach of genetic deletion of reactive species producing (pro‐oxidant) enzymes as well as by the overexpression of reactive species detoxifying (antioxidant) enzymes leading to a marked reduction of reactive oxygen and nitrogen species (RONS) and in parallel to an amelioration of the severity of diseases. Likewise, the development and progression of cardiovascular diseases is aggravated by overexpression of RONS producing enzymes as well as deletion of antioxidant RONS detoxifying enzymes. Thus, the consequences of the interaction (redox crosstalk) of superoxide/hydrogen peroxide produced by mitochondria with other ROS producing enzymes such as NADPH oxidases (Nox) are of outstanding importance and will be discussed including the consequences for endothelial nitric oxide synthase (eNOS) uncoupling as well as the redox regulation of the vascular function/tone in general (soluble guanylyl cyclase, endothelin‐1, prostanoid synthesis). Pathways and potential mechanisms leading to this crosstalk will be analysed in detail and highlighted by selected examples from the current literature including hypoxia, angiotensin II‐induced hypertension, nitrate tolerance, aging and others. The general concept of redox‐based activation of RONS sources via “kindling radicals” and enzyme‐specific “redox switches” will be discussed providing evidence that mitochondria represent key players and amplifiers of the burden of oxidative stress.
Linked Articles
This article is part of a themed section on Redox Biology and Oxidative Stress in Health and Disease. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.12/issuetoc
Abbreviations
- ΔΨm
mitochondrial membrane potential
- 5‐HD
5‐hydroxydecanoic acid
- ADMA
asymmetric dimethylarginine
- AT‐II
angiotensin‐II
- BH4
tetrahydrobiopterin
- cGMP
cyclic guanosine monophosphate
- COX
cyclooxygenase
- eNOS
endothelial nitric oxide synthase
- ET‐1
endothelin‐1
- GCH‐1
GTP cyclohydrolase‐1
- KATP,
ATP‐sensitive potassium channel
- MAO
monoamine oxidase (isoforms A and B)
- MAPK
mitogen‐activated protein kinases
- MitoSOX
triphenylphosphonium dihydroethidium (mitochondria‐targeted superoxide probe)
- MitoQ
triphenylphosphonium quinone (mitochondria‐targeted antioxidant)
- MnSOD
manganese superoxide dismutase
- mPTP
mitochondrial permeability transition pore
- mtKATP,
mitochondrial KATP
- mtROS
mitochondrial reactive oxygen species
- nNOS
neuronal nitric oxide synthase
- Nox
NADPH oxidase catalytic subunit (isoforms 1, 2 and 4)
- PGIS
prostacyclin
- PKC
protein kinase C
- RNS
reactive nitrogen species (accounts in the present review mostly for peroxynitrite and nitrogen dioxide)
- ROS
reactive oxygen species (accounts in the present review mostly for superoxide and hydrogen peroxide)
- sGC
soluble guanylyl cyclase
- SOD
superoxide dismutase
- Src (or cSrc)
tyrosine kinase
- XDH
xanthine dehydrogenase
- XO
xanthine oxidase
Tables of Links
| TARGETS |
|---|
| Enzymes a |
| ALDH2, aldehyde dehydrogenase 2 |
| eNOS, endothelial NOS |
| sGC, soluble guanylyl cyclase |
| MAO‐B, monoamine oxidase B |
| PGIS, prostacyclin synthase (CYP8A1) |
| Src kinase |
| Voltage‐gated ion channels b |
| KATP channels (Kir6.x) |
| LIGANDS |
|---|
| AT‐II, angiotensin II |
| Asymmetric dimethylarginine |
| Diazoxide |
| ET‐1, endothelin 1 |
| Glibenclamide |
| Hydrogen peroxide |
| NO |
| Pinacidil |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2006) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a bAlexander et al., 2014, 2015).
Introduction
Redox biology and impact of oxidative stress on disease progression
Since the discovery of the biological significance of reactive oxygen (ROS) and nitrogen species (RNS) (Sies, 1991), especially by identification of antioxidant enzymes such as superoxide dismutases (SODs) (McCord et al., 1971) or catalases (Stadie et al., 1945), much effort was made by the scientific community to elucidate the role of these reactive species in physiological and pathophysiological processes. The discovery and characterization of NADPH oxidases as superoxide or hydrogen peroxide producing enzymes provided another important milestone in the redox biology field (Griendling et al., 1994; Rajagopalan et al., 1996; Li and Shah, 2001; Takac et al., 2011) and further supported the concept that ROS and RNS can also fulfil physiological functions, by their participation in cellular redox signalling (Stamler, 1994; Ullrich and Kissner, 2006). Similar physiological functions have been demonstrated for mitochondria‐derived ROS (Sena and Chandel, 2012). There is growing body of evidence that ROS and RNS play a crucial role in cardiovascular (Cai and Harrison, 2000; Griendling and FitzGerald, 2003a, 2003b), neurodegenerative (Giasson et al., 2000; Ischiropoulos and Beckman, 2003) and inflammatory diseases (Szabo, 1996; Kooy et al., 1997). The underlying pathogenesis, however, and especially the physiological properties of ROS and RNS by conferring redox regulation are not fully understood.
Several clinical studies demonstrated beneficial effects of acute high‐dose vitamin C infusion into the forearm on flow‐mediated or acetylcholine‐induced vasodilation (Heitzer et al., 1996; Heitzer et al., 2001b; Heitzer et al., 2001a) clearly providing indirect evidence for increased oxidative stress in vessels from patients with hypercholesterolemia, diabetes type I and II or chronic smokers. Importantly, Heitzer et al. demonstrated that the degree of vitamin C effects on endothelial dysfunction assessed by forearm plethysmography in patients with coronary artery disease has a prognostic meaning (Heitzer et al., 2001a). Patients with a strong vitamin C (above the median) response had a worse prognosis and more cardiovascular events within the next 3 years including MI, death due to myocardial infarction, stroke or revascularization procedures as compared to patients having a vitamin C response below the median (Figure 1). The main conclusion of this study was that patients with increased oxidative stress in forearm vessels may also have increased oxidative stress in coronary arteries leading to increased cardiovascular events. Indeed, studies by Kathy Griendling's group, demonstrated in specimens from coronary artery disease (CAD) patients increased oxidative stress, especially in the shoulder of the plaque. This might explain why these patients are more prone to plaque ruptures and subsequent cardiovascular events (Sorescu et al., 2002).
Figure 1.
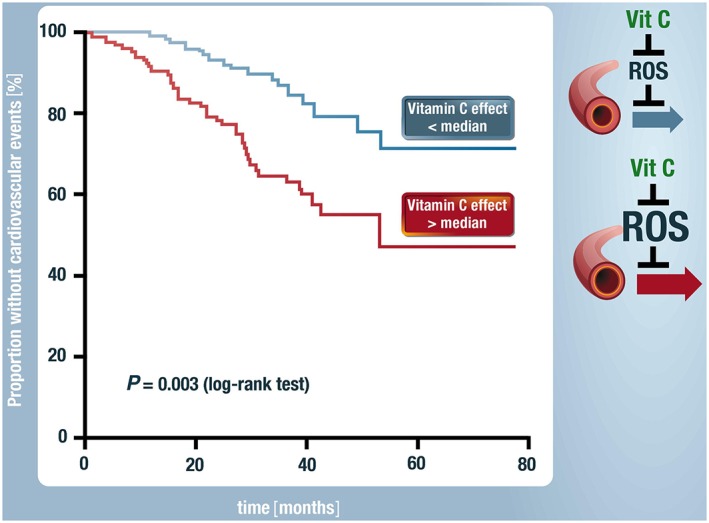
Indirect proof for the prognostic importance of oxidative stress. In patients with high oxidative stress in forearm vessels, compatible with a vitamin C response on endothelial function above the median, had within the next 5 years more cardiovascular events such as myocardial infarction, stroke, coronary and peripheral revascularization procedures as compared to patients with a vitamin C response below the median. Endothelial function was assessed by venous plethysmography and intraarterial infusion of the endothelium‐dependent vasodilator acetylcholine. Modified from Heitzer et al. and Münzel, Circulation 2001 (Heitzer et al., 2001a). With permission of Wolters Kluwer Health. Copyright 2001.
Despite these encouraging data from acute, parenteral therapy with vitamin C or other antioxidants, almost all huge clinical trials on oral antioxidant therapy were negative in respect of hard endpoints and clinical outcome (for review see (Gori and Munzel, 2011; Chen et al., 2012; Schmidt et al., 2015)). There are numerous reasons for this failure, discussed in the mentioned reviews, but most reasons might compromise off‐target effects of the antioxidants (e.g. interference with intrinsic redox‐triggered protective pathways) and the fact that ROS from the same source might have different effects due to concentration and timing.
Molecular proof of the involvement of oxidative stress in cardiovascular disease and concept of redox‐crosstalk of different sources of oxidants
A molecular proof of the involvement of oxidative stress in cardiovascular diseases is mainly based on animal experimental models with appreciable effects of genetic deletion or overexpression of NADPH oxidase subunits or of antioxidant enzymes on the disease phenotype (summarized in (Daiber et al., 2009; Chen et al., 2012; Karbach et al., 2013)). We summarized experimental data on deletion or overexpression as well as pharmacological inhibition of different ROS sources and their impact on progression and severity of hypertension (Table 1) or myocardial infarction (Table 2). However, it would be easy to extend such summary to almost all major diseases (e.g. neurodegenerative disease, inflammatory disease, metabolic syndrome/diabetes). The important role of MnSOD activity for carcinogenesis (Konzack and Kietzmann, 2014) and the impact of mitochondrial ROS on pathogenesis in brown adipose tissue in hyperlipidemia and hyperinsulinemia was recently reviewed in full detail (Markelic et al., 2013).
Table 1.
Important (causal) ROS sources in different hypertension models
| Model and ROS source | Ref. |
|---|---|
| Critical role of mitochondrial ROS for AT‐II induced hypertension. | (Doughan et al., 2008; Dikalova et al., 2010) |
| Critical role of the NADPH oxidase isoform Nox1 for AT‐II induced hypertension. | (Dikalova et al., 2005; Matsuno et al., 2005) |
| Critical role of the NADPH oxidase isoform Nox2 (gp91phox) for AT‐II induced hypertension. | (Bendall et al., 2007; Murdoch et al., 2011; Chrissobolis et al., 2012) |
| Critical role of the NADPH oxidase isoform Nox4 for AT‐II induced hypertension. | (Wingler et al., 2001; Lee et al., 2013) |
| Critical role of the NADPH oxidase subunit p47phox for AT‐II induced hypertension. | (Landmesser et al., 2002) |
| Critical role of the NADPH oxidase subunit p47phox for DOCA‐salt induced hypertension. | (Landmesser et al., 2003) |
| Critical role of xanthine oxidase for AT‐II induced hypertension. | (Landmesser et al., 2007) |
Table 2.
Important (causal) ROS sources in different myocardial infarction models.
| Model and ROS source | Ref. |
|---|---|
| Critical role of the NADPH oxidase subunit p47phox for MI damage. | (Doerries et al., 2007) |
| Critical role of the NADPH oxidase isoform Nox4 for MI damage. | (Infanger et al., 2010) |
| Critical role of the NADPH oxidase isoform Nox2 for MI damage. | (Looi et al., 2008; Somasuntharam et al., 2013) |
| Critical role of mitochondrial ROS for MI damage. | (Piot et al., 2008; Kloner et al., 2012; Somasuntharam et al., 2013) |
| Critical role of xanthine oxidase for MI damage. | (Akizuki et al., 1985; Werns et al., 1986; Engberding et al., 2004; Doerries et al., 2007) |
Here, we would like to put forward the concept that oxidative stress and redox signalling is based on a complex network of various ROS and RNS sources that may interact with and stimulate each other in a positive feedback fashion. These interactions make it very difficult to pharmacologically target oxidative stress as a cause of disease development and progression (Chen et al., 2012). The redox crosstalk of the major sources of ROS and RNS, mitochondria, NADPH oxidases, xanthine oxidase and eNOS, was previously reviewed in detail (Daiber, 2010; Dikalov, 2011; Schulz et al., 2014) (see Table 3 for more references).
Table 3.
Evidence for a redox crosstalk between different sources of ROS. Examples for mitochondria and NADPH oxidases.
| Model and mito/Nox signaling | Ref. |
|---|---|
| Mitochondrial ROS formation triggers the expression of Nox1 gene and protein expression in tumorigenesis. | (Desouki et al., 2005) |
| Cellular starvation activates NADPH oxidase and induces cell death through the mitochondrial ROS‐PI3K signaling axis in human 293 T cells. | (Lee et al., 2006) |
| First Evidence for a Crosstalk Between Mitochondrial and NADPH Oxidase‐Derived Reactive Oxygen Species in Nitroglycerin‐Triggered Vascular Dysfunction. | (Wenzel et al., 2008b) |
| Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2 +]i through the mitochondrial ROS‐PKCε signaling axis in pulmonary artery smooth muscle cells. | (Rathore et al., 2008) |
| Molecular Mechanisms of Angiotensin II–Mediated Mitochondrial Dysfunction. Linking Mitochondrial Oxidative Damage and Vascular Endothelial Dysfunction. | (Doughan et al., 2008) |
| Effects of mild mitochondrial dysfunction on angiotensin II – mediated increase in Nox isoform expression and activity in SMC. | (Wosniak et al., 2009) |
| RAGE‐Induced Cytosolic ROS Promote Mitochondrial Superoxide Generation in Diabetes. | (Coughlan et al., 2009) |
| Therapeutic Targeting of Mitochondrial Superoxide in Hypertension. | (Dikalova et al., 2010) |
| ROS‐induced ROS release in vascular biology. | (Zinkevich and Gutterman, 2011) |
| Nox2‐generated H2O2 regulates mitochondria‐derived superoxide production and cGMP nitration in response to lipopoly saccharide. | (Ahmed et al., 2012) |
| Interaction of mitochondria and phagocytic NADPH oxidase via ROS and calcium signaling in cultured lymphoblasts. | (Dikalov et al., 2012) |
| Interplay of Nox4 and mtROS in angiotensin‐II induced uncoupling of eNOS. | (Lee et al., 2013) |
| Nox2‐induced production of mitochondrial superoxide in angiotensin II ‐ mediated endothelial oxidative stress and hypertension. | (Dikalov and Nazarewicz, 2013) |
| Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. | (Nazarewicz et al., 2013) |
| Crosstalk of mitochondria and NADPH oxidases via ROS signaling in white blood cells and its relevance in the aging process, angiotensin II induced hypertension – aggravation by MnSOD deficiency and amelioration by cyclophilin D deficiency. | (Kroller‐Schon et al., 2014) |
| Interaction of mtROS with Nox2 but not Nox1, Nox4 or Nox5 in the angiotensin II‐induced hypertension model via hydrogen peroxide, sSrc, PKCε and mtKATP channels. | (Dikalov et al., 2014) |
| A role for mitochondrial oxidants in angiotensin II‐induced premature senescence of human vascular smooth muscle cells by NADPH oxidases. | (Mistry et al., 2013) |
| Mitochondrial regulation of NADPH oxidase in hindlimb unweighting rat cerebral arteries. | (Zhang et al., 2014) |
We will therefore focus on the most recent advances in this field with special emphasis on the cardiovascular system and discuss in particular the effects of the crosstalk between mitochondria and NADPH oxidases on vascular function by dysregulation/uncoupling of eNOS (Karbach et al., 2013). The network of these redox interactions is even more complex since recent data suggests that low‐grade inflammation (e.g. in conditions of psoriasis or rheumatoid arthritis) represents a risk factor for the development of arterial hypertension and cardiovascular disease (Harrison et al., 2011; Karbach et al., 2013). Also mental stress causes activation of the immune system with adverse effects on the cardiovascular system (Marvar and Harrison, 2012). Finally, inflammatory pathways themselves are redox regulated (Brune et al., 2013; Janko et al., 2014; Hwang et al., 2013). In the following paragraphs we will highlight the interaction of different sources of ROS and RNS and how this redox crosstalk affects the vascular system.
Redox regulation of vascular function by oxidative degradation of •NO, dysregulation/uncoupling of eNOS, desensitization of sGC, dysregulation of prostanoid synthesis and activation of the endothelin system
The first example of a redox regulation of the vascular tone was provided by Gryglewski and coworkers (Gryglewski et al., 1986). They demonstrated that the vasodilator capacity of the endothelium‐derived relaxing factor (EDRF, later being identified as nitric oxide) in isolated denuded aortic ring segments was decreased with increasing the diffusion distance of the perfusate from the cell culture to the endothelium‐devoid aortic ring segments and that upon addition of SOD vasodilation was strikingly improved. With these experiments, the authors demonstrated that superoxide is a direct antagonist of EDRF, even before its identity was widely accepted to be nitric oxide, and thereby provided the basis for a simple concept of how oxidative stress (increased superoxide formation) reduces the bioavailability of nitric oxide thereby leading to vascular dysfunction. Superoxide reacts with nitric oxide in a diffusion‐controlled reaction to produce peroxynitrite (Beckman et al., 1990; Beckman and Koppenol, 1996). Peroxynitrite in turn leads to endothelial(vascular) dysfunction by nitration of prostacyclin synthase (Zou and Ullrich, 1996; Zou et al., 1997) by inhibition of soluble guanylyl cyclase (Weber et al., 2001) and by causing eNOS uncoupling due to oxidation of the eNOS cofactor tetrahydrobiopterin (Kuzkaya et al., 2003; Schulz et al., 2008).
We have recently provided the concept of different “redox switches” in eNOS in order to explain its high sensitivity to oxidative stress conditions (Figure 2) (Karbach et al., 2013; Schulz et al., 2014). However, it should be noted that any of these switches described in Figure 2 can be activated in a certain disease condition, but usually they are simultaneously activated and one finds eNOS uncoupling, sGC desensitization and PGIS nitration along with increased COX activity at the same time (for review see (Bachschmid et al., 2005; Munzel et al., 2005)). Also up‐regulated ET‐1 activity is accompanied with oxidative stress conditions and by activation of other redox switches, which was observed in nitroglycerin‐induced nitrate tolerance (Munzel et al., 1995; Hink et al., 2003; Sayed et al., 2008; Knorr et al., 2011).
Figure 2.
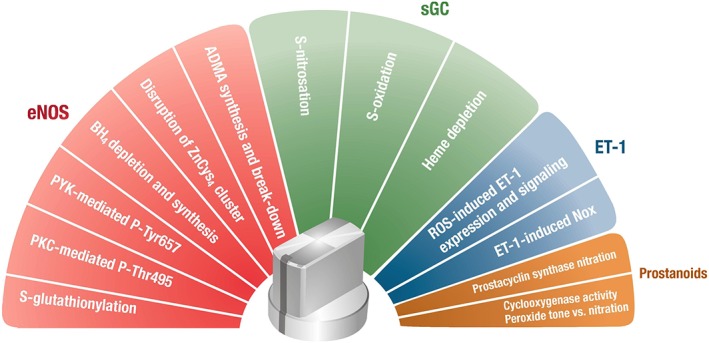
Redox regulation of the vascular tone. Potential „redox switches“in endothelial NO synthase (eNOS) are based on S‐glutathionylation of cysteines in the reductase domain, H2O2/ONOO− mediated activation of PKC and protein tyrosine kinase‐2 (PYK‐2) dependent eNOS phosphorylation, direct oxidative depletion of the eNOS cofactor BH4, oxidative disruption of the zinc‐sulphur‐complex in the dimer binding interface and regulation of ADMA synthesis/break‐down by oxidants. Potential „redox switches“in soluble guanylyl cyclase (sGC) are based on S‐nitrosation by species such as N2O3 or S‐oxidation of regulatory cysteines as well as oxidation (depletion) of the heme moiety. An additional “redox switch” comprises the endothelin‐1 (ET‐1) and NADPH oxidase (Nox) system since oxidants can lead to increased expression of ET‐1 and potentiate its vasoconstrictory properties, which can be further self‐amplified by ET‐1 dependent activation of NADPH oxidase. Finally, prostanoid synthesis (Cox and PGIS) represents a “redox switch” since the key enzyme of prostanoid synthesis, cyclooxygenase, is activated at low peroxide levels and nitrated/inactivated at high peroxynitrite or nitrite/H2O2 concentrations, whereas PGIS is nitrated/inactivated by peroxynitrite and both oxidative alterations may significantly shift the prostanoid profile from vasodilatory to vasoconstrictory conditions. Activation of all of these redox switches by ROS and RNS (most likely hydrogen peroxide, peroxynitrite or superoxide) alters endothelial and vascular function, which will favour vasoconstriction and/or the development of arterial hypertension. Any of these switches can be activated in a certain disease condition but usually they are simultaneously activated and one finds eNOS uncoupling, sGC desensitization and PGIS nitration along with increased COX activity or up‐regulated ET‐1 signalling at the same time.
Redox switches, eNOS uncoupling and impaired •NO signalling
The mechanisms underlying eNOS uncoupling including S‐glutathionylation of the eNOS reductase domain, eNOS phosphorylation, oxidation of the eNOS cofactor tetrahydrobiopterin, increased intracellular ADMA levels or oxidative disruption of the zinc‐sulphur complex (ZnCys4) have recently been reviewed (Forstermann and Munzel, 2006; Schulz et al., 2014). Therapeutic targeting of uncoupled eNOS was recently reviewed in detail (Risbano and Gladwin, 2013) (see also this virtual BJP issue Daiber et al.). Of note, also other NOS isoforms may uncouple and produce superoxide instead of •NO as shown for nNOS in models of metabolic syndrome and angiotensin‐II induced hypertension (Zhang et al., 2009; Wu et al., 2014). Moreover, a protective role of nNOS in preventing eNOS uncoupling was reported (Idigo et al., 2012). Increased oxidative stress also leads to an impaired •NO/cGMP‐signalling by oxidative inhibition of soluble guanylyl cyclase (sGC) via oxidation of Cys122 (Brune et al., 1990; Weber et al., 2001; Sayed et al., 2007), making the enzyme less sensitive to •NO (Figure 2). Under nitrosative stress conditions sGC may also undergo S‐nitrosation at Cys243 and Cys122 (Sayed et al., 2007). The discovery of compounds that are able to activate the enzyme in a •NO‐independent manner, such as heme‐dependent sGC stimulators and heme‐independent sGC activators has led to the development of several drugs for different clinical indications (for review see (Stasch et al., 2011) and this virtual BJP issue Daiber et al.).
Oxidative stress and the endothelin‐system
The vasoconstrictor endothelin‐1 (ET‐1) induces expression of NADPH oxidase (gp91phox or Nox2 dependent isoform) in endothelial cells (Duerrschmidt et al., 2000) and stimulates superoxide formation ex. vivo in isolated human arteries via a ET‐1 receptor dependent mechanism (Cerrato et al., 2012). ET‐1 also increases vascular superoxide formation via activation of the NADPH oxidase in mild hypertension models (Li et al., 2003a, 2003c). Vice versa, it was also shown that NADPH oxidase derived superoxide formation potentiates the vasoconstriction induced by ET‐1 (Li et al., 2003b). These findings provide evidence for a crosstalk comparable to that described above for the mitochondrial superoxide/hydrogen peroxide‐Nox2 axis (Figure 2). The demonstration that oxidative stress per se increases the expression of ET‐1 via induction of its promoter further substantiates the concept of a crosstalk involving mitochondrial superoxide/hydrogen peroxide, Nox2 and ET‐1 as important constituents (Kahler et al., 2000; Kahler et al., 2001). In addition, ET‐1 as well as AT‐II can directly trigger mitochondrial calcium uptake by mitochondrial calcium microdomains via IP3‐receptor (type 3) that co‐localises with mitochondria and subsequent activation of the mitochondrial ryanodine receptor (mRyR1) (Pacher et al., 2008). This leads to mPTP opening and release of mitochondrial superoxide/hydrogen peroxide (Seidlmayer et al., 2014). Likewise, mitochondrial superoxide/hydrogen peroxide formation was associated with increased ET‐1 secretion of pulmonary artery endothelial cells (Ouyang et al., 2012).
Oxidative stress and prostanoid synthesis
Prostanoid synthesis is also redox regulated by nitration of the prostacyclin synthase (Zou and Ullrich, 1996; Zou et al., 1997; Bachschmid et al., 2003) or by activation of cyclooxygenases in the presence of peroxides (“peroxide tone”) as well as inactivation of these enzymes by nitration of critical tyrosine residues (Figure 2) (Schildknecht et al., 2005; Schildknecht et al., 2006; Schildknecht et al., 2008; Schildknecht et al., 2012). Prostanoid synthesis and ROS generation can even interact with each other in a redox crosstalk fashion (Hernanz et al., 2014).
What are the biologically relevant sources of ROS and RNS in cardiovascular disease?
The failure of antioxidants to cure cardiovascular diseases was, as reported previously, likely due to their low reaction constants with the reactive species and their ineffectiveness in targeting a certain ROS/RNS source as well as their organ‐specific or even subcellular accumulation (Chen et al., 2012; Schmidt et al., 2015). Due to this spatial and target non‐specificity they may interfere on the other hand with important physiological cellular redox signalling pathways, important stress‐response systems or pathogen defence mechanisms of the organism. For instance, it has been shown that cell differentiation, proliferation and migration are redox‐regulated in various cell types and physiological or pathophysiological settings (Griendling and Ushio‐Fukai, 1998; Cappelletti et al., 2003; Schroder et al., 2007; Mofarrahi et al., 2008; Schroder et al., 2009; Hurd et al., 2012). Also pathogen defence by the immune system relies on the generation of large amounts of ROS and RNS in order to kill the pathogen/parasite and to ensure host survival (Miller and Britigan, 1997; Stolf et al., 2011), as was shown for Leishmania amazonensis infection (Giorgio et al., 1998; Linares et al., 2001) or Trypanosoma cruzi invasion (Denicola et al., 1993; Irigoin et al., 2008). The importance of the phagocytic NADPH oxidase for the immune system is also demonstrated by the high mortality of individuals with chronic granulomatous disease (genetic p22phox, p40phox, p47phox, p67phox or gp91phox mutation) (Kuhns et al., 2010). Finally, highly protective stress response mechanisms such as ischemic pre‐ or post‐conditioning rely on a tightly controlled and timely restricted formation of ROS and RNS (Das and Maulik, 2003; Dost et al., 2008; Di Lisa et al., 2011; Andreadou et al., 2015), which can be mimicked by certain drugs (e.g. organic nitrates) (Gori and Forconi, 2005; Gori and Parker, 2008) and inhibited by antioxidants (Thomas et al., 2007; Pechanova et al., 2015). Considering these highly beneficial actions of oxidants in the organism this may explain at least in part the failure of classical or non‐specific antioxidants so far but also stress the importance of discriminating between harmful and protective ROS and RNS sources (Figure 3A).
Figure 3.
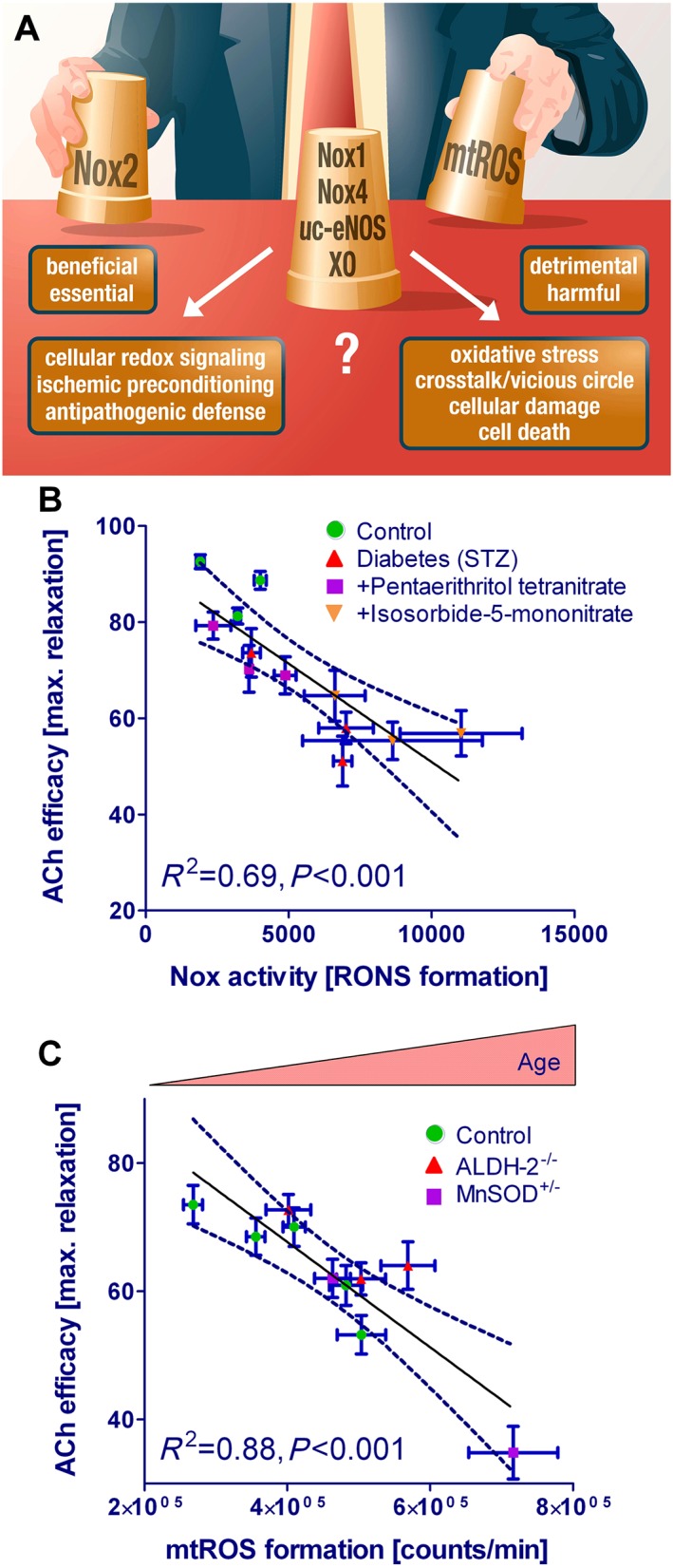
(A) Who's the bad guy – or which biological source of ROS formation is the most detrimental one? Likely candidates are mitochondrial ROS formation (mitochondrial superoxide/hydrogen peroxide), NADPH oxidases (Nox1, Nox2, Nox4), uncoupled eNOS (uc‐eNOS) or xanthine oxidase (XO). The most challenging task for the future is the discrimination between beneficial and detrimental effects of ROS formation and signalling. (A) Direct correlation between vascular ROS formation and endothelial function. Endothelial function (measured by isometric tension recording, efficacy in %) inversely correlates with NADPH oxidase activity (measured by lucigenin‐derived chemiluminescence [superoxide formation] in membrane fractions; expressed as RLU/min) in experimental type 1 diabetes mellitus and therapy with organic nitrates pentaerithrityl tetranitrate (PETN, beneficial effects) vs. isosorbide‐5‐mononitrate (ISMN, adverse effects). Type 1 diabetes mellitus is associated with activation of all ROS sources and organic nitrates such as isosorbide‐5‐mononitrate further aggravate these complications. Retraced from original data of Schuhmacher et al. and Daiber, Diabetes 2011 (Schuhmacher et al., 2011). (B) Endothelial function (measured by isometric tension recording, efficacy in %) inversely correlates with mitochondrial ROS formation (measured by L‐012‐derived chemiluminescence [peroxynitrite, superoxide or hydrogen peroxide formation] in isolated mitochondria; expressed as RLU/min) in aging mice with antioxidant enzyme deficiencies (mitochondrial aldehyde dehydrogenase [ALDH‐2]−/− and MnSOD+/−). ALDH‐2 deficiency leads to increased carbonyl stress since the enzyme is a major sink of toxic aldehydes. MnSOD deficiency increases mitochondrial superoxide levels and leads to increased cardiac fibrosis and impaired vascular function under oxidative stress conditions. RLU, relative light units. Retraced from original data of Wenzel et al. and Daiber, Cardiovasc. Res. 2008 (Wenzel et al., 2008c).
Disease‐relevant sources of oxidative stress
The identification of harmful ROS and RNS sources is further complicated by the fact that for almost any disease condition various enzymatic sources have been accused to cause the disease (Tables 1, 2). For example we have demonstrated an inverse correlation between mitochondrial ROS formation and endothelial dysfunction with increasing age (Wenzel et al., 2008c), whereas in type 1 diabetes loss of endothelial function inversely correlated with the activity of NADPH oxidase (Schuhmacher et al., 2011) (Figure 3B and C). Type 1 diabetes is a condition with activation of all cellular ROS sources, increased protein tyrosine nitration, eNOS uncoupling as well as low‐grade inflammation and impaired organic nitrate potency (nitrate resistance) (Wendt et al., 2005; Oelze et al., 2010). Aging per se causes increased mitochondrial oxidative stress that may be associated with increased Nox activity and low‐grade inflammation (Wenzel et al., 2008a; Oelze et al., 2014). ALDH‐2 and MnSOD are considered important antioxidant proteins for cardioprotection (Strassburger et al., 2005; Chen et al., 2008). In the setting of arterial hypertension an important pathophysiological role of the NADPH oxidase subunits p47phox, Nox1, Nox2 and Nox4 as well as mitochondrial ROS (mitochondrial superoxide/hydrogen peroxide) or xanthine oxidase was demonstrated (Table 1). For myocardial infarction it was shown that NADPH oxidase subunits such as p47phox, Nox2, Nox4, mitochondrial superoxide/hydrogen peroxide or xanthine oxidase contribute to the size and extension of MI (Table 2). Similar observations are made for metabolic (e.g. diabetes), neurodegenerative (e.g. Alzheimer's disease) and inflammatory disease (e.g. sepsis or liver steatosis) as well as the aging process itself (Ischiropoulos and Beckman, 2003; Loguercio and Federico, 2003; Griendling and FitzGerald, 2003b; Zou et al., 2004; Ceriello, 2006; Munzel et al., 2010; Chen et al., 2012; Kroller‐Schon et al., 2012; El Assar et al., 2013).
A logic explanation for the identification of various sources of ROS for specific diseases such as hypertension or myocardial infarction may be the interaction with each other in a crosstalk fashion. Thus, the elimination of only one constituent of this crosstalk network normalizes the pathophysiological condition since the overall oxidative stress is reduced below a certain threshold that can be managed by the cellular antioxidant defence system. In the following sections we will discuss the proposed crosstalk concept, however, without clear differentiation between different tissues and cells. While this might be no serious problem for most of the cytosolic proteins discussed, there might be substantial differences among mitochondria isolated form different cell types in different organs, especially at the level of subpopulations. We also have to stress that a major part of the studies on the nature of the crosstalk between different ROS sources and induction of oxidative stress in general was conducted in vitro due to lack of specific tools to address ROS signalling in vivo (for review see (Schmidt et al., 2015)).
Redox crosstalk of different sources of ROS and RNS
Redox crosstalk and vascular function – impact of NADPH oxidases and mitochondria
Vascular function is inversely correlated with activity of the membranous (particulate) NADPH oxidase activity in experimental type 1 diabetes mellitus (Figure 3B) (Schuhmacher et al., 2011). Because of the close spatial localization of eNOS and vascular NADPH oxidases or even phagocytic NADPH oxidase from infiltrated immune cells, this ROS source may easily trigger uncoupling of eNOS, may desensitize the NO target sGC and may activate other redox‐sensitive vasoactive systems such as the ET‐1 synthesis and receptor signalling pathways (Figure 2). The inverse correlation of vascular function and mitochondrial superoxide/hydrogen peroxide appears to be more complex (Figure 3C) (Wenzel et al., 2008c) since the release of superoxide produced in the matrix by complex I and II or more likely its dismutation product hydrogen peroxide is required for eliciting their detrimental effects on other cellular structures, such as induction of endothelial dysfunction or activation of other ROS sources. According to the concept of “kindling radicals” (or also “bonfire” hypothesis), initial formation of ROS (e.g., from NADPH oxidases or mitochondria) triggers the activation of secondary ROS sources (e.g. xanthine oxidase, uncoupled eNOS) (for review see (Daiber et al., 2014).
Amplification of cellular oxidative stress by mitochondria
The efflux from mitochondrial matrix is facilitated for hydrogen peroxide which can cross phospholipid membranes more easily. There are, however, also a number of mitochondrial pores (mPTP, aquaporins, inner membrane anion channel (IMAC) and others) that could be involved in this efflux of mitochondrial hydrogen peroxide from the matrix, especially for mitochondrial superoxide which cannot cross phospholipid membranes (Aon et al., 2003; Drose and Brandt, 2008; Almasalmeh et al., 2014; Kroller‐Schon et al., 2014) (Figure 4). Although the majority of mitochondrial superoxide is efficiently dismutated to hydrogen peroxide by MnSOD, it was recently shown that cyclosporine A significantly decreases flashes of superoxide formation (Wang et al., 2008; Jian et al., 2014) and mPTP directly stimulates superoxide release (Hou et al., 2014) suggesting a role of mPTP for mitochondrial superoxide efflux. Alternatively, Lustgarten et al. showed that mitochondrial superoxide is released from the matrix through voltage dependent anion channels (Lustgarten et al., 2012). Likewise, mitochondrial superoxide/hydrogen peroxide are directly produced in the intermembrane space facilitating their escape to the cytosol (e.g. complex III, p66shc, MAO) (Cadenas and Davies, 2000; Drose and Brandt, 2008; Gertz and Steegborn, 2010; Kaludercic et al., 2014b) (Figure 4). It is also unclear, whether mitochondrial superoxide/hydrogen peroxide can directly cause redox signalling in the cytosol without an amplification mechanism, which according to the crosstalk hypothesis is simply based on the ROS‐mediated activation of secondary ROS sources. There is also some evidence that the ROS involved in this crosstalk may include peroxynitrite since angiotensin‐II mediated activation of mitochondrial superoxide/hydrogen peroxide formation and endothelial dysfunction was prevented by the NOS inhibitor L‐NAME and the scavenger of peroxynitrite‐derived free radicals, uric acid (Doughan et al., 2008). There is evidence that almost any ROS source can interact with each other while the mitochondria could represent indeed a central amplification mechanism in this network since they are present in almost any cell and tissue (Figure 4). They also play an important role in oxygen sensing, energy metabolism and translation of redox signals (Dehne and Brune, 2014; Ullrich and Schildknecht, 2014; Yin et al., 2014). Mitochondrial superoxide/hydrogen peroxide formation and release by a certain dysfunctional/activated population of mitochondria can trigger mitochondrial superoxide/hydrogen peroxide formation and release from surrounding mitochondria initiating synchronized whole cell oscillations in mitochondrial metabolism (for review see (Zorov et al., 2014)).
Figure 4.
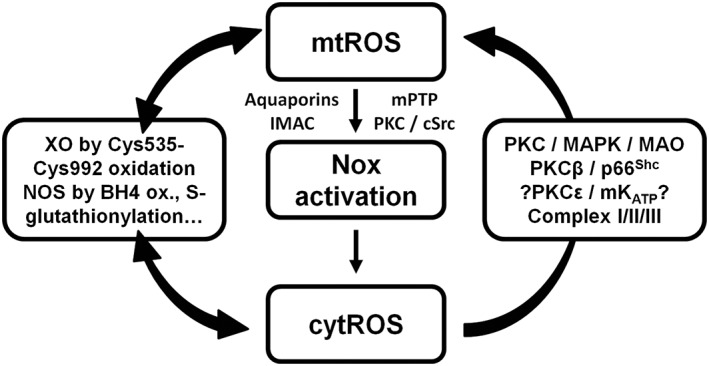
Crosstalk between different sources of ROS and RNS (mitochondria, NADPH oxidases, xanthine oxidase and NO synthase). Xanthine oxidase (XO) originates from oxidative stress‐mediated conversion of the xanthine dehydrogenase via oxidation of critical thiols in cysteine535/992. NO synthases (mainly eNOS) are uncoupled upon oxidative depletion of tetrahydrobiopterin (BH4), S‐glutathionylation (−SSG) and other redox switches (see Figure 2). Mitochondrial superoxide/hydrogen peroxide formation may is triggered by oxidative stress from all ROS sources (including other damaged/activated mitochondria) via redox‐activation of PKC, MAPK, other kinase pathways and potential involvement of redox‐sensitive mitochondrial ATP‐sensitive potassium channels (mtKATP) with subsequent p66Shc, monoamine oxidase (MAO), respiratory complex activation or impairment of mitochondrial antioxidant defence. Mitochondrial superoxide/hydrogen peroxide is released to the cytosol via mitochondrial pores and channels (e.g. redox‐sensitive mitochondrial permeability transition pore (mPTP), inner membrane anion channel (IMAC) or aquaporins) or by diffusion due to increased mitochondrial permeability under pro‐inflammatory conditions. In the cytosol these species (along with released calcium) cause activation of redox‐sensitive protein kinases (PKC) and tyrosine kinases (cSrc) with subsequent activation of NADPH oxidases and amplification of the cellular oxidative stress. Modified from Daiber, Biochim. Biophys. Acta 2010 (Daiber, 2010). With permission of Elsevier. Copyright 2010.
Redox activation of eNOS, xanthine oxidase and NADPH oxidase as superoxide sources
The redox‐dependent conversion of eNOS from a nitric oxide synthase to a superoxide generating enzyme and the redox‐triggered conversion of xanthine dehydrogenase to the oxidase, was described in detail in previous review articles (Munzel et al., 2005; Forstermann and Munzel, 2006; Schulz et al., 2008; Karbach et al., 2013; Schulz et al., 2014) (Figure 2). The most likely redox switches in Nox2 and to a minor extend Nox1 activation comprises the oxidation of a zinc‐cysteine complex in the phorbol ester and diacylglycerol binding domain of classical PKC isoforms (Lin and Takemoto, 2005), which will lead to its activation and phosphorylation of cytosolic regulatory subunits such as p47phox or the direct interaction of protein disulphide isomerases with these subunits (Laurindo et al., 2008) (summarized in (Schulz et al., 2014)). In addition, all Nox isoforms are redox regulated at the transcriptional level (Peshavariya et al., 2009; Lassegue and Griendling, 2010) (summarized in (Schulz et al., 2014)).
Redox activation of mitochondrial superoxide/hydrogen peroxide formation
Finally, the redox trigger for mitochondrial superoxide/hydrogen peroxide formation and release may compromise several pathways (Figures 4, 5): 1) Previous data has shown that H2O2 activates redox‐sensitive PKCβ leading to phosphorylation of p66Shc at serine 36 leading to mitochondrial accumulation of the protein, mtROS formation and subsequent mPTP opening leading to decreased mitochondrial calcium accumulation in the presence of ATP (Pinton et al., 2007). This sequence of events was prevented by cyclosporine A dependent inhibition of the mPTP, genetic deletion of p66Shc and aggravated by PKCβ overexpression; 2) Cytosolic superoxide/hydrogen peroxide formation directly activates redox‐sensitive PKC/MAPK signalling pathways (Lin and Takemoto, 2005; Liu et al., 2011) (or the inhibitory MAPK phosphatases are suppressed in a redox‐dependent fashion (Kim et al., 2012)) leading to activation of c‐Jun. and Egr‐1 transcription factors and increased expression of monoamine oxidase‐B (Wong et al., 2002). This will result in increased hydrogen peroxide production in the intermembrane space or in the cytosolic space (Kaludercic et al., 2014a). Of note, MAO‐B activation in a cardiac dysfunction model caused severe induction of mitochondrial hydrogen peroxide formation, loss in mitochondrial membrane potential and ATP synthase switches to the reverse mode in order to stabilize the proton gradient and membrane potential (Kaludercic et al., 2014b). In addition, genetic or pharmacological inhibition of both MAO isoforms were shown to prevent cardiac maladaptive remodelling induced by thoracic aortic constriction (Kaludercic et al., 2010; Kaludercic et al., 2014b) and endothelial dysfunction induced by lipopolysaccharide and angiotensin II (Sturza et al., 2013); 3) The involvement of redox‐sensitive mitochondrial KATP channels (Doughan et al., 2008; Dikalov et al., 2014). Of note, this pathway is only based on effects of pharmacological inhibitors so far (for details see below). 4) Depletion of mitochondrial defence system by shift from NADH/NADPH to NAD+/NADP+ in depolarized mitochondria (with low ΔΨm) and loss of glutathione peroxidase and peroxiredoxin systems that are coupled to NADPH‐dependent reduction leads to increased mitochondrial superoxide/hydrogen peroxide formation despite lower formation rate of these ROS by respiratory complexes (for details see below) (Nickel et al., 2014).
Figure 5.
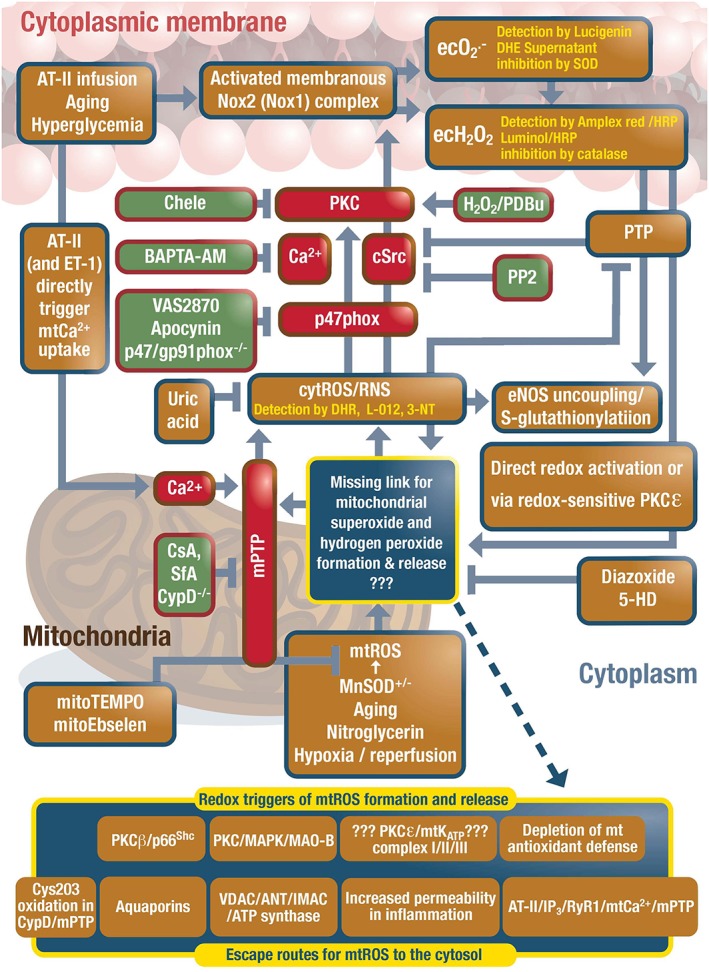
Redox crosstalk between mitochondria and NADPH oxidase through reactive oxygen and nitrogen species. Based on our own and others observations, we favour the crosstalk between mitochondria and Nox2 (in white blood cells and the vasculature). Mitochondrial superoxide/hydrogen peroxide formation is induced by the aging process, nitroglycerin treatment, hypoxia/reperfusion or stimulated by extramitochondrial ROS causing redox‐activation of PKC, MAPK, other kinase pathways and potential involvement of redox‐sensitive mitochondrial ATP‐sensitive potassium channels (mtKATP) with subsequent p66Shc, monoamine oxidase (MAO), respiratory complex activation or impairment of mitochondrial antioxidant defence. Evidence for involvement of redox‐sensitive mitochondrial permeability transition pore (mPTP) is based on its pharmacological and genetic inhibition and normalization of the phenotype in various disease and pharmacological complications (e.g. eNOS S‐glutathionylation and uncoupling). Previous data on redox‐sensitive cysteine residues in cyclophilin D, a regulatory subunit of the mPTP support opening of the pore under oxidative stress conditions. Other redox‐regulated parts of the pore may constitute of the voltage‐dependent anion channel (VDAC) and adenine nucleotide translocase (ANT) or it may be even formed by dimerised mitochondrial ATP synthase in the inner mitochondrial membrane. Direct angiotensin‐II (AT‐II) triggered mitochondrial calcium uptake could contribute to mPTP opening. Other routes for release of mitochondrial superoxide/hydrogen peroxide may involve the inner membrane anion channel (IMAC), aquaporins or diffusion due to increased mitochondrial permeability under pro‐inflammatory conditions. The contribution of mKATP channels to this crosstalk is so far only based on inhibitors and openers of mKATP channels (e.g. 5‐HD, glibenclamide and diazoxide). Evidence for involvement of redox‐sensitive protein kinases (PKC, cSrc) is based on pharmacological and genetic inhibition of these kinases and inhibition of the crosstalk and rescue of the adverse effects. The role of mitochondrial superoxide/hydrogen peroxide and calcium in this process was also shown by specific scavengers and chelators as well as MnSOD deficiency for mitochondrial superoxide importance. PTP means protein tyrosine phosphatase. This scheme includes the concepts of previous work on “ROS‐induced ROS” (for review see (Brandes, 2005; Di Lisa and Bernardi, 2006; Daiber, 2010; Dikalov, 2011; Nickel et al., 2014; Schulz et al., 2014; Zorov et al., 2014)). Modified from Kröller‐Schön et al. and Daiber, Antioxid. Redox Signal. 2014 (Kroller‐Schon et al., 2014). With permission of Mary Ann Liebert, Inc. Copyright 2014.
Concepts of increased mitochondrial permeability, reactivity and release pathways of mitochondrial oxidants
Once the superoxide is formed in the matrix it undergoes fast conversion to hydrogen peroxide, which may cross the mitochondrial membrane (Forman and Kennedy, 1974) or mitochondrial superoxide and hydrogen peroxide are released to the cytosol which could involve mitochondrial pores such as the permeability transition pore (mPTP) (Brandes, 2005; Di Lisa et al., 2011; Jian et al., 2014) and mPTP directly stimulates superoxide release (Hou et al., 2014). Alternatively, aquaporins were identified as mitochondrial channels that conduct small uncharged molecules such as water but also hydrogen peroxide facilitating the release of H2O2 to the cytosol (Almasalmeh et al., 2014). In addition, under inflammatory conditions such as sepsis mitochondrial permeability is largely increased allowing direct release of mitochondrial superoxide to the cytosol (Piskernik et al., 2008).
Although mitochondrial hydrogen peroxide can confer most of the redox chemical reactions required for the here described signalling pathways (e.g. oxidation of zinc‐sulphur complexes, thiol oxidations), superoxide (it remains to be elucidated whether formed from primary mitochondrial source or secondary crosstalk‐activated sources such as Nox) has at least two specific reactivities not shared by hydrogen peroxide (summarized in (Ullrich and Kissner, 2006)): 1) Interaction with metal centers as observed in the redox‐regulation of calcineurin. 2) Consumption of nitric oxide under formation of peroxynitrite. Without detailed discussion here about the higher chemical reactivity of peroxynitrite (and its derived free radicals) over hydrogen peroxide it should be kept in mind that a small portion of superoxide could elicit effects different from hydrogen peroxide (via peroxynitrite formation or direct interaction with transition metal centres), even when the latter is present at higher concentrations (for review see (Bachschmid et al., 2005; Ullrich and Kissner, 2006)).
Role for cyclophilin D, calcium and other redox‐regulated structures for increased mitochondrial permeability by the mPTP
In 2011, redox sensitive cysteine 203 in the regulator of mPTP, cyclophilin D, was shown to act as a redox switch of mPTP by conferring increased opening probability of the pore under oxidative stress conditions (Figure 5) (Nguyen et al., 2011). Transfection of cells with the C203S mutant of cyclophilin D decreased hydrogen peroxide‐dependent mPTP opening and cell death. Interestingly, S‐nitrosation of cysteine 203 also decreased H2O2‐induced mPTP opening to the level of cyclophilin D deficient cells. Many other redox‐sensitive regulatory structures related to mPTP have been suggested (e.g. nitration of the voltage‐dependent anion channel [VDAC] and oxidation of vicinal thiols in the adenine nucleotide translocase [ANT] summarized in (Radi et al., 2002; Daiber, 2010; Schulz et al., 2014)). For instance, peroxynitrite leads to mPTP opening (Vieira et al., 2001). However, even the pore forming subunits of mPTP are not finally identified, leaving the exact molecular nature of the mPTP elusive (Bernardi and Di Lisa, 2015).
According to very recent findings the IP3 agonists angiotensin‐II and endothelin‐1 cause activation of a sarcoplasmic IP3‐receptor (type 3) that colocalises with mitochondria and subsequent activation of the mitochondrial ryanodine receptor (mRyR1) to increase mitochondrial calcium levels causing opening of the mPTP in an ATP‐dependent fashion with subsequent, time‐dependent loss of mitochondrial calcium (Seidlmayer et al., 2014). Similar observations were made in ischemia reperfusion triggered mPTP opening (Seidlmayer et al., 2015). The involved receptors might be located in the so‐called mitochondrial calcium microdomain (Nickel et al., 2014).
Mitochondria‐associated membranes, ER calcium trafficking and impact on mPTP
Another emerging concept of mitochondrial calcium homeostasis is the “mitochondria‐associated membranes” one. Several contact sites (“microdomains”) between ER and mitochondria were identified and subsequent studies have identified an IP3R‐GRP75‐VDAC1 complex is localized in these microdomains and contributes to calcium homeostasis (Rizzuto et al., 1993; Szabadkai et al., 2006). IP3R1‐dependent Ca2 + release in the environment of mitochondria‐associated membranes is tightly regulated by the ER oxidoreductase Ero1‐Lα (Anelli et al., 2012) and contributes to the opening of mPTP since some of its subunits are probably localised in mitochondria‐associated membranes (Bonora et al., 2013). Interestingly, the content of p66Shc in mitochondria‐associated membranes increases with age (Lebiedzinska et al., 2009), a condition for which we have recently reported on a mitochondria‐Nox crosstalk in wild type and glutathione peroxidase‐1 knockout mice at different age (Oelze et al., 2014).
A more speculative redox pathway for increased mitochondrial permeability and oxidant formation – the mtKATP‐mPTP‐axis
Based on a more speculative hypothesis, the redox trigger for mitochondrial superoxide/hydrogen peroxide formation could involve the mitochondrial ATP‐sensitive potassium channels (mtKATP) upon direct activation by ROS and RNS (most probably via thiol oxidation/nitrosation) (Queliconi et al., 2011) or can be opened by redox‐based activation of PKC(ε) isoforms (Ohnuma et al., 2002). According to many reports opening of the mtKATP channel during ischemia/reperfusion may confer highly protective effects (e.g. in the setting of myocardial infarction) (Di Lisa et al., 2007). Therefore, it might be complicated to explain the opening of mPTP by ROS‐induced opening of mtKATP with simultaneous depolarization of the mitochondrial membrane potential and mitochondrial superoxide/hydrogen peroxide formation since previous work just showed the opposite (Costa et al., 2006). Decrease in ΔΨm would impair mitochondrial ATP synthesis leading to diminished calcium uptake, an essential activator of mPTP opening, which makes opening of the pore unlikely in deenergised mitochondria unless another stronger trigger such as redox‐driven mPTP opening would compensate for the lack of calcium (Bernardi and Di Lisa, 2015; Zorov et al., 2014). In summary of these reports in favour of a role for mtKATP in the redox crosstalk of NADPH oxidase and mitochondria one could say that activation of mitochondrial superoxide/hydrogen peroxide formation is sensitive to mtKATP inhibitors and openers.
Likewise, mPTP opening could be triggered in deenergised and depolarized mitochondria by alternative regulation of mitochondrial calcium levels (Saotome et al., 2005). In various reports, a collapse of ΔΨm was observed prior to mitochondrial swelling and opening of mPTP (reviewed in (Zorov et al., 2014)). Alternatively, the direct stimulation of mitochondrial calcium uptake by AT‐II via ryanodine receptors in the mitochondrial calcium microdomain may compensate for missing ATP (Seidlmayer et al., 2014). Likewise, the inhibitor of signal transducer and activator of transcription 3 (STAT3) stattic allowed mPTP opening at lower calcium levels, which was associated with increased mitochondrial hydrogen peroxide release (Boengler et al., 2013). Regulation by uncoupling proteins (Chen et al., 2015) and connexin 43 (Schulz et al., 2007) represent other important pathways associated with mPTP opening and mtROS release.
The concept of “redox‐optimized ROS balance” as a possible explanation of paradox in mitochondrial redox signalling
However, the concept of “redox‐optimized ROS balance” challenges the dogma that mitochondrial superoxide/hydrogen peroxide formation and release is a direct function of mitochondrial respiration and membrane potential with the general assumption that loss of ΔΨm results in decreased oxidative stress (for review see (Nickel et al., 2014)). Regulation of glutathione peroxidase and peroxiredoxin activity by NADH/NADPH ratio and nicotinamide nucleotide transhydrogenase (Nnt) activity plays a major role in this process. Whereas in isolated mitochondria uncoupled respiration was associated with decreased mitochondrial superoxide/hydrogen peroxide formation and release (Starkov and Fiskum, 2003), opposite observations were made in isolated cardiomyocytes with uncoupled mitochondria (Aon et al., 2003; Aon et al., 2010). Based on the concept of “ROS‐induced ROS release” extramitochondrial ROS can lead to loss of ΔΨm and consumption of NADH/NADPH with subsequent burst‐like release of mitochondrial superoxide to the cytosol associated with the opening of channels in the inner mitochondrial membrane, namely mPTP (Zorov et al., 2000) or IMAC (Aon et al., 2003).
Mechanism of redox crosstalk from mitochondria to NADPH oxidases and vice versa to induce vascular dysfunction
Evidence for this redox crosstalk
The interaction of mitochondrial superoxide/hydrogen peroxide with NADPH oxidases and vice versa may represent an essential redox axis for the pathogenesis of various disease (for review see (Kimura et al., 2005; Daiber, 2010; Dikalov, 2011; Nickel et al., 2014; Schulz et al., 2014)). The kindling radicals for initiation of the crosstalk can be formed by mitochondrial dysregulation in the aging process (Oelze et al., 2014), in response to nitroglycerin therapy, in the animal model of MnSOD deficiency (Wenzel et al., 2008c) or by angiotensin II‐dependent induction of the NADPH xoxidases (for summary see Table 3). Mitochondrial superoxide/hydrogen peroxide was also implicated in redox signalling events in response to β‐amyloid, lipopolysaccharide induced sepsis, or in the setting of hyperglycaemia, cancer as well as redox regulation of transcription (Schulz et al., 2014). Here we focus on recent publications and will provide the most actual overview on the detailed mechanism of this crosstalk.
Redox crosstalk from NADPH oxidases to mitochondria – role of mitochondrial pores and channels
This crosstalk can either start at the level of the NADPH oxidase (most likely the phagocytic isoform Nox2 or the Nox1, which can use the same cytosolic regulatory subunits as Nox2 and use the same redox triggers such as redox‐sensitive PKC or related kinases such as cSrc) or at the mitochondrial site – depending on the disease or stress condition (Figure 5) (Kimura et al., 2005; Wenzel et al., 2008b; Daiber, 2010; Kroller‐Schon et al., 2014; Schulz et al., 2014). Angiotensin II infusion activates Nox2 via formation of the classical PKC activator diacylglycerol. Likewise hyperglycaemia via advanced glycation end product (AGE)/AGE receptor (RAGE) signalling, activation of the renin‐angiotensin‐aldosterone system (RAAS), low‐grade inflammation and the aging process via similar mechanisms lead to activation of different NADPH oxidase isoforms, which will generate the kindling radicals. These initially formed ROS contribute either directly or indirectly via redox‐sensitive PKCε (and other isoforms) or MAPK to mitochondrial alkalinisation, impaired calcium haemostasis, change in mitochondrial membrane potential and finally increased mitochondrial superoxide/hydrogen peroxide formation/release (Heinzel et al., 2005; Andrukhiv et al., 2006; Di Lisa et al., 2007), which may involve a number of mitochondrial sources of ROS (Figure 5).
So far, the involvement of mtKATP channels was only indirectly proven by the effects of inhibitors such as glibenclamide or 5‐hydroxydecanoic acid (5‐HD) and openers such as diazoxide (Kimura et al., 2005; Doughan et al., 2008; Wenzel et al., 2008b; Dikalov et al., 2014). However, the mtKATP openers diazoxide or pinacidil increased mitochondrial ROS in a 5‐HD‐sensitive manner, whereas a potassium ionophore (valinomycin) increased mitochondrial ROS formation to a similar extent but without being affected by 5‐HD (Krenz et al., 2002). mtKATP opening and mitochondrial ROS formation in response to acetylcholine involved a tyrosine (Src) kinase (Oldenburg et al., 2003a). Likewise, the putative sarcolemma‐selective KATP channel opener P1075 caused mtKATP channel opening and mitochondrial ROS formation in isolated cardiac myocytes (Oldenburg et al., 2003b).
In contrast, mtKATP activation has been also shown to prevent cellular ROS formation and cell death in cultured neurons, and mtKATP opening directly and significantly prevented mitochondrial H2O2 release in isolated brain mitochondria (Fornazari et al., 2008), although a well‐known therapeutic feature of mtKATP opening in ischemic preconditioning in cardiac tissue is based on initial mild ROS formation in response to mtKATP opening leading to subsequent induction of redox‐sensitive transcription factor and up‐regulation of antioxidant and protective genes (Garlid, 2000; Philipp et al., 2006). In line with this observation, vitamin C therapy blocked cardioprotection by ischemic preconditioning in pigs supporting a role of ROS in this process (Skyschally et al., 2003). However, protection by mtKATP opening usually requires timely restricted activation of mitochondrial pores (e.g. by exact temporal periods of ischemia and reperfusion or repeated short‐time challenges with organic nitrates) (Hausenloy et al., 2004; Lim et al., 2007), which could represent a major difference to the adverse effects of continuous opening of this pore. Even tolerance to the protective effects of ischemic preconditioning has been described (Gori et al., 2010; Lisi et al., 2012). Likewise, the duration of ischemia has large impact on the role of mPTP opening for I/R damage (Ruiz‐Meana et al., 2011) and short‐term openings could have protective whereas long‐term openings could lead to detrimental effects (reviewed in (Bernardi and Di Lisa, 2015)). Recently, it was shown that hyperlipidemia leads to inhibition of cardioprotection by mtKATP channels (Csonka et al., 2014).
Signalling and damage by mitochondrial oxidants in the cytosol
Likewise, these kindling radicals can directly contribute to the dysregulation of eNOS, sGC, ET‐1 pathways or prostanoid synthesis (by the above described redox signalling events, see also Figure 2) with functional consequences (e.g. endothelial and cardiac dysfunction, increased vascular tone and hypertension). The vicious circle starts when the mitochondrial superoxide/hydrogen peroxide are released from the mitochondrial matrix to the cytosol, a process that is thought to be associated with a number of mitochondrial pores and channels or with increased mitochondrial permeability under pro‐inflammatory conditions (Figure 5). mPTP inhibition by cyclosporine A, sanglifehrin A or cyclophilin D deficiency, respectively, or mitochondria‐targeted antioxidants mitoTEMPO (superoxide scavenger) or mitoEbselen (hydrogen peroxide scavenger) prevented the activation of NADPH oxidases, normalized ROS formation and improved disease related complications (Doughan et al., 2008; Wenzel et al., 2008b; Dikalova et al., 2010; Dikalov et al., 2014; Kroller‐Schon et al., 2014). Once released to the cytosol these mitochondrial superoxide/hydrogen peroxide may activate secondary ROS sources such as Nox2 (Nox1) via PKC or tyrosine kinase cSrc (which is either up‐stream of PKC or more important for ROS‐triggered Nox2 activation than PKC (Dikalov et al., 2014; Kroller‐Schon et al., 2014)) mediated translocation of regulatory cytosolic Nox subunits, which was further substantiated by the effects of various Nox inhibitors, genetic deletions or silencing of Nox2 related subunits.
According to recent data, Src family kinase (SFK) Lyn functions as a redox sensor in leukocytes to detect H2O2 at wounds in zebrafish larvae (Yoo et al., 2011, 2012 ). The role of tyrosine kinases in the redox crosstalk between mitochondria and NADPH oxidases was also established in a model of cellular starvation by widespread increase in tyrosine kinase activity and impaired protein tyrosine phosphatase activity leading to a positive feedback loop involving generation of ROS by NADPH oxidase and mitochondria (Graham et al., 2012). The importance of calcium in these specific processes (e.g. for activation of protein kinases (Tsuda et al., 1986) or opening of mPTP (Di Lisa et al., 2007)) and the crosstalk in general is summarized in Table 4. According to a recent review by Zorov et al. calcium might play no role at all for mPTP opening in intact cells due to replacement by some other cytosolic factors but there seems to be an interconnection between ROS and calcium dependent down‐stream signalling (Juhaszova et al., 2004; Zorov et al., 2014).
Table 4.
Evidence for an essential role of calcium for the redox crosstalk between mitochondria and NADPH oxidases.
| Model and calcium signaling site | Ref. |
|---|---|
| Superoxide Flux in Endothelial Cells via the Chloride Channel‐3 Mediates Intracellular Calcium/ROS Signaling. | (Hawkins et al., 2007) |
| Hypoxia‐triggered Nox1 activation contributes to an increase in cytosolic calcium levels. | (Rathore et al., 2008) |
| Hypoxia‐triggered mtROS formation induces accumulation of cytoplasmic calcium via dissociation of FK506‐binding protein 12.6 from ryanodine receptor 2. | (Wang and Zheng, 2010; Liao et al., 2011) |
| Ischemia leads to closed mPTP and calcium accumulation that is released to the cytosol due to mtROS formation and pH increase with subsequent mPTP opening during the reperfusion phase with collapse of ΔΨm, matrix swelling and NAD+ release. | (Di Lisa et al., 2011) |
| ROS trigger oscillations of ΔΨm involving mitochondrial calcium uptake that can induce reperfusion arrhythmias and modulate the opening probability of the mPTP. | (Maack and Bohm, 2011) |
| mtROS increase cytosolic calcium levels and modulators of [Ca2 +]i such as BAPTA‐AM or thapsigargin impair the mtROS‐Nox2 crosstalk in human B lymphoblasts. | (Dikalov et al., 2012) |
| The chelator of intracellular calcium, BAPTA‐AM, completely prevented the mitochondrial superoxide/hydrogen peroxide‐Nox2 crosstalk in isolated leukocytes. | (Kroller‐Schon et al., 2014) |
Redox crosstalk from mitochondria to NADPH oxidases
The crosstalk can also originate from the mitochondrial site when mitochondrial superoxide/hydrogen peroxide are directly induced by the aging process (Wenzel et al., 2008c; Oelze et al., 2014), hypoxia/reperfusion, nitroglycerin treatment or manganese superoxide dismutase deficiency (Figure 5, Table 3). In this case the mitochondrial superoxide/hydrogen peroxide serve as kindling radicals and activate Nox2 (Nox1) leading to more potent cytosolic ROS formation by the NADPH oxidases and eventually xanthine oxidase (upon redox conversion from the xanthine dehydrogenase form) with subsequent dysregulation and uncoupling of eNOS (Kroller‐Schon et al., 2014). Indeed, we have recently shown that the aging process increases mitochondrial superoxide/hydrogen peroxide formation, induces low‐grade inflammation, leads to activation of NADPH oxidases and finally results in endothelial/vascular dysfunction, all of which is significantly aggravated in the absence of MnSOD (increased mitochondrial ROS) (Wenzel et al., 2008c) but also deficiency in the glutathione peroxide‐1 (increased cytosolic ROS) (Oelze et al., 2014).
Which NADPH oxidase is the target of this redox crosstalk?
The Nox2 isoform and to a minor extent also Nox1 isoform exert several redox switches (e.g. redox sensitive kinases, protein disulphide isomerases) making them a preferred target of redox regulation (Kroller‐Schon et al., 2014; Schulz et al., 2014). The Nox2 isoform of immune cells plays an important role for development of endothelial dysfunction and hypertension, an early vascular disturbance that leads to the development of atherosclerosis (Guzik et al., 2007; Wenzel et al., 2011). The silencing of Nox2, but not of Nox1, Nox4 and Nox5 normalizes angiotensin II‐induced mitochondrial and cytosolic ROS formation as well as cSrc activation in cultured endothelial cells and genetic Nox2 deletion improves hypertension in response to angiotensin II (Dikalov et al., 2014). Nox2 deficiency prevents eNOS S‐glutathionylation and uncoupling in aortic tissue upon in vivo angiotensin II treatment (Kroller‐Schon et al., 2014). Although Nox4 silencing normalized ROS formation and eNOS function as well as up‐regulation of Nox4 expression in mesangial cells treated with angiotensin II (Lee et al., 2013), a recent yeast two‐hybrid‐based study on the interaction of Nox4 with complex I revealed no relevance of Nox4 for basal mtROS formation (Hirschhauser et al., 2015). In addition, there is only limited evidence for direct redox‐based activation pathways of Nox4 (besides the transcriptional control) and for a role of mitochondrial superoxide/hydrogen peroxide in this process. Of note, microRNA‐mediated NOX4 upregulation in hyperlipidemia is responsible for oxidative and nitrosative stress and deterioration of cardiac function pointing towards epigenetic regulation of Nox4 expression (Varga et al., 2013).
Conclusions and clinical implications
The present review highlights the mechanisms underlying increased oxidative stress in cardiovascular disease. Although it is in general accepted that oxidative stress plays a crucial role in the initiation and continuation of the atherosclerotic process, therapies to fight oxidative stress such as supplementation with “classical antioxidants” such as vitamins yielded disappointing results. A so far underestimated reason may be among others the more complex nature of ROS formation e.g. via redox crosstalk mainly between the vascular and phagocytic NADPH oxidase and mitochondria resulting in the production of so called kindling radicals ultimately leading to eNOS uncoupling. More recently, the involvement of the immune system in the development of vascular dysfunction (Karbach et al., 2014) and the “ROS‐induced ROS concept” further contributed to the complexity of this crosstalk hypothesis but also provided new explanations on so far unanswered questions (Zorov et al., 2014). Of note, mitochondrial ROS were reported to activate the NLRP3 inflammasome (Zhou et al., 2011), promote the expression of proinflammatory cytokines in LPS‐induced sepsis (Bulua et al., 2011) and improve bactericidal activity (West et al., 2011) (for review see (Ortona et al., 2014)). Putting together these lines of evidence with recent data on redox activation of immune cells by mitochondrial superoxide/hydrogen peroxide formation (Dikalov et al., 2012; Kroller‐Schon et al., 2014) provides the basis for tight association between redox regulatory pathways and inflammation. The discovery of the players in this crosstalk, potential mechanisms and in addition the more and more complete understanding why and how eNOS uncoupling occurs will help to develop more specific and therefore more powerful antioxidants which may ultimately be more successful than current therapeutic approaches.
Conflict of interest
We certify that there is no conflict of interest with any financial organizations regarding the materials discussed in the manuscript.
Acknowledgements
We thank Philip Wenzel for helpful comments. We are also indebted to Thilo Weckmüller for graphical assistance. The present work was supported by the European Cooperation in Science and Technology (COST Action BM1203 / EU‐ROS to A.D. and F.D.L.), by generous financial support by the “Stiftung Mainzer Herz”, by the Centre of Translational Vascular Biology (CTVB), by “NMFZ” and “Stufe 1” funds from the Johannes Gutenberg University Mainz.
Daiber, A. , Di Lisa, F. , Oelze, M. , Kröller‐Schön, S. , Steven, S. , Schulz, E. , and Münzel, T. (2017) Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. British Journal of Pharmacology, 174: 1670–1689. doi: 10.1111/bph.13403.
References
- Ahmed KA, Sawa T, Ihara H, Kasamatsu S, Yoshitake J, Rahaman MM et al (2012). Regulation by mitochondrial superoxide and NADPH oxidase of cellular formation of nitrated cyclic GMP: potential implications for ROS signalling. Biochem J 441: 719–730. [DOI] [PubMed] [Google Scholar]
- Akizuki S, Yoshida S, Chambers DE, Eddy LJ, Parmley LF, Yellon DM et al (1985). Infarct size limitation by the xanthine oxidase inhibitor, allopurinol, in closed‐chest dogs with small infarcts. Cardiovasc Res 19: 686–692. [DOI] [PubMed] [Google Scholar]
- Almasalmeh A, Krenc D, Wu B, Beitz E (2014). Structural determinants of the hydrogen peroxide permeability of aquaporins. FEBS J 281: 647–656. [DOI] [PubMed] [Google Scholar]
- Andreadou I, Iliodromitis EK, Rassaf T, Schulz R, Papapetropoulos A, Ferdinandy P (2015). The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br J Pharmacol 172: 1587–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrukhiv A, Costa AD, West IC, Garlid KD (2006). Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am J Physiol Heart Circ Physiol 291: H2067–H2074. [DOI] [PubMed] [Google Scholar]
- Anelli T, Bergamelli L, Margittai E, Rimessi A, Fagioli C, Malgaroli A et al (2012). Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum‐mitochondria interface (MAM). Antioxid Redox Signal 16: 1077–1087. [DOI] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, O'Rourke B (2010). Redox‐optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta 1797: 865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Marban E, O'Rourke B (2003). Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem 278: 44735–44744. [DOI] [PubMed] [Google Scholar]
- Bachschmid M, Schildknecht S, Ullrich V (2005). Redox regulation of vascular prostanoid synthesis by the nitric oxide‐superoxide system. Biochem Biophys Res Commun 338: 536–542. [DOI] [PubMed] [Google Scholar]
- Bachschmid M, Thurau S, Zou MH, Ullrich V (2003). Endothelial cell activation by endotoxin involves superoxide/NO‐mediated nitration of prostacyclin synthase and thromboxane receptor stimulation. FASEB J 17: 914–916. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH (1996). Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271: C1424–C1437. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA (1990). Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A 87: 1620–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall JK, Rinze R, Adlam D, Tatham AL, de Bono J, Wilson N et al (2007). Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: studies in endothelial‐targeted Nox2 transgenic mice. Circ Res 100: 1016–1025. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Di Lisa F (2015). The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 78: 100–106. doi: 10.1016/j.yjmcc.2014.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boengler K, Ungefug E, Heusch G, Schulz R (2013). The STAT3 inhibitor stattic impairs cardiomyocyte mitochondrial function through increased reactive oxygen species formation. Curr Pharm Des 19: 6890–6895. [DOI] [PubMed] [Google Scholar]
- Bonora M, Bononi A, Di Marchi E, Giorgi C, Lebiedzinska M, Marchi S et al (2013). Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12: 674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes RP (2005). Triggering mitochondrial radical release: a new function for NADPH oxidases. Hypertension 45: 847–848. [DOI] [PubMed] [Google Scholar]
- Brune B, Schmidt KU, Ullrich V (1990). Activation of soluble guanylate cyclase by carbon monoxide and inhibition by superoxide anion. Eur J Biochem/FEBS 192: 683–688. [DOI] [PubMed] [Google Scholar]
- Brune B, Dehne N, Grossmann N, Jung M, Namgaladze D, Schmid T et al (2013). Redox control of inflammation in macrophages. Antioxid Redox Signal 19: 595–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY et al (2011). Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1‐associated periodic syndrome (TRAPS). J Exp Med 208: 519–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 29: 222–230. [DOI] [PubMed] [Google Scholar]
- Cai H, Harrison DG (2000). Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87: 840–844. [DOI] [PubMed] [Google Scholar]
- Cappelletti G, Maggioni MG, Tedeschi G, Maci R (2003). Protein tyrosine nitration is triggered by nerve growth factor during neuronal differentiation of PC12 cells. Exp Cell Res 288: 9–20. [DOI] [PubMed] [Google Scholar]
- Ceriello A (2006). Oxidative stress and diabetes‐associated complications. Endocr Pract 12: 60–62. [DOI] [PubMed] [Google Scholar]
- Cerrato R, Cunnington C, Crabtree MJ, Antoniades C, Pernow J, Channon KM et al (2012). Endothelin‐1 increases superoxide production in human coronary artery bypass grafts. Life Sci 91: 723–728. [DOI] [PubMed] [Google Scholar]
- Chen AF, Chen DD, Daiber A, Faraci FM, Li H, Rembold CM et al (2012). Free radical biology of the cardiovascular system. Clin Sci (Lond) 123: 73–91. [DOI] [PubMed] [Google Scholar]
- Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly‐Rosen D (2008). Activation of aldehyde dehydrogenase‐2 reduces ischemic damage to the heart. Science 321: 1493–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Liu J, Zheng Y, Wang J, Wang Z, Gu S et al (2015). Uncoupling protein 3 mediates H(2)O(2) preconditioning‐afforded cardioprotection through the inhibition of MPTP opening. Cardiovasc Res 105: 192–202. [DOI] [PubMed] [Google Scholar]
- Chrissobolis S, Banfi B, Sobey CG, Faraci FM (2012). Role of Nox isoforms in angiotensin II‐induced oxidative stress and endothelial dysfunction in brain. J Appl Physiol 113: 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AD, Jakob R, Costa CL, Andrukhiv K, West IC, Garlid KD (2006). The mechanism by which the mitochondrial ATP‐sensitive K+ channel opening and H2O2 inhibit the mitochondrial permeability transition. J Biol Chem 281: 20801–20808. [DOI] [PubMed] [Google Scholar]
- Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC et al (2009). RAGE‐induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol 20: 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csonka C, Kupai K, Bencsik P, Gorbe A, Paloczi J, Zvara A et al (2014). Cholesterol‐enriched diet inhibits cardioprotection by ATP‐sensitive K+ channel activators cromakalim and diazoxide. Am J Physiol Heart Circ Physiol 306: H405–H413. [DOI] [PubMed] [Google Scholar]
- Daiber A (2010). Redox signaling (cross‐talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta 1797: 897–906. [DOI] [PubMed] [Google Scholar]
- Daiber A, Oelze M, Wenzel P, Wickramanayake JM, Schuhmacher S, Jansen T et al (2009). Nitrate tolerance as a model of vascular dysfunction: Roles for mitochondrial aldehyde dehydrogenase and mitochondrial oxidative stress. Pharmacol Rep 61: 33–48. [DOI] [PubMed] [Google Scholar]
- Daiber A, Oelze M, Daub S, Steven S, Schuff A, Kroller‐Schon S et al (2014). Vascular Redox Signaling, Redox Switches in Endothelial Nitric Oxide Synthase and Endothelial Dysfunction In: {0} (ed)Laher I. Systems Biology of Free Radicals and Antioxidants. Springer‐Verlag: Berlin Heidelberg, pp. 1177–1211. [Google Scholar]
- Das DK, Maulik N (2003). Preconditioning potentiates redox signaling and converts death signal into survival signal. Arch Biochem Biophys 420: 305–311. [DOI] [PubMed] [Google Scholar]
- Dehne N, Brune B (2014). Sensors, transmitters, and targets in mitochondrial oxygen shortage‐a hypoxia‐inducible factor relay story. Antioxid Redox Signal 20: 339–352. [DOI] [PubMed] [Google Scholar]
- Denicola A, Rubbo H, Rodriguez D, Radi R (1993). Peroxynitrite‐mediated cytotoxicity to Trypanosoma cruzi . Arch Biochem Biophys 304: 279–286. [DOI] [PubMed] [Google Scholar]
- Desouki MM, Kulawiec M, Bansal S, Das GM, Singh KK (2005). Cross talk between mitochondria and superoxide generating NADPH oxidase in breast and ovarian tumors. Cancer Biol Ther 4: 1367–1373. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Bernardi P (2006). Mitochondria and ischemia‐reperfusion injury of the heart: fixing a hole. Cardiovasc Res 70: 191–199. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Canton M, Menabo R, Kaludercic N, Bernardi P (2007). Mitochondria and cardioprotection. Heart Fail Rev 12: 249–260. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Canton M, Carpi A, Kaludercic N, Menabo R, Menazza S et al (2011). Mitochondrial injury and protection in ischemic pre‐ and postconditioning. Antioxid Redox Signal 14: 881–891. [DOI] [PubMed] [Google Scholar]
- Dikalov S (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med 51: 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov SI, Nazarewicz RR (2013). Angiotensin II‐induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal 19: 1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov SI, Li W, Doughan AK, Blanco RR, Zafari AM (2012). Mitochondrial reactive oxygen species and calcium uptake regulate activation of phagocytic NADPH oxidase. Am J Physiol Regul Integr Comp Physiol 302: R1134–R1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov SI, Nazarewicz RR, Bikineyeva A, Hilenski L, Lassegue B, Griendling KK et al (2014). Nox2‐Induced Production of Mitochondrial Superoxide in Angiotensin II‐Mediated Endothelial Oxidative Stress and Hypertension. Antioxid Redox Signal 20: 281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S et al (2005). Nox1 overexpression potentiates angiotensin II‐induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112: 2668–2676. [DOI] [PubMed] [Google Scholar]
- Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W et al (2010). Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107: 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerries C, Grote K, Hilfiker‐Kleiner D, Luchtefeld M, Schaefer A, Holland SM et al (2007). Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ Res 100: 894–903. [DOI] [PubMed] [Google Scholar]
- Dost T, Cohen MV, Downey JM (2008). Redox signaling triggers protection during the reperfusion rather than the ischemic phase of preconditioning. Basic Res Cardiol 103: 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughan AK, Harrison DG, Dikalov SI (2008). Molecular mechanisms of angiotensin II‐mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 102: 488–496. [DOI] [PubMed] [Google Scholar]
- Drose S, Brandt U (2008). The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J Biol Chem 283: 21649–21654. [DOI] [PubMed] [Google Scholar]
- Duerrschmidt N, Wippich N, Goettsch W, Broemme HJ, Morawietz H (2000). Endothelin‐1 induces NAD(P)H oxidase in human endothelial cells. Biochem Biophys Res Commun 269: 713–717. [DOI] [PubMed] [Google Scholar]
- El Assar M, Angulo J, Rodriguez‐Manas L (2013). Oxidative stress and vascular inflammation in aging. Free Radic Biol Med 65: 380–401. [DOI] [PubMed] [Google Scholar]
- Engberding N, Spiekermann S, Schaefer A, Heineke A, Wiencke A, Muller M et al (2004). Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: a new action for an old drug? Circulation 110: 2175–2179. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Kennedy JA (1974). Role of superoxide radical in mitochondrial dehydrogenase reactions. Biochem Biophys Res Commun 60: 1044–1050. [DOI] [PubMed] [Google Scholar]
- Fornazari M, de Paula JG, Castilho RF, Kowaltowski AJ (2008). Redox properties of the adenoside triphosphate‐sensitive K+ channel in brain mitochondria. J Neurosci Res 86: 1548–1556. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Munzel T (2006). Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113: 1708–1714. [DOI] [PubMed] [Google Scholar]
- Garlid KD (2000). Opening mitochondrial K(ATP) in the heart–what happens, and what does not happen. Basic Res Cardiol 95: 275–279. [DOI] [PubMed] [Google Scholar]
- Gertz M, Steegborn C (2010). The Lifespan‐regulator p66Shc in mitochondria: redox enzyme or redox sensor? Antioxid Redox Signal 13: 1417–1428. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI et al (2000). Oxidative damage linked to neurodegeneration by selective alpha‐synuclein nitration in synucleinopathy lesions. Science 290: 985–989. [DOI] [PubMed] [Google Scholar]
- Giorgio S, Linares E, Ischiropoulos H, Von Zuben FJ, Yamada A, Augusto O (1998). In vivo formation of electron paramagnetic resonance‐detectable nitric oxide and of nitrotyrosine is not impaired during murine leishmaniasis. Infect Immun 66: 807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gori T, Forconi S (2005). The role of reactive free radicals in ischemic preconditioning–clinical and evolutionary implications. Clin Hemorheol Microcirc 33: 19–28. [PubMed] [Google Scholar]
- Gori T, Parker JD (2008). Nitrate‐induced toxicity and preconditioning: a rationale for reconsidering the use of these drugs. J Am Coll Cardiol 52: 251–254. [DOI] [PubMed] [Google Scholar]
- Gori T, Munzel T (2011). Oxidative stress and endothelial dysfunction: therapeutic implications. Ann Med 43: 259–272. [DOI] [PubMed] [Google Scholar]
- Gori T, Dragoni S, Di Stolfo G, Sicuro S, Liuni A, Luca MC et al (2010). Tolerance to nitroglycerin‐induced preconditioning of the endothelium: a human in vivo study. Am J Physiol Heart Circ Physiol 298: H340–H345. [DOI] [PubMed] [Google Scholar]
- Graham NA, Tahmasian M, Kohli B, Komisopoulou E, Zhu M, Vivanco I et al (2012). Glucose deprivation activates a metabolic and signaling amplification loop leading to cell death. Mol Syst Biol 8: 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griendling KK, Ushio‐Fukai M (1998). Redox control of vascular smooth muscle proliferation. J Lab Clin Med 132: 9–15. [DOI] [PubMed] [Google Scholar]
- Griendling KK, FitzGerald GA (2003a). Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation 108: 2034–2040. [DOI] [PubMed] [Google Scholar]
- Griendling KK, FitzGerald GA (2003b). Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo monitoring of ROS. Circulation 108: 1912–1916. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW (1994). Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74: 1141–1148. [DOI] [PubMed] [Google Scholar]
- Gryglewski RJ, Palmer RM, Moncada S (1986). Superoxide anion is involved in the breakdown of endothelium‐derived vascular relaxing factor. Nature 320: 454–456. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S et al (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR et al (2011). Inflammation, immunity, and hypertension. Hypertension 57: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy D, Wynne A, Duchen M, Yellon D (2004). Transient mitochondrial permeability transition pore opening mediates preconditioning‐induced protection. Circulation 109: 1714–1717. [DOI] [PubMed] [Google Scholar]
- Hawkins BJ, Madesh M, Kirkpatrick CJ, Fisher AB (2007). Superoxide flux in endothelial cells via the chloride channel‐3 mediates intracellular signaling. Mol Biol Cell 18: 2002–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel FR, Luo Y, Li X, Boengler K, Buechert A, Garcia‐Dorado D et al (2005). Impairment of diazoxide‐induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ Res 97: 583–586. [DOI] [PubMed] [Google Scholar]
- Heitzer T, Just H, Munzel T (1996). Antioxidant vitamin C improves endothelial dysfunction in chronic smokers. Circulation 94: 6–9. [DOI] [PubMed] [Google Scholar]
- Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T (2001a). Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 104: 2673–2678. [DOI] [PubMed] [Google Scholar]
- Heitzer T, Finckh B, Albers S, Krohn K, Kohlschutter A, Meinertz T (2001b). Beneficial effects of alpha‐lipoic acid and ascorbic acid on endothelium‐dependent, nitric oxide‐mediated vasodilation in diabetic patients: relation to parameters of oxidative stress. Free Radic Biol Med 31: 53–61. [DOI] [PubMed] [Google Scholar]
- Hernanz R, Briones AM, Salaices M, Alonso MJ (2014). New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo‐oxygenase in hypertension. Clin Sci (Lond) 126: 111–121. [DOI] [PubMed] [Google Scholar]
- Hink U, Oelze M, Kolb P, Bachschmid M, Zou MH, Daiber A et al (2003). Role for peroxynitrite in the inhibition of prostacyclin synthase in nitrate tolerance. J Am Coll Cardiol 42: 1826–1834. [DOI] [PubMed] [Google Scholar]
- Hirschhauser C, Bornbaum J, Reis A, Bohme S, Kaludercic N, Menabo R et al (2015). NOX4 in Mitochondria: Yeast Two‐Hybrid‐Based Interaction with Complex I Without Relevance for Basal Reactive Oxygen Species? Antioxid Redox Signal 23: 1106–1112. doi: 10.1089/ars.2014.6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Ghosh P, Wan R, Ouyang X, Cheng H, Mattson MP et al (2014). Permeability transition pore‐mediated mitochondrial superoxide flashes mediate an early inhibitory effect of amyloid beta1‐42 on neural progenitor cell proliferation. Neurobiol Aging 35: 975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd TR, Degennaro M, Lehmann R (2012). Redox regulation of cell migration and adhesion. Trends Cell Biol 22: 107–115. doi: 10.1016/j.tcb.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JW, Yao H, Caito S, Sundar IK, Rahman I (2013). Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic Biol Med 61C: 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idigo WO, Reilly S, Zhang MH, Zhang YH, Jayaram R, Carnicer R et al (2012). Regulation of endothelial nitric‐oxide synthase (NOS) S‐glutathionylation by neuronal NOS: evidence of a functional interaction between myocardial constitutive NOS isoforms. J Biol Chem 287: 43665–43673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infanger DW, Cao X, Butler SD, Burmeister MA, Zhou Y, Stupinski JA et al (2010). Silencing nox4 in the paraventricular nucleus improves myocardial infarction‐induced cardiac dysfunction by attenuating sympathoexcitation and periinfarct apoptosis. Circ Res 106: 1763–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irigoin F, Cibils L, Comini MA, Wilkinson SR, Flohe L, Radi R (2008). Insights into the redox biology of Trypanosoma cruzi: Trypanothione metabolism and oxidant detoxification. Free Radic Biol Med 45: 733–742. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Beckman JS (2003). Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest 111: 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janko C, Filipovic M, Munoz LE, Schorn C, Schett G, Ivanovic‐Burmazovic I et al (2014). Redox Modulation of HMGB1‐Related Signaling. Antioxid Redox Signal 20: 1075–1085. doi: 10.1089/ars.2013.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian C, Hou T, Yin R, Cheng H, Wang X (2014). Regulation of superoxide flashes by transient and steady mitochondrial calcium elevations. Sci China Life Sci 57: 495–501. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW et al (2004). Glycogen synthase kinase‐3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113: 1535–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahler J, Mendel S, Weckmuller J, Orzechowski HD, Mittmann C, Koster R et al (2000). Oxidative stress increases synthesis of big endothelin‐1 by activation of the endothelin‐1 promoter. J Mol Cell Cardiol 32: 1429–1437. [DOI] [PubMed] [Google Scholar]
- Kahler J, Ewert A, Weckmuller J, Stobbe S, Mittmann C, Koster R et al (2001). Oxidative stress increases endothelin‐1 synthesis in human coronary artery smooth muscle cells. J Cardiovasc Pharmacol 38: 49–57. [DOI] [PubMed] [Google Scholar]
- Kaludercic N, Mialet‐Perez J, Paolocci N, Parini A, Di Lisa F (2014a). Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol 73C: 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D et al (2010). Monoamine oxidase A‐mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res 106: 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaludercic N, Carpi A, Nagayama T, Sivakumaran V, Zhu G, Lai EW et al (2014b). Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxid Redox Signal 20: 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbach S, Wenzel P, Waisman A, Munzel T, Daiber A (2013). eNOS Uncoupling in Cardiovascular Diseases ‐ the Role of Oxidative Stress and Inflammation. Curr Pharm Des . [DOI] [PubMed] [Google Scholar]
- Karbach S, Wenzel P, Waisman A, Munzel T, Daiber A (2014). eNOS uncoupling in cardiovascular diseases–the role of oxidative stress and inflammation. Curr Pharm Des 20: 3579–3594. [DOI] [PubMed] [Google Scholar]
- Kim HS, Ullevig SL, Zamora D, Lee CF, Asmis R (2012). Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment. Proc Natl Acad Sci U S A 109: E2803–E2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY et al (2005). Mitochondria‐derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension 45: 438–444. [DOI] [PubMed] [Google Scholar]
- Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, Koomalsingh KJ et al (2012). Reduction of ischemia/reperfusion injury with bendavia, a mitochondria‐targeting cytoprotective Peptide. J Am Heart Assoc 1: e001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr M, Hausding M, Kroller‐Schuhmacher S, Steven S, Oelze M, Heeren T et al (2011). Nitroglycerin‐Induced Endothelial Dysfunction and Tolerance Involve Adverse Phosphorylation and S‐Glutathionylation of Endothelial Nitric Oxide Synthase: Beneficial Effects of Therapy With the AT1 Receptor Blocker Telmisartan. Arterioscler Thromb Vasc Biol 31: 2223–2231. [DOI] [PubMed] [Google Scholar]
- Konzack A, Kietzmann T (2014). Manganese superoxide dismutase in carcinogenesis: friend or foe? Biochem Soc Trans 42: 1012–1016. [DOI] [PubMed] [Google Scholar]
- Kooy NW, Lewis SJ, Royall JA, Ye YZ, Kelly DR, Beckman JS (1997). Extensive tyrosine nitration in human myocardial inflammation: evidence for the presence of peroxynitrite. Crit Care Med 25: 812–819. [DOI] [PubMed] [Google Scholar]
- Krenz M, Oldenburg O, Wimpee H, Cohen MV, Garlid KD, Critz SD et al (2002). Opening of ATP‐sensitive potassium channels causes generation of free radicals in vascular smooth muscle cells. Basic Res Cardiol 97: 365–373. [DOI] [PubMed] [Google Scholar]
- Kroller‐Schon S, Knorr M, Hausding M, Oelze M, Schuff A, Schell R et al (2012). Glucose‐independent improvement of vascular dysfunction in experimental sepsis by dipeptidyl‐peptidase 4 inhibition. Cardiovasc Res 96: 140–149. [DOI] [PubMed] [Google Scholar]
- Kroller‐Schon S, Steven S, Kossmann S, Scholz A, Daub S, Oelze M et al (2014). Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species‐studies in white blood cells and in animal models. Antioxid Redox Signal 20: 247–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE et al (2010). Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med 363: 2600–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzkaya N, Weissmann N, Harrison DG, Dikalov S (2003). Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric‐oxide synthase. J Biol Chem 278: 22546–22554. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H et al (2002). Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 40: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM et al (2003). Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111: 1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmesser U, Spiekermann S, Preuss C, Sorrentino S, Fischer D, Manes C et al (2007). Angiotensin II induces endothelial xanthine oxidase activation: role for endothelial dysfunction in patients with coronary disease. Arterioscler Thromb Vasc Biol 27: 943–948. [DOI] [PubMed] [Google Scholar]
- Lassegue B, Griendling KK (2010). NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30: 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurindo FR, Fernandes DC, Amanso AM, Lopes LR, Santos CX (2008). Novel role of protein disulfide isomerase in the regulation of NADPH oxidase activity: pathophysiological implications in vascular diseases. Antioxid Redox Signal 10: 1101–1113. [DOI] [PubMed] [Google Scholar]
- 2Lebiedzinska M, Duszynski J, Rizzuto R, Pinton P, Wieckowski MR (2009). Age‐related changes in levels of p66Shc and serine 36‐phosphorylated p66Shc in organs and mouse tissues. Arch Biochem Biophys 486: 73–80. [DOI] [PubMed] [Google Scholar]
- Lee DY, Wauquier F, Eid AA, Roman LJ, Ghosh‐Choudhury G, Khazim K et al (2013). Nox4 NADPH oxidase mediates peroxynitrite‐dependent uncoupling of endothelial nitric‐oxide synthase and fibronectin expression in response to angiotensin II: role of mitochondrial reactive oxygen species. J Biol Chem 288: 28668–28686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Bae IH, Bae YS, Um HD (2006). Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem 281: 36228–36235. [DOI] [PubMed] [Google Scholar]
- Li JM, Shah AM (2001). Differential NADPH‐ versus NADH‐dependent superoxide production by phagocyte‐type endothelial cell NADPH oxidase. Cardiovasc Res 52: 477–486. [DOI] [PubMed] [Google Scholar]
- Li L, Chu Y, Fink GD, Engelhardt JF, Heistad DD, Chen AF (2003a). Endothelin‐1 stimulates arterial VCAM‐1 expression via NADPH oxidase‐derived superoxide in mineralocorticoid hypertension. Hypertension 42: 997–1003. [DOI] [PubMed] [Google Scholar]
- Li L, Watts SW, Banes AK, Galligan JJ, Fink GD, Chen AF (2003b). NADPH oxidase‐derived superoxide augments endothelin‐1‐induced venoconstriction in mineralocorticoid hypertension. Hypertension 42: 316–321. [DOI] [PubMed] [Google Scholar]
- Li L, Fink GD, Watts SW, Northcott CA, Galligan JJ, Pagano PJ et al (2003c). Endothelin‐1 increases vascular superoxide via endothelin(A)‐NADPH oxidase pathway in low‐renin hypertension. Circulation 107: 1053–1058. [DOI] [PubMed] [Google Scholar]
- Liao B, Zheng YM, Yadav VR, Korde AS, Wang YX (2011). Hypoxia induces intracellular Ca2+ release by causing reactive oxygen species‐mediated dissociation of FK506‐binding protein 12.6 from ryanodine receptor 2 in pulmonary artery myocytes. Antioxid Redox Signal 14: 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SY, Davidson SM, Hausenloy DJ, Yellon DM (2007). Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovasc Res 75: 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Takemoto DJ (2005). Oxidative activation of protein kinase Cgamma through the C1 domain. Effects on gap junctions. J Biol Chem 280: 13682–13693. [DOI] [PubMed] [Google Scholar]
- Linares E, Giorgio S, Mortara RA, Santos CX, Yamada AT, Augusto O (2001). Role of peroxynitrite in macrophage microbicidal mechanisms in vivo revealed by protein nitration and hydroxylation. Free Radic Biol Med 30: 1234–1242. [DOI] [PubMed] [Google Scholar]
- Lisi M, Oelze M, Dragoni S, Liuni A, Steven S, Luca MC et al (2012). Chronic protection against ischemia and reperfusion‐induced endothelial dysfunction during therapy with different organic nitrates. Clin Res Cardiol 101: 453–459. [DOI] [PubMed] [Google Scholar]
- Liu H, Xiao Y, Xiong C, Wei A, Ruan J (2011). Apoptosis induced by a new flavonoid in human hepatoma HepG2 cells involves reactive oxygen species‐mediated mitochondrial dysfunction and MAPK activation. Eur J Pharmacol 654: 209–216. [DOI] [PubMed] [Google Scholar]
- Loguercio C, Federico A (2003). Oxidative stress in viral and alcoholic hepatitis. Free Radic Biol Med 34: 1–10. [DOI] [PubMed] [Google Scholar]
- Looi YH, Grieve DJ, Siva A, Walker SJ, Anilkumar N, Cave AC et al (2008). Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 51: 319–325. [DOI] [PubMed] [Google Scholar]
- Lustgarten MS, Bhattacharya A, Muller FL, Jang YC, Shimizu T, Shirasawa T et al (2012). Complex I generated, mitochondrial matrix‐directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem Biophys Res Commun 422: 515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maack C, Bohm M (2011). Targeting mitochondrial oxidative stress in heart failure throttling the afterburner. J Am Coll Cardiol 58: 83–86. [DOI] [PubMed] [Google Scholar]
- Markelic M, Velickovic K, Golic I, Klepal W, Otasevic V, Stancic A et al (2013). The origin of lipofuscin in brown adipocytes of hyperinsulinaemic rats: the role of lipid peroxidation and iron. Histol Histopathol 28: 493–503. [DOI] [PubMed] [Google Scholar]
- Marvar PJ, Harrison DG (2012). Stress‐dependent hypertension and the role of T lymphocytes. Exp Physiol . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M et al (2005). Nox1 is involved in angiotensin II‐mediated hypertension: a study in Nox1‐deficient mice. Circulation 112: 2677–2685. [DOI] [PubMed] [Google Scholar]
- McCord JM, Keele BB Jr, Fridovich I (1971). An enzyme‐based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proc Natl Acad Sci U S A 68: 1024–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Britigan BE (1997). Role of oxidants in microbial pathophysiology. Clin Microbiol Rev 10: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry Y, Poolman T, Williams B, Herbert KE (2013). A role for mitochondrial oxidants in stress‐induced premature senescence of human vascular smooth muscle cells. Redox Biol 1: 411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mofarrahi M, Brandes RP, Gorlach A, Hanze J, Terada LS, Quinn MT et al (2008). Regulation of proliferation of skeletal muscle precursor cells by NADPH oxidase. Antioxid Redox Signal 10: 559–574. [DOI] [PubMed] [Google Scholar]
- Munzel T, Daiber A, Ullrich V, Mulsch A (2005). Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP‐dependent protein kinase. Arterioscler Thromb Vasc Biol 25: 1551–1557. [DOI] [PubMed] [Google Scholar]
- Munzel T, Gori T, Bruno RM, Taddei S (2010). Is oxidative stress a therapeutic target in cardiovascular disease? Eur Heart J 31: 2741–2748. [DOI] [PubMed] [Google Scholar]
- Munzel T, Giaid A, Kurz S, Stewart DJ, Harrison DG (1995). Evidence for a role of endothelin 1 and protein kinase C in nitroglycerin tolerance. Proc Natl Acad Sci 92: 5244–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch CE, Alom‐Ruiz SP, Wang M, Zhang M, Walker S, Yu B et al (2011). Role of endothelial Nox2 NADPH oxidase in angiotensin II‐induced hypertension and vasomotor dysfunction. Basic Res Cardiol 106: 527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarewicz RR, Dikalova AE, Bikineyeva A, Dikalov SI (2013). Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am J Physiol Heart Circ Physiol 305: H1131–H1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E (2011). Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem 286: 40184–40192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel A, Kohlhaas M, Maack C (2014). Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol 73C: 26–33. [DOI] [PubMed] [Google Scholar]
- Oelze M, Schuhmacher S, Daiber A (2010). Organic nitrates and nitrate resistance in diabetes: the role of vascular dysfunction and oxidative stress with emphasis on antioxidant properties of pentaerithrityl tetranitrate. Exp Diabetes Res 2010: 213176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelze M, Kroller‐Schon S, Steven S, Lubos E, Doppler C, Hausding M et al (2014). Glutathione peroxidase‐1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension 63: 390–396. [DOI] [PubMed] [Google Scholar]
- Ohnuma Y, Miura T, Miki T, Tanno M, Kuno A, Tsuchida A et al (2002). Opening of mitochondrial K(ATP) channel occurs downstream of PKC‐epsilon activation in the mechanism of preconditioning. Am J Physiol Heart Circ Physiol 283: H440–H447. [DOI] [PubMed] [Google Scholar]
- Oldenburg O, Critz SD, Cohen MV, Downey JM (2003a). Acetylcholine‐induced production of reactive oxygen species in adult rabbit ventricular myocytes is dependent on phosphatidylinositol 3‐ and Src‐kinase activation and mitochondrial K(ATP) channel opening. J Mol Cell Cardiol 35: 653–660. [DOI] [PubMed] [Google Scholar]
- Oldenburg O, Yang XM, Krieg T, Garlid KD, Cohen MV, Grover GJ et al (2003b). P1075 opens mitochondrial K(ATP) channels and generates reactive oxygen species resulting in cardioprotection of rabbit hearts. J Mol Cell Cardiol 35: 1035–1042. [DOI] [PubMed] [Google Scholar]
- Ortona E, Maselli A, Delunardo F, Colasanti T, Giovannetti A, Pierdominici M (2014). Relationship between redox status and cell fate in immunity and autoimmunity. Antioxid Redox Signal 21: 103–122. [DOI] [PubMed] [Google Scholar]
- Ouyang JS, Li YP, Li CY, Cai C, Chen CS, Chen SX et al (2012). Mitochondrial ROS‐K+ channel signaling pathway regulated secretion of human pulmonary artery endothelial cells. Free Radic Res 46: 1437–1445. [DOI] [PubMed] [Google Scholar]
- Pacher P, Sharma K, Csordas G, Zhu Y, Hajnoczky G (2008). Uncoupling of ER‐mitochondrial calcium communication by transforming growth factor‐beta. Am J Physiol Renal Physiol 295: F1303–F1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechanova O, Varga ZV, Cebova M, Giricz Z, Pacher P, Ferdinandy P (2015). Cardiac NO signalling in the metabolic syndrome. Br J Pharmacol 172: 1415–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peshavariya H, Jiang F, Taylor CJ, Selemidis S, Chang CW, Dusting GJ (2009). Translation‐linked mRNA destabilization accompanying serum‐induced Nox4 expression in human endothelial cells. Antioxid Redox Signal 11: 2399–2408. [DOI] [PubMed] [Google Scholar]
- Philipp S, Cui L, Ludolph B, Kelm M, Schulz R, Cohen MV et al (2006). Desferoxamine and ethyl‐3,4‐dihydroxybenzoate protect myocardium by activating NOS and generating mitochondrial ROS. Am J Physiol Heart Circ Physiol 290: H450–H457. [DOI] [PubMed] [Google Scholar]
- Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M et al (2007). Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life‐span determinant p66Shc. Science 315: 659–663. [DOI] [PubMed] [Google Scholar]
- Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N et al (2008). Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 359: 473–481. [DOI] [PubMed] [Google Scholar]
- Piskernik C, Haindl S, Behling T, Gerald Z, Kehrer I, Redl H et al (2008). Antimycin A and lipopolysaccharide cause the leakage of superoxide radicals from rat liver mitochondria. Biochim Biophys Acta 1782: 280–285. [DOI] [PubMed] [Google Scholar]
- Queliconi BB, Wojtovich AP, Nadtochiy SM, Kowaltowski AJ, Brookes PS (2011). Redox regulation of the mitochondrial K(ATP) channel in cardioprotection. Biochim Biophys Acta 1813: 1309–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R, Cassina A, Hodara R, Quijano C, Castro L (2002). Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med 33: 1451–1464. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK et al (1996). Angiotensin II‐mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Investig 97: 1916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathore R, Zheng YM, Niu CF, Liu QH, Korde A, Ho YS et al (2008). Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS‐PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med 45: 1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risbano MG, Gladwin MT (2013). Therapeutics targeting of dysregulated redox equilibrium and endothelial dysfunction. Handb Exp Pharmacol 218: 315–349. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T (1993). Microdomains with high Ca2+ close to IP3‐sensitive channels that are sensed by neighboring mitochondria. Science 262: 744–747. [DOI] [PubMed] [Google Scholar]
- Ruiz‐Meana M, Inserte J, Fernandez‐Sanz C, Hernando V, Miro‐Casas E, Barba I et al (2011). The role of mitochondrial permeability transition in reperfusion‐induced cardiomyocyte death depends on the duration of ischemia. Basic Res Cardiol 106: 1259–1268. [DOI] [PubMed] [Google Scholar]
- Saotome M, Katoh H, Satoh H, Nagasaka S, Yoshihara S, Terada H et al (2005). Mitochondrial membrane potential modulates regulation of mitochondrial Ca2+ in rat ventricular myocytes. Am J Physiol Heart Circ Physiol 288: H1820–H1828. [DOI] [PubMed] [Google Scholar]
- Sayed N, Baskaran P, Ma X, van den Akker F, Beuve A (2007). Desensitization of soluble guanylyl cyclase, the NO receptor, by S‐nitrosylation. Proc Natl Acad Sci U S A 104: 12312–12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed N, Kim DD, Fioramonti X, Iwahashi T, Duran WN, Beuve A (2008). Nitroglycerin‐induced S‐nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ Res 103: 606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildknecht S, Bachschmid M, Ullrich V (2005). Peroxynitrite provides the peroxide tone for PGHS‐2‐dependent prostacyclin synthesis in vascular smooth muscle cells. FASEB J 19: 1169–1171. [DOI] [PubMed] [Google Scholar]
- Schildknecht S, van der Loo B, Weber K, Tiefenthaler K, Daiber A, Bachschmid MM (2008). Endogenous peroxynitrite modulates PGHS‐1‐dependent thromboxane A2 formation and aggregation in human platelets. Free Radic Biol Med 45: 512–520. [DOI] [PubMed] [Google Scholar]
- Schildknecht S, Heinz K, Daiber A, Hamacher J, Kavakli C, Ullrich V et al (2006). Autocatalytic tyrosine nitration of prostaglandin endoperoxide synthase‐2 in LPS‐stimulated RAW 264.7 macrophages. Biochem Biophys Res Commun 340: 318–325. [DOI] [PubMed] [Google Scholar]
- Schildknecht S, Karreman C, Daiber A, Zhao C, Hamacher J, Perlman D et al (2012). Autocatalytic nitration of prostaglandin endoperoxide synthase‐2 by nitrite inhibits prostanoid formation in rat alveolar macrophages. Antioxid Redox Signal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HH, Stocker R, Vollbracht C, Paulsen G, Riley DP, Daiber A et al (2015). Antioxidants in Translational Medicine. Antioxid Redox Signal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Wandzioch K, Helmcke I, Brandes RP (2009). Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol 29: 239–245. [DOI] [PubMed] [Google Scholar]
- Schroder K, Helmcke I, Palfi K, Krause KH, Busse R, Brandes RP (2007). Nox1 mediates basic fibroblast growth factor‐induced migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 27: 1736–1743. [DOI] [PubMed] [Google Scholar]
- Schuhmacher S, Oelze M, Bollmann F, Kleinert H, Otto C, Heeren T et al (2011). Vascular Dysfunction in Experimental Diabetes Is Improved by Pentaerithrityl Tetranitrate but not Isosorbide‐5‐Mononitrate Therapy. Diabetes 60: 2608–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz E, Wenzel P, Munzel T, Daiber A (2014). Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid Redox Signal 20: 308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz E, Jansen T, Wenzel P, Daiber A, Munzel T (2008). Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid Redox Signal 10: 1115–1126. [DOI] [PubMed] [Google Scholar]
- Schulz R, Boengler K, Totzeck A, Luo Y, Garcia‐Dorado D, Heusch G (2007). Connexin 43 in ischemic pre‐ and postconditioning. Heart Fail Rev 12: 261–266. [DOI] [PubMed] [Google Scholar]
- Seidlmayer L, Kuhn J, Ritter O (2014). Mitochondrial metabolism is altered via IP3‐mediated calcium release. Clin Res Cardiol 103: V530. [Google Scholar]
- Seidlmayer LK, Juettner VV, Kettlewell S, Pavlov EV, Blatter LA, Dedkova EN (2015). Distinct mPTP activation mechanisms in ischaemia‐reperfusion: contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc Res 106: 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Chandel NS (2012). Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48: 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H. (ed.). (1991). Oxidative Stress: Oxidants and Antioxidants. Academic Press: London, UK. [DOI] [PubMed] [Google Scholar]
- Skyschally A, Schulz R, Gres P, Korth HG, Heusch G (2003). Attenuation of ischemic preconditioning in pigs by scavenging of free oxyradicals with ascorbic acid. Am J Physiol Heart Circ Physiol 284: H698–H703. [DOI] [PubMed] [Google Scholar]
- Somasuntharam I, Boopathy AV, Khan RS, Martinez MD, Brown ME, Murthy N et al (2013). Delivery of Nox2‐NADPH oxidase siRNA with polyketal nanoparticles for improving cardiac function following myocardial infarction. Biomaterials 34: 7790–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP et al (2002). Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 105: 1429–1435. [DOI] [PubMed] [Google Scholar]
- Stadie WC, Riggs BC, Haugaard N (1945). Oxygen poisoning; the effect of high oxygen pressure upon enzymes; pepsin, catalase, cholinesterase, and carbonic anhydrase. J Biol Chem 161: 175–180. [PubMed] [Google Scholar]
- Stamler JS (1994). Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell 78: 931–936. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G (2003). Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem 86: 1101–1107. [DOI] [PubMed] [Google Scholar]
- Stasch JP, Pacher P, Evgenov OV (2011). Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 123: 2263–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolf BS, Smyrnias I, Lopes LR, Vendramin A, Goto H, Laurindo FR et al (2011). Protein disulfide isomerase and host‐pathogen interaction. Sci World J 11: 1749–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassburger M, Bloch W, Sulyok S, Schuller J, Keist AF, Schmidt A et al (2005). Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic Biol Med 38: 1458–1470. [DOI] [PubMed] [Google Scholar]
- Sturza A, Leisegang MS, Babelova A, Schroder K, Benkhoff S, Loot AE et al (2013). Monoamine oxidases are mediators of endothelial dysfunction in the mouse aorta. Hypertension 62: 140–146. [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D et al (2006). Chaperone‐mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175: 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C (1996). The pathophysiological role of peroxynitrite in shock, inflammation, and ischemia‐reperfusion injury. Shock 6: 79–88. [DOI] [PubMed] [Google Scholar]
- Takac I, Schroder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD et al (2011). The E‐loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem 286: 13304–13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GR, DiFabio JM, Gori T, Parker JD (2007). Once daily therapy with isosorbide‐5‐mononitrate causes endothelial dysfunction in humans: evidence of a free‐radical‐mediated mechanism. J Am Coll Cardiol 49: 1289–1295. [DOI] [PubMed] [Google Scholar]
- Tsuda T, Hamamori Y, Yamashita T, Fukumoto Y, Takai Y (1986). Involvement of three intracellular messenger systems, protein kinase C, calcium ion and cyclic AMP, in the regulation of c‐fos gene expression in Swiss 3 T3 cells. FEBS Lett 208: 39–42. [DOI] [PubMed] [Google Scholar]
- Ullrich V, Kissner R (2006). Redox signaling: bioinorganic chemistry at its best. J Inorg Biochem 100: 2079–2086. [DOI] [PubMed] [Google Scholar]
- Ullrich V, Schildknecht S (2014). Sensing hypoxia by mitochondria: a unifying hypothesis involving s‐nitrosation. Antioxid Redox Signal 20: 325–338. [DOI] [PubMed] [Google Scholar]
- Varga ZV, Kupai K, Szucs G, Gaspar R, Paloczi J, Farago N et al (2013). MicroRNA‐25‐dependent up‐regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia‐induced oxidative/nitrative stress and subsequent dysfunction in the heart. J Mol Cell Cardiol 62: 111–121. [DOI] [PubMed] [Google Scholar]
- Vieira HL, Belzacq AS, Haouzi D, Bernassola F, Cohen I, Jacotot E et al (2001). The adenine nucleotide translocator: a target of nitric oxide, peroxynitrite, and 4‐hydroxynonenal. Oncogene 20: 4305–4316. [DOI] [PubMed] [Google Scholar]
- Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J et al (2008). Superoxide flashes in single mitochondria. Cell 134: 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Zheng YM (2010). ROS‐dependent signaling mechanisms for hypoxic Ca(2+) responses in pulmonary artery myocytes. Antioxid Redox Signal 12: 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Lauer N, Mulsch A, Kojda G (2001). The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radic Biol Med 31: 1360–1367. [DOI] [PubMed] [Google Scholar]
- Wendt MC, Daiber A, Kleschyov AL, Mulsch A, Sydow K, Schulz E et al (2005). Differential effects of diabetes on the expression of the gp91(phox) homologues nox1 and nox4. Free Radic Biol Med 39: 381–391. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Muller J, Zurmeyer S, Schuhmacher S, Schulz E, Oelze M et al (2008a). ALDH‐2 deficiency increases cardiovascular oxidative stress–evidence for indirect antioxidative properties. Biochem Biophys Res Commun 367: 137–143. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Mollnau H, Oelze M, Schulz E, Wickramanayake JM, Muller J et al (2008b). First evidence for a crosstalk between mitochondrial and NADPH oxidase‐derived reactive oxygen species in nitroglycerin‐triggered vascular dysfunction. Antioxid Redox Signal 10: 1435–1447. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Schuhmacher S, Kienhofer J, Muller J, Hortmann M, Oelze M et al (2008c). Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age‐dependent vascular dysfunction. Cardiovasc Res 80: 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S et al (2011). Lysozyme M‐positive monocytes mediate angiotensin II‐induced arterial hypertension and vascular dysfunction. Circulation 124: 1370–1381. [DOI] [PubMed] [Google Scholar]
- Werns SW, Shea MJ, Mitsos SE, Dysko RC, Fantone JC, Schork MA et al (1986). Reduction of the size of infarction by allopurinol in the ischemic‐reperfused canine heart. Circulation 73: 518–524. [DOI] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument‐Bromage H, Tempst P et al (2011). TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472: 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler K, Wunsch S, Kreutz R, Rothermund L, Paul M, Schmidt HH (2001). Upregulation of the vascular NAD(P)H‐oxidase isoforms Nox1 and Nox4 by the renin‐angiotensin system in vitro and in vivo. Free Radic Biol Med 31: 1456–1464. [DOI] [PubMed] [Google Scholar]
- Wong WK, Ou XM, Chen K, Shih JC (2002). Activation of human monoamine oxidase B gene expression by a protein kinase C MAPK signal transduction pathway involves c‐Jun and Egr‐1. J Biol Chem 277: 22222–22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosniak J Jr, Santos CX, Kowaltowski AJ, Laurindo FR (2009). Cross‐talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II‐mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal 11: 1265–1278. [DOI] [PubMed] [Google Scholar]
- Wu KL, Chao YM, Tsay SJ, Chen CH, Chan SH, Dovinova I et al (2014). Role of nitric oxide synthase uncoupling at rostral ventrolateral medulla in redox‐sensitive hypertension associated with metabolic syndrome. Hypertension 64: 815–824. [DOI] [PubMed] [Google Scholar]
- Yin F, Boveris A, Cadenas E (2014). Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal 20: 353–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SK, Starnes TW, Deng Q, Huttenlocher A (2011). Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature 480: 109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SK, Freisinger CM, LeBert DC, Huttenlocher A (2012). Early redox, Src family kinase, and calcium signaling integrate wound responses and tissue regeneration in zebrafish. J Cell Biol 199: 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang GX, Kimura S, Murao K, Shimizu J, Matsuyoshi H, Takaki M (2009). Role of neuronal NO synthase in regulating vascular superoxide levels and mitogen‐activated protein kinase phosphorylation. Cardiovasc Res 81: 389–399. [DOI] [PubMed] [Google Scholar]
- Zhang R, Ran HH, Peng L, Xu F, Sun JF, Zhang LN et al (2014). Mitochondrial regulation of NADPH oxidase in hindlimb unweighting rat cerebral arteries. PLoS One 9: e95916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225. [DOI] [PubMed] [Google Scholar]
- Zinkevich NS, Gutterman DD (2011). ROS‐induced ROS release in vascular biology: redox‐redox signaling. Am J Physiol Heart Circ Physiol 301: H647–H653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ (2014). Mitochondrial Reactive Oxygen Species (ROS) and ROS‐Induced ROS Release. Physiol Rev 94: 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ (2000). Reactive oxygen species (ROS)‐induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 192: 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou M, Martin C, Ullrich V (1997). Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. Biol Chem 378: 707–713. [DOI] [PubMed] [Google Scholar]
- Zou MH, Ullrich V (1996). Peroxynitrite formed by simultaneous generation of nitric oxide and superoxide selectively inhibits bovine aortic prostacyclin synthase. FEBS Lett 382: 101–104. [DOI] [PubMed] [Google Scholar]
- Zou MH, Cohen R, Ullrich V (2004). Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium 11: 89–97. [DOI] [PubMed] [Google Scholar]