

Abstract
This review describes recent developments in the search for effective therapeutic agents that target redox homeostasis in neurodegenerative disease. The disruption to thiol redox homeostasis in Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis and multiple sclerosis is discussed, together with the experimental strategies that are aimed at preventing, or at least minimizing, oxidative damage in these diseases. Particular attention is given to the potential of increasing antioxidant capacity by targeting the Nrf2 pathway, the development of inhibitors of NADPH oxidases that are likely candidates for clinical use, together with strategies to reduce nitrosative stress and mitochondrial dysfunction. We describe the shortcomings of compounds that hinder their progression to the clinic and evaluate likely avenues for future research.
Linked Articles
This article is part of a themed section on Redox Biology and Oxidative Stress in Health and Disease. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.12/issuetoc
Abbreviations
- 6(OH)DA
6‐hydroxydopamine
- AD
Alzheimer's disease
- ADME(T)
absorption, distribution, metabolism and excretion/toxicity
- ALS
amyotrophic lateral sclerosis
- APP
amyloid precursor protein
- ARE
antioxidant response element
- ASSNAC
S‐allylmercapto‐N‐acetyl cysteine
- Aβ
amyloid‐β
- CA
carnosic acid
- DMF
dimethylfumarate
- EAE
experimental autoimmune encephalomyelitis
- EpRE
electrophile response element
- GCL
glutamate cysteine ligase
- GR
glutathione reductase
- Grx
glutaredoxin
- GSK‐3β
glycogen synthase kinase‐3β
- Keap1
Kelch‐like ECH‐associated protein‐1
- MMF
monomethylfumarate
- MPTP
1‐methyl, 4‐phenyl‐1,2,3,6‐tetrahydropyridine
- MS
multiple sclerosis
- MPO
myeloperoxidase
- NAC
N‐acetylcysteine
- NACA
N‐acetylcysteine amide
- NOX
NADPH oxidase
- NQO1
NAD(P)H quinone oxidoreductase‐1
- Nrf2
nuclear factor (erythroid‐derived 2)‐like 2
- PD
Parkinson's disease
- PS1
pre‐senelin‐1
- RNS
reactive nitrogen species
- SN
substantia nigra
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- xCT
functional subunit of the xc − exchanger
Tables of Links
| TARGETS | |
|---|---|
| Enzymes a | Other proteins b |
| Akt | Keap1, Kelch‐like ECH‐associated protein‐1 |
| GSK‐3β, glycogen synthase kinase‐3β | Transporters c |
| HO‐1, haemoxygenase‐1 | xCT‐ cystine‐glutamate exchanger, SLC7A11 |
| Myeloperoxidase | Ligand‐gated ion channels d |
| NO synthases | P2X7 purinergic receptor |
| P13K |
| LIGANDS | |
|---|---|
| Bardoxolone | Levetiracetam |
| Chlorpromazine | Resveratrol |
| Curcumin | Sulfasalazine |
| DMF, dimethyl fumarate | Sulforaphane |
| DPI, diphenyleneiodonium chloride | Zonisamide |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,dAlexander et al., 2015a, 2015b, 2015c, 2015d).
Disruption of cerebral redox homeostasis is a common occurrence in a range of human neurodegenerative disorders, and although much is understood of mechanistic dysfunction, the gap between knowledge and the availability of effective therapies remains wide. In this review, we focus on the potential for developing therapeutic agents that promote the availability of thiol redox antioxidants or boost the antioxidant capacity of cells via stimulation of the transcription factor, nuclear factor (erythroid‐derived 2)‐like 2 (Nrf2). In addition, we discuss options for reducing ROS production by inhibition of NADPH oxidases (NOXs), limiting nitrative stress or targeting mitochondrial dysfunction. Other potential strategies for limiting oxidative stress in neurodegenerative disease include nutrient and vitamin supplementation and metal chelators, which are excluded from the discussion. These approaches are the subject of several recent reviews, to which the reader is referred for more information (Bigford and Del Rossi, 2014; Casamenti et al., 2015; Di Domenico et al., 2015; Fernando et al., 2015; Gess et al., 2015; Harrison et al., 2014; La Fata et al., 2014).
We describe the current understanding of the basis for thiol redox imbalance in neurodegenerative disease and discuss the potential of thiol redox‐based therapies for the treatment of disorders that include Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS). In many instances, while the results from in vitro experiments are encouraging, the absence of good animal models that fully replicate the human condition hampers a thorough evaluation of antioxidant‐based therapies. In other cases, the absence of early disease biomarkers adds to the difficulty in establishing successful therapeutic strategies. Moreover, many antioxidants showed no efficacy in clinical trials for reasons that include absence of specific targets, difficulty in gaining access to the brain or an inappropriate time‐course of action that may not map to a late‐onset and slowly progressing disease. Nonetheless, the search for potential therapies to combat disruption of thiol‐redox homeostasis is ongoing, and we report on the most promising developments in this field.
The strategy of targeting Nrf2 in neurodegenerative disorders has much to offer. It is anticipated that modulation of Nrf2 activity may provide two advantages over direct antioxidants. First, the induction of NADPH, glutathione (GSH) and thioredoxin (Trx) metabolism is a natural system which may boost antioxidant activity in places where needed, whilst leaving physiological ROS signalling intact. Second, because proteins have a longer half‐life than low MW activators, the effect on the antioxidant defence may be more prolonged. Many Nrf2 inducers are electrophilic compounds that react with the cysteine residues in Kelch‐like ECH‐associated protein‐1 (Keap1). Nevertheless, it has been recently shown that several molecules are capable of inhibiting the protein–protein interaction between Keap1 and Nrf2.
The NOXs, as a key source of reactive oxygen species (ROS) in neurodegenerative diseases, are increasingly recognized as potential therapeutic targets. Recent developments in the design and synthesis of NOX subtype‐specific ligands bring the likelihood of therapeutic application closer to reality.
Activation of glial cells (microglia and astrocytes) is a common characteristic of neurodegenerative diseases and is accompanied by an increased production of NO through upregulation of inducible nitric oxide synthase (iNOS). It is well established that NO release from activated glial cells has the capacity to cause extensive neurodegeneration, for example, via inhibition of mitochondrial cytochrome oxidase (Brown and Cooper, 1994) or release of glutamate causing hyperactivation of NMDA receptors (Bal‐Price and Brown, 2001). The mechanisms of microglial cell activation are the subject of recent reviews and will not be discussed here (Rojo et al., 2014; Vilhardt et al., 2017). However, the oxidative effects of increased NO production are pertinent to this discussion and are viewed as a potential target for redox‐based therapeutics.
There is increasing recognition that mitochondrial oxidative stress is a significant component in neurodegenerative disease (Ruszkiewicz and Albrecht, 2015). For example, one of the correlates of increased oxidant production in PD is mitochondrial dysfunction in dopaminergic neurons, leading to neurodegeneration (Dranka et al., 2012). Moreover, mitochondrial neuronal nitric oxide synthase (mtNOS) generates NO in immediate proximity to high concentrations of superoxide, thus favouring formation of peroxynitrite, leading to nitrative stress. With the emergence of molecules with improved membrane permeability, the possibility of selectively targeting mitochondrial respiration is closer to reality.
Thiol‐based redox therapies in neurodegenerative disease
GSH is the principal thiol‐based antioxidant in the brain and is present in millimolar concentrations in both neurons and glial cells. GSH readily oxidizes to GSSG, but is maintained in the reduced form due to transfer of electrons from NADPH in a reaction catalysed by glutathione reductase (GR). Astrocytes and microglial cells are responsible for de novo synthesis of GSH, whereas neurons rely on precursors that are supplied by astrocytes (Dringen et al., 1999). Cysteine and glutamate are co‐substrates of the rate‐limiting step of GSH synthesis that is catalysed by γ‐glutamate cysteine ligase (GCL). In astrocytes, transulphuration of methionine to cysteine makes a minor contribution to GSH synthesis (Vitvitsky et al., 2006; Kandil et al., 2010; McBean, 2012), but in these cells and microglia, the majority of cysteine comes from uptake of its oxidized form, cystine, from the extracellular space. The inward transport of cystine is mediated by the xc − cystine‐glutamate exchanger located in the plasma membrane that was first characterized in the 1980s (Bannai, 1984). The exchanger is comprised of two subunits, 4F2hc, which stabilizes the protein to the membrane and xCT that confers substrate selectivity. The two subunits are joined by a disulphide bridge (Sato et al., 1999). One of the most interesting characteristics of the xc − exchanger is that the export of glutamate provides the driving force for cystine uptake (Bannai, 1984). This occurs as a 1:1 exchange, and it is increasingly recognized that extracellular glutamate derived from the exchanger has both physiological and pathological functions (Lewerenz et al., 2013). Glutamate released from the exchanger is re‐cycled into astrocytes by the high affinity glutamate transporters (Piani and Fontana, 1994; Sato et al., 1999). Changes in expression and function of the xc − exchanger in response to disease have a significant bearing on thiol redox balance and antioxidant status. Moreover, the inextricable association between the need to import cystine for GSH and the concurrent release of glutamate via the exchanger gives rise to a potentially excitotoxic concentration of glutamate (see McBean et al., 2015). Thus, while such a change in extracellular glutamate does not directly involve redox homeostasis, it is part of the bigger picture and cannot be entirely excluded from this discussion.
In PD, a deficiency in complex 1 of the electron transport chain is regarded as one of the contributing factors in the development of the disease (Papa and De Rasmo, 2013). On the one hand, it increases O2 − production, but, on the other, it decreases ATP. In turn, a fall in ATP leads to a decline in the GSH/GSSG redox potential. Brain tissue taken post mortem from PD patients shows a 40% reduction in GSH in the substantia nigra (SN), compared with normal tissue, but no change in the rest of the brain (Perry and Yong, 1986). Additionally, GSH in the SN of other neurodegenerative diseases affecting this region, such as multiple system atrophy and supranuclear palsy, is unchanged. Expression of the functional subunit (xCT) of the xc − exchanger in the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) mouse model of PD is increased in the striatum, but reduced in the SN. However, MPTP‐associated depletion of dopamine in the SN is unaffected by loss of the exchanger (Bentea et al., 2015a).
Neurodegenerative disorders that entail activation of microglial cells commonly display enhanced activity of the xc − exchanger and elevated levels of extracellular glutamate. In AD, senile plaques contain reactive microglial cells that contribute to neurotoxicity by releasing glutamate via the xc − exchanger (Qin et al., 2006). Similarly, injection of amyloid‐β (Aβ) into the hippocampus of adult mice promotes expression of the exchanger subunit xCT in activated microglia (Qin et al., 2006). Increased expression of xCT and elevated extracellular glutamate in the cortex in 18 month old AβPP23 mice has also been identified (Schallier et al., 2011b). In ALS, the xc − exchanger is expressed in spinal cord samples taken post mortem from ALS patients that correlate positively with the extent of inflammation (Mesci et al., 2015). Similarly, in the SOD1‐G93 A mouse model of ALS, there is increased protein expression of the xc − exchanger in spinal cord and isolated microglial cells. In acute spinal cord slices of 70 day old SOD1 G93 A mice, xc −‐mediated cystine uptake was increased, but was unchanged at 50, 100 or 130 days (Albano et al., 2013).
In MS, the elevated ROS originate from activated microglia and invading macrophages, due to increased expression of NOXs, myeloperoxidases (MPOs) and inducible nitric oxide synthase (iNOS) (Carvalho et al., 2014). In addition, thiol redox homeostasis is disrupted: GSH levels, as measured by magnetic resonance imaging, are reduced in the CSF of MS patients, whereas blood GSH levels are increased, possibly due to heightened activity of GR, compared with normal controls (Carvalho et al., 2014). Increased expression of the xc − exchanger has been reported in activated human monocytes that are accompanied by enhanced release of glutamate. Expression of the xc − exchanger is likewise elevated in the experimental autoimmune encephalomyelitis (EAE) mouse model of MS, as well as in CNS and peripheral blood samples from patients with the disease (Pampliega et al., 2011). Accordingly, elevated concentrations of glutamate have been recorded in CSF and plasma of MS patients.
GSH precursors, analogues and conjugates
Several attempts have been made to potentiate the antioxidant capacity of brain tissue in animal models of neurodegenerative disease. For example, strategies to reduce neurodegeneration in PD include both direct and indirect approaches to restore GSH levels, on the premise that such action would boost the antioxidant capacity and therefore, reduce the rate of neurodegeneration (Johnson et al., 2012; Smeyne and Smeyne, 2013; Johnson et al., 2015a). Efforts to supply GSH directly have failed, due to limits of solubility, absorption, stability and the fact that GSH has a short half‐life in human plasma. For example, Zeevalk and colleagues observed a dose‐dependent increase in GSH using a monoethylester of GSH (GEE) in rat mesencephalic cultures (Zeevalk et al., 2007). However, peripheral administration of GEE to rats (0.1–50 mg·kg−1·day−1 for 28 days) failed to produce a significant elevation in brain GSH, whereas direct intracerebral delivery was effective.
The recent emergence of GSH derivatives may provide an opportunity for development of more effective therapeutic agents. Other approaches to assess delivery of GSH as a therapeutic option in PD include liposomes or nanoparticles, co‐drugs (GSH: dopamine or GSH:L‐dopamine decarboxylase conjugates) and GSH analogues or other hybrid compounds (Zeevalk et al., 2007; Zeevalk et al., 2010; Cacciatore et al., 2012; Smeyne and Smeyne, 2013). Overall, the results are variable and minimal improvement in intracellular GSH levels has been observed. Ultimately, nanoparticle‐associated GSH delivery may prove to be the best option, as this technique permits longer‐term drug delivery and better brain penetration than comparable approaches.
In a comparative study on the effects of antioxidants on the activity of cerebrocortical Mn superoxide dismutase (MnSOD) in mitochondria isolated from post mortem human AD brain, GSH was ineffective, whereas synthetic GSH analogues (for example, 4‐methoxy‐L‐tyrosinyl‐L‐γ‐glutamyl‐L‐cysteinylglycine) effectively increased MnSOD activity (Kairane et al., 2014). However, further work is required to determine whether off‐target effects of GSH analogues limit their potential as therapeutic agents.
Direct administration of cysteine to increase GSH is not a viable option, because of poor absorption and the toxicity of high doses (Johnson et al., 2012). In experimental systems, N‐acetylcysteine (NAC) can be substituted for cysteine as a GSH precursor and has been used many times to boost GSH levels (Figures 1 and 2). NAC is readily taken up in vitro and potentiates intracellular GSH synthesis, promotes GSH‐mediated detoxification mechanisms and scavenges ROS. In vivo, the picture is less clear; NAC is well absorbed by the intestine, and dietary supplementation is effective for increasing GSH (Atkuri et al., 2007). However, NAC is only effective in situations in which GSH is depleted, and it does not work independently of GSH as an antioxidant (Schmitt et al., 2015). Moreover, dietary NAC may be metabolized to hepatic GSH before reaching the brain (Schmitt et al., 2015). N‐acetylcysteine amide (NACA) has greater lipophilicity, membrane permeability and antioxidant capacity, compared with NAC. The blood brain barrier (BBB) permeability of NACA has been demonstrated using cell lines and rodent models of PD and AD (Sunitha et al., 2013). NACA has recently been shown to prevent neuronal degeneration following focal penetrating traumatic brain injury in rats (Günther et al., 2015) and may ultimately prove to be a more effective therapy than NAC for neurodegenerative disorders.
Figure 1.
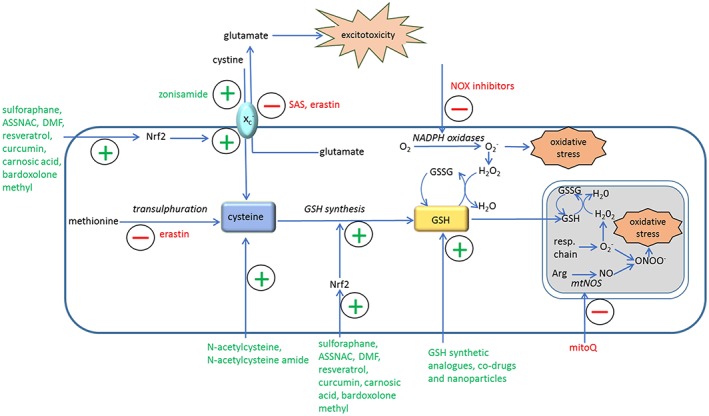
Schematic diagram of redox‐based therapeutic strategies in neurodegenerative disease models. Arg, L‐arginine; ASSNAC, S‐allyl‐mercapto‐N‐acetyl cysteine; GSSG, oxidised glutathione; DMF, dimethylfumarate; mtNOS, mitochondrial nitric oxide synthase; resp.chain, mitochondrial respiratory chain; SAS, sulfasalazine; xc −, cystine‐glutamate exchanger.
Figure 2.
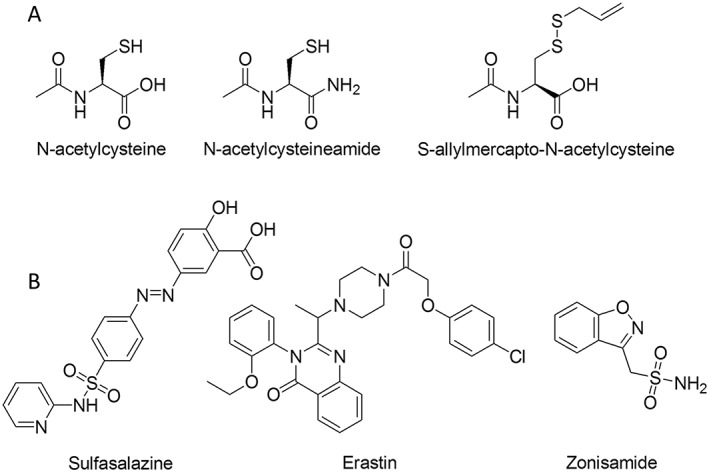
A, chemical structures of low MW thiols that increase cysteine or GSH and B, compounds that target the xc − cystine glutamate exchanger.
Another option is S‐allylmercapto‐N‐acetylcysteine (ASSNAC) (Savion et al., 2014). In the model of MS, mice with experimental autoimmune encephalomyelitis (EAE), spinal cord and brain GSH increased by 54 and 47%, respectively, following administration of ASSNAC (200 mg·kg−1·day−1)and the clinical symptoms showed greater improvement than with NAC. ASSNAC activates Nrf2 and increases the expression of phase II detoxifying enzymes that include the xc − exchanger and GCL (Savion et al., 2014).
In summary, the effectiveness of compounds that increase GSH production through provision of cysteine in the brain may be limited by hepatic metabolism, and/or the fact that they can only restore depletion of GSH to normal levels. It is possible that, in the future, a stimulation of GSH beyond normal levels may be achieved by coupling GSH precursors with Nrf2 activators to upregulate enzymes that control GSH synthesis.
The xc −cystine‐glutamate exchanger
The xc − exchanger is viewed by many as a potential therapeutic target, particularly in instances wherein limitation of cysteine availability and GSH synthesis is deemed advantageous for patient survival, such as in the treatment of brain tumours (McBean, 2012). Considerable effort has been devoted to the search for selective inhibitors of the xc − exchanger (De Groot and Sontheimer, 2011; Bridges et al., 2012; Sontheimer and Bridges, 2012). Sulfasalazine (2‐hydroxy‐5‐[(E)‐2‐{4‐[(pyridin‐2‐yl)sulfamoyl]phenyl}diazen‐1‐yl]benzoic acid) is one of the most promising, but in vivo use (tested for treatment of glioma patients) has been of limited value. This is due to lack of selectivity and a high rate of metabolism by intestinal bacteria (Gout et al., 2001). More recently, erastin (2‐[1‐[4‐[2‐(4‐chlorophenoxy)acetyl]‐1‐piperazinyl]ethyl]‐3‐(2‐ethoxyphenyl)‐4(3H)‐quinazolinone) has been shown to inhibit both the xc − exchanger and cystathionine‐γ‐lyase, regulatory enzyme of the transsulphuration pathway, in glioma cells (Chen et al., 2015) (Figure 2). Its effectiveness in models of neurodegenerative disease remains to be determined.
Experiments on manipulation of the xc − exchanger in PD models have revealed conflicting results. Repeated injections of the anti‐epileptic agent, zonisamide (benzo[d]isoxazol‐3‐ylmethanesulfonamide), into 6‐hydroxydopamine (6(OH)DA)‐treated mice improve the cardinal symptoms of PD (Asanuma et al., 2010) by increasing GSH through enhanced activity of the xc − exchanger. Similar results using levetiracetam have recently been reported (Miyazaki et al., 2016). On the other hand, Massie and colleagues observed that mice lacking the xCT subunit of the xc − exchanger were less susceptible to toxicity by 6(OH)DA, possibly because of reduced glutamate release (Massie et al., 2011). Contrary to expectation, GSH levels in the SN were not reduced in xCT ‐/‐ mice (Gout et al., 2001; De Groot and Sontheimer, 2011; Bridges et al., 2012; Sontheimer and Bridges, 2012; Sunitha et al., 2013; Savion et al., 2014; Chen et al., 2015).
New information on the potential of blockade of the xc − exchanger for ALS therapy has recently been published (Mesci et al., 2015). Deletion of the xCT subunit in mice confirms that the source of glutamate release from activated microglia is via the xc − exchanger. Furthermore, xCT deletion reduces production of microglial pro‐inflammatory and neurotoxic factors, including NO, TNFα, IL6, whereas production of anti‐inflammatory markers was increased (Mesci et al., 2015). In ALS mice, xCT deletion unexpectedly led to earlier symptom onset, but, ultimately, a significantly slowed disease phase and more surviving motor neurons were observed. It is proposed that this could be used as a clinical approach to slow disease progression after symptoms have appeared.
In summary, the xc − exchanger remains an attractive target in thiol redox‐based therapy, but much remains to be understood regarding cysteine metabolism in the brain before the full implications of intervention in cysteine homeostasis can be understood. For example, the fact that the transsulfuration pathway is upregulated in response to GSH depletion (Kandil et al., 2010) implies that provision of cysteine from methionine may compensate for a reduction in cystine import via the xc − exchanger. A multitargeted approach, such as that used by Chen et al. (2015) may ultimately prove more effective. The problem also remains of how to selectively target affected cells whilst avoiding damage to normal sulphur metabolism.
Protein thiol redox state as a potential target in neurodegenerative disease
Alterations in the oxidation state of redox‐sensitive proteins are frequently associated with disease‐related neurodegeneration. Free thiol groups on proteins are either protected from oxidation by S‐glutathionylation or, if oxidized to sulphenic acid (Pr‐SOH), are converted back to the reduced state (Pr‐SH) by protein thiol reductases. Exposure of proteins to high levels of ROS, as may occur in neurodegenerative disease, will generate irreversibly‐oxidized sulphinic (Pr‐SO2H) or sulphonic (Pr‐SO3H) acids that cannot be reduced (see Johnson et al., 2015a; McBean et al., 2015; Rojo et al., 2014).
The GSH/GSSG redox couple operates in concert with glutathione peroxidase, glutathione reductase, the thioredoxin system, glutaredoxin and peroxiredoxin to maintain protein thiol redox homeostasis (McBean et al., 2015; Rojo et al., 2014). The thioredoxin system incorporates Trx and its reducing enzyme, thioredoxin reductase (TrxR). Trx has two principal isoforms, Trx1 (cytosolic) and Trx2 (mitochondrial) that associate with their respective reductases, TrxR1 and TrxR2. The Trx system is expressed throughout the brain, particularly in regions of high energy demand and a high rate of production of oxidized metabolites, such as in the SN and subthalamic nucleus. Post mortem tissue from AD patients displays a reduction in Trx, but upregulation of TrxR, most likely in compensation for increased ROS production (Akterin et al., 2006). In vitro, overexpression of Trx1 protects a neuronal cell line and primary hippocampal neurons from Aβ‐induced toxicity. In the MPP+ model of PD, the expression of Trx1 and 2 is decreased (Silva‐Adaya et al., 2014). The content of Grx1 is similarly decreased in post mortem PD brain and, in the Caenorhabditis elegans model of PD, loss of Grx1 enhances the symptoms of PD (Johnson et al., 2015b). Contrastingly, glutathione peroxidase 3 and glutathione peroxidase 4 levels from PD patients are elevated compared with control subjects and cortical samples from PD patients. To date, notwithstanding the increasing body of work on the antioxidant or neuroprotective effects of the Trx and Grx systems, no therapeutic agents that have the potential to arrest protein oxidation in AD or PD have been developed.
Nrf2‐mediated antioxidant action as a therapeutic target in neurodegenerative disease
Nrf2 is considered the master regulator of cell homeostasis that regulates the expression of antioxidant and cytoprotective genes that contain a specific enhancer sequence in their regulatory regions known as antioxidant response elements (ARE) or the electrophile responsive element (EpRE). Nrf2 controls both the basal expression of genes under non‐stressed homeostatic conditions and the inducible expression of genes upon redox perturbation. The number of genes regulated by Nrf2 account for more than 1% of the human genome, of which the best known are those related to GSH synthesis, redox regulation and drug metabolism (Ishii et al., 2000; Thimmulappa et al., 2002). However, it is now recognized that Nrf2 may play a role in regulation of genes that contribute to NADPH generation, lipid metabolism, glucose/glycogen metabolism and hydrogen sulphide (H2S) production (Yates et al., 2009; Wu et al., 2011; Hourihan et al., 2013; Uruno et al., 2013; Hayes and Dinkova‐Kostova, 2014). In addition to its capacity to afford cytoprotection against endogenous and environmental stressors, Nrf2 seems to control metabolic reprogramming during stress, thereby contributing to adaptation by upregulating the repair and degradation of macromolecules. Furthermore, Nrf2 modulates intermediary metabolism by virtue of the fact that its targets include other transcription factors (Hayes and Dinkova‐Kostova, 2014).
Under basal normal conditions, the level of Nrf2 in the cytosol is low, due to constitutive synthesis and degradation of the protein. There are essentially three regulatory mechanisms that control Nrf2: (a) Keap1, (b) regulation by kinases and (c) epigenetic control. The main regulatory mechanism is achieved by the repressor protein, Keap1, which is located mainly in the cytoplasm. Under normal conditions, Keap1 binds to an E3 ubiquitin ligase complex (Rbx‐1) via cullin‐3, a protein adaptor (Kobayashi et al., 2004). This complex promotes Nrf2 ubiquitination and proteasome degradation (Rushmore et al., 1990; Dhakshinamoorthy and Jaiswal, 2001). However, under cellular stress conditions, oxidative/electrophilic molecules modify the cysteine residues of Keap1 (at least, Cys151, Cys273 and Cys288), and the cullin‐3/Rbx‐1‐dependent poly‐ubiquitination of Nrf2 assisted by Keap1 is blocked, leading to release of Nrf2 and translocation to the nucleus, where it heterodimerizes with small MAF or JUN proteins. This complex then binds to the EpRE of target genes (Rushmore et al., 1990). The second mechanism of Nrf2 regulation is based on signalling cascades that include various kinases influenced by oxidative stress molecules. For instance, protein kinase RNA – like endoplasmic reticulum kinase and PKA, activated by misfolded proteins or phorbol esters, respectively, as well as PKC, phosphorylate Nrf2, thereby disrupting its association with Keap1 (Huang et al., 2002; Cullinan et al., 2003). Other kinases, such as CK2, Fyn or MAPKs phosphorylate Nrf2 without altering its interaction with Keap1. Interestingly, physiological activation of glycogen synthase kinase‐3β (GSK‐3β) phosphorylates Fyn, which then phosphorylates Nrf2, triggering its nuclear export and proteasomal degradation (Jain and Jaiswal, 2007). Furthermore, GSK‐3β (which is abnormally active in Alzheimer's disease), inhibits Nrf2 by nuclear exclusion. In addition, GSK‐3β phosphorylates Nrf2 and activates an alternative degradation pathway that is dependent on a different E3‐ligase, named β‐TrCP (see Rojo et al., 2014). The physiological relevance of the inhibition of Nrf2 by this kinase is illustrated by two studies in which GSK‐3β decreased Nrf2 activity, thereby sensitising neurons against oxidative damage. Interestingly, stimulation of P13K activates Akt, which in turn inhibits GSK‐3β and represses Nrf2‐driven and ARE‐driven genes (Innamorato et al., 2008; Rojo et al., 2008). Furthermore, the kinase MAPK p38 stabilizes the interaction between Keap1 and Nrf2 and promotes breakdown of Nrf2 (Keum et al., 2006). The third regulatory mechanism for Nrf2 includes epigenetic modifications. For example, binding of Nrf2 to EpRE is increased by direct acetylation of Nrf2 by CBP/p300 (Sun et al., 2009). In contrast, methylation of cytosine‐guanine islands within the Nrf2 promotor suppresses its expression (Su et al., 2013). Lastly, a number of miRNAs, such as miR153, miR27a, miR142‐5p and miR144, have been shown to reduce Nrf2 activation (Narasimhan et al., 2012).
Nrf2 and neurodegenerative disease
The most compelling evidence in favour of a role of Nrf2 in prevention or protection from disease comes from studies with Nrf2‐knockout mice. Loss of Nrf2 signalling increases susceptibility to acute toxicity, inflammation and carcinogenesis, due to the inability to mount adaptive responses. With increasing age, these mice show chronic pathologies related to oxidant and inflammatory stress including cognitive deficits (Jo et al., 2014), depressive disorder (Martin‐de‐Saavedra et al., 2013b) and increased incidence of lupus‐like autoimmune nephritis (Ma et al., 2006). There is evidence in humans where single nucleotide polymorphisms have been identified in the coding and non‐coding regions of the gene encoding Nrf2, NFE2L2, and epidemiological studies have revealed significant associations of NFE2L2 haplotypes with risks in pulmonary, gastrointestinal, autoimmune and neurodegenerative diseases (Cho, 2013).
Regarding AD, reduced NAD(P)H quinone oxidoreducase‐1 (NQO1), glutathione synthetic enzymes and Nrf2 levels in hippocampal neurons were found in the mouse model that expresses mutated human amyloid precursor protein (APP) and pre‐senelin‐1 (PS1) genes (APP/PS1 mice) (Kanninen et al., 2008); furthermore, the Nrf2‐EpRE pathway was attenuated at the time of Aβ deposition. Importantly, in vitro Nrf2 over‐expression protected against Aβ‐mediated neurotoxicity, as shown by increased expression of Nrf2 target genes and the consequent reduction of oxidative stress. Post mortem brains from AD patients show reduced nuclear levels of Nrf2 within hippocampal neurons (Ramsey et al., 2007).
A relationship between Nrf2 and PD has been documented by the following observations: (i) Nrf2 activity decreases with age, and age is one of the main risk factors for PD, (ii) in post mortem brains from PD patients, Nrf2 has been localized in nuclei although the induction of phase II enzymes is insufficient to protect neurons, indicating a dysregulation of this pathway (Ramsey et al., 2007), (iii) there is a protective Nrf2 haplotype which delays PD onset, (iv) Nrf2 KO mice are more susceptible to 6(OH)DA and MPTP PD models, and (v) Nrf2 activation protects against different PD models (Lou et al., 2014).
In ALS, studies with post mortem tissues have demonstrated that the expression of Nrf2 related genes was diminished in motor neurons of mice expressing mutant SOD1 (Pehar et al., 2005) and in spinal cord of ALS patients (Sarlette et al., 2008). Also, Keap1 was co‐localized with intracellular inclusions in motor neurons in the spinal cord of ALS patients (Tanji et al., 2013). In samples of motor cortex of ALS patients, Nrf2 mRNA and protein levels were reduced, whereas Keap1 mRNA expression was increased (Sarlette et al., 2008), suggesting a dysfunction of the Nrf2‐EpRE pathway in this disease.
In MS, the expression of DJ‐1 (an important protein for Nrf2 stabilization) and Nrf2 are increased in cerebral spinal fluid of patients with relapsing – remitting MS (Hirotani et al., 2008; van Horssen et al., 2010). In fact, increased levels of Nrf2‐regulated antioxidant proteins, such as NAD(P)H dehydrogenase (quinone) 1 (NQO1) and haemoxygenase‐1 (HO‐1) are present in active demyelinating MS lesions, mostly associated with infiltrating macrophages and reactive astrocytes (van Horssen et al., 2010; van Horssen et al., 2008). Furthermore, induction of EAE in Nrf2‐knockout mice showed increased severity of the disease (Johnson et al., 2010). It is believed that low levels of Nrf2 or impaired Nrf2 activation in oligodendrocytes may be a cause of selective susceptibility under neuroinflammatory conditions, due to their high degree of vulnerability to oxidative stress (Juurlink et al., 1998).
Overall, Nrf2 inducers are emerging as a valuable tool for treatment of different neurodegenerative conditions where oxidative stress plays a major role. Details of a selection of drugs, including curcumin, carnosic acid, resveratrol, bardoxolone methyl and dimethylfumarate (Figure 3) are discussed in the following text in relation to different neurodegenerative conditions. Most information to date comes from pre‐clinical data. The exception is dimethylfumarate (DMF), which has been approved for clinical use in MS patients.
Figure 3.
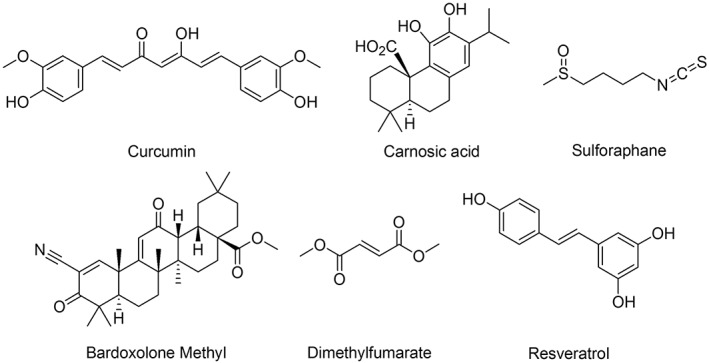
Chemical structures of various Nrf2 inducers.
Nrf2 inducers for the treatment of neurodegenerative disease
Curcumin
Curcumin is the main curcuminoid present in the rhizomes of Curcuma longa and is the active ingredient of herbs and spices used in traditional Asian cuisine. Curcumin contains electrophilic α,β unsaturated carbonyl groups that selectively react with nucleophiles, such as cysteine‐thiols present at Keap1, thus releasing Nrf2 (Balogun et al., 2003). Curcumin is a highly lipophilic natural compound that crosses the blood–brain barrier and concentrates mainly in the hippocampus (Tsai et al., 2011). In AD related models, curcumin inhibits Aβ oligomers and restored neurite damage (Yang et al., 2005; Garcia‐Alloza et al., 2007). Most interesting is that epidemiological studies have demonstrated that populations with a high level of consumption of curcumin show lower incidence of AD and better performance in Mini‐Mental State Examination (Chandra et al., 2001; Ng et al., 2006). Based on these promising findings, curcumin has been tested in AD patients (Ono et al., 2004). Several clinical trials have been performed, or are currently recruiting patients. The results have been limited, most probably because of its poor bioavailability (Anand et al., 2007). Therefore, new curcumin derivatives are being synthesized in order to improve its pharmacological properties (Hu et al., 2011; Orlando et al., 2012; Pulido‐Moran et al., 2016).
In regard to ALS, preclinical data indicate that curcumin increases Nrf2 nuclear translocation, thereby inducing the expression of its target genes HO‐1, NQO1 and GCLc in primary spinal cord astrocytes (Jiang et al., 2011). Also, curcumin has been shown to reduce oxidative stress and mitochondrial dysfunction in motoneurons expressing mutant TDP‐43 (Dong et al., 2014).
Curcumin has also been tested as a potential treatment for MS. Its immunomodulatory and anti‐inflammatory properties have been assessed in several models and are mostly mediated by its ability to inhibit COX‐2, iNOS and the NF‐κB transcription pathway (Menon and Sudheer, 2007). In the EAE model, curcumin showed behavioural improvement and reduction in the number of inflammatory cells infiltrating the spinal cord (Xie et al., 2009). Furthermore, curcumin reduced IL‐17 and TNFα production and up‐regulated IL‐4, IL‐10 and CD4 + CD25 + −Foxp3+ Treg cells in the EAE model. These results suggest that curcumin augments Th2/Treg responses in EAE in the CNS (Kanakasabai et al., 2012). In order to improve curcumin's bioavailability, a new polymerized form of nano‐curcumin was evaluated in the EAE model; the results showed corrected balance of pro‐inflammatory and anti‐inflammatory gene expression, decreased oxidative stress markers, improved myelination and increased progenitor cell markers (Mohajeri et al., 2015).
Carnosic acid
Carnosic acid (CA) is an active ingredient in the extract of Rosmarinus officinalis that improves long‐term memory performance in rats (Ozarowski et al., 2013). CA crosses the blood–brain barrier to up‐regulate endogenous antioxidant enzymes via Nrf2 activation and promotes over‐expression of phase II enzymes, GCL and HO‐1 (Satoh et al., 2008). In relation to AD, CA reduced Aβ1–42 secretion by 71% in human U373MG astrocytoma cells and by 61% in SH‐SY5Y neuroblastoma cells (Meng et al., 2013; Yoshida et al., 2014). Application of CA (10 mg·kg−1) to an AD animal model in vivo reduced cell death in the CA1 region of the hippocampus. With regard to PD, CA provided neuroprotection of human neuroblastoma SH‐SY5Y cells against 6(OH)DA‐mediated toxicity (Wu et al., 2015) and against dieldrin‐induced toxicity in the neuronal dopaminergic SN4741 cell line (Park et al., 2008b). This activity was further confirmed in vivo by the improvement of behavioural performance in rats intrastriatally injected with 6‐OHDA (Wu et al., 2015). In the ALS transgenic mice model G93 A‐hSOD1, administration of rosemary extract, in which rosmarinic and carnosic acid are present, led to improved motor performance and prolonged mean survival, compared with control conditions (Shimojo et al., 2010).
Resveratrol
Resveratrol is a natural polyphenolic compound especially abundant in the skin of red grapes, which activates the Nrf2‐EpRE pathway (Hsieh et al., 2006). Resveratrol has shown beneficial effects in in vitro and in vivo models related to AD (Ma et al., 2014; Savaskan et al., 2003). In humans, a phase III Clinical Trial of resveratrol has been performed to study its effect in mild–moderate AD. Additional clinical trials are presently recruiting participants. Although resveratrol has the ability to cross the blood brain barrier to reach the brain, it metabolizes very rapidly, which reduces its half‐life and bioavailability (Davinelli et al., 2012). A variety of approaches have been taken to bypass this problem: one is synthesis of new resveratrol derivatives with improved pharmacokinetics, such as pterostilbene, which shows a greater efficiency in modulating cellular stress and cognition than its parent compound (Joseph et al., 2008; Chang et al., 2012). Another is use of resveratrol‐loaded polysorbate 80‐coated poly(lactide) nanoparticles. The latter was evaluated in the MPTP in vivo model of PD, where it showed significant neuroprotection and improved behavioural and neurochemical indices (da Rocha Lindner et al., 2015b).
Bardoxolone methyl
Bardoxolone methyl is a triterpenoid derivative, 2 cyano‐3,12‐dioxooleana‐1,9‐dien‐28‐oic acid methyl ester (CDDO‐Me), with potent Nrf2‐inducing activity. It was first studied as a treatment for diabetic nephropathy (Wang et al., 2014). The initial excitement about this compound was hampered by the observation that in the phase III clinical trial, BEACON, there was a small yet significant increase in heart failure in the treated group compared to the control group. There is no clear explanation for these adverse events; it was speculated that fluid retention, increased afterload and higher heart rate could have contributed to the heart failure in the bardoxolone‐treated group, although direct toxic effects cannot be discounted (Chin et al., 2014). Despite these adverse effects, bardoxolone methyl is now being tested in pulmonary arterial hypertension, melanoma and Friedreich's ataxia.
In the context of AD, bardoxolone methyl showed improved spatial memory retention and decreased microgliosis, oxidative stress and Aβ burden in the TG19959 AD transgenic mouse, signifying its potential as a therapeutic agent in this disease (Dumont et al., 2009).
The mechanism of action of bardoxolone consists of causing a reversible Michael addition of SH groups in Keap‐1 to attenuate Nrf2 degradation; as mentioned earlier, this is one of the main defence systems against redox stress. Some compounds, such as RTA 408, a novel synthetic triterpenoid related to bardoxolone, are cytotoxic in cancer cells by this mechanism (Probst et al., 2015). It is therefore postulated that some of these compounds could act as ‘chemical irritants’ to activate body defences.
Sulforaphane
Another interesting compound is sulforaphane, which is found in cruciferous vegetables, such as broccoli. Due to its electrophilic structure, sulforaphane interacts with specific redox sensitive cysteine residues in Keap1, which disrupts the Keap1/Nrf2 complex and leads to the increase in Nrf2 protein levels and transcriptional activity (Dinkova‐Kostova and Talalay, 2008). Sulforaphane (5 μM) exerts protection against Aβ‐toxicity via activation of the phase II antioxidant response in vitro (Lee et al., 2013) and ameliorates cognitive deficit in AD animal models (Kim et al., 2013; Zhang et al., 2014). In the AD‐model induced by the combination of D‐galactose and aluminium, sulforaphane reduced the number of lesions, decreased the levels of aluminium in the brain and reduced spatial memory deficits and amyloid plaques (Kim et al., 2013). In PD, sulforaphane protected against MPTP‐induced toxicity by increasing Nrf2 protein levels in basal ganglia, upregulating HO‐1 and NQO1 enzymes and decreasing astrogliosis, microgliosis and the release of pro‐inflammatory cytokines (Jazwa et al., 2011). In MS, sulforaphane treatment inhibits progression of disease in EAE mice, again via activation of the Nrf2 pathway and reduction in oxidative stress and inflammation. Furthermore, it reduced demyelination and CNS infiltration and inhibited production of MMP‐9 (metalloproteinase that degrades extracellular matrix proteins), which contributes to BBB preservation (Li et al., 2013).
Most clinical studies performed with sulforaphane have used a highly standardized formulation of broccoli sprout extracts. In contrast to dimethylfumarate (DMF) and bardoxolone methyl, two major drawbacks for sulforaphane for clinical use are the absence of a pure formulation and the lack of commercial value that could attract significant investment by pharmaceutical companies. Nevertheless, several attempts are being made to provide proof of concept that Nrf2 targeting with sulforaphane has therapeutic value. Currently, at least 32 studies are assessing its clinical efficacy in chronic diseases such as cancer, asthma, autism, schizophrenia, chronic kidney disease, type 2 diabetes and cystic fibrosis.
Dimethylfumarate (DMF)
The most successful case reported so far of an indirect antioxidant targeting Nrf2 is the dimethyl ester derivative of fumaric acid, DMF. As early as 1994, topical and oral administration of DMF (commercial name, ‘Fumaderm’) was used to treat psoriasis (Altmeyer et al., 1994). In the EAE model, DMF inhibits progression of MS symptoms, including macrophage inflammation in the spinal cord and decreased expression of the pro‐inflammatory cytokine IL‐1β (Schilling et al., 2006). In Nrf2‐deficient murine motoneuron cultures, the protective effect of DMF is completely abolished (Linker et al., 2011a), supporting the role of Nrf2 in its mechanism of action. DMF crosses the gastrointestinal barrier, after which it is converted into its active metabolite, monomethyl fumarate (MMF), which binds to Keap1. This binding disrupts the Keap1/Nrf2 interaction and leads to up‐regulation of the transcriptional Nrf2 signature of antioxidant genes. MMF is more potent than DMF in activating Nrf2, but is probably metabolized more rapidly. In an attempt to allow DMF to bypass gastric metabolism, it has been packaged in an oral delayed release formulation (BG‐12, known commercially as Tecfidera ®) (Bomprezzi, 2015). It has shown positive results in two clinical trials and has been recently approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) as first‐line therapy for relapsing–remitting MS (Xu et al., 2015). A new formulation of MMF (‘ALKS 8700’) has completed a phase 1 trial as a pro‐drug for MS (NIH Trial number NCT02201849), but results have not been posted. The immune modulatory effect of DMF has been exploited more recently for other autoimmune diseases such as lupus erythematosus (Tsianakas et al., 2014) and asthma (Seidel and Roth, 2013).
Despite these promising results, its use seems to be limited by significant adverse effects and regular monitoring requirement (Dubey et al., 2016) due to the fact that some patients with MS, chronically treated with DMF, have shown reduction of total leukocyte and lymphocyte counts (Spencer et al., 2015), and progressive multifocal leukoencephalopathy (Faulkner, 2015).
In conclusion, Nrf2 inducers can be of potential interest for neurodegenerative diseases because of their antioxidant and antinflammatory effects. However, their use in clinic has been hampered by different circumstances. For example, curcumin has poor bioavailability and bardoxolone has major side effects. Until today, the only drug with this mechanism of action used clinically is DMF. However, long term treatment with this drug exhibits, in a subset of patients, severe side effects such as lymphopenia and progressive multifocal leukoencephalopathy. Therefore new derivatives of these compounds are being synthesized and developed with the hope of improving their pharmacokinetic and safety profile.
NOX‐based therapies in neurodegenerative disease
The NOX family of enzymes are a major source of ROS in the brain, and there is now little doubt of their association with pathological ROS production in a range of neurodegenerative disorders. The NOX enzymes use NADPH as electron donor in the reduction of molecular oxygen to the superoxide anion, O2 −. Of the seven mammalian subtypes of the enzyme, four isoforms (NOX1 through NOX4) have received greatest attention in the context of physiological and pathological function of the CNS. All four isoforms are expressed in neurons, mostly as inducible enzymes associated with disease states. NOX4, and to a lesser extent NOX1 and NOX2, is expressed in astrocytes and is particularly evident in glioma cells. The expression of NOX2 is high in activated microglial cells, but low in the inactivated state. NOX1 and NOX4 are also expressed in microglia. However, full evaluation of the cellular, regional and inter‐species distribution of NOX's is hampered by the absence of reliable antibodies (Nayernia et al., 2014). Full details of the subunit structure, subtypes and cellular localisation of the enzymes can be found in a number of excellent and informative review articles (Infanger et al., 2006; Sorce and Krause, 2009; Sorce et al., 2012; Nayernia et al., 2014).
The association of NOX with a wide spectrum of neurodegenerative disorders constitutes a compelling case for development of therapeutic strategies that modulate their activity. The therapeutic opportunities presented by targeting NOX in diseases of the CNS have been discussed comprehensively elsewhere (Sorce et al., 2012; Nayernia et al., 2014). Here, we provide an update on recent research and the development of NOX‐based therapeutic agents. There is a growing body of evidence illustrating the beneficial effects of targeting NOX in several animal models of neurodegenerative disorders. However, advances into the arena of clinical intervention are still at an early stage.
In PD, loss of dopamine in the SN correlates with an increase in expression of NOX in SN neurons and microglial cells. The rat dopaminergic N27 cell line displays constitutive expression of most NOX isoforms, but a selective increase in NOX1 is observed following treatment of cells with 6(OH)DA (Choi et al., 2012). NOX1 expression was also identified in the nucleus of dopaminergic SN neurons of PD patients, post mortem. Hernandes et al. assessed the association between NOX activation with PD progression using the 6(OH)DA mouse model of the disease (Hernandes et al., 2013). Mice lacking the catalytic subunit of NOX2 (gp91(phox −/−)) were protected from dopaminergic cell loss, and microglial cell activation was abolished. The results support a role for NOX2 in the 6(OH)DA‐induced degeneration of DA neurons. Taken together, these studies suggest that inhibition of NOX1 and/or NOX2 could be viable targets for arresting ROS production in PD.
Other recent developments in the PD/NOX field come from exploring the influence of angiotensin receptor activation on NOX‐derived ROS production. Following from rodent studies, in which dopamine loss and changes in NOX activity in the SN correlate with expression of the angiotensin AT1 receptor, Zawada and colleagues measured the distribution of AT1 receptors and NOX4 in different nigral compartments in post mortem samples from PD patients (Zawada et al., 2015). The ratio of nuclear/total neuron AT1 expression increased according to disease progression and was associated with increased levels of NOX4. It is proposed that AT1 receptor antagonists offer a viable approach to modify NOX‐mediated ROS production and slow the progression of the disease. On the other hand, and in line with the opposing effects of AT2 receptors, the AT2 agonist, CGP42112, inhibits NOX activation and protects against oxidative stress in the rotenone model of PD in vitro (Lu et al., 2015). These results are promising but further work is required to clarify the therapeutic potential of targeting the renin‐angiotensin system in PD.
A new focus in the search for antioxidant approaches for treatment of PD is the enzyme MPO. This is a ROS‐generating enzyme that is expressed in microglia. In recent work, Jucaite and co‐workers used a selective and irreversible MPO inhibitor, AZD3241, to assess the effect of MPO blockade on microglial activity in PD. Patients received 600 mg orally twice daily for 8 weeks, during which time microglial activity was determined using positron emission tomography to map binding of a probe to a microglial marker translocator protein. The drug was well tolerated and, by the end of the study, a reduction in the binding of the probe to the translocator protein was recorded. Following this confirmation that MPO blockade affects microglial activity, further studies on the efficacy of this treatment in neurodegenerative disorders are warranted (Jucaite et al., 2015).
In regard to ALS, the focus is more clearly on the therapeutic benefits of inactivation of NOX. For example, NOX2 activity in 83 ALS patients was assessed by measuring neutrophil oxidative burst in fresh blood samples by flow cytometry (Marrali et al., 2014). Although the activity of the enzyme was independent of gender and disease duration, a lower NOX2 level correlated with a longer survival time from disease onset. It was concluded that modulation of NOX2 may have value in arresting the rate of progression of the disease. Similar conclusions have been drawn from work on the SOD1 G93 A mouse model of ALS. Mutant SOD1 protein directly activates NOX2 in microglia, due to protein misfolding (Apolloni et al., 2013). Furthermore, stimulation of the P2X7 purinergic receptor with 2′3′‐O‐(benzoyl‐benzoyl) ATP enhanced NOX2 activity by stimulating the translocation of the p67phox subunit to the membrane, with a consequent increase in ROS production. This process was inhibited following P2X7 receptor knock‐out, or by using pharmacological antagonists of the receptor, signifying that P2X7 receptors may be a promising target to limit oxidative stress in the disease (Apolloni et al., 2013).
It should be noted that NOX derived ROS production is a very conserved response mechanism towards microbial infections and other stress signals (Aguirre and Lambeth, 2010). It is therefore important that possible negative effects on resistance to infection are taken into account when using NOX inhibitors, especially so in AD, where there is a connection between infections and cognition (Honjo et al., 2013), although this connection may very well be ROS‐mediated (Trumbull et al., 2012).
NOX inhibitors
In the past decade, NOX inhibitors have attracted increasing interest and researchers are reaching a point where they may be on the verge of clinical applications (Altenhöfer et al., 2014b). The range of NOX inhibitors has been extensively reviewed previously (Kim et al., 2014b, 2012, 2011). Peptide‐based inhibitors such as gp91ds‐tat (Rey et al., 2001) have been much used in experimental studies. However, due to the non‐optimal absorption, distribution, metabolism and excretion/toxicity (ADME(T)) properties of peptides, they are less attractive as treatment options. However, continuous progress in novel methods for peptide delivery and targeting may increase their viability in future but, for now, low MW NOX inhibitors are closer to market. A selection of inhibitors is described in Figure 4 and Table 1.
Figure 4.
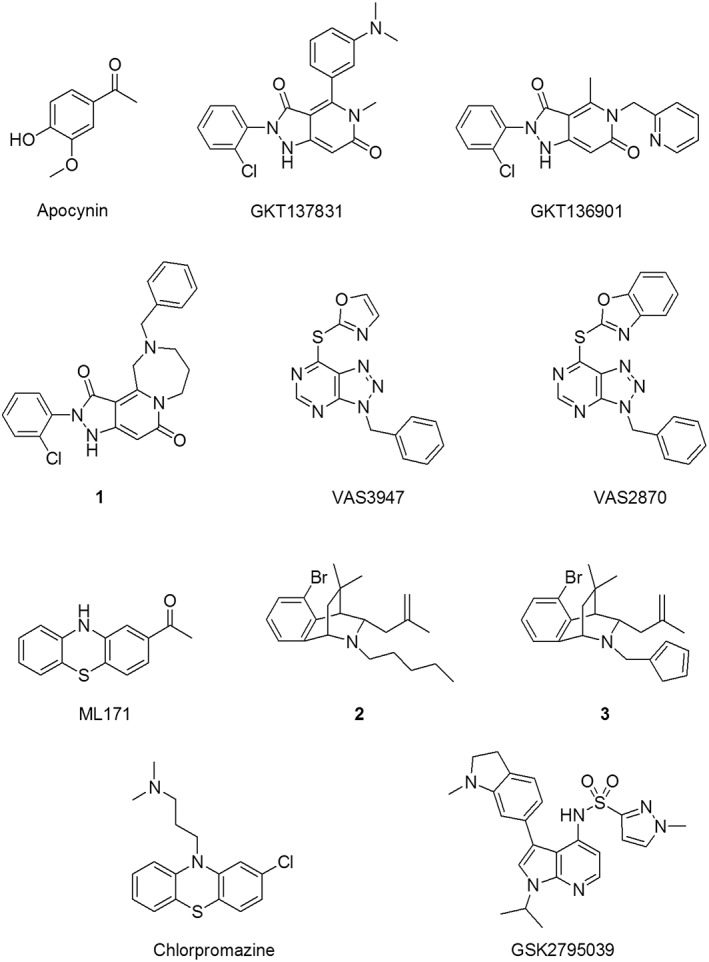
Selected NOX inhibitors.
Table 1.
Summary of potency, specificity and ADME(T) properties of selected NOX inhibitors
| Compound | Potencya | NOX specificityb | ADME(T)/Off target effectsc | Reference |
|---|---|---|---|---|
| Apocynin | ~30 μM (IC50; neutrophils) | – | H2O2 scavenging | (Stolk et al., 1994) |
| GKT137831 | 110 nM (Ki; NOX4) | NOX1,NOX4 > NOX5 > NOX2 | Good ADME(T), no known off target effects | (Aoyama et al., 2012) |
| GKT136901 | 160 nM (Ki; NOX1) | NOX1,NOX4> > NOX2 | Good ADME(T), no known off target effects | (Laleu et al., 2010) |
| 1 | 72 nM (Ki; NOX4) | NOX1,NOX4 > NOX5 > NOX2 | Good ADME(T), no known off target effects | (Gaggini et al., 2011) |
| VAS2870 | 77 nM (IC50; neutrophils) | – | No effect on XO and eNOS | (Gatto et al., 2013a) |
| VAS3947 | 2 μM (IC50; HL60 cells) | NOX2 > NOX1,NOX4 | No effect on XO and eNOS | (Wind et al., 2010) |
| ML171 | 0.25 μM (IC50; NOX1) | NOX1> > NOX3,NOX2,NOX4 | No or low effects on XO and several CNS targets | (Gianni et al., 2010) |
| 2 | 20 μM (IC50; NOX2) | NOX2 > NOX1,NOX4,NOX5 | No effect on XO or ROS scavenging | (Cifuentes‐Pagano et al., 2012) |
| 3 | 38 μM (IC50; NOX2) | NOX2 > NOX1,NOX4,NOX5 | No effect on XO or ROS scavenging | (Cifuentes‐Pagano et al., 2012) |
| GSK2795039 | 0.3 μM (IC50; NOX2) | NOX2> > NOX1,NOX3,NOX4,NOX5 | No or very low effect on a set of secondary assays, such as XO and reduction potential | (Hirano et al., 2015) |
| Chlorpromazine | 4.3 μM (IC50; NOX2) | NOX2 > NOX1, NOX3 > NOX4,NOX5 | Inhibition of dopamine D2 receptors | (Seredenina et al., 2015) |
Potency is reported as Ki or IC50 values for the isoform or cell type where the potency is highest.
The NOX isoforms are ordered according to compound potency. Isoforms separated with comma have similar potency, > indicates 3–5 fold selectivity, > > indicates a selectivity over 10 fold. A dash indicates that the selectivities have not been reported.
Summary of the reported effects on non‐NOX enzymes. XO, xanthine oxidase
Traditionally, NOX activity has been inhibited with compounds such as diphenyleneiodonium, a non‐specific inhibitor of flavin‐containing enzymes, and apocynin (Stolk et al., 1994), a compound believed to interfere with p47‐phox binding to other components of the NOX complex. However, neither of these are specific (Aldieri et al., 2008a) and are not generally considered as treatment options for NOX inhibition. In addition, apocynin appears to require oxidative activation before inhibiting NOX enzymes, and both apocynin and the analogue diapocynin are more likely to act as ROS scavengers, at least in vitro (Heumuller et al., 2008). It should, however, be mentioned that both apocynin and diapocynin have been used successfully in vivo (for example, Dranka et al., 2013 and Hernandes et al., 2013), and some researchers have called for clinical studies (Drummond & Sobey, 2014). However, it is not known whether their effects were based on NOX inhibition (Drummond and Sobey, 2014). More recently, several laboratories have identified more specific NOX inhibitors that are more promising as therapeutic agents. For instance, GenKyoTex has developed several series of pyrazolopyridine diones (Laleu et al., 2010; Page et al., 2010; Gaggini et al., 2011; Brandes et al., 2013) and pyrazolo piperidine (Page et al., 2011) derivatives with varying NOX specificity. Recently, a phase II study of the most advanced compound, GKT137831, was marked as completed, although the results have not yet been released (2016). Although the primary endpoint was not met, it showed a good safety profile and patients with diabetic kidney disease had significantly reduced level of circulating liver enzymes and inflammatory markers (NIH Trials number NCT 02010242). GKT137831 has a NOX1/4 inhibitory effect with a Ki of around 100 nM and a 10‐fold or 4‐fold selectivity over NOX2 and NOX5, respectively, and is essentially inactive towards xanthine oxidase (Aoyama et al., 2012). Also, GKT136901 (Laleu et al., 2010) and compound 1, described in Gaggini et al., (2011), absorption, distribution, metabolism and excretion/toxicity (ADME(T)) properties. GKT136901 has been shown to scavenge peroxynitrite (Schildknecht et al., 2014). The mechanism of the scavenging is not known except for the fact that GKT136901 is consumed in the process. The phenomenon could very likely be present also for the other compounds in this series.
Vasopharm Biotech has described a series of triazolopyrimidine derivatives (Tegtmeier et al., 2005), with special focus on VAS2870 (ten Freyhaus et al., 2006; Gatto et al., 2013a) and VAS3947 (Wind et al., 2010), which are both considered to be pan‐NOX inhibitors with limited off‐target effects, even though some have been reported (Laleu et al., 2010; Sun et al., 2012). However, the broad‐spectrum NOX effects of these compounds will render them less likely to be used in a clinical setting. ML171 is a NOX1 specific inhibitor with nanomolar potency and 12‐fold to 20‐fold specificity over the other NOX isoforms and minimal off target effects (Gianni et al., 2010). However, these results have recently been questioned by Serendenina et al. (2015) in a study that combined a multitude of readouts to minimize assay interference effects. In contrast, they describe N‐substituted phenothiazines as NOX inhibitors with varying specificities. Some of these were independently found to inhibit NOX2 by Zielonka et al. (2016). One of the compounds discussed in both these papers is chlorpromazine that, interestingly enough, was used as a negative control in the work on ML171 (Gianni et al., 2010). The phenothiazines, like chlorpromazine, are used as antipsychotics and are known to pass the blood brain barrier, which increases their potential suitability for repurposing, in the event that the observed effects translate to human use. In other work, researchers from the University of Pittsburgh have developed NOX2‐specific tetrahydroisoquinoline inhibitors, such as compounds 2 and 3 (Cifuentes‐Pagano et al., 2012; Pagano et al., 2014) that do not inhibit xanthine oxidase or scavenge ROS. Very recently, GSK2795039 was described as a selective NOX2 inhibitor with more than 30‐fold selectivity over other NOX enzymes and good PK properties (Hirano et al., 2015). In addition to these inhibitors, there is a plethora of reported NOX inhibitors targeting different isozymes. However, less is known about the specificities of these compounds within the NOX family, as well as towards other off‐target effects.
Taken together, the future of NOX inhibiting compounds looks very interesting, with several groups making progress on novel, more specific inhibitors and clinical results showing good safety profiles, together with some positive results. Of high importance is also the fact that known CNS active drugs have been shown to inhibit NOX enzymes, raising hopes that repurposing will become a reality.
Reactive nitrogen species as a therapeutic target in neurodegenerative disease
NO‐mediated S‐nitrosylation (the chemical reaction is S‐nitrosation, but the ‘ylation’ suffix is commonly used in biology) entails the addition or transfer of a nitroso group to a free sulphydryl group in a protein, forming S‐nitrosocysteine. This happens as part of normal cellular signalling, but dysregulation of S‐nitrosylation (termed ‘nitrosative stress’) is being increasingly linked to human neurodegenerative disorders (Chung, 2006; Cobb and Cole, 2015; Lee et al., 2016).
Another significant reaction involving NO is nitration of protein tyrosine residues to 3‐nitro‐tyrosine. Frequently, this occurs when excess ROS and NO form peroxynitrite (ONOO−) and has been deemed a biomarker of reactive nitrogen species (RNS)‐mediated cellular damage (Radi, 2013). In this case, the process is referred to as ‘nitrative stress’ and commonly occurs in neurodegenerative disorders because both ROS and RNS are in oversupply. In some instances, ‘nitroxidative stress’ is used as a collective term for nitrosative, nitrative and oxidative stress (Lancaster, 2006).
Information is accumulating on the association between nitrative stress and neurodegenerative disease, but using this knowledge to design therapeutic strategies is at an early stage of development. For example, α‐synuclein accumulation in PD increases both ROS and RNS. In fact, exogenous α‐synuclein increases NO synthesis via NMDA receptor‐induced activation of iNOS and increased NO in rat brain experiments (Wilkaniec et al., 2013). Concurrent stimulation of superoxide by α‐synuclein increases production of ONOO−. Lee and colleagues tested several antioxidants, including superoxide dismutase, catalase, 2,2,6,6‐tetramethyl‐1‐piperidinoxyl (TEMPO), NAC, dimethylthiourea and uric acid in preventing protein tyrosine nitration caused by ROS and NO in primary neuronal cultures (Lee et al., 2016). Of these, TEMPO was the most effective at promoting clearance of ROS/RNS and inhibition of stress signalling pathways. It was concluded that TEMPO offers potential for development of combined reduction in ROS and RNS in neurodegenerative disease.
Experimentally‐induced nitrative stress using the peroxynitrite donor, 3‐morpholinosyndnonimine, was examined in SH‐SY5Y neuroblastoma cells and mouse motor neuron – neuroblastoma fusion NSC‐34 cell lines (Kupershmidt et al., 2010). Notably, Fe III and Mn III corroles (metal complexes structurally similar to porphyrin compounds) increased cell survival. The water solubility, low effective concentration and non‐toxic properties of the metallocorroles rendered them attractive as therapeutic agents able to decompose ROS and RNS and limit neuronal loss in a number of neurodegenerative disorders. Okun and colleagues undertook further investigation of the structure–activity relationships of the antioxidant properties of metallocorroles, concluding that the C(10)‐anysil‐substituted complex was most effective (Okun and Gross, 2012). To date, the metallocorroles, together with other peroxynitrite decomposition catalysts, have demonstrated preclinical efficacy, oral availability and reduced toxicity risk (Slosky and Vanderah, 2015). Work is ongoing to generate compounds with greater oral availability and peroxynitrite selectivity, with the aim of providing safe, effective and selective modulators of nitrative stress (Okun and Gross, 2012).
An alternative strategy to combat the toxic effects of RNS is to limit synthesis of NO by inhibition of iNOS, for example, using aminoguanidine (Díaz et al., 2014). Systemic administration of aminoguanidine to rats (100 mg·kg−1/day for 4 days) improved the spatial memory deficits caused by Aβ injection into the temporal cortex. The effect was accompanied by decreased levels of pro‐inflammatory cytokines and nitrite, plus a reduction in reactive gliosis. At the time of writing, the most recent report on aminoguanidine relates to its use in two animal models of schizophrenia (Lafioniatis et al., 2016). In this study, aminoguanidine (25 and 50 mg·kg−1) attenuated the extinction of recognition memory that is characteristic of the disorder. However, aminoguanidine had no effect on ketamine‐induced social isolation in the animals, even at the higher dose of 100 mg·kg−1. These observations led to the conclusion that aminoguanidine may be effective in reducing memory impairments in patients with neurodegenerative disease.
Another approach to reduce nitrative stress in neurodegenerative disease is to use cerium oxide nanoparticles, known as nanoceria. These particles scavenge superoxide anions, hydrogen peroxide and peroxide by switching between their Ce3+ and Ce4 + states (Dowding et al., 2014). The potential of nanoceria to reduce oxidative and nitrative stress in neurodegenerative disease is considerable: they are taken up by neurons and accumulate at both the plasma membrane and the outer mitochondrial membrane and have been shown to reduce endogenous peroxinitrite formation, Aβ‐induced mitochondrial fragmentation and neuronal death in experimental systems (Dowding et al., 2014).
Other work, likely to be the focus of future investigations, has examined cross‐talk between inflammatory cytokine production, astrocyte activation and generation of NO. For instance, fingolimod (Gilenya®, Novartis; a sphingosine‐1‐phosphate receptor antagonist approved as oral therapy in MS) inhibits astrocyte activation and NO production following administration to the EAE mouse model of MS, thereby providing neuroprotection (Colombo et al., 2014). Similar neuroprotective effects of fingolimod have been observed in a mouse model of AD (Aytan et al., 2016). However, use of fingolimod in clinical situations for AD may be some way off, due to recent evidence of lesions in the brain of patients taking fingomilod for MS, as identified by magnetic resonance imaging (Fragoso and Sato, 2016).
Other redox components as therapeutic targets in neurodegenerative disease
Li and colleagues explored the potential of inhibition of MPO as a redox‐based therapeutic target in neurodegenerative disease (Li et al., 2015). MPO catalyses the reaction between hydrogen peroxide and halides that generates hypochlorite. Hypochlorite is a powerful oxidant and chlorinator of tyrosine residues in proteins, forming 3‐chloro‐tyrosine. MPO is expressed in microglia and has been implicated in a number of diseases, including AD and PD. However, the development of inhibitors to target MPO in neurodegenerative disease is at an early stage. For example, Li and colleagues synthesized and tested over 50 low MW MPO inhibitors and a number were identified with IC50 values in the high nanomolar range. For example, AZD3241 is a novel selective and irreversible inhibitor of MPO that has undergone a phase 2a randomized placebo controlled study on PD patients (Jucaite et al., 2015). Using positron emission tomography, it was confirmed that AZD3241 targets microglial cells. Jucaite and colleagues concluded that further studies on the efficacy of AZD3241 in neurodegenerative disorders were warranted.
New developments in understanding the pathogenesis of AD include the ‘Inverse Warburg Hypothesis’, which is a bioenergetics model that places metabolic dysfunction as a central element in development of the disease (Demetrius et al., 2014). If this proves to be the case, as seems likely, then therapeutic agents that are based upon modulating the availability of oxidisable substrates, on the one hand, and ROS homeostasis on the other, may be most beneficial. Finally, it is also worth mentioning that, although exercise is an excellent generator of radicals, it has been proven to have the greatest effect in preventing progression of mild cognitive impairment to AD (Fiatarone Singh et al., 2014).
Mitochondrial dysfunction as a source of ROS and RNS
Dranka and colleagues sought to determine the relationship between mitochondrial dysfunction and oxidant production in a number of PD disease models – finding that mitochondrial dysfunction was a more significant cause of degeneration than oxidative damage in these models (Dranka et al., 2012). Similarly, Haddad and co‐workers recently reviewed the evidence that mitochondrial stressors may cause PD and reached the conclusion that more information is needed to understand how mitochondria contribute to the pathogenesis of PD (Haddad and Nakamura, 2015). Nonetheless, mitochondria‐targeting antioxidants are emerging as new therapeutic agents in treatments for neurodegenerative disease. Chief amongst these is MitoQ (mitoquinone mesylate: [10‐(4,5‐dimethoxy‐2‐methyl‐3,6‐dioxo‐1,4‐cycloheexadienl‐yl) decyl triphenylphosphonium methanesulfonate]), which is ubiquinone covalently bound to a triphenylphosphonium cation. MitoQ readily permeates the blood brain barrier and neuronal membranes, where it concentrates within mitochondria. MitoQ was assessed for prevention of AD pathology in mouse cortical neurons in culture and a triple transgenic mouse model of AD (McManus et al., 2011). MitoQ successfully attenuated Aβ‐induced toxicity in cortical neurons and prevented the loss in mitochondrial membrane potential. Moreover, treatment of mice for 5 months with MitoQ provided protection against neurodegeneration in the AD mouse model in vivo (McManus et al., 2011). MitoQ was also tested in the 6(OH) DA model of PD (SH‐SY56Y cell cultures) (Solesio et al., 2013c). Again, pre‐treatment with MitoQ prevented redox‐related mitochondrial dysfunction.
Using the same rationale of boosting the antioxidant capacity of mitochondria, Dranka and colleagues tested a mitochondrial‐permeable form of the antioxidant apocynin (mito‐apocynin) in the transgenic mouse model of PD (Dranka et al., 2014). Significant deterioration in physical ability and increased hyposmia in the mice was prevented by treatment with mito‐apocynin (3 mg·kg−1) three times a week.
Acetyl‐L‐carnitine and α‐lipoic acid are two compounds that target mitochondria and have been tested in their capacity as antioxidants to improve mitochondrial function in a PD animal model (Zaitone et al., 2012). Acetyl‐L‐carnitine is an essential component in the α‐oxidation of fatty acids to generate ATP. α‐Lipoic acid is a co‐enzyme, but also has antioxidant properties and readily crosses the blood brain barrier. Treatment of rats with acetyl‐L‐carnitine and α‐lipoic acid, either alone or in combination, significantly improved rotenone‐induced neurotoxicity (Zaitone et al., 2012). It was concluded that these agents are promising candidates for neuroprotection in the early stages of PD.
Collectively, the results from experiments with mitochondrial antioxidants suggest that mitochondria‐targeting therapeutic agents may have a place in neurodegenerative disease treatment. Onyango and colleagues provide an excellent review on mitochondrial dysfunction in AD and discuss the rationale behind the search for bioenergetics‐based therapies. A number of investigations are ongoing that include potentiation of energy production, scavenging ROS and curtailing oxidative damage (Onyango et al., 2016). Several of these avenues of experimentation are promising and progression beyond pre‐clinical experiments is eagerly awaited.
Conclusion and future perspectives
Much progress has been made in designing strategies to combat the loss of redox homeostasis in neurodegenerative disorders. It is becoming increasingly clear that several diseases share common pathways of ROS‐related damage and much can be gained from comparative studies that incorporate a range of disorders. As the origins and targets of ROS are better understood, therapies must evolve that address the deficiencies in ROS signalling more effectively. In this review, we have chosen to discuss, on the one hand, three major strategies that we believe best address gaps in knowledge and, on the other, more effective therapies. Nonetheless, the challenges are considerable, particularly in the arena of polypharmacy and improved drug delivery. Therapeutic approaches that will address both the inflammatory and oxidative‐nitrosative pathways in the neuropathogenesis of age‐related neurodegeneration are urgently needed (Daulatzai, 2016). Similarly, improvements in drug delivery and targeting will greatly assist in enhancing bioavailablity and aid in the development of more effective therapeutic agents. For example, Hagl and colleagues review improvements in the bioavailability of curcumin compounds as potential treatments for neurodegenerative disease (Hagl et al., 2015). Curcumin micelles had improved effectiveness in protecting PC12 cells from nitrosative stress, compared with curcumin alone. It is expected that similar developments using other antioxidants will emerge in the near future. There are still significant gaps in our understanding of brain thiol redox biochemistry. It is only relatively recently accepted that the brain contains a functional transsulphuration pathway, but its significance in generating sulphur‐based compounds with antioxidant properties remains to be fully understood. In their well‐focussed and timely review, Hensley and Denton describe how the broad‐spectrum enzymes of the transsulphuration pathway generate a range of compounds, including H2S and the unusual amino acid, lanthionine (Hensley & Denton, 2015). Derivatives of the transamination product of this amino acid, lanthionine ketimine, have potent antioxidant, neuroprotective, neurotrophic and antioxidant actions. The opportunities for development of thiol‐based therapeutics for treatment of neurodegenerative disorders based on these compounds must be considerable.
The Nrf2‐EpRE pathway is an intrinsic mechanism of defence against oxidative stress which acts by inducing the expression of a wide variety of cytoprotective and detoxificant genes. There is much evidence for the protective role of the Nrf2‐EpRE pathway in different neurodegenerative conditions, as it reduces oxidative stress and neuroinflammation. The proof of concept that Nrf2 induction can be a good strategy to develop drugs for neurodegenerative conditions is exemplified by DMF, which is already in the clinic for the treatment of MS. Therefore, the development of Nrf2 inducers opens a new direction for the treatment of patients with neurodegenerative diseases.
With the emergence of clinical data for NOX inhibitors, interest is increasing. In addition, novel, highly selective inhibitors have been shown to have effect in a variety of animal models. Together, these results describe a future where detrimental ROS can be inhibited at the production stage, without disturbing essential ROS signalling.
Conflict of interest
F. K. W. is an employee of Redoxis AB, a Swedish company that develops low MW NOX modulators.
Acknowledgements
M. G. L. acknowledges the support of the Spanish Ministry of Economy and Competence grant ref. SAF2015‐63935R.
All authors were supported by the European Cooperation in Science and Technology (COST Action BM1203/EU‐ROS).
McBean, G. J. , López, M. G. , and Wallner, F. K. (2017) Redox‐based therapeutics in neurodegenerative disease. British Journal of Pharmacology, 174: 1750–1770. doi: 10.1111/bph.13551.
References
- Aguirre J, Lambeth JD (2010). Nox enzymes from fungus to fly to fish and what they tell us about Nox function in mammals. Free Radic Biol Med 49: 1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akterin S, Cowburn RF, Miranda‐Vizuete A, Jimenez A, Bogdanovic N, Winblad B et al. (2006). Involvement of glutaredoxin‐1 and thioredoxin‐1 in beta‐amyloid toxicity and Alzheimer's disease. Cell Death Differ 13: 1454–1465. [DOI] [PubMed] [Google Scholar]
- Albano R, Liu X, Lobner D (2013). Regulation of system x(c)‐ in the SOD1‐G93 A mouse model of ALS. Exp Neurol 250: 69–73. [DOI] [PubMed] [Google Scholar]
- Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I et al. (2008a). Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr Drug Metab 9: 686–696. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenhöfer S, Radermacher KA, Kleikers PW, Wingler K, Schmidt HH (2014b). Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid Redox Signal 23: 406–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmeyer PJ, Matthes U, Pawlak F, Hoffmann K, Frosch PJ, Ruppert P et al. (1994). Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double‐blind study in 100 patients. J Am Acad Dermatol 30: 977–981. [DOI] [PubMed] [Google Scholar]
- Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB (2007). Bioavailability of curcumin: problems and promises. Mol Pharm 4: 807–818. [DOI] [PubMed] [Google Scholar]
- Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso‐Cartier L et al. (2012). Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatol 56: 2316–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apolloni S, Parisi C, Pesaresi MG, Rossi S, Carri MT, Cozzolino M et al. (2013). The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1‐G93 A microglia model of amyotrophic lateral sclerosis. J Immunol 190: 5187–5195. [DOI] [PubMed] [Google Scholar]
- Asanuma M, Miyazaki I, Diaz‐Corrales FJ, Kimoto N, Kikkawa Y, Takeshima M et al. (2010). Neuroprotective effects of zonisamide target astrocyte. Ann Neurol 67: 239–249. [DOI] [PubMed] [Google Scholar]
- Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA (2007). N‐Acetylcysteine‐‐a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol 7: 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytan N, Choi JK, Carreras I, Brinkmann V, Kowall NW, Jenkins BG et al. (2016). Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer's disease. Sci Rep 6: 24939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal‐Price A, Brown GC (2001). Inflammatory neurodegeneration mediated by nitric oxide from activated glia‐inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci 21: 6480–6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogun E, Hoque M, Gong P, Killeen E, Green CJ, Foresti R et al. (2003). Curcumin activates the haem oxygenase‐1 gene via regulation of Nrf2 and the antioxidant‐responsive element. Biochem J 371: 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai S (1984). Induction of cystine and glutamate transport activity in human fibroblasts by diethylmaleate and other electrophilic agents. J Biol Chem 259: 2435–2440. [PubMed] [Google Scholar]
- Bentea E, Sconce MD, Churchill MJ, Van J, Sato H, Meshul CK et al. (2015a). MPTP‐induced parkinsonism in mice alters striatal and nigral xCT expression but is unaffected by the genetic loss of xCT. Neurosci Lett 593: 1–6. [DOI] [PubMed] [Google Scholar]
- Bigford GE, Del Rossi G (2014). Supplemental substances derived from foods as adjunctive therapeutic agents for treatment of neurodegenerative diseases and disorders. Adv Nutr 5: 394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomprezzi R (2015). Dimethyl fumarate in the treatment of relapsing–remitting multiple sclerosis: an overview. Ther Adv Neurol Disord 8: 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes R, Schroeder K, Page P, Laleu B, Gaggini F (2013). NADPH oxidase 4 inhibitors for prevention of osteoporosis or osteoglastogenesis dysfunction. PCT Int Appl Johann Wolfgang Goethe‐Universitaet, Germany.
- Bridges R, Lutgen V, Lobner D, Baker DA (2012). Thinking outside the cleft to understand synaptic activity: contribution of the cystine‐glutamate antiporter (system xc −) to normal and pathological glutamatergic signaling. Pharmacol Rev 64: 780–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Cooper CE (1994). Nanomolar concentrations of nitric oxide inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett 356: 295–298. [DOI] [PubMed] [Google Scholar]
- Cacciatore I, Baldassarre L, Fornasari E, Mollica A, Pinnen F (2012). Recent advances in the treatment of neurodegenerative diseases based on GSH delivery systems. Oxid Med Cell Longev 2012: 240146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AN, Lim JL, Nijland PG, Witte ME, Van J (2014). Glutathione in multiple sclerosis: more than just an antioxidant? Mult Scler 20: 1425–1431. [DOI] [PubMed] [Google Scholar]
- Casamenti F, Grossi C, Rigacci S, Pantano D, Luccarini I, Stefani M (2015). Oleuropein aglycone: a possible drug against degenerative conditions. In vivo evidence of its effectiveness against Alzheimer's disease. J Alzheimers Dis 45: 679–688. [DOI] [PubMed] [Google Scholar]
- Chandra V, Pandav R, Dodge HH, Johnston JM, Belle SH, DeKosky ST et al. (2001). Incidence of Alzheimer's disease in a rural community in India: the Indo‐US study. Neurology 57: 985–989. [DOI] [PubMed] [Google Scholar]
- Chang J, Rimando A, Pallas M, Camins A, Porquet D, Reeves J et al. (2012). Low‐dose pterostilbene, but not resveratrol, is a potent neuromodulator in aging and Alzheimer's disease. Neurobiol Aging 33: 2062–2071. [DOI] [PubMed] [Google Scholar]
- Chen L, Li X, Liu L, Yu G, Xue Y, Liu Y (2015). Erastin sentizes glioblastoma cells to temozolomide by restraining xCT and cystathionine‐g‐lyase function. Oncol Rep 33: 1456–1474. [DOI] [PubMed] [Google Scholar]
- Chin MP, Reisman SA, Bakris GL, O'Grady M, Linde PG, McCullough PA et al. (2014). Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am J Nephrol 39: 499–508. [DOI] [PubMed] [Google Scholar]
- Cho HY (2013). Genomic structure and variation of nuclear factor (erythroid‐derived 2)‐like 2. Oxid Med Cell Longev 2013: 286524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DH, Cristovao AC, Guhathakurta S, Lee J, Joh TH, Beal MF et al. (2012). NADPH oxidase 1‐mediated oxidative stress leads to dopamine neuron death in Parkinson's disease. Antioxid Redox Signal 16: 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK (2006). Say NO to neurodegeneration: role of S‐nitrosylation in neurodegenerative disorders. Neurosignals 15: 307–313. [DOI] [PubMed] [Google Scholar]
- Cifuentes‐Pagano E, Csanyi G, Pagano PJ (2012). NADPH oxidase inhibitors: a decade of discovery from Nox2ds to HTS. Cell Mol Life Sci 69: 2315–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb CA, Cole MP (2015). Oxidative and nitrative stress in neurodegeneration. Neurobiol Dis 84: 4–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo E, Di M, Capitolo E, Chaabane L, Newcombe J, Martino G et al. (2014). Fingolimod may support neuroprotection via blockade of astrocyte nitric oxide. Ann Neurol 76: 325–337. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA (2003). Nrf2 is a direct PERK substrate and effector of PERK‐dependent cell survival. Mol Cell Biol 23: 7198–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daulatzai MA (2016). Fundamental role of pan‐inflammation and oxidative‐nitrosative pathways in neuropathogenesis of Alzheimer's disease. Am J Neurodegener Dis 5: 1–28. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Davinelli S, Sapere N, Zella D, Bracale R, Intrieri M, Scapagnini G (2012). Pleiotropic protective effects of phytochemicals in Alzheimer's disease. Oxid Med Cell Longev 2012: 386527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot JF, Sontheimer H (2011). Glutamate and the biology of the gliomas. Glia 59: 1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetrius LA, Magistretti PJ, Pellerin L (2014). Alzheimer's disease: the amyloid hypothesis and the Inverse Warburg effect. Front Physiol 5: 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhakshinamoorthy S, Jaiswal AK (2001). Functional characterization and role of INrf2 in antioxidant response element‐mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene 20: 3906–3917. [DOI] [PubMed] [Google Scholar]
- Di F, Barone E, Perluigi M, Butterfield DA (2015). Strategy to reduce free radical species in Alzheimer's disease: an update of selected antioxidants. Expert Rev Neurother 15: 19–40. [DOI] [PubMed] [Google Scholar]
- Díaz A, Rojas K, Espinosa B, Chavez R, Zenteno E, Limon D et al. (2014). Aminoguanidine treatment ameliorates inflammatory responses and memory impairment induced by amyloid‐beta 25‐35 injection in rats. Neuropeptides 48: 153–159. [DOI] [PubMed] [Google Scholar]
- Dinkova‐Kostova AT, Talalay P (2008). Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res 52 (Suppl 1): S128–S138. [DOI] [PubMed] [Google Scholar]
- Dong H, Xu L, Wu L, Wang X, Duan W, Li H et al. (2014). Curcumin abolished mutant TDP‐43 induced excitability in a motoneuro‐like cellular model of ALS. Neurosci 272: 141–153. [DOI] [PubMed] [Google Scholar]
- Dowding JM, Song W, Bossy K, Karakoti A, Kumar A, Kim A et al. (2014). Cerium oxide nanoparticles protect against Abeta‐induced mitochondrial fragmentation and neuronal cell death. Cell Death Differ 21: 1622–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Gifford A, Ghosh A, Zielonka J, Joseph J, Kanthasamy AG et al. (2013). Diapocynin prevents early Parkinson's disease symptoms in the leucine‐rich repeat kinase 2 (LRRK2R(1)(4)(4)(1)G) transgenic mouse. Neurosci Lett 549: 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Gifford A, McAllister D, Zielonka J, Joseph J, O'Hara CL et al. (2014). A novel mitochondrially‐targeted apocynin derivative prevents hyposmia and loss of motor function in the leucine‐rich repeat kinase 2 (LRRK2(R1441G)) transgenic mouse model of Parkinson's disease. Neurosci Lett 583: 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Zielonka J, Kanthasamy AG, Kalyanaraman B (2012). Alterations in bioenergetic function induced by Parkinson's disease mimetic compounds: lack of correlation with superoxide generation. J Neurochem 122: 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, Pfeiffer B, Hamprecht B (1999). Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci 19: 562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GR, Sobey CG (2014). Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol Metab 25: 452–463. [DOI] [PubMed] [Google Scholar]
- Dubey D, Cano CA, Stuve O (2016). Update on monitoring and adverse effects of approved second‐generation disease‐modifying therapies in relapsing forms of multiple sclerosis. Curr Opin Neurol 29: 278–285. [DOI] [PubMed] [Google Scholar]
- Dumont M, Wille E, Calingasan NY, Tampellini D, Williams C, Gouras GK et al. (2009). Triterpenoid CDDO‐methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer's disease. J Neurochem 109: 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner M (2015). Risk of progressive multifocal leukoencephalopathy in patients with multiple sclerosis. Expert Opin Drug Saf 14: 1737–1748. [DOI] [PubMed] [Google Scholar]
- Fernando WM, Martins IJ, Goozee KG, Brennan CS, Jayasena V, Martins RN (2015). The role of dietary coconut for the prevention and treatment of Alzheimer's disease: potential mechanisms of action. Br J Nutr 114: 1–14. [DOI] [PubMed] [Google Scholar]
- Fiatarone Singh MA, Gates N, Saigal N, Wilson GC, Meiklejohn J, Brodaty H et al. (2014). The Study of Mental and Resistance Training (SMART) study‐resistance training and/or cognitive training in mild cognitive impairment: a randomized, double‐blind, double‐sham controlled trial. J Am Med Dir Assoc 15: 873–880. [DOI] [PubMed] [Google Scholar]
- Fragoso YD, Sato HK (2016). Catastrophic magnetic resonance images in the central nervous system of patients undergoing treatment with Fingolimod. CNS Neurosci Ther. [DOI] [PMC free article] [PubMed]
- Gaggini F, Laleu B, Orchard M, Fiorasi‐Cartier L, Cagnon L, Houngninou‐Molango S et al. (2011). Design, synthesis and biological activity of oridinal pyrazolo‐pyrido‐diazepine, −pyrazine and ‐oxazine dione derivatives as novel dual Nox4/Nox1 inhibitors. Bioorg Med Chem 19: 6989–6999. [DOI] [PubMed] [Google Scholar]
- Garcia‐Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ (2007). Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem 102: 1095–1104. [DOI] [PubMed] [Google Scholar]
- Gatto GJ Jr, Ao Z, Kearse MG, Zhou M, Morales CR, Daniels E et al. (2013a). NADPH oxidase‐dependent and ‐independent mechanisms of reported inhibitors of reactive oxygen generation. J Enzyme Inhib Med Chem 28: 95–104. [DOI] [PubMed] [Google Scholar]
- Gess B, Baets J, De Jonghe P, Reilly MM, Pareyson D, Young P (2015). Ascorbic acid for the treatment of Charcot–Marie‐Tooth disease. Cochrane Database Syst Rev 12: Cd011952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni D, Taulet N, Zhang H, DerMardirossian C, Kister J, Martinez L et al. (2010). A novel and specific NADPH oxidase‐1 (Nox1) small‐molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem Biol 5: 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout PW, Kang YJ, Buckley DJ, Bruchovsky N, Buckley AR (2001). Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the xc‐ cystine transporter: a new action for an old drug. Leukemia 15: 1633–1640. [DOI] [PubMed] [Google Scholar]
- Günther M, Davidsson J, Plantman S, Norgren S, Mathiesen T, Risling M (2015). Neuroprotective effects of N‐acetylcysteine amide on experimental focal penetrating brain injury in rats. J Clin Neurosci 22: 1477–1483. [DOI] [PubMed] [Google Scholar]
- Haddad D, Nakamura K (2015). Understanding the susceptibility of dopamine neurons to mitochondrial stressors in Parkinson's disease. FEBS Lett 589: 3702–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagl S, Kocher A, Schiborr C, Kolesova N, Frank J, Eckert GP (2015). Curcumin micelles improve mitochondrial function in neuronal PC12 cells and brains of NMRI mice ‐ Impact on bioavailability. Neurochem Int 89: 234–242. [DOI] [PubMed] [Google Scholar]
- Harrison FE, Bowman GL, Polidori MC (2014). Ascorbic acid and the brain: rationale for the use against cognitive decline. Nutrients 6: 1752–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, Dinkova‐Kostova AT (2014). The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39: 199–218. [DOI] [PubMed] [Google Scholar]
- Hensley K, Denton TT (2015). Alternative functions of the brain transsulfuration pathway represent an underappreciated aspect of brain redox biochemistry with significant potential for therapeutic engagement. Free Radic Biol Med 78: 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandes MS, Cafe‐Mendes CC, Britto LR (2013). NADPH oxidase and the degeneration of dopaminergic neurons in parkinsonian mice. Oxid Med Cell Longev 2013: 157857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heumuller S, Wind S, Barbosa‐Sicard E, Schmidt HH, Busse R, Schroder K et al. (2008). Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51: 211–217. [DOI] [PubMed] [Google Scholar]
- Hirano K, Chen WS, Chueng AL, Dunne AA, Seredenina T, Filippova A et al. (2015). Discovery of GSK2795039, a novel small molecule NADPH oxidase 2 inhibitor. Antioxid Redox Signal 23: 358–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotani M, Maita C, Niino M, Iguchi‐Ariga S, Hamada S, Ariga H et al. (2008). Correlation between DJ‐1 levels in the cerebrospinal fluid and the progression of disabilities in multiple sclerosis patients. Mult Scler 14: 1056–1060. [DOI] [PubMed] [Google Scholar]
- Hourihan JM, Kenna JG, Hayes JD (2013). The gasotransmitter hydrogen sulfide induces nrf2‐target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys‐226 and cys‐613. Antioxid Redox Signal 19: 465–481. [DOI] [PubMed] [Google Scholar]
- Hsieh TC, Lu X, Wang Z, Wu JM (2006). Induction of quinone reductase NQO1 by resveratrol in human K562 cells involves the antioxidant response element ARE and is accompanied by nuclear translocation of transcription factor Nrf2. Medicinal Chem 2: 275–285. [DOI] [PubMed] [Google Scholar]
- Hu X, Weng Z, Chu CT, Zhang L, Cao G, Gao Y et al. (2011). Peroxiredoxin‐2 protects against 6‐hydroxydopamine‐induced dopaminergic neurodegeneration via attenuation of the apoptosis signal‐regulating kinase (ASK1) signaling cascade. J Neurosci 31: 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB (2002). Phosphorylation of Nrf2 at Ser‐40 by protein kinase C regulates antioxidant response element‐mediated transcription. J Biol Chem 277: 42769–42774. [DOI] [PubMed] [Google Scholar]
- Infanger DW, Sharma RV, Davisson RL (2006). NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal 8: 1583–1596. [DOI] [PubMed] [Google Scholar]
- Innamorato NG, Rojo AI, Garcia‐Yague AJ, Yamamoto M, de Caballos ML, Cuadrado A (2008). The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol 181: 680–689. [DOI] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y et al. (2000). Transcription factor Nrf2 coordinately regulates a group of oxidative stress‐inducible genes in macrophages. J Biol Chem 275: 16023–16029. [DOI] [PubMed] [Google Scholar]
- Jain AK, Jaiswal AK (2007). GSK‐3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF‐E2 related factor 2. J Biol Chem 282: 16502–16510. [DOI] [PubMed] [Google Scholar]
- Jazwa A, Rojo AI, Innamorato NG, Hesse M, Fernandez‐Ruiz J, Cuadrado A (2011). Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental Parkinsonism. Antioxid Redox Signal 14: 2347–2360. [DOI] [PubMed] [Google Scholar]
- Jiang H, Tian X, Guo Y, Duan W, Bu H, Li C (2011). Activation of nuclear factor erythroid 2‐related factor 2 cytoprotective signaling by curcumin protect primary spinal cord astrocytes against oxidative toxicity. Biol Pharm Bull 34: 1194–1197. [DOI] [PubMed] [Google Scholar]
- Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GV (2014). Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun 5: 3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA (2010). The absence of the pro‐antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol Sci 114: 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WM, Wilson‐Delfosse AL, Chen SG, Mieyal JJ (2015a). The roles of redox enzymes in Parkinson's disease: focus on glutaredoxin. Ther Targets Neurol Dis 2: e790. [PMC free article] [PubMed] [Google Scholar]
- Johnson WM, Wilson‐Delfosse AL, Mieyal JJ (2012). Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 4: 1399–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WM, Yao C, Siedlak SL, Wang W, Zhu X, Caldwell GA et al. (2015b). Glutaredoxin deficiency exacerbates neurodegeneration in C. elegans models of Parkinson's disease. Hum Mol Genet 24: 1322–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JA, Fisher DR, Cheng V, Rimando AM, Shukitt‐Hale B (2008). Cellular and behavioral effects of stilbene resveratrol analogues: implications for reducing the deleterious effects of aging. J Agric Food Chem 56: 10544–10551. [DOI] [PubMed] [Google Scholar]
- Jucaite A, Svenningsson P, Rinne JO, Cselenyi Z, Varnas K, Johnstrom P et al. (2015). Effect of the myeloperoxidase inhibitor AZD3241 on microglia: a PET study in Parkinson's disease. Brain 138: 2687–2700. [DOI] [PubMed] [Google Scholar]
- Juurlink BH, Thorburne SK, Hertz L (1998). Peroxide‐scavenging deficit underlies oligodendrocyte susceptibility to oxidative stress. Glia 22: 371–378. [DOI] [PubMed] [Google Scholar]
- Kairane C, Mahlapuu R, Ehrlich K, Zilmer M, Soomets U (2014). The effects of different antioxidants on the activity of cerebrocortical MnSOD and Na,K‐ATPase from post mortem Alzheimer's disease and age‐matched normal brains. Curr Alzheimer Res 11: 79–85. [DOI] [PubMed] [Google Scholar]
- Kanakasabai S, Casalini E, Walline CC, Mo C, Chearwae W, Bright JJ (2012). Differential regulation of CD4(+) T helper cell responses by curcumin in experimental autoimmune encephalomyelitis. J Nutr Biochem 23: 1498–1507. [DOI] [PubMed] [Google Scholar]
- Kandil S, Brennan L, McBean GJ (2010). Glutathione depletion causes a JNK and p38MAPK‐mediated increase in expression of cystathione‐g‐lyase and upregulation of the transulfuration pathway in C6 glioma cells. Neurochem Int 56: 611–619. [DOI] [PubMed] [Google Scholar]
- Kanninen K, Malm TM, Jyrkkanen HK, Goldsteins G, Keksa‐Goldsteine V, Tanila H et al. (2008). Nuclear factor erythroid 2‐related factor 2 protects against beta amyloid. Mol Cell Neurosci 39: 302–313. [DOI] [PubMed] [Google Scholar]
- Keum YS, Yu S, Chang PP, Yuan X, Kim JH, Xu C et al. (2006). Mechanism of action of sulforaphane: inhibition of p38 mitogen‐activated protein kinase isoforms contributing to the induction of antioxidant response element‐mediated heme oxygenase‐1 in human hepatoma HepG2 cells. Cancer Res 66: 8804–8813. [DOI] [PubMed] [Google Scholar]
- Kim HV, Kim HY, Ehrlich HY, Choi SY, Kim DJ, Kim Y (2013). Amelioration of Alzheimer's disease by neuroprotective effect of sulforaphane in animal model. Amyloid 20: 7–12. [DOI] [PubMed] [Google Scholar]
- Kim JA, Neupane GP, Lee ES, Jeong BS, Park BC, Thapa P (2011). NADPH oxidase inhibitors: a patent review. Expert Opin Ther Pat 21: 1147–1158. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T et al. (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3‐based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24: 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupershmidt L, Okun Z, Amit T, Mandel S, Saltsman I, Mahammed A et al. (2010). Metallocorroles as cytoprotective agents against oxidative and nitrative stress in cellular models of neurodegeneration. J Neurochem 113: 363–373. [DOI] [PubMed] [Google Scholar]
- La G, Weber P, Mohajeri MH (2014). Effects of vitamin E on cognitive performance during ageing and in Alzheimer's disease. Nutrients 6: 5453–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafioniatis A, Orfanidou MA, Papadopoulou ES, Pitsikas N (2016). Effects of the inducible nitric oxide synthase inhibitor aminoguanidine in two different rat models of schizophrenia. Behav Brain Res 309: 14–21. [DOI] [PubMed] [Google Scholar]
- Laleu B, Gaggini F, Orchard M, Fiorasi‐Cartier L, Cagnon L, Hougninou‐Molango S et al. (2010). First in class, potent and orally bioavailable NADPH oxidase isoform 4 (NOX4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem 53: 7715–7730. [DOI] [PubMed] [Google Scholar]
- Lancaster JR Jr (2006). Nitroxidative, nitrosative, and nitrative stress: kinetic predictions of reactive nitrogen species chemistry under biological conditions. Chem Res Toxicol 19: 1160–1174. [DOI] [PubMed] [Google Scholar]
- Lee C, Park GH, Lee SR, Jang JH (2013). Attenuation of beta‐amyloid‐induced oxidative cell death by sulforaphane via activation of NF‐E2‐related factor 2. Oxid Med Cell Longev 2013: 313510–313522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CT, Yu LE, Wang JY (2016). Nitroxide antioxidant as a potential strategy to attenuate the oxidative/nitrosative stress induced by hydrogen peroxide plus nitric oxide in cultured neurons. Nitric Oxide 54: 38–50. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Hewett S, Huang Y, Lambros M, Gout PW, Kalivas PW et al. (2013). The cystine/glutamate antiporter system xc‐ in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal 18: 522–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Cui W, Liu J, Li R, Liu Q, Xie XH et al. (2013). Sulforaphane ameliorates the development of experimental autoimmune encephalomyelitis by antagonizing oxidative stress and Th17‐related inflammation in mice. Exp Neurol 250: 239–249. [DOI] [PubMed] [Google Scholar]
- Li Y, Ganesh T, Diebold BA, Zhu Y, McCoy JW, Smith SM et al. (2015). Thioxo‐dihydroquinazolin‐one compounds as novel inhibitors of myeloperoxidase. ACS Med Chem Lett 6: 1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linker RA, Lee DH, Ryan S, Dam AM, Conrad R, Bista P et al. (2011a). Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 134: 678–692. [DOI] [PubMed] [Google Scholar]
- Lou H, Jing X, Wei X, Shi H, Ren D, Zhang X (2014). Naringenin protects against 6‐OHDA‐induced neurotoxicity via activation of the Nrf2/ARE signaling pathway. Neuropharmacology 79: 380–388. [DOI] [PubMed] [Google Scholar]
- Lu J, Wu L, Jiang T, Wang Y, Zhao H, Gao Q et al. (2015). Angiotensin AT2 receptor stimulation inhibits activation of NADPH oxidase and ameliorates oxidative stress in rotenone model of Parkinson's disease in CATH.a cells. Neurotoxicol Teratol 47: 16–24. [DOI] [PubMed] [Google Scholar]
- Ma Q, Battelli L, Hubbs AF (2006). Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant‐activated transcription factor Nrf2. Am J Pathol 168: 1960–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Tan MS, Yu JT, Tan L (2014). Resveratrol as a therapeutic agent for Alzheimer's disease. Biomed Res Int 2014: 350516–350529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrali G, Casale F, Salamone P, Fuda G, Caorsi C, Amoroso A et al. (2014). NADPH oxidase (NOX2) activity is a modifier of survival in ALS. J Neurol 261: 2178–2183. [DOI] [PubMed] [Google Scholar]
- Martin‐de‐Saavedra MD, Budni J, Cunha MP, Gomez‐Rangel V, Lorrio S, Del L et al. (2013b). Nrf2 participates in depressive disorders through an anti‐inflammatory mechanism. Psychoneuroendocrinology 38: 2010–2022. [DOI] [PubMed] [Google Scholar]
- Massie A, Schallier A, Sim SW, Fernando R, Kobayashi S, Beck H et al. (2011). Dopaminergic neurons of system xc‐ ‐deficient mice are highly protected against 6‐hydroxydopamine‐induced neurotoxicity. FASEB J 25: 1359–1369. [DOI] [PubMed] [Google Scholar]
- McBean GJ (2012). The transslfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids 42: 199–205. [DOI] [PubMed] [Google Scholar]
- McBean GJ, Aslan M, Griffiths HR, Torrão R (2015). Thiol redox homeostasis in neurodegenerative disease. Redox Biol 5: 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus MJ, Murphy MP, Franklin JL (2011). The mitochondria‐targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease. J Neurosci 31: 15703–15715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng P, Yoshida H, Matsumiya T, Imaizumi T, Tanji K, Xing F et al. (2013). Carnosic acid suppresses the production of amyloid‐beta 1‐42 by inducing the metalloprotease gene TACE/ADAM17 in SH‐SY5Y human neuroblastoma cells. Neurosci Res 75: 94–102. [DOI] [PubMed] [Google Scholar]
- Menon VP, Sudheer AR (2007). Antioxidant and anti‐inflammatory properties of curcumin. Adv Exp Med Biol 595: 105–125. [DOI] [PubMed] [Google Scholar]
- Mesci P, Zaïdi S, Lobsiger C, Millecamps S, Escartin C, Seilhean D et al. (2015). System xc‐ is a mediator of microglial function and its deletion slows symptoms in amyotrophic lateral sclerosis mice. Brain 138: 53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki I, Murakami S, Torigoe N, Kitamura Y, Asanuma M (2016). Neuroprotective effects of levetiracetam target xCT in astrocytes in parkinsonian mice. J Neurochem 136: 194–204. [DOI] [PubMed] [Google Scholar]
- Mohajeri M, Sadeghizadeh M, Najafi F, Javan M (2015). Polymerized nano‐curcumin attenuates neurological symptoms in EAE model of multiple slerosis through down regulation of inflammatory and oxidative processes and enhancing neuroprotection and myelin repair. Neuropharmacol 99: 156–167. [DOI] [PubMed] [Google Scholar]
- Narasimhan M, Patel D, Vedpathak D, Rathinam M, Henderson G, Mahimainathan L (2012). Identification of nocel microRNAs in post‐transcriptional control of Nrf2 expression and redox homeostasis in neuronal SH‐SY5Y eclls. PLoS One 7: e51111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayernia Z, Jaquet V, Krause KH (2014). New insights on NOX enzymes in the central nervous system. Antioxid Redox Signal 20: 2815–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng TP, Chiam PC, Lee T, Chua HC, Lim L, Kua EH (2006). Curry consumption and cognitive function in the elderly. Am J Epidemiol 164: 898–906. [DOI] [PubMed] [Google Scholar]
- Okun Z, Gross Z (2012). Fine tuning the reactivity of corrole‐based catalytic antioxidants. Inorg Chem 51: 8083–8090. [DOI] [PubMed] [Google Scholar]
- Ono K, Hasegawa K, Naiki H, Yamada M (2004). Curcumin has potent anti‐amyloidogenic effects for Alzheimer's beta‐amyloid fibrils in vitro . J Neurosci Res 75: 742–750. [DOI] [PubMed] [Google Scholar]
- Onyango IG, Dennis J, Khan SM (2016). Mitochondrial dysfunction in Alzheimer's disease and the rationale for bioenergetics based therapies. Aging Dis 7: 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando RA, Gonzales AM, Royer RE, Deck LM, Vander Jagt DL (2012). A chemical analog of curcumin as an improved inhibitor of amyloid Abeta oligomerization. PLoS One 7: e31869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozarowski M, Mikolajczak PL, Bogacz A, Gryszczynska A, Kujawska M, Jodynis‐Liebert J et al. (2013). Rosmarinus officinalis L. leaf extract improves memory impairment and affects acetylcholinesterase and butyrylcholinesterase activities in rat brain. Fitoterapia 91: 261–271. [DOI] [PubMed] [Google Scholar]
- Pagano PJ, Wipf P, Cifuentes‐Pagano ME, Skoda EM (2014). Preparation of bridged tetrahydroisoquinolines as selective NADPH oxidase 2 inhibitors. PCT Int Appl. [DOI] [PMC free article] [PubMed]
- Page P, Gaggini F, Laleu B (2010). Pyrazoline dione derivatives as NADPH oxidase inhibitors. US Patent Office.
- Page P, Laleu B, Gaggini F, Orchard M (2011). Preparation of pyrazolo piperidine derivatives as NADPH oxidase inhibitors. Eur Pat Appl GenKyoTex SA, Switzerland.
- Pampliega O, Domercq M, Soria FN, Villoslada P, Rodríguez‐Antigüedad A, Matute C (2011). Increased expression of cystine/glutamate antiporter in multiple sclerosis. J Neuroinflamm 8: 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa S, De D (2013). Complex I deficiencies in neurological disorders. Trends Mol Med 19: 61–69. [DOI] [PubMed] [Google Scholar]
- Park JA, Kim S, Lee SY, Kim CS, Kim K, Kim SJ et al. (2008b). Beneficial effects of carnosic acid on dieldrin‐induced dopaminergic neuronal cell death. Neuroreport 19: 1301–1304. [DOI] [PubMed] [Google Scholar]
- Pehar M, Vargas MR, Cassina P, Barbeito AG, Beckman JS, Barbeito L (2005). Complexity of astrocyte‐motor neuron interactions in amyotrophic lateral sclerosis. Neurodegener Dis 2: 139–146. [DOI] [PubMed] [Google Scholar]
- Perry TL, Yong VW (1986). Idiopathic Parkinson's disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci Lett 67: 269–274. [DOI] [PubMed] [Google Scholar]
- Piani D, Fontana A (1994). Involvement of the cystine transport system xc‐ in the macrophage‐induced glutamate‐dependent cytotoxicity to neurons. J Immunol 152: 3578–3585. [PubMed] [Google Scholar]
- Probst BL, Trevino I, McCauley L, Bumeister R, Dulubova I, Wigley WC et al. (2015). RTA 408, a novel synthetic triterpenoid with broad anticancer and anti‐inflammatory activity. PLoS One 10: e0122942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido‐Moran M, Moreno‐Fernandez J, Ramirez‐Tortosa C, Ramirez‐Tortosa M (2016). Curcumin and Health. Molecules 21: E264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M (2006). System xc‐ and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid‐beta peptide 1‐40. J Neurosci 26: 3345–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R (2013). Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects. Acc Chem Res 46: 550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA et al. (2007). Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol 66: 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ (2001). Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2‐ and systolic blood pressure in mice. Circ Res 89: 408–414. [DOI] [PubMed] [Google Scholar]
- Rocha Lindner G, Bonfanti Santos D, Colle D, Gasnhar Moreira EL, Daniel Prediger R, Farina M et al. (2015b). Improved neuroprotective effects of resveratrol‐loaded polysorbate 80‐coated poly(lactide) nanoparticles in MPTP‐induced Parkinsonism. Nanomedicine (Lond) 10: 1127–1138. [DOI] [PubMed] [Google Scholar]
- Rojo AI, McBean GJ, Cindric M, Egea J, López MG, Rada P et al. (2014). Redox control of microglial function: molecular mechanisms and functional significance. Antioxid Redox Signal 21: 1766–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo AI, Sagarra MR, Cuadrado A (2008). GSK‐3beta down‐regulates the transcription factor Nrf2 after oxidant damage: relevance to exposure of neuronal cells to oxidative stress. J Neurochem 105: 192–202. [DOI] [PubMed] [Google Scholar]
- Rushmore TH, King RG, Paulson KE, Pickett CB (1990). Regulation of glutathione S‐transferase Ya subunit gene expression: identification of a unique xenobiotic‐responsive element controlling inducible expression by planar aromatic compounds. Proc Natl Acad Sci U S A 87: 3826–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruszkiewicz J, Albrecht J (2015). Changes in the mitochondrial antioxidant systems in neurodegenerative diseases and acute brain disorders. Neurochem Int 88: 66–72. [DOI] [PubMed] [Google Scholar]
- Sarlette A, Krampfl K, Grothe C, Neuhoff N, Dengler R, Petri S (2008). Nuclear erythroid 2‐related factor 2‐antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 67: 1055–1062. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Ishii T, Bannai S (1999). Cloning and expression of a plasma membane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 274: 11455–11458. [DOI] [PubMed] [Google Scholar]
- Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y et al. (2008). Carnosic acid, a catechol‐type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S‐alkylation of targeted cysteines on Keap1. J Neurochem 104: 1116–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savaskan E, Olivieri G, Meier F, Seifritz E, Wirz‐Justice A, Muller‐Spahn F (2003). Red wine ingredient resveratrol protects from beta‐amyloid neurotoxicity. Gerontol 49: 380–383. [DOI] [PubMed] [Google Scholar]
- Savion N, Izigov N, Morein M, Pri‐Chen S, Kotev‐Emeth S (2014). S‐Allylmercapto‐N‐acetylcysteine (ASSNAC) protects cultured nerve cells from oxidative stress and attenuates experimental autoimmune encephalomyelitis. Neurosci Lett 583: 108–113. [DOI] [PubMed] [Google Scholar]
- Schallier A, Smolders I, Van D, Loyens E, De PP, Michotte A et al. (2011b). Region‐ and age‐specific changes in glutamate transport in the AbetaPP23 mouse model for Alzheimer's disease. J Alzheimers Dis 24: 287–300. [DOI] [PubMed] [Google Scholar]
- Schildknecht S, Weber A, Gerding HR, Pape R, Robotta M, Drescher M et al. (2014). The NOX1/4 inhibitor GKT136901 as selective and direct scavenger of peroxynitrite. Curr Med Chem 21: 365–376. [DOI] [PubMed] [Google Scholar]
- Schilling S, Goelz S, Linker R, Luehder F, Gold R (2006). Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin Exp Immunol 145: 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt B, Vicenzi M, Garrel C, Denis FM (2015). Effects of N‐acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: a comparative crossover study. Redox Biol 6: 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel P, Roth M (2013). Anti‐inflammatory dimethylfumarate: a potential new therapy for asthma? Mediators Inflamm 2013: 875403–875413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seredenina T, Chiriano G, Filippova A, Nayernia Z, Mahiout Z, Fioraso‐Cartier L et al. (2015). A subset of N‐substituted phenothiazines inhibits NADPH oxidases. Free Radic Biol Med 86: 239–249. [DOI] [PubMed] [Google Scholar]
- Shimojo Y, Kosaka K, Noda Y, Shimizu T, Shirasawa T (2010). Effect of rosmarinic acid in motor dysfunction and life span in a mouse model of familial amyotrophic lateral sclerosis. J Neurosci Res 88: 896–904. [DOI] [PubMed] [Google Scholar]
- Silva‐Adaya D, Gonsebatt ME, Guevara J (2014). Thioredoxin system regulation in the central nervous system: experimental models and clinical evidence. Oxid Med Cell Longev 2014: 590808–590825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slosky LM, Vanderah TW (2015). Therapeutic potential of peroxynitrite decomposition catalysts: a patent review. Expert Opin Ther Pat 25: 443–466. [DOI] [PubMed] [Google Scholar]
- Smeyne M, Smeyne RJ (2013). Glutathione metabolism and Parkinson's disease. Free Radic Biol Med 62: 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solesio ME, Prime TA, Logan A, Murphy MP, Del M, Jordan J et al. (2013c). The mitochondria‐targeted anti‐oxidant MitoQ reduces aspects of mitochondrial fission in the 6‐OHDA cell model of Parkinson's disease. Biochim Biophys Acta 1832: 174–182. [DOI] [PubMed] [Google Scholar]
- Sontheimer H, Bridges RJ (2012). Sulfasalazine for brain cancer fits. Expert Opin Investig Drugs 21: 575–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorce S, Krause KH (2009). NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal 11: 2481–2504. [DOI] [PubMed] [Google Scholar]
- Sorce S, Krause KH, Jaquet V (2012). Targeting NOX enzymes in the central nervous system: therapeutic opportunities. Cell Mol Life Sci 69: 2387–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CM, Crabtree‐Hartman EC, Lehmann‐Horn K, Cree BA, Zamvil SS (2015). Reduction of CD8(+) T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm 2: e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ (1994). Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy‐substituted catechol. Am J Respir Cell Mol Biol 11: 95–102. [DOI] [PubMed] [Google Scholar]
- Su ZY, Shu L, Khor TO, Lee JH, Fuentes F, Kong AN (2013). A perspective on dietary phytochemicals and cancer chemoprevention: oxidative stress, Nrf2 and epigenomics. Top Curr Chem 329: 133–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QA, Hess DT, Wang B, Miyagi M, Stamler JS (2012). Off‐target thiol alkylation by the NADPH oxidase inhibitor 3‐benzyl‐7‐(2‐benzoxazolyl)thio‐1,2,3‐triazolo[4,5‐d]pyrimidine (VAS2870). Free Radic Biol Med 52: 1897–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Chin YE, Zhang DD (2009). Acetylation of Nrf2 by p300/CBP augments promoter‐specific DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol 29: 2658–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunitha K, Hemshekhar M, Thushara RM, Santhosh MS, Yariswamy M, Kemparaju K et al. (2013). N‐Acetylcysteine amide: a derivative to fulfill the promises of N‐Acetylcysteine. Free Radic Res 47: 357–367. [DOI] [PubMed] [Google Scholar]
- Tanji KA, Maruyame S, Odagiri F, Mori K, Itoh A, Kakita H et al. (2013). Keap1 is localized in neuronal and glial cytoplasmic inclusions in various neurodegenerative diseases. J Neuropathol Exp Neurol 72: 18–28. [DOI] [PubMed] [Google Scholar]
- Tegtmeier F, Walter U, Schinzel R, Wingler K, Scheurer P, Schmidt H (2005). Compounds containing a N‐heteroaryl moeity linked to fused ring moeities for the inhibition of NAD(P)H oxidases and platelet activation. European Patent Application EP1598354.
- ten H, Huntgeburth M, Wingler K, Schnitker J, Baumer AT, Vantler M et al. (2006). Novel NOX inhibitor VAS2870 attenuates PDGF‐dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res 71: 331–341. [DOI] [PubMed] [Google Scholar]
- Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S (2002). Identification of Nrf2‐regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 62: 5196–5203. [PubMed] [Google Scholar]
- Trials.gov C (2015). Safety and Efficacy of oral GKT137831 in patient with type 2 diabetes and albuminuria. Available from https://clinicaltrialsgov/ct2/show/study/NCT02010242.
- Trumbull KA, McAllister D, Gandelman MM, Fung WY, Lew T, Brennan L et al. (2012). Diapocynin and apocynin administration fails to significantly extend survival in G93 A SOD1 ALS mice. Neurobiol Dis 45: 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YM, Chien CF, Lin LC, Tsai TH (2011). Curcumin and its nano‐formulation: the kinetics of tissue distribution and blood–brain barrier penetration. Int J Pharm 416: 331–338. [DOI] [PubMed] [Google Scholar]
- Tsianakas A, Herzog S, Landmann A, Patsinakidis N, Perusquia Ortiz AM, Bonsmann G et al. (2014). Successful treatment of discoid lupus erythematosus with fumaric acid esters. J Am Acad Dermatol 71: e15–e17. [DOI] [PubMed] [Google Scholar]
- Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T et al. (2013). The Keap1‐Nrf2 system prevents onset of diabetes mellitus. Mol Cell Biol 33: 2996–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Horssen J, Schreibelt G, Drexhage J, Hazes T, Dijkstra CD, van der Valk P et al (2008). Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic Biol Med 45: 1729-1737. [DOI] [PubMed] [Google Scholar]
- van Horssen J, Drexhage JA, Flor T, Gerritsen W, Valk P, Vries HE (2010). Nrf2 and DJ1 are consistently upregulated in inflammatory multiple sclerosis lesions. Free Radic Biol Med 49: 1283–1289. [DOI] [PubMed] [Google Scholar]
- Vilhardt F, Haslund‐Vinding J, Jaquet V, McBean G (2017). Microglia antioxidant systems and redox signalling. Brit J Pharmacol 174: 1719–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitvitsky V, Thomas M, Ghorpade A, Gendelman HE, Banerjee RA (2006). A functional transsulfuration pathway in the brain links to GSH homeostasis. J Biol Chem 281: 25785–35793. [DOI] [PubMed] [Google Scholar]
- Wang YY, Yang YX, Zhe H, He ZX, Zhou SF (2014). Bardoxolone methyl (CDDO‐Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties. Drug Des Devel Ther 8: 2075–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkaniec A, Strosznajder JB, Adamczyk A (2013). Toxicity of extracellular secreted alpha‐synuclein: its role in nitrosative stress and neurodegeneration. Neurochem Int 62: 776–783. [DOI] [PubMed] [Google Scholar]
- Wind S, Beuerlein K, Eucker T, Muller H, Scheurer P, Armitage ME et al. (2010). Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol 161: 885–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CR, Tsai CW, Chang SW, Lin CY, Huang LC, Tsai CW (2015). Carnosic acid protects against 6‐hydroxydopamine‐induced neurotoxicity in in vivo and in vitro model of Parkinson's disease: involvement of antioxidative enzymes induction. Chem Biol Interact 225: 40–46. [DOI] [PubMed] [Google Scholar]
- Wu KC, Cui JY, Klaassen CD (2011). Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci 123: 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Li XK, Funeshima‐Fuji N, Kimura H, Matsumoto Y, Isaka Y et al. (2009). Amelioration of experimental autoimmune encephalomyelitis by curcumin treatment through inhibition of IL‐17 production. Int Immunopharmacol 9: 575–581. [DOI] [PubMed] [Google Scholar]
- Xu Z, Zhang F, Sun F, Gu K, Dong S, He D (2015). Dimethyl fumarate for multiple sclerosis. Cochrane Database Syst Rev 4: Cd011076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS et al. (2005). Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo . J Biol Chem 280: 5892–5901. [DOI] [PubMed] [Google Scholar]
- Yates MS, Tran QT, Dolan PM, Osburn WO, Shin S, McCulloch CC et al. (2009). Genetic versus chemoprotective activation of Nrf2 signaling: overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid‐treated mice. Carcinogenesis 30: 1024–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Meng P, Matsumiya T, Tanji K, Hayakari R, Xing F et al. (2014). Carnosic acid suppresses the production of amyloid‐beta 1‐42 and 1‐43 by inducing an alpha‐secretase TACE/ADAM17 in U373MG human astrocytoma cells. Neurosci Res 79: 83–93. [DOI] [PubMed] [Google Scholar]
- Zaitone SA, Abo‐Elmatty DM, Shaalan AA (2012). Acetyl‐L‐carnitine and alpha‐lipoic acid affect rotenone‐induced damage in nigral dopaminergic neurons of rat brain, implication for Parkinson's disease therapy. Pharmacol Biochem Behav 100: 347–360. [DOI] [PubMed] [Google Scholar]
- Zawada WM, Mrak RE, Biedermann J, Palmer QD, Gentleman SM, Aboud O et al. (2015). Loss of angiotensin II receptor expression in dopamine neurons in Parkinson's disease correlates with pathological progression and is accompanied by increases in Nox4‐ and 8‐OH guanosine‐related nucleic acid oxidation and caspase‐3 activation. Acta Neuropathol Commun 3: 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevalk GD, Bernard LP, Guilford FT (2010). Liposomal‐glutathione provides maintenance of intracellular glutathione and neuroprotection in mesencephalic neuronal cells. Neurochem Res 35: 1575–1587. [DOI] [PubMed] [Google Scholar]
- Zeevalk GD, Manzino L, Sonsalla PK, Bernard LP (2007). Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: relevance to Parkinson's disease. Exp Neurol 203: 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Miao QW, Zhu CX, Zhao Y, Liu L, Yang J et al. (2014). Sulforaphane ameliorates neurobehavioral deficits and protects the brain from amyloid beta deposits and peroxidation in mice with Alzheimer‐like lesions. Am J Alzheimers Dis Other Demen 30: 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielonka J, Zielonka M, VerPlank L, Cheng G, Hardy M, Ouari O et al. (2016). Mitigation of NADPH oxidase 2 activity as a strategy to inhibit peroxynitrite formation. J Biol Chem 291: 7029–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]