

Abstract
Although reactive oxygen species (ROS) act as crucial factors in the onset and progression of a wide array of diseases, they are also involved in numerous signalling pathways related to cell metabolism, growth and survival. ROS are produced at various cellular sites, and it is generally agreed that mitochondria generate the largest amount, especially those in cardiomyocytes. However, the identification of the most relevant sites within mitochondria, the interaction among the various sources, and the events responsible for the increase in ROS formation under pathological conditions are still highly debated, and far from being clarified. Here, we review the information linking the adaptor protein p66Shc with cardiac injury induced by ischaemia and reperfusion (I/R), including the contribution of risk factors, such as metabolic syndrome and ageing. In response to several stimuli, p66Shc migrates into mitochondria where it catalyses electron transfer from cytochrome c to oxygen resulting in hydrogen peroxide formation. Deletion of p66Shc has been shown to reduce I/R injury as well as vascular abnormalities associated with diabetes and ageing. However, p66Shc‐induced ROS formation is also involved in insulin signalling and might contribute to self‐endogenous defenses against mild I/R injury. In addition to its role in physiological and pathological conditions, we discuss compounds and conditions that can modulate the expression and activity of p66Shc.
Linked Articles
This article is part of a themed section on Redox Biology and Oxidative Stress in Health and Disease. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.12/issuetoc
Abbreviations
- ATG
DNA codon for methionine
- CAD
coronary artery disease
- CH
collagen homologues
- Δψm
mitochondrial membrane potential
- ETC
electron transport chain
- GSH/GSSG
GSH in its reduced or oxidized form
- IGF‐1
insulin‐like growth factor 1
- PTP
permeability transition pore
- PBM
peripheral blood monocyte
- Ras
rat sarcoma
- SH2
sarcoma homologous type 2
- Sirtuin
silent mating type information regulation 2 homolog
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Enzymes c |
| Bcl‐2 | Akt (PKB) |
| GPCRs b | Arginase II |
| α1‐adrenoceptor | ATP synthase |
| PAR1 | Caspase‐3 |
| Endothelial NOS | |
| ERK1 | |
| ERK2 | |
| JNK1 | |
| MMP2 | |
| mTOR | |
| PKCβ | |
| PTEN | |
| Sirtuin 1 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a, 2015b, 2015c).
Cardiac injury induced by ischaemia and reperfusion
The maintenance of cardiac structure and function depends on the continuous supply of ATP resulting from the mitochondrial coupling of substrate oxidation with ATP synthesis, termed as oxidative phosphorylation. The strict dependence of the heart on aerobic metabolism is indicated by the large fraction (i.e. >30%) of cardiomyocyte volume occupied by mitochondria. Therefore, it is hardly surprising that mitochondrial dysfunction and cardiac diseases are inevitably associated. This concept is perfectly exemplified by cardiac injury induced by ischaemia and reperfusion.
Because more than 95% of oxygen is utilized by the terminal reaction of the respiratory chain, namely, cytochrome oxidase, anoxia or ischaemia is established when oxygen availability is no longer sufficient for the activity of cytochrome oxidase. Therefore, within any cell, ischaemic injury is determined primarily by mitochondrial dysfunction.
The arrest of electron flow causes inevitably a drop in mitochondrial membrane potential (Δψm) that is the driving force for ATP synthesis by Fo–F1 ATP synthase. Not only is ATP synthesis curtailed, but also its hydrolysis is stimulated to maintain Δψm. To this aim, the Fo–F1 ATP synthase inverts its physiological operation, becoming an ATPase that pumps protons from the matrix into the intermembrane space at the expense of ATP hydrolysis. In this way, mitochondria change from ATP producers into avid utilizers that hydrolyze not only intramitochondrial ATP but also cytosolic ATP produced by anaerobic glycolysis. These biochemical changes have profound functional consequences. On the one hand, the stimulation of glycolysis resulting in lactate formation causes intracellular acidification that is responsible for the asystole occurring in the first minutes of ischaemia. On the other hand, the drop in ATP content causes rigour contracture (Di Lisa et al., 1998).
If coronary flow is re‐established after a short ischaemic episode, viability is maintained, and contractility slowly recovers (Bolli and Marbán, 1999). However, when reperfusion occurs after a prolonged ischaemia (i.e. more than 20 min of no flow ischaemia in crystalloid‐perfused isolated hearts), viability and contractility are no longer restored (Jennings et al., 1975; Di Lisa et al., 1998; Yellon and Hausenloy, 2007). In fact, under these conditions reperfusion seems to cause additional injury, because cardioprotection can be obtained by means of intervention applied at the onset of reperfusion (Ovize et al., 2010).
Mitochondrial dysfunction is likely to be pivotal in determining the very rapid transition towards irreversible injury occurring upon reperfusion. In this respect, a seminal observation was the presence of calcium precipitates within swollen mitochondria in irreversibly‐injured cardiomyocytes (Shen and Jennings, 1972). This finding implies that the readmission of oxygen allows mitochondria to recover Δψm required for mitochondrial Ca2 + uptake. In this process, mitochondria remove the excess Ca2 + in the cytosol that might activate proteases, phospholipases and signalling pathways jeopardizing cell survival. However, a large increase in Ca2+ uptake hampers ATP synthesis, because both these processes depend on Δψm (Bernardi, 1999). The resulting lack of recovery of ATP content in the presence of an elevated intracellular [Ca2 +] is likely to promote hypercontracture eventually leading to sarcolemma rupture (Altschuld et al., 1985; Siegmund et al., 1991; Silverman and Stern, 1994). The dependence of this deleterious sequence of events on mitochondrial function is indicated by the attenuation of reperfusion‐induced cell death induced by respiratory chain inhibitors or uncouplers of oxidative phosphorylation (Ganote et al., 1976; Elz and Nayler, 1988). This notion indicates that during reperfusion, mitochondria are paradoxically essential for both functional recovery after a short ischaemic episode and cell death after a prolonged ischaemia.
Besides decreasing ATP synthesis, a large elevation in matrix [Ca2 +] further contributes to ATP depletion and mitochondrial de‐energization by promoting the opening of the mitochondrial permeability transition pore (PTP) (Bernardi and Di Lisa, 2015). The elevation of intramitochondrial [Ca2 +] per se might be not sufficient to trigger PTP opening. In fact, in isolated mitochondria, PTP opening is obtained in the presence of [Ca2 +] >0.1 M, which is hardly attained in viable cells. Therefore, within intact cells, the PTP sensitivity to Ca2 + is likely to be increased by additional factors or processes among which ROS appear extremely relevant (Bernardi and Di Lisa, 2015). Post‐ischaemic reperfusion causes a rapid elevation in ROS levels (Zweier, 1988; Bolli and Marbán, 1999) that has been recently demonstrated to occur within mitochondria in vivo (Chouchani et al., 2013). ROS favours PTP opening that increases ROS formation generating a vicious cycle. This process termed as ROS‐induced ROS release is likely to exacerbate the injury within a cardiomyocyte and spread it to adjacent cardiomyocytes (Zorov et al., 2014).
Although the relevance of mitochondria in ROS formation is well established, especially under pathological conditions, it is far from clear which mitochondrial enzymes provide the largest contribution to oxidative stress.
Mitochondrial ROS formation
Mitochondria contain enzymes that catalyse hydrogen peroxide (H2O2) generation as the essential product along with systems generating ROS in sporadic, possibly undesired, reactions. This is especially the case with the electron transport chain (ETC) (Murphy, 2009; Chen and Zweier, 2014; Zorov et al., 2014). Indeed, a minor fraction (about 0.1%) of the electrons flowing through the ETC is thought to cause the partial reduction of oxygen (O2) into superoxide that is then rapidly reduced to H2O2 by superoxide dismutases (SODs) (Murphy, 2009). Therefore, H2O2 is the main ROS produced. The removal of H2O2 by mitochondrially targeted catalase has been shown to be cardioprotective (Schriner et al., 2005; Anderson et al., 2009; Dai et al., 2011). Besides superoxide dismutation, peroxide handling is carried out by a thiol redox system centred on glutathione (GSH and GSSG in its reduced and oxidized form, respectively) and thioredoxin (Berndt et al., 2007; Murphy, 2012; Forman et al., 2014; Nickel et al., 2014).
The physiological role of mitochondrial ROS generation in a wide variety of cardiomyocyte functions is likely to depend on post‐translational modifications of proteins, especially at the level of cysteine residues (Finkel, 2012). Long‐lasting changes are obtained by the effects of mitochondrial ROS on transcriptional factors, such as hypoxia‐inducible factor (HIF) and nuclear factor erythroid 2‐related factor 2 (Nrf2) (Hayes and Dinkova‐Kostova, 2014; Semenza, 2014; Yun and Finkel, 2014). The detrimental effects on proteins, lipids, carbohydrates and nucleotides obtained in the presence of high levels of ROS explain the large body of evidence linking a high mitochondrial formation of ROS with every cardiac disease. This is especially the case with ischaemia–reperfusion injury.
Besides respiratory chain complexes, several other mitochondrial enzymes, such as glycerol‐3‐phosphate and 2‐oxoglutarate dehydrogenase, have been described as potential ROS producers. The list of dedicated enzymes for ROS formation in mitochondria might include nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4; Ago et al., 2010), although results from a recent study argued against its mitochondrial localization in the normoxic heart (Hirschhäuser et al., 2015).
However, the contribution of the respiratory chain and mitochondrial dehydrogenases to the overall ROS production of mitochondria in vivo cannot be defined precisely, because their deletion would inevitably compromise functions other than ROS generation. The demonstration that mitochondria generate ROS in vivo is provided by studies on other mitochondrial enzymes, such as p66Shc and monoamine oxidases that generate H2O2 as a direct and obligatory product. While the relevance of monoamine oxidase (MAO) in cardiac diseases has been covered in a recent review (Kaludercic et al., 2014), the role of p66Shc in cardiovascular pathophysiology is detailed in the following sections.
p66Shc structure, function and expression
Biochemical features
p66Shc is a ubiquitously expressed vertebrate protein. It sustains the intracellular concentration of ROS by catalysing their formation from the mitochondrial respiratory chain, by triggering plasma membrane oxidases and by suppressing ROS scavenging (see for review Trinei et al., 2009; Trinei et al., 2013).
P66Shc is one of the three isoforms encoded by the human and mouse ShcA locus. ShcA was identified in 1992 by low‐stringency hybridization to human cDNA libraries, using a sarcoma homologous type 2 (SH2) coding sequence as a probe (Pelicci et al., 1992). It is located on chromosome 1q21. The originally isolated ShcA transcript displayed two in‐frame ATGs and was shown to encode two ubiquitously expressed polypeptides: p52Shc and p46Shc.
Members of the spontaneous human combustion (Shc) family are characterized by the phosphotyrosine binding domain (PTB) –collagen homologues 1 (CH1)–SH2 modularity where CH1 is a glycine/proline rich region, containing two major phosphorylation sites (tyrosine 239–240 and tyrosine 317), SH2 is the C‐terminal domain involved in protein–protein interaction and the formation of multi‐protein signalling complexes and PTB is able to bind non‐phosphorylated tyrosine‐containing peptides. p66Shc contains an additional amino‐terminal proline rich region, named CH2.
Relations with signalling pathways and H2O2 production
Isoforms p46Shc and p52Shc serve as phosphotyrosine adaptor molecules in various receptor‐mediated signalling pathways. Once phosphorylated, they recruit the growth factor receptor‐bound protein 2 (Grb2)/son of sevenless (SOS) complex to the plasma membrane (Ravichandran, 2001) and subsequently activate the MAPK cascade. p52Shc and p46Shc function as initiators of the rat sarcoma (Ras) signalling cascade in various non‐neuronal systems (Migliaccio et al., 1997). p66Shc is a target of receptor tyrosine kinases (Okada et al., 1997) and is able to bind the Grb2/SOS complex. However, p66Shc is not involved in Ras signalling regulation, and its overexpression has a negative effect on the Ras–MAPK–fos pathway in response to EGF. On the contrary, p66Shc converts stress signals into apoptosis (Migliaccio et al., 1999).
A fraction of p66Shc has been observed within the mitochondrial intermembrane space, although the import mechanism of p66Shc into mitochondrial intermembrane space has not been clearly identified. During apoptosis, p66Shc levels in mitochondria increase. Some stress kinases such as JNK‐1 and PKCß phosphorylate p66Shc on serine 36, and peptidyl‐prolyl cis–trans isomerase‐1 induces its prolyl‐isomerization (Pinton et al., 2007); these post‐translational modifications are involved in p66Shc translocation into mitochondria. In basal conditions, mitochondrial p66Shc associates to a high molecular weight complex of about 670 kDa and to heat shock protein 70 (Orsini et al., 2006) and components of the transporter of the outer and inner mitochondrial membrane complex (Cosentino et al., 2008). Notably, treatment of cells with pro‐apoptotic stimuli such as ultraviolet light or H2O2 induces the dissociation of these complexes and the release of monomeric p66Shc, which reacts with cytochrome c (Orsini et al., 2006) sequestering electrons from the ETC to generate H2O2 (Giorgio et al., 2005; Trinei et al., 2013). Through this process, p66Shc appears to provide an important contribution to mitochondrial ROS formation that has been highlighted in a wide array of physiological and pathological conditions (Figure 1).
Figure 1.
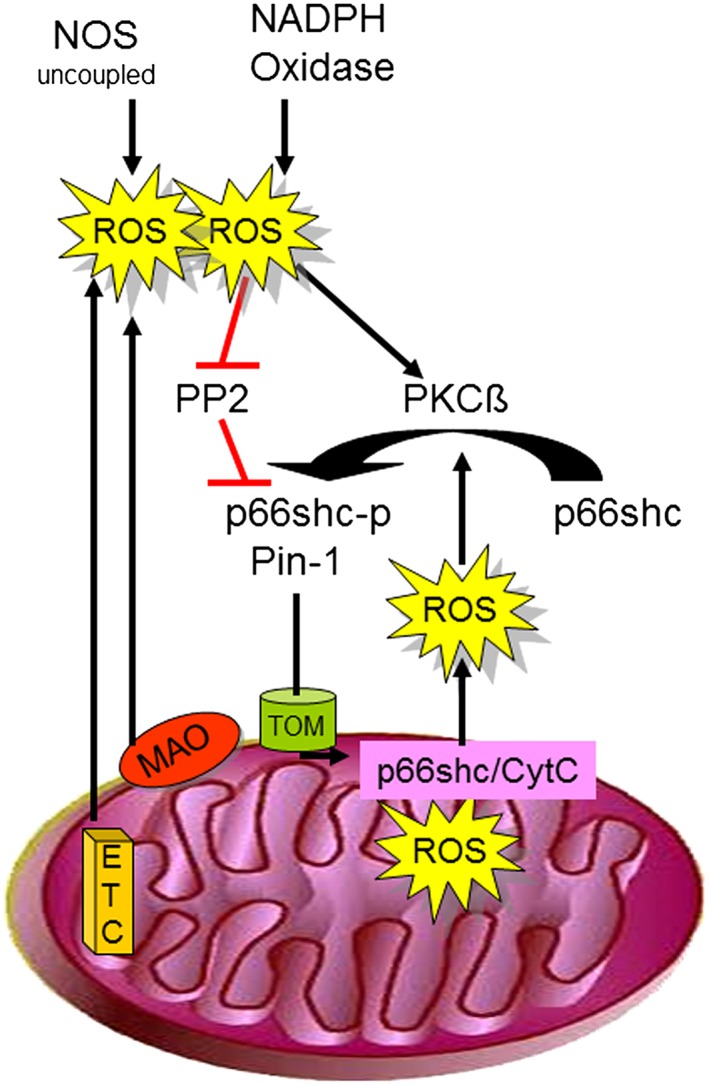
ROS derived from uncoupled NOS, NAPDH oxidase, monoamine oxidases (MAO) or the ETC activate PKCß and – at the same time – inhibit protein phosphate 2 (PP2). Both PKCß activation and PP2 inhibition contribute to increased p66Shc phosphorylation, which then translocate in the presence of peptidyl‐propyl cis–trans isomerase 1 (Pin‐1) into the mitochondrial intermembrane space using the translocator of the outer mitochondrial membrane (TOM). p66Shc then catalyses electron transfer from cytochrome c to oxygen, resulting in increased ROS formation.
Modulation of p66Shc expression in the heart
Compared with wild‐type littermates, p66Shc knockout mice had similar blood pressure, heart rate and left ventricular wall thickness. However, cardiomyocyte number was increased in p66Shc knockout mice (Graiani et al., 2005). One potential explanation for a decreased cardiomyocyte number at a similar myocardial wall thickness and cardiac weight (implying hypertrophied cardiomyocytes) in wild‐type mice relates to the expression of p66Shc that was detected only in neonatal, but not in adult cardiomyocytes (Obreztchikova et al., 2006). In cardiomyocyte cultures from 2‐day‐old rats, an α1‐adrenoceptor ROS‐dependent p66Shc–Akt–forkhead box O3 (FOXO3) phosphorylation pathway decreased SOD2 expression, thereby impairing ROS detoxification and increasing cardiomyocyte apoptosis. Furthermore, in these cardiomyocytes, p66Shc acted as a negative regulator of hypertrophy (Guo et al., 2009), potentially explaining the above differences found in in vivo hearts.
Ageing, comparing 6‐ and 36‐month‐old Fischer 344xBN rats, was associated with decrements in cardiac mitochondrial content and respiratory function. Mitochondria from aged hearts demonstrated a greater enrichment of p66Shc (Ljubicic et al., 2010). Similarly, intense exercise activated p66Shc; in male Wistar rats, swimming for 3 h per day increased p66Shc phosphorylation, surprisingly without a significant increase in either mitochondrial ROS release or mitochondrial oxidative stress markers or the antioxidant enzyme activities (SOD and catalase) (Ziolkowski et al., 2015). Thus, regular exercise and ageing affect cardiac p66Shc expression and/or activity; however, the question whether such an increase in p66Shc expression and/or activity relates to cardiomyocyte mitochondria or to mitochondria obtained from other cell types (such as fibroblasts) cannot be resolved using whole cardiac homogenates and remains to be answered using isolated cell preparations.
p66Shc expression appears to be low in cardiomyocytes under physiological conditions, yet it can be induced by various stimuli. In vitro experiments showed that angiotensin II caused apoptotic death of cardiomyocytes isolated from wild‐type but not p66Shc knockout mice hearts (Graiani et al., 2005). Consistent with the in vitro results, infusion of a subpressor dose of angiotensin II (300 nmol·kg−1 body weight daily for 28 days) caused left ventricular hypertrophy and apoptotic death of cardiomyocytes and endothelial cells in wild‐type but not p66Shc knockout mice (Graiani et al., 2005).
Thrombin activates protease‐activated receptor‐1 (PAR1) and engages signalling pathways that influence the growth and survival of cardiomyocytes as well as extracellular matrix remodelling by cardiac fibroblasts. Thrombin increased p66Shc phosphorylation at serine 36 in cardiac fibroblasts and cardiomyocytes, the latter requiring increased PKC and MAPK/ERK activities. Pasteurella multocida toxin, a Gαq agonist, also promoted p66Shc expression and cardiomyocyte hypertrophy (Obreztchikova et al., 2006).
p66Shc and cardiac pathologies
Ischaemia/reperfusion injury
Initial evidence for p66Shc involvement in ischaemia/reperfusion injury was obtained by showing that in hindlimb ischaemia, p66Shc deletion prevented the decrease in both capillary density and tissue viability (Zaccagnini et al., 2004). p66Shc deletion in satellite muscle and endothelial cells protected against apoptosis by reducing ROS formation. Cardiac protection was demonstrated in isolated mouse hearts undergoing ischaemia and reperfusion. p66Shc deletion resulted in a decrease in both lactate dehydrogenase release reflecting viability maintenance and parameters of oxidative stress, such as malondialdehyde formation and tropomyosin oxidation (Carpi et al., 2009). Interestingly, this cardioprotective efficacy associated with p66Shc ablation was comparable with that of other antioxidant interventions and could not be increased by antioxidant co‐administration, thus suggesting that p66Shc is downstream of other pathways involved in ROS formation.
In isolated guinea pig hearts, phosphorylation of p66Shc and its translocation into mitochondria increased during reperfusion after 20 and 30 min ischaemia, but not during ischaemia only, or during 5 or 10 min ischaemia followed by 20 min reperfusion. Amobarbital, a complex I blocker, or hispidin, a PKCß inhibitor, reduced p66Shc phosphorylation and its mitochondrial translocation induced by 30 min ischaemia and 20 min reperfusion. Decreased phosphorylation of p66Shc by amobarbital or hispidin led to better functional recovery and less infarction during reperfusion (Yang et al., 2014).
In contrast to the above finding, when reperfusion was established after 30 min of ischaemia, p66Shc knockout was associated with larger infarcts compared with wild‐type mice. p66Shc inhibition was not associated with modifications in post‐infarction inflammation, oxidative burst, cardiac vessel density or structure. Thus, genetic deletion of p66Shc increased susceptibility to myocardial injury in response to reperfusion after short‐term ischaemia (Akhmedov et al., 2015). Notably, in the in vivo mice study, infarct size following 30 ischaemia reperfusion after 30 min of ischaemia was associated with an infarct size much lower than that commonly detected in other laboratories. Furthermore, the difference in infarct size between wild‐type and p66Shc knockout mice was minimal, if not negligible, casting doubts on its biological/clinical importance. Finally, p66Shc phosphorylation or translocation to mitochondria in wild‐type mice was not assessed.
Besides significant technical concerns of the above study, there is indeed the potential of p66Shc being protective during short periods of ischaemia. p66Shc might increase the resistance to shorter periods of ischaemia by activating Akt (as part of the so‐called reperfusion injury salvage kinase pathway) through increased phosphorylation. Akt phosphorylation can be enhanced by inhibition of protein phosphatases, among which protein tyrosine phosphatases are inactivated by ROS via oxidation of critical cysteinyl residues. Indeed in fibroblasts, p66Shc deletion decreased oxidation and thus inactivation of protein phosphatases associated with a reduced activation of ERK and Akt (Frijhoff et al., 2014).
In p66Shc knockout mice, activation of the protective and anti‐apoptotic reperfusion injury salvage kinase (RISK) and survivor activating factor enhancement pathways following 30 min ischaemia and reperfusion were blunted, and mitochondrial swelling and cellular apoptosis via the caspase‐3 pathway increased compared with wild type mice (Akhmedov et al., 2015).
Therefore, p66Shc‐induced ROS formation could contribute to endogenous self‐defenses triggered by a short/mild ischaemic episode, yet no information is available on the role of p66Shc in the protection attributed to conditioning stimuli. However, p66Shc‐induced ROS formation would become detrimental when exacerbated upon reperfusion after a long/severe ischaemia or prolonged over time as appears to be the case with hypertensive or diabetic cardiomyopathy (Figure 2).
Figure 2.
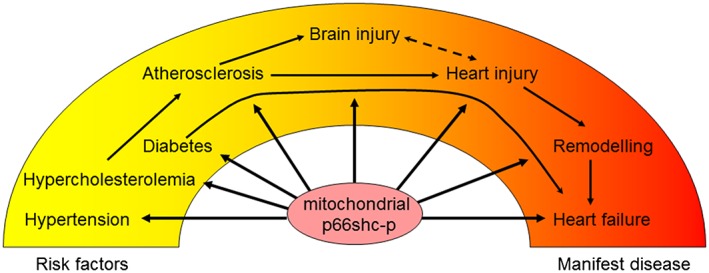
Increased translocation of p66Shc into mitochondria results in increased ROS formation. Indeed, p66Shc activation contributes to several cardiovascular pathologies including diabetes, atherosclerosis, acute brain and heart injury as well as cardiac remodelling and failure. Thus, p66Shc may be an interesting target to attenuate cardiovascular disease progression.
Surprisingly, little attention has been given to the role of p66Shc in post‐myocardial infarction changes. Recently, evidence has been provided that p66Shc deletion improves myocardial healing and reduces cardiac fibrosis (Baysa et al., 2015). In particular, the absence of p66Shc was associated with a striking decrease in the incidence of cardiac rupture. The obvious elimination of the ruptured hearts from functional evaluation might explain the lack of a significant difference in contractile function between post‐myocardial infarction hearts from p66Shc knockout mice and wild‐type littermates. The study of the underlying mechanism highlighted fibroblast activation along with increased collagen formation and reduced activation of MMP2 in p66Shc knockout hearts. These findings are likely to explain the protection against adverse remodelling in p66Shc knockout hearts. Interestingly, in isolated fibroblasts, p66Shc deletion was associated with a decrease in MMP2 expression, while an increase was observed when p66Shc was overexpressed. Nevertheless, regulation of oxidative activation of MMP2 by p66Shc may suggest its involvement in ischaemia/reperfusion injury and endogenous cardioprotection (Lalu et al., 2002).
Myocardial hypertrophy and failure
The observation that p66Shc expression and/or activity was induced by angiotensin II, thrombin and a Gα(q) agonist leading to cardiomyocyte hypertrophy and apoptosis (see Modulation of p66Shc expression in the heart section) suggests the involvement of p66Shc in cardiac hypertrophy and failure.
Indeed, alcohol‐ and diabetes‐induced cardiomyopathy has been linked to p66Shc. High‐dose alcohol induced apoptosis in cardiomyocytes, which was associated with a dose‐dependent phosphorylation of p66Shc and ROS formation. Exposure to alcohol also led to the loss of Δψm and cytochrome c release. Depletion of p66Shc and inhibition of PKCß successfully reversed these effects and suppressed the alcohol‐induced apoptosis in cardiomyocytes (Wang et al., 2015). While alcohol appeared to act directly on cardiomyocytes, in a model of insulin‐dependent diabetes mellitus, the generation of ROS led to telomeric shortening and apoptosis of cardiac progenitor cells, thereby impairing the growth reserve of the heart. Ablation of the p66Shc gene prevented these negative adaptations of the cardiac progenitor cell compartment, interfering with the development of heart failure in diabetes (Rota et al., 2006). Through p66Shc and sirtuin 1, diabetes and sympathectomy elevated the expression of various adhesion molecules on stem and progenitor cells (including CD62 antigen‐like family member L), thereby inhibiting their mobilization. Knockout of CD62 antigen‐like family member L (CD62L) partially restored the defective stem/progenitor cell mobilization (Albiero et al., 2014). More general information on the relationship between p66Shc and diabetes is provided in the P66Shc and cerebral pathologies section.
Taken together, these finding indicate that there is no doubt that with prolonged stresses, p66Shc contributes to cardiac pathologies, such as the development of heart failure. Regarding cardiac ischaemia/reperfusion injury, p66Shc might be either be protective or deleterious depending upon the duration and severity of ischaemia. At present, data are sparse, and no evidence exists whether or not p66Shc is involved in endogenous cardioprotection.
P66Shc and vascular abnormalities
Endothelial dysfunction and cardiovascular risk factors
Increased expression of p66Shc was reported to be associated with several stimuli or conditions leading to endothelial dysfunction, while p66Shc deletion resulted in beneficial effects. Major examples are as follows.
Angiotensin II and pressure overload. Aortic segments from mice exposed to increased blood pressure secondary to transaortic constriction showed a decreased phosphorylation of endothelial NOS, an increased p66Shc phosphorylation and superoxide production (Lee et al., 2008). The increase in p66Shc expression was most likely a consequence of the activation of the renin–angiotensin system secondary to transaortic constriction, because angiotensin II per se elicited an increase in p66Shc phosphorylation in cultured mice endothelial cells (Lee et al., 2008).
Hypercholesterolaemia up‐regulated human endothelial cell p66Shc expression leading to a dysfunctional endothelial cell surface with pro‐adhesive and procoagulant features (Kim et al., 2012). In mice in vivo, high‐fat diet induced endothelial dysfunction that was associated with increased endothelial Wnt3a, dephosphorylated β‐catenin and phosphorylated p66Shc expression. High‐fat diet‐induced dephosphorylation of endothelial β‐catenin was diminished in p66Shc knockout mice (Vikram et al., 2014). Hypercholesterolaemia also led to a prothrombotic phenotype on platelets. In wild‐type mice on high‐fat diet, surface P‐selectin expression on platelets and platelet aggregation induced by thrombin were increased. These exaggerated platelet responses induced by high‐fat diet were significantly blunted in p66Shc siRNA‐treated mice (Kumar et al., 2014).
Diabetes. p66Shc‐induced ROS formation is involved not only in the disorders caused by diabetes but also in its onset. This notion is hardly surprising, because p66Shc is critical in maintaining insulin‐dependent signalling and glucose homeostasis in a variety of tissues (Tomilov et al., 2011). In particular, in adipocytes, insulin induces the phosphorylation of p66Shc (serine 36). The consequent p66Shc‐catalysed ROS production causes the oxidation of specific phosphatases, resulting in their inactivation. Major examples are given by phosphatase and tensin homolog (PTEN; Berniakovich et al., 2008) and protein tyrosine phosphatase 1B (Frijhoff et al., 2014) that regulate the intracellular insulin transduction cascade. Consequently, p66Shc potentiates insulin signalling and regulates insulin‐induced gene expression and triglyceride accumulation in adipocytes (Berniakovich et al., 2008).
Regarding its involvement in diabetes, p66Shc is activated by hyperglycaemia (Pagnin et al., 2005), and the consequent H2O2 generated by p66Shc within mitochondria promotes the loss of viability in tissues of diabetic mice (Menini et al., 2007; Fadini et al., 2010; Cheng et al., 2013) or patients (Albiero et al., 2014).
Mechanistically, p66Shc appears to antagonize insulin and mammalian target of rapamycin (mTOR) signalling, which limits glucose uptake and metabolism (for review, see Soliman et al., 2014).
The data available support a model in which adipose PKCß activation is among the initiating events that disrupt mitochondrial function through interaction with p66Shc and amplify fat accumulation and adipose dysfunction with systemic consequences. Manipulation of PKCß levels, activity or signalling could provide a therapeutic approach to combat obesity and associated metabolic disorders (for review, see Mehta and Mehta, 2014, as well as Diogo et al., 2013).
Remarkably, p66Shc knockout mice are protected from diet‐induced obesity, the associated pro‐inflammatory state and decrease in glucose tolerance (Berniakovich et al., 2008), although they are more sensitive to cold stress and starvation (Giorgio et al., 2012). Also, in lepOb/Ob mice, an established model of obesity and insulin resistance, p66Shc deletion was found to improve glucose tolerance without affecting (hyper)insulinaemia and independently of body weight (Ranieri et al., 2010). However, more recently in the same model, a decrease in body weight was not associated with changes in glucose tolerance, insulin resistance and adipose tissue remodelling (Ciciliot et al., 2015).
In diabetic mice, up‐regulation of p66Shc contributed to impaired vascular reactivity of the cavernosal tissue (Cheng et al., 2013), and p66Shc was shown to be involved in the delayed skin wound healing process in the setting of diabetes and ischaemia (Fadini et al., 2010).
In patients, body mass index was correlated with p66Shc protein levels derived from adipose tissue. An additional clinical report correlated the higher level of p66Shc mRNA in peripheral mononuclear blood cells from type 2 diabetic patients with elevated markers of oxidative stress in plasma (Pagnin et al., 2005).
Oxidative stress and lipoproteins. LDL cholesterol becomes modified by ROS, and oxidized LDL cholesterol binding to its receptor (lectin‐like oxidized LDL receptor‐1) further increases ROS formation. Incubation of human aortic endothelial cells with oxidized LDL cholesterol increased p66Shc phosphorylation at serine 36 (Shi et al., 2011; Shi et al., 2014a). Blockade of the lectin‐like oxidized LDL receptor‐1 prevented p66Shc phosphorylation as did inhibition of PKCßII and JNK. p66Shc silencing blunted oxidized LDL cholesterol induced ROS production (Shi et al., 2011). Apurinic/apyrimidinic endonuclease 1 suppressed oxidized LDL cholesterol‐induced p66Shc activation in endothelial cells by inhibiting PKCß‐mediated p66Shc phosphorylation and subsequently prevented vasoconstriction induced by activation of PKC (Lee et al., 2011).
NOS. Further supporting the importance of p66Shc for endothelial cell (dys)function, overexpression of p66Shc inhibited endothelial NOS‐dependent NO production (Yamamori et al., 2005), in part by uncoupling of endothelial NOS (Shi et al., 2014a). siRNA‐mediated down‐regulation of endogenous p66Shc activated Akt kinase and subsequently increased serine 1177 phosphorylation of endothelial NOS. In rat aortic rings, down‐regulation of p66Shc suppressed the vasoconstrictor response to phenylephrine that was prevented by treatment with the NOS inhibitor L‐NAME, and enhanced vasodilation induced by submaximal doses of acetylcholine (Yamamori et al., 2005).
Finally, ageing affects p66Shc expression and vascular function. Endothelium‐dependent relaxation was age‐dependently impaired in aortic rings (Francia et al., 2004) and cerebral arteries, but not in femoral arteries of wild‐type mice (Shi et al., 2014b). This process was paralleled by an increase in ROS production and was mediated by the p66Shc gene since p66Shc knockout abolished the age‐dependent decrease in endothelial function.
Thus, many of the known cardiovascular risk factors increase p66Shc expression and/or activity and subsequently impair endothelial cell function and vascular reactivity (for details, see Lizama‐Manibusan and McLaughlin, 2013; Magenta et al., 2014; Grimaldi et al., 2015).
Plaque development
Subsequent to endothelial dysfunction, plaque development occurs leading to coronary artery disease (CAD). This well‐established sequence of events appears to both involve p66Shc and be prevented by its deletion. Major lines of evidence are summarized as follows.
High‐fat diet. The atherosclerotic lesion area resulting from chronic high‐fat diet was reported to increase more in wild‐type than p66Shc knockout mice. Early lesions from p66Shc knockout mice had fewer macrophage‐derived foam cells and apoptotic vascular cells in comparison with those from wild‐type mice (Napoli et al., 2003). Similar results were obtained in hypercholesterolaemic apolipoprotein E knockout mice in which p66Shc knockout attenuated atherosclerotic development caused by a high fat diet (Martin‐Padura et al., 2008).
IGF‐1. This endocrine and autocrine/paracrine growth factor circulates at high levels in the plasma and is expressed in most cell types (for review, see Higashi et al., 2012). p66Shc inhibited IGF‐I signalling, leading to attenuation of IGF‐I‐stimulated vascular smooth muscle cell proliferation and migration (Xi et al., 2008).
In vascular smooth muscle cells overexpressing wild‐type arginase II, mitochondrial dysfunction and cell apoptosis occurred, which were abrogated by p66Shc silencing. The activation of p66Shc by arginase II was dependent on ERK and sequential activation of 40S ribosomal protein S6 kinase 1 – JNKs (Xiong et al., 2013).
Thus, deletion of p66Shc protected against endothelial dysfunction and atherosclerotic plaque formation in mice fed a high‐fat diet (Martin‐Padura et al., 2008). Moreover, P66Shc overexpression has been shown to mediate platelet activation and aggregation in hypercholesterolaemia in both mouse and human platelets (Kumar et al., 2014).
CAD. The p66Shc mRNA and protein expression levels in peripheral blood leukocytes were significantly higher in CAD patients compared with control patients. Interestingly, the expression of p66Shc positively correlated with the serum homocysteine level. The mechanisms linking p66Shc and homocysteine are not yet elucidated. However, the homocysteine‐p66Shc connection appears to involve CpG methylation in the p66Shc promoter. In fact, plasma homocysteine levels showed a significant difference between CAD patients with a high or low degree of CpG methylation in peripheral blood leukocytes. Thus, homocysteine appears to up‐regulate human p66Shc expression via hypomethylation of specific CpG dinucleotides in the p66Shc promoter (Kim et al., 2011).
Besides changes in promoter methylation, p66Shc RNA levels were increased in peripheral blood monocytes (PBMs) of acute coronary syndrome patients as compared with stable CAD patients and controls. Furthermore, oxidative stress (malondialdehyde levels) increased in plasma of acute coronary syndrome patients, and levels of malondialdehyde correlated positively with p66Shc (Franzeck et al., 2012). Thus, p66Shc gene expression level in PBMs may be viewed as a marker of CAD in humans (Noda et al., 2010).
Flow‐mediated dilatation was lower and the carotid intima‐media thickness higher in coronary heart disease patients. Notably, in a multiple linear regression analysis, flow‐mediated dilatation was inversely correlated with p66Shc mRNA expression, again pointing to a pivotal role for the expression of p66Shc in endothelial dysfunction (Miao et al., 2015).
Taken together, these findings provide clear evidence that p66Shc contributes to vascular pathologies such as endothelial dysfunction or plaque formation and that silencing of p66Shc through improvement of the NO/ROS balance exerts protective effects.
P66Shc and cerebral pathologies
Ischaemia/reperfusion injury
A role for p66Shc in cardiac susceptibility to ischaemia/reperfusion injury and other pathological conditions is also relevant in neuronal cells.
In wild‐type mice, post‐ischaemic p66Shc knockdown preserved blood–brain barrier integrity resulting in improved stroke outcome, as identified by smaller lesion volumes, decreased neurological deficits and increased survival (Spescha et al., 2015). Thus, p66Shc appears to be involved in irreversible brain injury following ischaemia/reperfusion. Similarly, p66Shc knockout protected against experimental autoimmune encephalomyelitis in mice without affecting the overall immune response; reduced neuronal cell death most likely occurred through the delayed opening of the PTP in p66Shc knockout mice (Savino et al., 2013).
Similar to CAD, in stroke patients, p66Shc gene expression in PBM was transiently increased, and this increase correlated with short‐term neurological outcome (Spescha et al., 2015).
In an in vitro neuronal culture model, p66Shc activation occurred within 1 h of preconditioning ischaemia. Phosphorylated p66Shc rapidly relocalized to the mitochondria, and preconditioned cells experienced increased oxidative stress. Inhibiting p66Shc activation during preconditioning blocked this neuroprotection, suggesting that p66Shc was critical for preconditioning protection (Brown et al., 2010). In human neuroblastoma cells, a brief preconditioning stress induced by serum deprivation for 2 h mediated tolerance against subsequent lethal oxidative stress. Preconditioning stress concomitantly up‐regulated the expression of the anti‐apoptotic B‐cell lymphoma‐2 (Bcl‐2) protein and down‐regulated the p66Shc adaptor protein during the lethal stress period (Andoh et al., 2000). However, there is still a lack of in vivo brain conditioning studies.
Other pathologies
In mouse hippocampal HT22 cells, angiotensin II up‐regulated the expression of PKCßII and induced p66Shc serine 36 phosphorylation and facilitated p66Shc mitochondrial translocation resulting in increased ROS formation, mitochondrial cytochrome c release, caspase‐3 activation and the inhibition of cell viability. Interestingly, propofol inhibited the angiotensin II‐induced PKCßII expression, p66Shc mitochondrial translocation, ROS formation, mitochondrial cytochrome c release, caspase‐3 activation and finally improved cell viability (Zhu et al., 2014).
In rat glioblastoma cells, β‐amyloid‐induced ROS production was observed in the presence of p66Shc leading to cell death. ROS scavengers and p66Shc knockdown decreased ROS formation and cell death (Bashir et al., 2014).
Taken together, these data from in vitro and in vivo experiments indicate that p66Shc contributes to several brain pathologies but that it might also be involved in neuroprotection depending on the strength of the stimulus.
Therapeutic potential of p66Shc inhibition
Several exogenous factors including synthetic chemicals and plant compounds have been demonstrated to boost or suppress p66Shc gene expression or protein function (Table 1). Consequently, both the activation and inhibition of p66Shc activity might be pursued as therapeutic strategies. A reduced expression and/or activity should be imposed when the loss of tissue viability has to be prevented, such as in the case of ischaemic syndromes, while anticancer therapies are likely to be potentiated by enhancing p66Shc‐induced ROS formation.
Table 1.
p66Shc modulation by compounds and interventions of therapeutic interest
| Compounds or stimuli | Overall effect | P66Shc expression | P66Shc Ser36 phosphorylation | P66Shc translocation to mitochondria | Oxidative changes of p66Shc | References |
|---|---|---|---|---|---|---|
| Ethanol | Activation | ↑ | ↑ | Wang et al., 2015 | ||
| Nicotine | Activation | ↑ | ↑ | Arany et al., 2013a | ||
| Taxanes | Activation | ↑ | ↑ | Arany et al., 2013b | ||
| Hydroxydopamine | Activation | ↑ | ↑ | Yamamori et al., 2011 | ||
| Steroids (in transformed cells) | Activation | ↑ | Kumar et al., 2011 | |||
| Steroids (in non‐transformed cells) | Inhibition | ↓ | La Colla et al., 2015 | |||
| Caloric restriction and fasting | Inhibition | ↓ | ↓ | Giorgio et al., 2012 | ||
| Exercise | Inhibition | ↓ | ↓ | Santos‐Alves et al., 2014, 2015 | ||
| Exposure to cold | Activation | ↑ | Giorgio et al., 2012 | |||
| High‐fat diet | Activation | ↑ | Berniakovich et al., 2008; Tomilov et al., 2011 | |||
| Palmitate | Activation | ↑ | Favre et al., 2015 | |||
| Polyunsaturated fatty acids | Inhibition | ↓ | Jing et al., 2014 | |||
| Resveratrol | Activation | ↑ | Fabbrocini et al., 2010 | |||
| Salvianolic acid A | Inhibition | ↓ | Xu et al., 2013 | |||
| PKC inhibitors | Inhibition | ↓ | Fuller et al., 2012; Pinton et al., 2007; Song et al., 2014 | |||
| Amobarbital | Inhibition | ↓ | Yang et al., 2014 | |||
| Hispidin | Inhibition | ↓ | Yang et al., 2014 | |||
| Sotrastaurin | Inhibition | ↓ | Fuller et al., 2012 | |||
| H2S | Inhibition | ↓ | ↑ | Xie et al., 2014 | ||
| Trichostatin | Inhibition | ↓ | Kang et al., 2015 | |||
| Polysaccharide from Ganoderma lucidum | Inhibition | ↓ | ↓ | Kirmani et al., 2013 | ||
| Ascorbic acid | Inhibition | ↓ | ↓ | Kirmani et al., 2013 | ||
| SHetA2 | Inhibition | ↓ |
Activators
The activators of p66Shc belong to diverse chemical classes and share the ability to increase intracellular oxidative stress (Orsini et al., 2006).
Different toxic chemicals that trigger cell death increase p66Shc levels or activate it by stimulating Ser36 phosphorylation including ethanol (Wang et al., 2015), nicotine (Arany et al., 2013a), anticancer taxanes (Arany et al., 2013b) and the neurotoxin hydroxydopamine (Yamamori et al., 2011). Notably, p66Shc levels do not always correlate with increased cell death. In fact, although in ovarian and prostate cancer cells, p66Shc is up‐regulated by steroids as a result of decreased ubiquitination, the increase in p66Shc levels correlates with cell proliferation (Kumar et al., 2011). Conversely, in non‐transformed cells, oestradiol was reported to inhibit p66Shc translocation to mitochondria. This effect might explain the ability of oestradiol to protect against mitochondria‐generated apoptosis in several non‐reproductive tissues (La Colla et al., 2015).
Among known activators of sirtuin 1, resveratrol was found to induce p66Shc expression and Ser36 phosphorylation in a process dependent on ERK1/2 activation (Fabbrocini et al., 2010). Salvianolic acid A extracted from Salvia miltiorrhiza, another polyphenol that increases sirtuin 1, inhibited p66Shc transcription in hepatocytes (Xu et al., 2013).
Inhibitors
The addition of palmitate increased the expression of p66Shc in different cell types (Favre et al., 2015). However, polyunsaturated fatty acids that activate the AMP kinase/sirtuin 1 pathway suppressed p66Shc (serine36) phosphorylation (Jing et al., 2014).
The PKC inhibitors hispidin (Pinton et al., 2007), rottlerin (Song et al., 2014) and sotrastaurin (Fuller et al., 2012) were found to suppress p66Shc phosphorylation. Notably, amobarbital, hispidin (Yang et al., 2014) or sotrastaurin (Fuller et al., 2012) prevented the mitochondrial translocation of p66Shc occurring in cardiomyocytes upon ischaemia/reperfusion, reducing injury and improving recovery after myocardial infarction. More recently, an additional link with signalling pathways has been described relating p66Shc with hydrogen sulphide (H2S) (Xie et al., 2014). In particular, sulfhydration of Cys59 has been shown to disrupt p66Shc interaction with PKCβII, eventually hampering its phosphorylation at Ser36 and the consequent translocation into mitochondria. p66Shc sulfhydration is likely to play a relevant role in both the antioxidant property of H2S and its cardioprotective effects (Andreadou et al., 2015).
Trichostatin was also found to inhibit p66Shc phosphorylation, especially in the presence of angiotensin II (Kang et al., 2015). Less clear is how the polysaccharide extract from Ganoderma lucidum or ascorbic acid attenuated the expression and phosphorylation of p66Shc (Kirmani et al., 2013).
Finally, the only compound known to directly bind p66Shc, thus preventing its association with heat shock protein 70 and mitochondrial import, is the synthetic heteroarotinoid SHetA2 (Benbrook et al., 2014). However, effects on tissue degeneration have not yet been reported.
In addition to chemical compounds, lifestyle interventions also affect p66Shc expression/activity. Caloric restriction, fasting (Giorgio et al., 2012) and exercise (Santos‐Alves et al., 2014, 2015) were reported to reduce p66Shc gene expression and p66Shc (serine 36) phosphorylation in mouse liver, fat and muscle. In contrast, cold exposure or obesogenic high fat diet increase p66Shc protein levels in mouse fat and muscle (Berniakovich et al., 2008; Tomilov et al., 2011; Giorgio et al., 2012).
Some open questions relating to p66Shc
Does p66Shc decrease or increase irreversible tissue injury following ischaemia/reperfusion?
In the heart, in particular, the data are controversial with p66Shc knockdown either decreasing or increasing infarct size following short periods of ischaemia/reperfusion (see above). While methodological and/or species differences might contribute to these divergent findings, p66Shc could indeed induce organ damage or act as a protectant, through increased ROS formation or mast cell stabilization respectively. Degranulation of mast cells occurs following myocardial ischaemia/reperfusion leads to an interstitial increase in chymase (Zheng et al., 2014) and renin concentration, the latter contributing to activation of the local cardiac renin–angiotensin system. Indeed, mast cell modulators (Jaggi et al., 2007), including adenosine receptor agonists (Rork et al., 2008), reduced ischaemia/reperfusion injury. p66Shc limits the basal release of granule contents from mast cells by inhibiting microvesicle budding from the plasma membrane through regulation of mast cell actin dynamics (Masi et al., 2014).
Does p66Shc activation contribute to endogenous organ protective interventions such as pre‐conditioning, post‐conditioning and remote conditioning in vivo?
The involvement of p66Shc in endogenous protection induced by ischaemic preconditioning has been suggested from studies on neuronal cells (see discussion earlier). However, in vivo studies and studies in other organs – such as the heart – are lacking.
Again, one interesting observation comes from studies in pig hearts in vivo in which the reduction in infarct size induced by ischaemic preconditioning was attenuated by pretreatment with ascorbic acid. This loss of protection might involve scavenging of ROS during the preconditioning ischaemia/reperfusion episode (Skyschally et al., 2003). Interestingly, p66Shc‐mediated ROS up‐regulation was significantly decreased in the presence of ascorbic acid, which decreases p66Shc expression by increasing its ubiquitination (Kirmani et al., 2013).
Does inhibition of p66Shc activation following prolonged ischaemia/reperfusion present a new therapeutic option to reduce irreversible tissue injury?
As detailed above, data on p66Shc knockout suggest that organ damage following ischaemia/reperfusion might be reduced. However, data on pharmacological interventions modifying p66Shc expression and/or activity are sparce and need to be extended. Furthermore, existing pharmacological interventions for cardiac protection should be revisited to investigate their effect on p66Shc expression and activity.
Conclusion
The evidence so far reported and herein discussed supports the concept that pharmacological modulation of p66Shc expression and activity may be an effective target for the treatment of atherosclerotic vascular disease, as well as a means of modifying the effects of hypertrophic, inflammatory and neurohormonal stimuli in the overloaded heart. The importance of p66Shc for ischaemia/reperfusion injury in the heart, as well as in the brain, requires further elucidation, and the role of p66Shc in the signalling cascade of endogenous protective mechanisms needs to be clarified.
Conflict of interest
R. S. received research grants from Zealand Pharma and honoraria for lectures and advisory boards from AstraZeneca, Recordati, Sanofi and Servier. P. F. is a founder and CEO of Pharmahungary Group.
Acknowledgements
R. S. received grants from the German Research Foundations (Schu 843/9‐1). The work in F D L's laboratory is supported by CARIPARO Foundation (Progetti di Eccellenza) and Progetto Strategico “Dycendi” of the University of Padova. Peter Ferdinandy and R. S. received a collaborative grant from the European Foundation for the Study of Diabetes. P. F. received grants from the Hungarian Scientific Research Fund (OTKA K 109737 and OTKA ANN 107803). The MS was elaborated in the scope of the European COST EU‐ROS network (BM1203).
Di Lisa, F. , Giorgio, M. , Ferdinandy, P. , and Schulz, R. (2017) New aspects of p66Shc in ischaemia reperfusion injury and other cardiovascular diseases. British Journal of Pharmacology, 174: 1690–1703. doi: 10.1111/bph.13478.
References
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J (2010). Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res 106: 1253–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmedov A, Montecucco F, Braunersreuther V, Camici GG, Jakob P, Reiner MF et al. (2015). Genetic deletion of the adaptor protein p66Shc increases susceptibility to short‐term ischaemic myocardial injury via intracellular salvage pathways. Eur Heart J 36: 516–526. [DOI] [PubMed] [Google Scholar]
- Albiero M, Poncina N, Tjwa M, Ciciliot S, Menegazzo L, Ceolotto G et al. (2014). Diabetes causes bone marrow autonomic neuropathy and impairs stem cell mobilization via dysregulated p66Shc and Sirt1. Diabetes 63: 1353–1365. [DOI] [PubMed] [Google Scholar]
- Altschuld RA, Wenger WC, Lamka KG, Kindig OR, Capen CC, Mizuhira V et al. (1985). Structural and functional properties of adult rat heart myocytes lysed with digitonin. J Biol Chem 260: 14325–14334. [PubMed] [Google Scholar]
- Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT et al. (2009). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh T, Lee SY, Chiueh CC (2000). Preconditioning regulation of bcl‐2 and p66Shc by human NOS1 enhances tolerance to oxidative stress. FASEB J 14: 2144–2146. [DOI] [PubMed] [Google Scholar]
- Andreadou I, Iliodromitis EK, Rassaf T, Schulz R, Papapetropoulos A, Ferdinandy P (2015). The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br J Pharmacol 172: 1587–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany I, Clark J, Reed DK, Juncos LA (2013a). Chronic nicotine exposure augments renal oxidative stress and injury through transcriptional activation of p66Shc. Nephrol Dial Transplant 28: 1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany I, Clark JS, Reed D, Szabó I, Ember I, Juncos LA (2013b). The role of p66Shc in taxol‐ and dichloroacetic acid‐dependent renal toxicity. Anticancer Res 33: 3119–3122. [PubMed] [Google Scholar]
- Bashir M, Parray AA, Baba RA, Bhat HF, Bhat SS, Mushtaq U et al. (2014). beta‐Amyloid‐evoked apoptotic cell death is mediated through MKK6‐p66Shc pathway. Neuromolecular Med 16: 137–149. [DOI] [PubMed] [Google Scholar]
- Baysa A, Sagave J, Carpi A, Zaglia T, Campesan M, Dahl CP et al. (2015). The p66ShcA adaptor protein regulates healing after myocardial infarction. Basic Res Cardiol 110: 13. [DOI] [PubMed] [Google Scholar]
- Benbrook DM, Nammalwar B, Long A, Matsumoto H, Singh A, Bunce RA et al. (2014). SHetA2 interference with mortalin binding to p66Shc and p53 identified using drug‐conjugated magnetic microspheres. Invest New Drugs 32: 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P (1999). Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 79: 1127–1155. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Di Lisa F (2015). The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 78: 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt C, Lillig CH, Holmgren A (2007). Thiol‐based mechanisms of the thioredoxin and glutaredoxin systems: Implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol 292: H1227–H1236. [DOI] [PubMed] [Google Scholar]
- Berniakovich I, Trinei M, Stendardo M, Migliaccio E, Minucci S, Bernardi P et al. (2008). p66Shc‐generated oxidative signal promotes fat accumulation. J Biol Chem 283: 34283–34393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolli R, Marbán E (1999). Molecular and cellular mechanisms of myocardial stunning. Physiol Rev 79: 609–634. [DOI] [PubMed] [Google Scholar]
- Brown JE, Zeiger SL, Hettinger JC, Brooks JD, Holt B, Morrow JD et al. (2010). Essential role of the redox‐sensitive kinase p66Shc in determining energetic and oxidative status and cell fate in neuronal preconditioning. J Neurosci 30: 5242–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpi A, Menabò R, Kaludercic N, Pelicci P, Di Lisa F, Giorgio M (2009). The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta 1787: 774–780. [DOI] [PubMed] [Google Scholar]
- Chen YR, Zweier JL (2014). Cardiac mitochondria and reactive oxygen species generation. Circ Res 114: 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YS, Cong XD, Dai DZ, Zhang Y, Dai Y (2013). Argirein alleviates corpus cavernosum dysfunction by suppressing pro‐inflammatory factors p66Shc and ER stress chaperone Bip in diabetic rats. J Pharm Pharmacol 65: 94–101. [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S et al. (2013). Cardioprotection by S‐nitrosation of a cysteine switch on mitochondrial complex I. Nat Med 19: 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciciliot S, Albiero M, Menegazzo L, Poncina N, Scattolini V, Danesi A et al. (2015). p66Shc deletion or deficiency protects from obesity but not metabolic dysfunction in mice and humans. Diabetologia 58: 2352–2360. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Francia P, Camici GG, Pelicci PG, Lüscher TF, Volpe M (2008). Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein. Arterioscler Thromb Vasc Biol 28: 622–628. [DOI] [PubMed] [Google Scholar]
- Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves‐Cintron M, Chen T et al. (2011). Mitochondrial oxidative stress mediates angiotensin II‐induced cardiac hypertrophy and galphaq overexpression‐induced heart failure. Circ Res 108: 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lisa F, Menabò R, Canton M, Petronilli V (1998). The role of mitochondria in the salvage and the injury of the ischemic myocardium. Biochim Biophys Acta 1366: 69–78. [DOI] [PubMed] [Google Scholar]
- Diogo CV, Suski JM, Lebiedzinska M, Karkucinska‐Wieckowska A, Wojtala A, Pronicki M et al. (2013). Cardiac mitochondrial dysfunction during hyperglycemia–the role of oxidative stress and p66Shc signalling. Int J Biochem Cell Biol 45: 114–122. [DOI] [PubMed] [Google Scholar]
- Elz JS, Nayler WG (1988). Calcium gain during postischemic reperfusion. The effect of 2,4‐dinitrophenol. Am J Pathol 131: 137–145. [PMC free article] [PubMed] [Google Scholar]
- Fabbrocini G, Kisslinger A, Iannelli P, Vitale N, Procaccini C, Sparaneo G et al. (2010). Resveratrol regulates p66Shc activation in HaCaT cells. Exp Dermatol 19: 895–903. [DOI] [PubMed] [Google Scholar]
- Fadini GP, Albiero M, Menegazzo L, Boscaro E, Pagnin E, Iori E et al. (2010). The redox enzyme p66Shc contributes to diabetes and ischemia‐induced delay in cutaneous wound healing. Diabetes 59: 2306–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre J, Yıldırım C, Leyen TA, Chen WJ, van Genugten RE, van Golen LW et al. (2015). Palmitic acid increases pro‐oxidant adaptor protein p66Shc expression and affects vascularization factors in angiogenic mononuclear cells: action of resveratrol. Vascul Pharmacol 2015 .pii: S1537‐1891(15)00179‐2 [DOI] [PubMed] [Google Scholar]
- Finkel T (2012). From sulfenylation to sulfhydration: what a thiolate needs to tolerate. Sci Signal 5 : pe10. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Ursini F, Maiorino M (2014). An overview of mechanisms of redox signalling. J Mol Cell Cardiol 73: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia P, Delli GC, Bachschmid M, Martin‐Padura I, Savoia C, Migliaccio E et al. (2004). Deletion of p66Shc gene protects against age‐related endothelial dysfunction. Circulation 110: 2889–2895. [DOI] [PubMed] [Google Scholar]
- Franzeck FC, Hof D, Spescha RD, Hasun M, Akhmedov A, Steffel J et al. (2012). Expression of the aging gene p66Shc is increased in peripheral blood monocytes of patients with acute coronary syndrome but not with stable coronary artery disease. Atherosclerosis 220: 282–286. [DOI] [PubMed] [Google Scholar]
- Frijhoff J, Dagnell M, Augsten M, Beltrami E, Giorgio M, Östman A (2014). The mitochondrial reactive oxygen species regulator p66Shc controls PDGF‐induced signalling and migration through protein tyrosine phosphatase oxidation. Free Radic Biol Med 68: 268–277. [DOI] [PubMed] [Google Scholar]
- Fuller TF, Kusch A, Chaykovska L, Catar R, Pützer J, Haller M et al. (2012). Protein kinase C inhibition ameliorates posttransplantation preservation injury in rat renal transplants. Transplantation 94: 679–686. [DOI] [PubMed] [Google Scholar]
- Ganote CE, Worstell J, Kaltenbach JP (1976). Oxygen‐induced enzyme release after irreversible myocardial injury. Effects of cyanide in perfused rat hearts. Am J Pathol 84: 327–350. [PMC free article] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C et al. (2005). Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122: 221–233. [DOI] [PubMed] [Google Scholar]
- Giorgio M, Berry A, Berniakovich I, Poletaeva I, Trinei M, Stendardo M et al. (2012). The p66Shc knocked out mice are short lived under natural condition. Aging Cell 11: 162–168. [DOI] [PubMed] [Google Scholar]
- Graiani G, Lagrasta C, Migliaccio E, Spillmann F, Meloni M, Madeddu P et al. (2005). Genetic deletion of the p66Shc adaptor protein protects from angiotensin II‐induced myocardial damage. Hypertension 46: 433–440. [DOI] [PubMed] [Google Scholar]
- Grimaldi V, Vietri MT, Schiano C, Picascia A, De Pascale MR, Fiorito C et al. (2015). Epigenetic reprogramming in atherosclerosis. Curr Atheroscler Rep 17: 476. [DOI] [PubMed] [Google Scholar]
- Guo J, Gertsberg Z, Ozgen N, Steinberg SF (2009). p66Shc links alpha1‐adrenergic receptors to a reactive oxygen species‐dependent AKT–FOXO3A phosphorylation pathway in cardiomyocytes. Circ Res 104: 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, Dinkova‐Kostova AT (2014). The nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39: 199–218. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Sukhanov S, Anwar A, Shai SY, Delafontaine P (2012). Aging, atherosclerosis, and IGF‐1. J Gerontol A Biol Sci Med Sci 67: 626–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhäuser C, Bornbaum J, Reis A, Böhme S, Kaludercic N, Menabò R et al. (2015). NOX4 in mitochondria: yeast two‐hybrid‐based interaction with complex I without relevance for basal reactive oxygen species? Antioxid Redox Signal 23: 1106–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggi AS, Singh M, Sharma A, Singh D, Singh N (2007). Cardioprotective effects of mast cell modulators in ischemia–reperfusion‐induced injury in rats. Methods Find Exp Clin Pharmacol 29: 593–600. [DOI] [PubMed] [Google Scholar]
- Jennings RB, Ganote CE, Reimer KA (1975). Ischemic tissue injury. Am J Pathol 81: 179–198. [PMC free article] [PubMed] [Google Scholar]
- Jing H, Yao J, Liu X, Fan H, Zhang F, Li Z et al. (2014). Fish‐oil emulsion (omega‐3 polyunsaturated fatty acids) attenuates acute lung injury induced by intestinal ischemia–reperfusion through adenosine 5'‐monophosphate‐activated protein kinase‐sirtuin1 pathway. J Surg Res 187: 252–261. [DOI] [PubMed] [Google Scholar]
- Kaludercic N, Mialet‐Perez J, Paolocci N, Parini A, Di Lisa F (2014). Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol 73: 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Lee YR, Joo HK, Park MS, Kim CS, Choi S et al. (2015). Trichostatin A modulates angiotensin ii‐induced vasoconstriction and blood pressure via inhibition of p66Shc activation. Korean J Physiol Pharmacol 19: 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CS, Kim YR, Naqvi A, Kumar S, Hoffman TA, Jung SB et al. (2011). Homocysteine promotes human endothelial cell dysfunction via site‐specific epigenetic regulation of p66Shc. Cardiovasc Res 92: 466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YR, Kim CS, Naqvi A, Kumar A, Kumar S, Hoffman TA et al. (2012). Epigenetic upregulation of p66Shc mediates low‐density lipoprotein cholesterol‐induced endothelial cell dysfunction. Am J Physiol Heart Circ Physiol 303: H189–H196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmani D, Bhat HF, Bashir M, Zargar MA, Khanday FA (2013). P66Shc‐rac1 pathway‐mediated ROS production and cell migration is downregulated by ascorbic acid. J Recept Signal Transduct Res 33: 107–113. [DOI] [PubMed] [Google Scholar]
- Kumar S, Kumar S, Rajendran M, Alam SM, Lin FF, Cheng PW et al. (2011). Steroids up‐regulate p66Shc longevity protein in growth regulation by inhibiting its ubiquitination. PLoS One 6 :e15942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Vikram A, Kim YR, Jacobs S, Irani K (2014). P66Shc mediates increased platelet activation and aggregation in hypercholesterolemia. Biochem Biophys Res Commun 449: 496–501. [DOI] [PubMed] [Google Scholar]
- La Colla A, Boland R, Vasconsuelo A (2015). 17β‐Estradiol abrogates apoptosis inhibiting PKCδ, JNK, and p66Shc activation in C2C12 cells. J Cell Biochem 116: 1454–1465. [DOI] [PubMed] [Google Scholar]
- Lalu MM, Csonka C, Giricz Z, Csont T, Schulz R, Ferdinandy P (2002). Preconditioning decreases ischemia/reperfusion‐induced release and activation of matrix metalloproteinase‐2. Biochem Biophys Res Commun 296: 937–941. [DOI] [PubMed] [Google Scholar]
- Lee SK, Chung JI, Park MS, Joo HK, Lee EJ, Cho EJ et al. (2011). Apurinic/apyrimidinic endonuclease 1 inhibits protein kinase C‐mediated p66Shc phosphorylation and vasoconstriction. Cardiovasc Res 91: 502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Kim HS, Song YJ, Joo HK, Lee JY, Lee KH et al. (2008). Alteration of p66Shc is associated with endothelial dysfunction in the abdominal aortic coarctation of rats. FEBS Lett 582: 2561–2566. [DOI] [PubMed] [Google Scholar]
- Lizama‐Manibusan B, McLaughlin B (2013). Redox modification of proteins as essential mediators of CNS autophagy and mitophagy. FEBS Lett 587: 2291–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubicic V, Menzies KJ, Hood DA (2010). Mitochondrial dysfunction is associated with a pro‐apoptotic cellular environment in senescent cardiac muscle. Mech Ageing Dev 131: 79–88. [DOI] [PubMed] [Google Scholar]
- Magenta A, Greco S, Capogrossi MC, Gaetano C, Martelli F (2014). Nitric oxide, oxidative stress, and p66Shc interplay in diabetic endothelial dysfunction. Biomed Res Int 2014: ID 193095, 16 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Padura I, de Nigris F, Migliaccio E, Mansueto G, Minardi S, Rienzo M et al. (2008). p66Shc deletion confers vascular protection in advanced atherosclerosis in hypercholesterolemic apolipoprotein E knockout mice. Endothelium 15: 276–287. [DOI] [PubMed] [Google Scholar]
- Masi G, Mercati D, Vannuccini E, Paccagnini E, Riparbelli MG, Lupetti P et al. (2014). p66Shc regulates vesicle‐mediated secretion in mast cells by affecting F‐actin dynamics. J Leukoc Biol 95: 285–292. [DOI] [PubMed] [Google Scholar]
- Mehta NK, Mehta KD (2014). Protein kinase C‐beta: an emerging connection between nutrient excess and obesity. Biochim Biophys Acta 1841: 1491–1497. [DOI] [PubMed] [Google Scholar]
- Menini S, Iacobini C, Ricci C, Oddi G, Pesce C, Pugliese F et al. (2007). Ablation of the gene encoding p66Shc protects mice against AGE‐induced glomerulopathy by preventing oxidant‐dependent tissue injury and further AGE accumulation. Diabetologia 50: 1997–2007. [DOI] [PubMed] [Google Scholar]
- Miao Q, Wang Q, Dong L, Wang Y, Tan Y, Zhang X (2015). The expression of p66Shc in peripheral blood monocytes is increased in patients with coronary heart disease and correlated with endothelium‐dependent vasodilatation. Heart Vessels 30: 451–457. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, Mele S, Salcini AE, Pelicci G, Lai KM, Superti‐Furga G et al. (1997). Opposite effects of the p52shc/p46shc and p66Shc splicing isoforms on the EGF receptor‐MAP kinase‐fos signalling pathway. EMBO J 16: 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP et al. (1999). The p66Shc adaptor protein controls oxidative stress response and life span in mammals. Nature 402: 309–313. [DOI] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2012). Mitochondrial thiols in antioxidant protection and redox signalling: distinct roles for glutathionylation and other thiol modifications. Antioxid Redox Signal 16: 476–495. [DOI] [PubMed] [Google Scholar]
- Napoli C, Martin‐Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P et al. (2003). Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high‐fat diet. Proc Natl Acad Sci U S A 100: 2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel A, Kohlhaas M, Maack C (2014). Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol 73: 26–33. [DOI] [PubMed] [Google Scholar]
- Noda Y, Yamagishi S, Matsui T, Ueda S, Ueda S, Jinnouchi Y et al. (2010). The p66Shc gene expression in peripheral blood monocytes is increased in patients with coronary artery disease. Clin Cardiol 33: 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obreztchikova M, Elouardighi H, Ho M, Wilson BA, Gertsberg Z, Steinberg SF (2006). Distinct signalling functions for Shc isoforms in the heart. J Biol Chem 281: 20197–20204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Kao AW, Ceresa BP, Blaikie P, Margolis B, Pessin JE (1997). The 66‐kDa Shc isoform is a negative regulator of the epidermal growth factor‐stimulated mitogen‐activated protein kinase pathway. J Biol Chem 272: 28042–28049. [DOI] [PubMed] [Google Scholar]
- Orsini F, Moroni M, Contursi C, Pelicci PG, Giorgio M, Migliaccio E (2006). Mitochondrial regulation of p66Shc mitochondrial function. Biol Chem 387: 1405–1410. [DOI] [PubMed] [Google Scholar]
- Ovize M, Baxter GF, Di Lisa F, Ferdinandy P, Garcia‐Dorado D, Hausenloy DJ et al. (2010). Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res 87: 406–423. [DOI] [PubMed] [Google Scholar]
- Pagnin E, Fadini G, de Toni R, Tiengo A, Calo L, Avogaro A (2005). Diabetes induces p66Shc gene expression in human peripheral blood mononuclear cells: relationship to oxidative stress. J Clin Endocrinol Metab 90: 1130–1136. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G et al. (1992). A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 70: 93–104. [DOI] [PubMed] [Google Scholar]
- Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M et al. (2007). Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life‐span determinant p66Shc. Science 315: 659–663. [DOI] [PubMed] [Google Scholar]
- Ranieri SC, Fusco S, Panieri E, Labate V, Mele M, Tesori V et al. (2010). Mammalian life‐span determinant p66ShcA mediates obesity‐induced insulin resistance. Proc Natl Acad Sci U S A 107: 13420–13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran KS (2001). Signalling via Shc family adapter proteins. Oncogene 20: 6322–6330. [DOI] [PubMed] [Google Scholar]
- Rork TH, Wallace KL, Kennedy DP, Marshall MA, Lankford AR, Linden J (2008). Adenosine A2A receptor activation reduces infarct size in the isolated, perfused mouse heart by inhibiting resident cardiac mast cell degranulation. Am J Physiol Heart Circ Physiol 295: H1825–H1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota M, LeCapitaine N, Hosoda T, Boni A, De AA, Padin‐Iruegas ME et al. (2006). Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66Shc gene. Circ Res 99: 42–52. [DOI] [PubMed] [Google Scholar]
- Santos‐Alves E, Marques‐Aleixo I, Coxito P, Balça MM, Rizo‐Roca D, Rocha‐Rodrigues S et al. (2014). Exercise mitigates diclofenac‐induced liver mitochondrial dysfunction. Eur J Clin Invest 44: 668–677. [DOI] [PubMed] [Google Scholar]
- Santos‐Alves E, Marques‐Aleixo I, Rizo‐Roca D, Torrella JR, Oliveira PJ, Magalhães J et al. (2015). Exercise modulates liver cellular and mitochondrial proteins related to quality control signalling. Life Sci 135: 124–130. [DOI] [PubMed] [Google Scholar]
- Savino C, Pelicci P, Giorgio M (2013). The P66Shc/mitochondrial permeability transition pore pathway determines neurodegeneration. Oxid Med Cell Longev 2013: ID 719407, 7 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M et al. (2005). Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308: 1909–1911. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2014). Hypoxia‐inducible factor 1 and cardiovascular disease. Annu Rev Physiol 76: 39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen AC, Jennings RB (1972). Kinetics of calcium accumulation in acute myocardial ischemic injury. Am J Pathol 67: 441–452. [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Cosentino F, Camici GG, Akhmedov A, Vanhoutte PM, Tanner FC et al. (2011). Oxidized low‐density lipoprotein activates p66Shc via lectin‐like oxidized low‐density lipoprotein receptor‐1, protein kinase C‐beta, and c‐Jun N‐terminal kinase kinase in human endothelial cells. Arterioscler Thromb Vasc Biol 31: 2090–2097. [DOI] [PubMed] [Google Scholar]
- Shi Y, Luscher TF, Camici GG (2014a). Dual role of endothelial nitric oxide synthase in oxidized LDL‐induced, p66Shc‐mediated oxidative stress in cultured human endothelial cells. PLoS One 9 : e107787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Savarese G, Perrone‐Filardi P, Luscher TF, Camici GG (2014b). Enhanced age‐dependent cerebrovascular dysfunction is mediated by adaptor protein p66Shc. Int J Cardiol 175: 446–450. [DOI] [PubMed] [Google Scholar]
- Siegmund B, Klietz T, Schwartz P, Piper HM (1991). Temporary contractile blockade prevents hypercontracture in anoxic‐reoxygenated cardiomyocytes. Am J Physiol 260: H426–H435. [DOI] [PubMed] [Google Scholar]
- Silverman HS, Stern MD (1994). Ionic basis of ischaemic cardiac injury: insights from cellular studies. Cardiovasc Res 28: 581–597. [DOI] [PubMed] [Google Scholar]
- Skyschally A, Schulz R, Gres P, Korth HG, Heusch G (2003). Attenuation of ischemic preconditioning in pigs by scavenging of free oxyradicals with ascorbic acid. Am J Physiol Heart Circ Physiol 284: H698–H703. [DOI] [PubMed] [Google Scholar]
- Soliman MA, Abdel Rahman AM, Lamming DW, Birsoy K, Pawling J, Frigolet ME et al. (2014). The adaptor protein p66Shc inhibits mTOR‐dependent anabolic metabolism. Sci Signal 7 : ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Yang S, Xiao L, Xu X, Tang C, Yang Y et al. (2014). PKCδ promotes high glucose induced renal tubular oxidative damage via regulating activation and translocation of p66Shc. Oxid Med Cell Longev 2014: ID 746531, 11 pages. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Spescha RD, Klohs J, Semerano A, Giacalone G, Derungs RS, Reiner MF et al. (2015). Post‐ischaemic silencing of p66Shc reduces ischaemia/reperfusion brain injury and its expression correlates to clinical outcome in stroke. Eur Heart J 36: 1590–1600. [DOI] [PubMed] [Google Scholar]
- Tomilov AA, Ramsey JJ, Hagopian K, Giorgio M, Kim KM, Lam A et al. (2011). The Shc locus regulates insulin signalling and adiposity in mammals. Aging Cell 10: 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinei M, Berniakovich I, Beltrami E, Migliaccio E, Fassina A, Pelicci PG et al. (2009). P66Shc signals to age. Aging 1: 503–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinei M, Migliaccio E, Bernardi P, Paolucci F, Pelicci P, Giorgio M (2013). Mitochondria, and the generation of reactive oxygen species. p66Shc. Methods Enzymol 528: 99–110. [DOI] [PubMed] [Google Scholar]
- Vikram A, Kim YR, Kumar S, Naqvi A, Hoffman TA, Kumar A et al. (2014). Canonical Wnt signalling induces vascular endothelial dysfunction via p66Shc‐regulated reactive oxygen species. Arterioscler Thromb Vasc Biol 34: 2301–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhao J, Yang W, Bi Y, Chi J, Tian J et al. (2015). High‐dose alcohol induces reactive oxygen species‐mediated apoptosis via PKC‐beta/p66Shc in mouse primary cardiomyocytes. Biochem Biophys Res Commun 456: 656–661. [DOI] [PubMed] [Google Scholar]
- Xi G, Shen X, Clemmons DR (2008). p66Shc negatively regulates insulin‐like growth factor I signal transduction via inhibition of p52shc binding to Src homology 2 domain‐containing protein tyrosine phosphatase substrate‐1 leading to impaired growth factor receptor‐bound protein‐2 membrane recruitment. Mol Endocrinol 22: 2162–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie ZZ, Shi MM, Xie L, Wu ZY, Li G, Hua F et al. (2014). Sulfhydration of p66Shc at cysteine59 mediates the antioxidant effect of hydrogen sulfide. Antioxid Redox Signal 21: 2531–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Yu Y, Montani JP, Yang Z, Ming XF (2013). Arginase‐II induces vascular smooth muscle cell senescence and apoptosis through p66Shc and p53 independently of its l‐arginine ureahydrolase activity: implications for atherosclerotic plaque vulnerability. J Am Heart Assoc 2 : e000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Hu Y, Zhai X, Lin M, Chen Z, Tian X et al. (2013). Salvianolic acid A preconditioning confers protection against concanavalin A‐induced liver injury through SIRT1‐mediated repression of p66Shc in mice. Toxicol Appl Pharmacol 273: 68–76. [DOI] [PubMed] [Google Scholar]
- Yamamori T, White AR, Mattagajasingh I, Khanday FA, Haile A, Qi B et al. (2005). P66Shc regulates endothelial NO production and endothelium‐dependent vasorelaxation: implications for age‐associated vascular dysfunction. J Mol Cell Cardiol 39: 992–995. [DOI] [PubMed] [Google Scholar]
- Yamamori T, Mizobata A, Saito Y, Urano Y, Inanami O, Irani K et al. (2011). Phosphorylation of p66Shc mediates 6‐hydroxydopamine cytotoxicity. Free Radic Res 45: 342–350. [DOI] [PubMed] [Google Scholar]
- Yang M, Stowe DF, Udoh KB, Heisner JS, Camara AK (2014). Reversible blockade of complex I or inhibition of PKCbeta reduces activation and mitochondria translocation of p66Shc to preserve cardiac function after ischemia. PLoS One 9 : e113534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellon DM, Hausenloy DJ (2007). Myocardial reperfusion injury. N Engl J Med 357: 1121–1135. [DOI] [PubMed] [Google Scholar]
- Yun J, Finkel T (2014). Mitohormesis. Cell Metab 19: 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccagnini G, Martelli F, Fasanaro P, Magenta A, Gaetano C, Di Carlo A et al. (2004). p66ShcA modulates tissue response to hindlimb ischemia. Circulation 109: 2917–2923. [DOI] [PubMed] [Google Scholar]
- Zheng J, Wei CC, Hase N, Shi K, Killingsworth CR, Litovsky SH et al. (2014). Chymase mediates injury and mitochondrial damage in cardiomyocytes during acute ischemia/reperfusion in the dog. PLoS One 9 : e94732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Chen J, Wen M, Sun Z, Sun X, Wang J et al. (2014). Propofol protects against angiotensin II‐induced mouse hippocampal HT22 cells apoptosis via inhibition of p66Shc mitochondrial translocation. Neuromolecular Med 16: 772–781. [DOI] [PubMed] [Google Scholar]
- Ziolkowski W, Flis DJ, Halon M, Vadhana DM, Olek RA, Carloni M et al. (2015). Prolonged swimming promotes cellular oxidative stress and p66Shc phosphorylation, but does not induce oxidative stress in mitochondria in the rat heart. Free Radic Res 49: 7–16. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ (2014). Mitochondrial reactive oxygen species (ros) and ros‐induced ros release. Physiol Rev 94: 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier JL (1988). Measurement of superoxide‐derived free radicals in the reperfused heart. Evidence for a free radical mechanism of reperfusion injury. J Biol Chem 263: 1353–1357. [PubMed] [Google Scholar]