

ABSTRACT
The L protein of the single-stranded RNA phage MS2 causes lysis of Escherichia coli without inducing bacteriolytic activity or inhibiting net peptidoglycan (PG) synthesis. To find host genes required for L-mediated lysis, spontaneous Ill (insensitivity to L lysis) mutants were selected as survivors of L expression and shown to have a missense change of the highly conserved proline (P330Q) in the C-terminal domain of DnaJ. In the dnaJP330Q mutant host, L-mediated lysis is completely blocked at 30°C without affecting the intracellular levels of L. At higher temperatures (37°C and 42°C), both lysis and L accumulation are delayed. The lysis block at 30°C in the dnaJP330Q mutant was recessive and could be suppressed by L overcomes dnaJ (Lodj) alleles selected for restoration of lysis. All three Lodj alleles lack the highly basic N-terminal half of the lysis protein and cause lysis ∼20 min earlier than full-length L. DnaJ was found to form a complex with full-length L. This complex was abrogated by the P330Q mutation and was absent with the Lodj truncations. These results suggest that, in the absence of interaction with DnaJ, the N-terminal domain of L interferes with its ability to bind to its unknown target. The lysis retardation and DnaJ chaperone dependency conferred by the nonessential, highly basic N-terminal domain of L resembles the SlyD chaperone dependency conferred by the highly basic C-terminal domain of the E lysis protein of ϕX174, suggesting a common theme where single-gene lysis can be modulated by host factors influenced by physiological conditions.
IMPORTANCE Small single-stranded nucleic acid lytic phages (Microviridae and Leviviridae) lyse their host by expressing a single “protein antibiotic.” The protein antibiotics from two out of three prototypic small lytic viruses have been shown to inhibit two different steps in the conserved PG biosynthesis pathway. However, the molecular basis of lysis caused by L, the lysis protein of the third prototypic virus, MS2, is unknown. The significance of our research lies in the identification of DnaJ as a chaperone in the MS2 L lysis pathway and the identification of the minimal lytic domain of MS2 L. Additionally, our research highlights the importance of the highly conserved P330 residue in the C-terminal domain of DnaJ for specific protein interactions.
KEYWORDS: Escherichia coli, bacteriophage genetics, bacteriophage lysis, protein chaperone
INTRODUCTION
The single-stranded RNA phage MS2 is one of the simplest viruses, encoding just four proteins. Of the four proteins, three, RNA-dependent RNA replicase (Rep), major capsid protein (Coat), and maturation protein (A), are involved in viral replication and assembly. The fourth protein is the lysis protein (L), which causes lysis of the host and thus controls the length of the infection cycle (8). L was the first gene shown to be embedded in two different genes, the Coat- and Rep-encoding genes, in this case, in the +1 reading frame of each gene (2) (Fig. 1). Expression of L from a plasmid is necessary and sufficient to elicit lysis (3). L is one of the three canonical “single gene lysis” (SGL) systems used by small phages to effect lysis, the other two being E from the single-stranded DNA (ssDNA) phage ϕX174, representing the ubiquitous members of the family Microviridae, and A2 from single-stranded RNA (ssRNA) phage Qβ, representing the family Alloleviviridae (8). Genetic and molecular analyses revealed that both E and A2 inhibit specific steps in the peptidoglycan (PG) biosynthesis pathway; A2 inhibits MurA, which catalyzes the first committed step, and E inhibits MraY, which catalyzes the formation of the first lipid-linked intermediate (4–6). In both cases, the isolation of dominant mutations conferring resistance to the lethal function of the lysis protein and the mapping of these mutations to the gene encoding the biosynthetic enzyme were the keys to deciphering the lytic mechanism. In the case of E, the first and most common mutations conferring resistance to lysis mapped to a different gene, slyD, encoding a cytoplasmic, FKBP-type cis-trans peptidyl-prolyl isomerase (7). These mutations were recessive, however, and subsequent investigation revealed that SlyD was required for the stability of E, which has five Pro residues in its 91-amino-acid (aa) length. Suppressor mutations in E were all uptranslation alleles compensating for the instability of the E protein (8).
FIG 1.
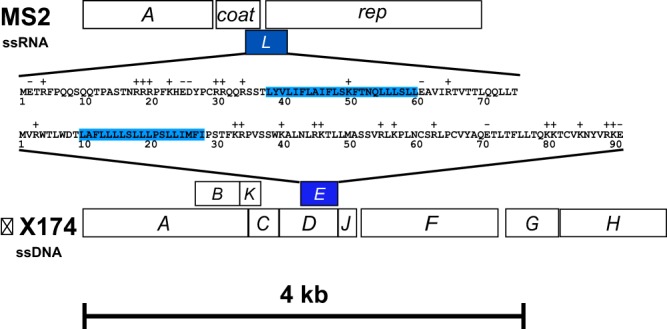
Genome organization of ϕX174 and MS2 phages and similarities between their lysis proteins. Shown are maps of the MS2 and linearized ϕX174 genomes, drawn to scale. The lysis genes of the two phages are shaded blue. Lysis gene L of MS2 encodes a 75-aa lysis protein, and lysis gene E of ϕX174 encodes a 91-aa protein. The residues spanning the transmembrane domain are highlighted in blue. Basic and acidic residues are indicated above the primary structure of the lysis proteins.
In contrast to E and A2, which have been called “protein antibiotics” because of their functional resemblance to cell wall antibacterial agents (8), no clear conceptual framework exists for the lytic function of the 75-aa L protein (Fig. 1). L has a hydrophilic N-terminal domain dominated by multiple basic residues and a hydrophobic C-terminal domain, presenting an interesting comparison with ϕX174 E (3). Genetic analysis had shown that the lytic function of E requires only the N-terminal hydrophobic domain and that the highly charged, basic C-terminal domain could be replaced with unrelated sequences, including β-galactosidase and green fluorescent protein (4, 9). Drawing on this comparison, the van Duin group showed that expression of N-terminal deletions of L retaining as few as 42 C-terminal residues were fully lytic and, indeed, truncations retaining only the last 27 residues had partial function (3). Thus, E and L, although lacking any sequence similarity, seem to have mirror image organization of functional domains. However, unlike that of E, induction of L did not lead to a block of PG synthesis, as assessed by incorporation of [3H]diaminopimelate (10). Moreover, a synthetic peptide corresponding to the C-terminal 25 aa was reported to dissipate the proton motive force of Escherichia coli inverted membrane vesicles and cause fluorescent dye leakage in reconstituted liposomes (11). Interestingly, despite the nonessential character of the N-terminal domain and the absence of any other secretory or membrane localization signals, it was reported that L was primarily localized in the periplasmic zones of adhesion between the inner membrane and outer membrane, also known as Bayer's patches (12). Moreover, biochemical analysis of the murein from cells that had undergone L-mediated lysis was reported to have a slightly decreased average chain length of glycan strands and slightly altered cross-linking between them. In addition, L-mediated lysis was blocked in cells grown at low pH, where penicillin-induced autolysis was also inhibited. Taken together, these results were interpreted as support for a general model where L somehow activates host autolytic enzymes, such as lytic transglycoslyases and dd-endopeptidases (13). Unfortunately, in the nearly 3 decades since thosse studies, no molecular link between the putative autolytic response or, indeed, any host protein and L has been established.
With the aim of identifying host factors involved in L-mediated lysis, a genetic approach was undertaken. Here, we report the identification of one such factor, the host chaperone DnaJ, and the results are discussed in terms of a model of L function and regulation.
(Methods, results, and analysis pertaining to the role of pcnB in resistance to plasmid-borne L expression have been previously reported in the dissertation of Brenley McIntosh [14]).
RESULTS
Selection of Ill mutants.
As our first attempt to identify host factors involved in L-mediated lysis, we cloned the L gene into a medium-copy-number vector under the control of the lambda late promoter, pR′, which in turn is driven by the inducible supply of the lambda late activator, Q, from a low-copy-number lac-ara vector. Induction of cells carrying these two plasmids (pQ and pRE-L) results in lysis in approximately the same time as infection by phage MS2, with comparable levels of L synthesis (Fig. 2A and 3A). HfrH cells carrying these plasmids were mutagenized with ethyl methanesulfonate and subjected to two rounds of induced lysis before plating for survivor colonies. Of 3,300 colonies screened, 5 were found to be Lac+, Q+, MS2r, and M13s (14). However, Hfr mapping and P1 transduction revealed that all five were recessive missense alleles of pcnB, encoding the poly(A) polymerase of E. coli (Fig. 2B). As expected, the pcnB defect reduced the copy number of the pRE-L plasmid to approximately single copy (not shown) (15), which accounted for the survival of these mutants in our inducible-plasmid system. Interestingly, these pcnB missense mutants were clustered in the poly(A) polymerase active site (Fig. 3B) and conferred dominant resistance to MS2 (Fig. 2C), suggesting a heretofore unsuspected role for PcnB in the MS2 infection cycle.
FIG 2.
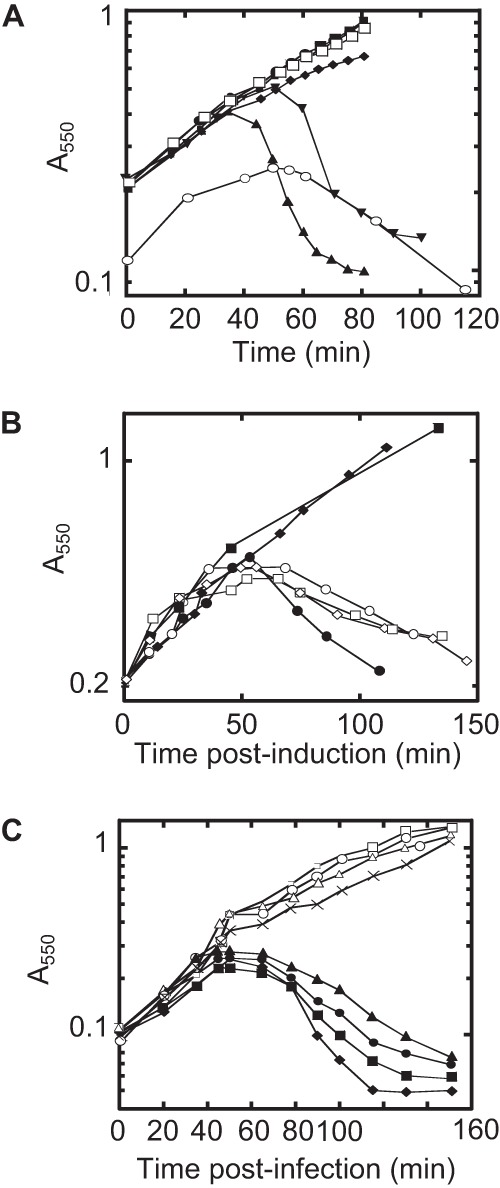
Expression of L from the pQ pRE-L dual-plasmid system is sufficient for lysis. (A) Lysis profile from induction of the dual-plasmid system with MS2 L under the control of the pR′ promoter with 1 mM IPTG or 1 mM IPTG and 0.2% arabinose (Ara) in RY15177 compared to lysis by MS2 infections of RY15177 at an MOI of 5. Cultures were grown at 37°C. Symbols: ●, pQ plus IPTG; ■, pRE-L plus IPTG; ◆, pQ pRE plus IPTG; ▼, pQ pRE-L plus IPTG; ▲, pQ pRE-L plus IPTG plus Ara; □, pQ pRE-L, uninduced; ○, MS2. (B) pcnB alleles are recessive. Induction of plasmid-borne L with IPTG (final concentration of 1 mM) at time zero. Host and plasmid symbols: ●, pcnB+ and vector; ○, pcnB+/pCA24N-pcnB; ■, BMC1 and vector; □, BMC1/pCA24N-pcnB; ◆, BMC2 and vector; ♢, BMC2/pCA24N-pcnB. (C) pcnB alleles confer dominant resistance to MS2 infection. Infection of pcnB alleles with MS2 at an MOI of 5 at time zero. Symbols: ●, pcnB+ and vector; ■, pcnB+/pCA24N-pcnB; ◆, pcnB and vector; ▲, pcnB/pCA24N-pcnB; □, BMC1 and vector; ○, BMC1/pCA24N-pcnB; Δ, BMC2 and vector; ×, BMC2/pCA24N-pcnB. The BMC1 and BMC2 pcnB mutations are defined in the legend to Fig. 3. This image was adapted from reference 14 with permission.
FIG 3.
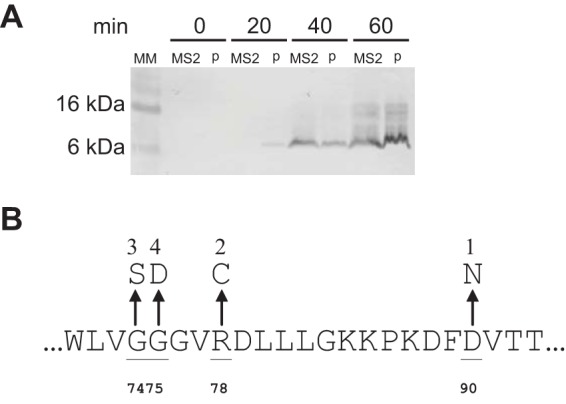
Plasmid-borne L is expressed at a level comparable to that attained in MS2-infected cells. MM represents the molecular mass marker, with sizes indicated on the left. Samples were collected at the times indicated after induction of the plasmids with IPTG (lanes p) or infection with MS2 at an MOI of 5 (lanes MS2). (B) The mutations of pcnB that confer dominant MS2 insensitivity are shown as missense changes in the region defined by residues 72 through 93. Strains BMC1 and BMC2 in Fig. 2 have the allelic changes marked 1 and 2 in panel B. This image was adapted from reference 14 with permission.
To avoid host mutants like pcnB that reduce either the plasmid copy number or expression of the L gene, we constructed a modified L expression vector, pKC11, with L and the lacZα gene in tandem under the control of an ara promoter. This allowed screening for blue survivor colonies after L induction. Expression of L from pKC11 was found to cause lysis at ∼45 min after induction. From approximately 200 survivor colonies screened, 2 blue colonies with irregular morphology were isolated on 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal)–arabinose plates. Both isolates exhibited an absolute plating defect for MS2 (see Fig. S1 in the supplemental material). We next tested the kinetics of lysis in liquid culture in comparison with the parental host and found that the mutants displayed a 40- to 50-min delay in the onset of lysis (Fig. 4A). To eliminate the possibility that the mutants confer nonspecific resistance to lysis, we tested the lysis by E of ϕX174 and A2 of Qβ in the mutant hosts and found no significant delay in either case (not shown). Thus, the mutants confer partial resistance specific to L-mediated lysis, enough to delay its onset, and were designated Ill mutants (insensitivity to L lysis).
FIG 4.
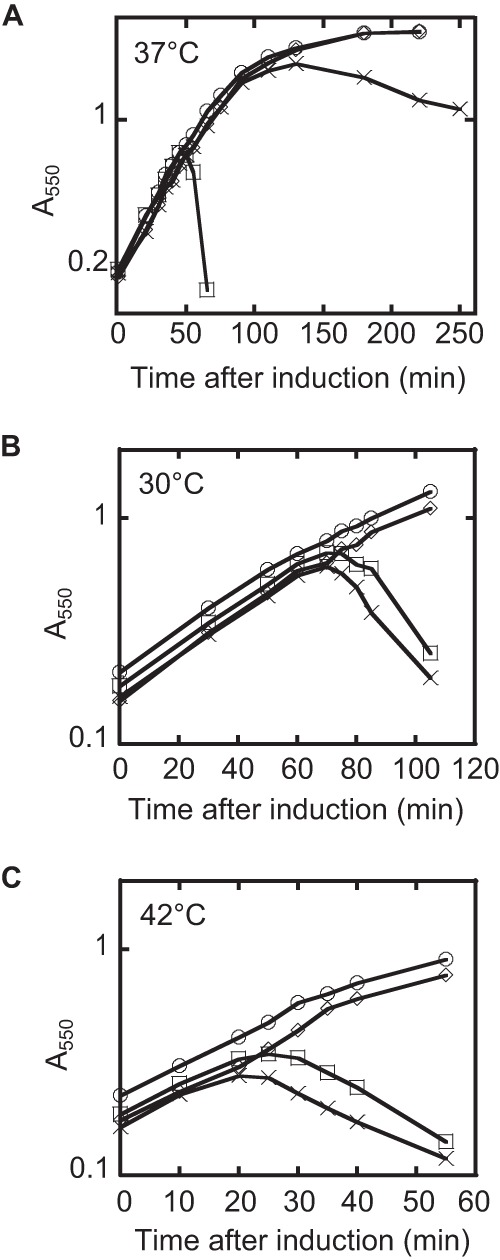
L-mediated lysis in a dnaJP330Q muant background is delayed, and the allele is recessive to the wild type. (A) The lysis profile of L in the wild-type and dnaJP330Q mutant backgrounds at 37°C. Cultures were grown to an A550 of ∼0.2 and induced with arabinose at a 0.4% (wt/vol) final concentration. Symbols: ○, TB28 and empty vector; ♢, dnaJP330Q and empty vector; □, TB28 and L; X, dnaJP330Q and L. (B, C) Same as panel A, except that the strains are merodiploid for dnaJ and the lysis profiles were taken at 30°C (B) and 42°C (C).
The Ill phenotype is due to a dnaJ mutation.
To assign the Ill phenotype to a specific host gene, genomic DNA from two Ill mutants was purified and subjected to whole-genome sequencing. An analysis of the genomic data revealed that both the Ill mutants, which may have been siblings, had a P330Q missense mutation in dnaJ. To determine whether the dnaJ mutation was the sole factor responsible for the lysis phenotype, the mutation was transduced to a new background. The newly constructed dnaJP330Q mutant retained the observed blue-colony survivor phenotype of the parental mutant (not shown). Moreover, the dnaJP330Q mutation exhibited strictly recessive behavior, with L-mediated lysis fully restored in a merodiploid (Fig. 4B and C).
Since DnaJ is a heat shock protein (16), we examined the effect of temperature on the kinetics of L-mediated lysis in the wild-type and mutant dnaJ backgrounds. As shown in Fig. 5A, the dnaJP330Q allele exhibited an absolute lysis defect at 30°C, as well as significant lysis delays at higher temperatures (Fig. 5B). In addition, the ΔdnaJ allele exhibited a more modest delay in lysis than the lysis profile of the Ill mutant (Fig. 5C). Immunoblot analysis showed that L accumulation paralleled the lysis defect at 37°C and 42°C but was normal at 30°C in the wild-type and dnaJP330Q mutant backgrounds (Fig. 6). Irrespective of the temperature dependence of the phenotype, the lysis defect of the P330Q allele is not due to an appreciable defect in the DnaJ/DnaK folding pathway, as assessed by the ability of phage lambda to propagate (see Fig. S2).
FIG 5.
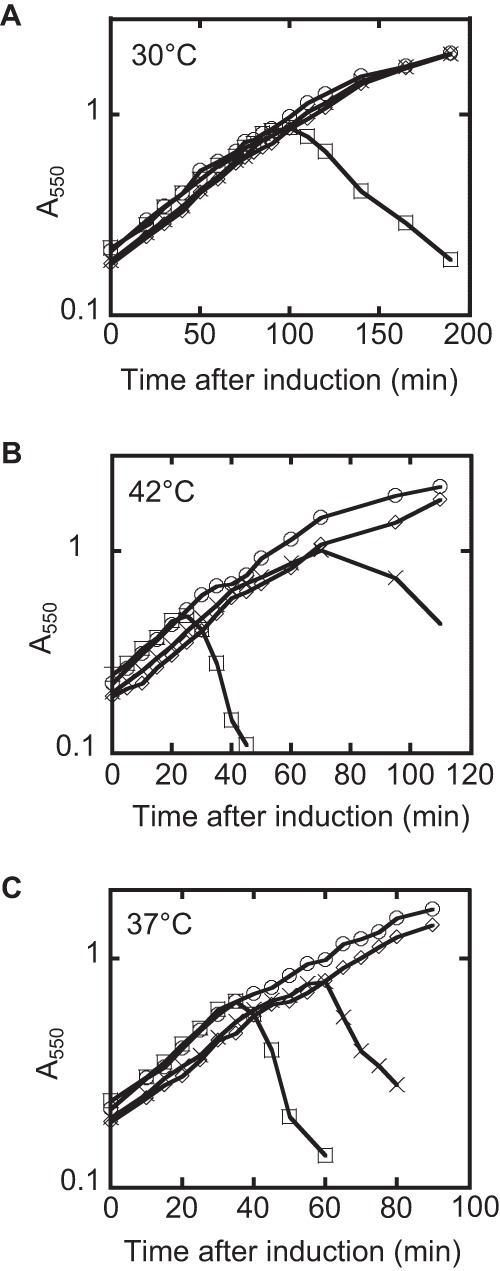
L-mediated lysis in the dnaJP330Q and ΔdnaJ mutant backgrounds. (A) The lysis profiles of L in the wild-type and dnaJP330Q mutant backgrounds at 30°C. Cultures were grown to an A550 of ∼0.2 and induced with arabinose at a 0.4% (wt/vol) final concentration. (B) Same as panel A, except that the lysis profiles were taken at 42°C. (C) Lysis profile of L in the wild-type and ΔdnaJ mutant backgrounds at 37°C. Symbols: ○, TB28 and empty vector; ♢, dnaJP330Q or ΔdnaJ mutant and empty vector; □, TB28 and L; X, dnaJP330Q or ΔdnaJ mutant and L.
FIG 6.
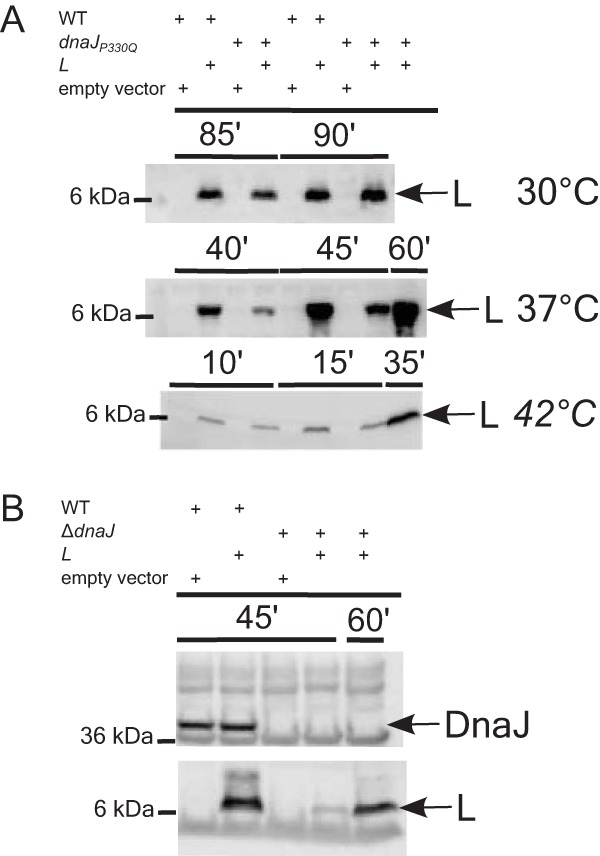
L accumulation in dnaJ mutants. (A) L accumulation is delayed in a dnaJP330Q mutant background compared to the wild-type (WT) background at 37°C and 42°C but not at 30°C. (B) L accumulation in the wild type and a ΔdnaJ mutant at 37°C (bottom). The absence of DnaJ in the ΔdnaJ mutant is shown by blotting with anti-DnaJ antibody (top). The bands corresponding to L and DnaJ are indicated with an arrow on the right. The molecular mass standard is shown on the left. The sample collection time points are shown directly above the blot in minutes.
The Lodj mutants bypass DnaJ.
To examine L dependence on DnaJ, we sought suppressors in L that could overcome the absolute lysis defect at 30°C in the dnaJP330Q mutant host. To achieve this, we used the plasmid release technique (see Materials and Methods) to enrich for L alleles with restored lytic competence in the mutant background. Plasmid DNAs prepared from individual lytic transformants, designated Lodj (L overcomes dnaJ) alleles, were sequenced. Each Lodj mutant plasmid was found to have a single nucleotide deletion that created an L gene encoding a protein in which the entire N-terminal half of L was replaced with a few N-terminal residues of the Rep protein (Fig. 7A). To verify that Lodj alleles were indeed lytically functional, we cloned the new truncated L genes into an inducible plasmid and showed that the lysis timing supported by the Lodj alleles was comparable and early in both parental and dnaJP330Q mutant hosts at 30°C, as well as 37°C (Fig. 7B). To rule out the possibility that it is the presence of the first few amino acid residues of Rep that confers the ability to cause lysis in the dnaJP330Q background, we used site-directed mutagenesis (SDM) to replace the first 36 codons of L with a His tag (Fig. 7A) and showed that this construct also functions as a Lodj allele (not shown). Taken together, these results indicate that it is the presence of the dispensable, N-terminal, highly basic domain of L that confers the requirement for DnaJ and that the N-terminal truncations supported significantly early lysis compared to the full-length allele.
FIG 7.

The Lodj alleles suppress lysis block in dnaJP330Q mutants. (A) Primary structure of L aligned with three Lodj proteins and His6-L37–75. Amino acids that are different from those of wild-type L are highlighted. (B, C) The lysis profiles of L and Lodj1 to Lodj3 in the wild-type (WT) and dnaJP330Q mutant backgrounds at 30°C (B) and 37°C (C). Symbols: ○, TB28 and empty vector; □, TB28 and L; quartered squares, dnaJP330Q and L; ♢, TB28 and Lodj1; ×, TB28 and Lodj2; ◆, TB28 and Lodj3; ▲, dnaJP330Q and Lodj1; Δ, dnaJP330Q and Lodj2; ■, dnaJP330Q and Lodj3.
DnaJ interacts with MS2 L.
To further investigate the role of DnaJ in L-mediated lysis, we asked if DnaJ and L form a complex. We constructed both His6-L and His6-Lodj, encoding L and Lodj proteins tagged with an N-terminal hexahistidine sequence, and showed that both were fully functional (not shown). Cultures carrying these plasmid-borne alleles were induced, and membrane extracts were prepared from samples taken immediately before lysis. Pulldown assays with Dynabeads (see Materials and Methods) showed that DnaJ was associated with L in the parental cells but not in the dnaJP330Q Ill mutant (Fig. 8); this association was abrogated for the Lodj mutant in both the parental and Ill mutant backgrounds. Taken together, these results indicate that L and DnaJ form a membrane-associated complex in vivo and that the complex depends on the nonessential N-terminal segment of L interacting with the C-terminal domain of DnaJ.
FIG 8.

DnaJ interacts with L but not with Lodj. Membrane extracts containing His6-L or His6-Lodj1 were solubilized in detergent and bound to anti-His Dynabeads, and the proteins bound to the beads were analyzed by Western blotting with anti-DnaJ (top) and anti-His (bottom) antibodies. The bands corresponding to DnaJ, His6-L, and His6-Lodj1 are indicated on the right. The molecular mass standards are shown on the left. WT, wild type.
DISCUSSION
The Leviviridae, among the most ancient and possibly the simplest known viruses, effect rapid and efficient lysis of the host through the action of the L protein; this occurs without inhibition of cell wall biosynthesis (10). For Gram-negative bacteria, this is the only known lysis phenomenon mediated by antibiotics or by phage that does not involve either inhibition of PG synthesis or elaboration of a muralytic enzyme by the phage. Understanding how L triggers an autolytic response and identifying the host factors involved in the response might reveal an entirely new perspective for novel antibiotics.
Prior perspectives on L function.
Most of the extant work on L-mediated lysis involved a combination of biochemical and electron microscopy analyses of the murein structure in both intact and L-lysed bacteria. A model was proposed in which L protein localized to the periplasmic zones of adhesion and caused inappropriate activation of cellular autolytic functions such as lytic transglycosylases and dd-endopeptidases (12, 13). Moreover, it was shown that a defect in membrane-derived oligosaccharide (MDO) biosynthesis provided resistance to L lysis, possibly by impeding appropriate localization of the lysis protein (17). However, this model lacked genetic evidence and afforded no clear molecular framework that could lead to mechanistic understanding. In addition, the very existence of zones of adhesion in growing cells has become controversial, since they have not been observed in cryo-electron microscopic images but only in cells that have undergone dehydration and fixation for transmission electron microscopy (18). Importantly, the MDO dependency was not specific to L-mediated lysis, as lysis protein E of ssDNA phage ϕX174 displayed a similar dependency (17). At the time, E was also thought to induce autolysis (19). However, we have since shown unambiguously that E is a specific inhibitor of MraY and causes lysis by inhibiting the biosynthesis of lipid I (4, 6). Thus, it seems likely that the MDO sensitivity of these lysis pathways, one of which involves blockage of PG synthesis and the other does not, is indirect and nonspecific.
The role of DnaJ in L-mediated lysis.
Here we have taken a genetic approach to identify host factors required for L lysis, by the conceptually simple method of selecting for host mutants resistant to L function. The results show that the host chaperone DnaJ is one such factor. We have shown that the P330Q missense change in DnaJ, although preserving the essential heat shock function of DnaJ, confers a defect in L-mediated lysis, absolute at 30°C and partial at 37°C. Proline 330 is the most conserved residue in the C-terminal domain of DnaJ, present in 689 of the 690 full-length DnaJ sequences from Proteobacteria (http://eggnog.embl.de/version_3.0/index.html). The one exception, from a 2003 genomic sequence of Shigella flexneri, is probably a sequencing error, since more recent S. flexneri genomes do not show this variance. Little is known about the biological function of the C terminus. Proline 330 was chosen as the end of the last DnaJ subdomain before a putative extreme C-terminal domain that was shown to be required for dimerization of DnaJ (20). Although the block in lysis at 30°C is sufficiently strict to allow the isolation of the Lodj suppressor mutations, it is unclear why the lysis-defective phenotype is leakier at 37°C and 42°C. It has been estimated that at 37°C there are <500 copies of DnaJ per cell and the rate of synthesis of new DnaJ molecules at 30°C is ∼10-fold lower than that at 37°C. (16). It is possible that at higher temperatures (37°C and 42°C), higher levels of DnaJP330Q and/or the induction of other heat shock proteins contribute to partial rescue of the lysis defect.
The lysis-defective phenotype of the dnaJP330Q Ill mutant is L specific, with no effect on lysis by E or A2, the other two prototypic SGL proteins. Since the delayed lysis phenotype is specific for L-mediated lysis and not a general defect, the simplest notion is that the missense change in DnaJ abrogates an important interaction between DnaJ and L. This idea is supported by the finding that DnaJ, but not DnaJP330Q, copurifies with His-tagged L.
What role does DnaJ play in L lysis? It is clearly not the target of L, since deletions of the N-terminal domain of L relieve the dependency on DnaJ and restore normal L-mediated lysis, indeed, the Lodj derivatives, expressed from isogenic plasmid environments, evoke lysis much faster than the parental full-length proteins. The simplest idea is that DnaJ is required for proper folding of full-length L but not of the truncated Lodj proteins. Figure 9 shows a cartoon rendition of the model. In the wild-type host, DnaJ interacts with the improperly folded N-terminal hydrophilic domain, thus avoiding a steric clash with a putative cytoplasmic domain of the (still unknown) target protein. Once the complex is formed, DnaJ can dissociate and catalyze further L target events. In support of this notion, there is a precedent for DnaJ acting as a chaperone independently of DnaK-ATPase heat shock activity, in stabilizing a Lys/Arg-rich C-terminal domain of TorI, the recombination directionality factor of the KplE1 cryptic prophage of E. coli (21). In the ΔdnaJ mutant host, compensatory elevations in other, less specific, chaperones would lead to similar folding events, although with slower kinetics in the absence of the specific interaction. In contrast, the Lodj truncations lack the N-terminal domain and thus do not suffer from the potential steric clash with the target. This model of DnaJ-mediated activation of L assumes that L has a protein target (Fig. 9). However, the same arguments could be used for the role of DnaJ activating L by converting it to a conformation allowing its oligomerization, a notion more compatible with the model mentioned above, in which Goessens et al. (11) implicated the C-terminal 25 residues of L in the formation of oligomeric pores in the cytoplasmic membrane. While this scenario cannot be ruled out, we favor a general model of L having a protein target based on comparisons to the other lysis proteins of single-stranded nucleic acid phages (8) and to the fact that, to date, no association between membrane depolarization and autolysis in E. coli has been reported.
FIG 9.
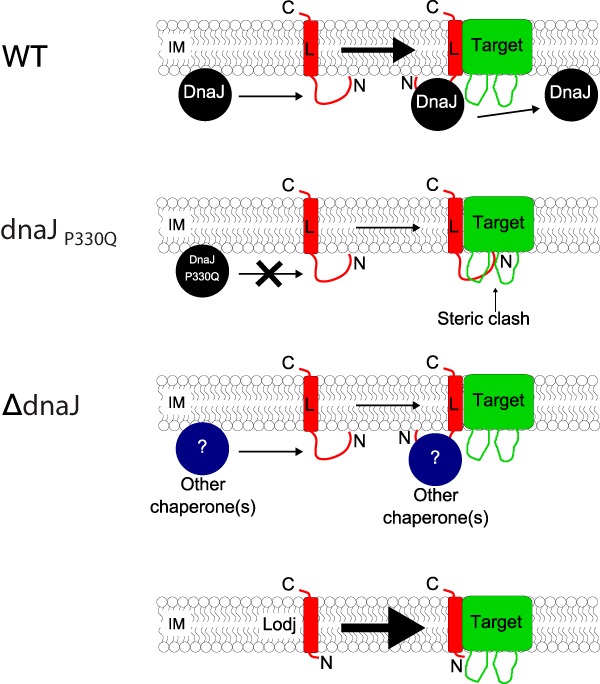
Model of the role of DnaJ in L-mediated lysis. DnaJ interacts with the highly basic N-terminal domain of L at the membrane, possibly to remove steric constraints inhibiting the interaction of L (red) with its target (green). The DnaJP330Q variant loses its ability to interact with L, leading to a less stable interaction between L and its target, resulting in a delayed and gradual onset of lysis. However, in the complete absence of DnaJ, compensatory cellular chaperone activities stabilize the L-target interaction, resulting in a modest delay in lysis. The products of the Lodj alleles lack the N-terminal domain and thus bypass the requirement for DnaJ. The thickness of the arrows indicates favorability for complex formation; i.e., the thicker the arrow, the more favorable the interaction between L and its target. WT, wild type.
From this perspective, the parallel with lysis protein E and the cytoplasmic chaperone SlyD is striking. SlyD is absolutely required for the proteolytic stability of E, which, like L, has a large dispensable domain rich in charged and hydrophilic residues. Like that of L, removal of this dispensable domain also abrogates the chaperone dependence of E, although the position of the dispensable domain, at the C terminus in E and the N terminus in L, is opposite in the two lysis proteins. We suggest that these dispensable domains, evidently requiring chaperone activity for proper folding, have evolved as regulatory “damping” features of the two lysis proteins. Among general phage functions like genome replication and virion morphogenesis, lysis is distinct in that maximum efficiency and speed are undesirable. Moreover, in these simple phages with highly constrained genome sizes, both the E and L lysis genes were forced to evolve in alternate reading frames of essential genes. It is logical that the smallest possible lytic domain would emerge from such an evolutionary constraint, and indeed, the essential segments of E and L are the N-terminal 32 and C-terminal 30 residues, respectively. In these severely restricted out-of-frame contexts, it would be much less challenging to evolve a highly charged region lacking secondary structure and thus compromised in terms of unassisted folding. The acquisition of the crippled domain conferred a requirement for interaction with a cytoplasmic chaperone and thus provided a context for retarding lysis to allow time for the assembly of progeny virions. Indeed, this scenario would also confer potential for physiological regulation of lysis timing in the E and L systems via the level or activity of the respective chaperone. In passing, we note that the distinctive domain structure of MS2 L and its homologs is not common to all ssRNA phage lysis proteins. For example, in the coliphage M and the Caulobacter phage Cb5, the predicted membrane domains are at the N terminus, more akin to the arrangement in ϕX174 E. In these cases, a similar regulatory interaction with a chaperone protein might be involved in lysis timing, as proposed for the FKBP-like cytoplasmic chaperone SlyD and E (1).
The results reported here thus complement the extensive studies by van Duin and colleagues on the regulation of the L gene at the level of translation (22, 23). Those studies revealed that sequestration of the Shine-Dalgarno sequence and start codon of L by the formation of a stable RNA hairpin secondary structure effectively represses L expression. L translation is thought to require that ribosomes that terminate at the Coat-encoding gene stop codon randomly backtrack and reinitiate translation at the L start codon. On the basis of the relative levels of Coat monomers, levels of L protein, and random probability of drifting ribosomes, it is estimated that only 5% of the ribosomes backtrack to reinitiate at L (24). This was proposed to ensure that L accumulates gradually in the cell and thus provides time for progeny maturation. Our results suggest that the function of L is further regulated by a posttranslational regulator, DnaJ. It may be interesting to explore whether levels of DnaJ activity can be correlated with the length and fecundity of MS2 infection cycles under different physiological conditions.
MATERIALS AND METHODS
Culture growth, antibodies, and chemicals.
Unless indicated otherwise, LB broth and agar were used as growth medium. When indicated, medium was supplemented with ampicillin (Amp), kanamycin (Kan), chloramphenicol (Cam), and tetracycline (Tet) at concentrations of 100, 40, 10, and 10 μg ml−1, respectively. Growth in liquid cultures and lysis were monitored by measuring the A550 as previously described (25). When indicated, isopropyl-β-d-thiogalactopyranoside (IPTG; Research Products International), X-Gal, and arabinose were added at final concentrations of 1 mM, 10 μg/ml, and 0.4%, respectively. Primary antibodies against MS2 L peptide TPASTNRRRPFKHEDC were raised in a rabbit (Sigma Genosys), mouse anti-His antibodies were purchased from Sigma, and rabbit anti-DnaJ antibodies were purchased from Enzo Life Sciences. Horseradish peroxidase-conjugated secondary goat anti-mouse and goat anti-rabbit antibodies were purchased from Thermo Scientific. Unless otherwise indicated, all chemicals were purchased from Sigma-Aldrich.
Bacterial strains and bacteriophages.
The bacterial strains and bacteriophages used in this study are described in Table 1, and the primers used are described in Table S1 in the supplemental material. The dnaJP330Q allele was moved from the Ill1 mutant into the threonine auxotroph RY34314 (TB28 ΔthrC::kan) by P1 transduction (26), selecting for growth on M9 minimal agar supplemented with 0.2% glucose (27). The dnaJ locus was amplified from Thr+ transductants with primers KC130 and KC131 and sequenced to confirm the mutation. The dnaJ merodiploids were constructed by mating the RY15784 (F′104 thr-leu ΔleuA::cat) strain with either TB28 or RY34356 (TB28 thrC+ dnaJP330Q) and selecting exconjugants on LB supplemented with both Kan and Cam. Similarly, RY34155 was constructed by mating RY34154 with XL1-Blue and selecting exconjugants on LB supplemented with both Amp and Tet. Strain RY34179 was constructed by P1 transduction of the ΔdnaJ::kan marker from JW0014.
TABLE 1.
Strains, phages, and plasmids used in this study
| Strain, phage, or plasmid | Relevant genotype or description | Reference or source |
|---|---|---|
| E. coli strains | ||
| XL1-Blue | recA endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′::Tn10 proAB+ lacIq Δ(lacZ)M15] | Stratagene |
| MG1655 | ilvG rfb-50 rph-1 | 30 |
| TB28 | MG1655 lacIZYA <frt> | 31 |
| JW0003 | ΔthrC::kan | 32 |
| JW0014 | ΔdnaJ::kan | 32 |
| RY15784 | F′104 thr-leu ΔleuA::cat | Laboratory strain |
| RY15177 | HfrH lacIq fhuA::Tn10 | 14 |
| RY34179 | TB28 ΔdnaJ::kan | This study |
| RY34314 | TB28 ΔthrC::kan | This study |
| RY34356 | RY34314 thrC+ dnaJP330Q | This study |
| RY34154 | TB28/pKC11 | This study |
| RY34155 | RY34154 [F′::Tn10 proAB+ lacIq Δ(lacZ)M15] | This study |
| Phages | ||
| MS2 | ssRNA phage MS2 | Laboratory stock |
| λcI857bor::kan | dsDNA phage lambda with thermosensitive cI repressor; nonessential bor gene replaced with Kanr-encoding gene | Laboratory stock |
| Plasmids | ||
| pBAD24 | bla araC Para | 33 |
| pRE-L | L gene from MS2 cloned under control of lambda late promoter pR′ | 14 |
| pUC57-Lsyn lacZα | Synthetic tandem L (Lsyn) and lacZα genes cloned between EcoRI and HindIII sites | GenScript |
| pKC11 | bla araC Para::Lsyn lacZα | This study |
| pKC12 | bla araC Para::His6-Lsyn | This study |
| pKC13 | bla araC Para::Lodj1 | This study |
| pKC14 | bla araC Para::Lodj2 | This study |
| pKC15 | bla araC Para::Lodj3 | This study |
| pKC16 | bla araC Para::His6-Lodj1 | This study |
Plasmids.
The plasmids used in this study are listed in Table 1. A DNA fragment containing synthetic tandem L (Lsyn) and lacZα genes was cloned under the control of the pBAD promoter in plasmid pBAD24, resulting in plasmid construct pKC11. In this construct, both the Lsyn and lacZα genes were codon optimized for E. coli expression (Codon Optimization Tool; Integrated DNA Technologies, Coralville, IA) and the synthetic DNA with flanking EcoRI and HindIII sites at the 5′ and 3′ ends was cloned into pUC57 at GenScript. Using the same restriction sites, the synthetic fragment was moved into pBAD24 by standard techniques. Plasmid pKC12 was constructed by subcloning His6-L from pRE-His6-L (14). The DNA of His6-L was PCR amplified with primers KC18 and KC19, gel purified, digested with EcoRI and HindIII, and cloned into pBAD24. Plasmids pKC13, pKC14, and pKC15 were obtained through selection for Lodj alleles (see below). pKC16 was constructed by SDM of pKC12 with primer KC149.
MS2 infection.
Cultures of male strains were grown to an A550 of ∼0.4 and then diluted to an A550 of 0.1 in prewarmed LB medium. MS2 lysate was then added to the freshly diluted cultures at the MOIs (multiplicities of infection) indicated.
Ill mutant selection.
Cultures (25 ml) of RY34155 were grown to an A550 of 0.2 and induced with arabinose. After ∼2 h, the lysate was harvested by centrifugation at 10,000 × g for 10 min. The pellet was washed once with PBS (phosphate-buffered saline, pH 7.2), and the survivors were allowed to recover overnight in 5 ml of LB with appropriate antibiotics. The induction procedure was repeated, and the survivors from the second round of induction were serially diluted in PBS and plated on LB agar plates supplemented with appropriate antibiotics, IPTG, X-Gal, and arabinose. A total of six blue colonies were isolated and screened for MS2 phage resistance by using cross streaks as previously described (4). Phage-insensitive isolates were studied further by monitoring lysis phenotypes in induced cultures as described above. To quantify the expression of L in the mutants, a 1-ml sample was mixed with 111 μl of cold 100% trichloroacetic acid as previously described (28). The precipitates were collected by centrifugation at 13,000 rpm for 10 min in a microcentrifuge. The pellets were washed with 1 ml of cold acetone and air dried. The dried pellets were resuspended in 2× sample loading buffer with β-mercaptoethanol and boiled for 10 min. Equivalent amounts of protein were analyzed by SDS-PAGE and Western blotting as previously described (29). Both primary and secondary antibodies were used at a 1:3,000 dilution.
DNA sequencing and analysis.
Sequencing to check cloning and strain construction was done by Eton Biosciences (San Diego, CA). For whole-genome sequencing, genomic DNA extracted with the Qiagen genomic DNA kit was used to prepare 250-bp paired-end libraries with the Illumina TruSeq Nano DNA LT Library kit (set A) in accordance with the manufacturer's instructions. Whole-genome sequencing was done at the DNA sequencing facility of the Institute for Cellular and Molecular Biology at the University of Texas at Austin. The raw sequencing data were processed on Mutation Analysis Beta 1 (https://cpt.tamu.edu/galaxy-pub/workflow). Briefly, Bowtie2 was used to align the trimmed reads to the MG1655 reference sequence (accession no. NC_000913.3). To facilitate the visual display of single nucleotide polymorphisms (SNPs) or insertions/deletions (indels), the BAM output from Bowtie2 was processed through Mpileup and BCFtools to generate Variant Call Format, a standardized text file format. The SNP/indel variants with QUAL scores of >100 in both parental (RY34155) and Ill mutants were scored as positive. The positive variants present in Ill mutants but absent from the parental strain were further characterized.
Error-prone PCR mutagenesis and selection for L overcomes dnaJ (Lodj) mutants.
Error-prone PCR mutagenesis of the L gene was done with the GeneMorph II Random Mutagenesis kit (Agilent Technologies) in accordance with the instructions provided in the kit. Briefly, ∼900 ng of the L gene template (amplified from pRE-L) was used in the reaction mixture with primers KC30 and KC31 to provide ∼1 bp of change per double-stranded DNA molecule. The randomly mutagenized PCR product was gel purified, digested with EcoRI and HindIII, ligated into pBAD24 digested with the same enzymes, and electroporated into XL1-Blue. The transformants were pooled, and the plasmid DNA was extracted with a Qiagen mini-prep kit to generate a library of L mutants. Plasmids carrying Lodj alleles that restored lysis in the dnaJP330Q mutant background were obtained by plasmid release. Briefly, ∼100 ng of the mutant library was electroporated into RY34356. The transformants were pooled, diluted 1:20,000, grown in 25 ml of LB supplemented with appropriate antibiotics to an A550 of ∼0.2, and induced with arabinose. After 50 min, the culture medium was collected by centrifugation at 10,000 × g. A 20-ml volume of the supernatant medium was filtered through a 0.22-μm syringe filter (VWR) and then passed through a QIAprep 2.0 Spin Column (Qiagen). The bound DNA was eluted in 20 μl of sterile water. Ten microliters of the released plasmids was retransformed into the same strain, and the procedure was repeated for another two rounds. At the end of two rounds of amplification, the plasmid DNA was extracted and sequenced.
Pulldown of L and Lodj proteins.
LB cultures (500 ml) of TB28 with appropriate plasmids were grown to an A550 of ∼0.4, induced with arabinose at time zero, and harvested at allele-specific times by rapid cooling in ice and centrifugation at 10,000 × g for 10 min, 20 min (His6-Lodj1 allele), 40 min (L in TB28), or 60 min (L in dnaJP330Q). The cell pellets were resuspended in ∼3 ml of PBS supplemented with the P8849 Protease Inhibitor Cocktail (Sigma; 1 μl/35 A550 U of original culture) and passed through an Aminco French pressure cell at 16,000 lb/in2 three times to lyse the cells. The lysate was centrifuged at 10,000 × g for 10 min to remove intact cells. The cleared supernatant was centrifuged at 100,000 × g in a TLA 100.3 rotor (Beckman TL-100 centrifuge) to collect membrane fractions. The membrane fraction was then resuspended in 1 ml of STE (50 mM Tris [pH 8.0], 300 mM NaCl, 1% Empigen BB [Fluka] [pH 8.0]) and incubated overnight at 4°C with gentle mixing. The detergent extract was centrifuged at 100,000 × g to separate the detergent-solubilized proteins from detergent-insoluble components. The supernatant (∼900 μl) was collected, mixed with 50 μl of Dynabeads His tag beads, and incubated for 5 min at room temperature on a roller drum. The binding and elution protocol was followed in accordance with the manufacturer's instructions, except that the beads were washed five times in STE. A 20-μl volume of each elution was mixed with 20 μl of 2× sample loading buffer, heated at 100°C for 10 min, and then analyzed by Western blotting.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant GM27099 and by the Center for Phage Technology at Texas A&M University, jointly sponsored by Texas A&M AgriLife. Additional funding for this project and support of J. S. Tran was provided by the Beckman Scholars Program administered by the Arnold and Mabel Beckman Foundation.
We thank Catrina Reed for important constructs and advice. The clerical assistance of Daisy Wilbert is highly appreciated.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00058-17.
REFERENCES
- 1.Bernhardt TG, Roof WD, Young R. 2002. The Escherichia coli FKBP-type PPIase SlyD is required for the stabilization of the E lysis protein of bacteriophage ϕX174. Mol Microbiol 45:99–108. doi: 10.1046/j.1365-2958.2002.02984.x. [DOI] [PubMed] [Google Scholar]
- 2.Atkins JF, Steitz JA, Anderson CW, Model P. 1979. Binding of mammalian ribosomes to MS2 phage RNA reveals an overlapping gene encoding a lysis function. Cell 18:247–256. doi: 10.1016/0092-8674(79)90044-8. [DOI] [PubMed] [Google Scholar]
- 3.Berkhout B, de Smit MH, Spanjaard RA, Blom T, van Duin J. 1985. The amino terminal half of the MS2-coded lysis protein is dispensable for function: implications for our understanding of coding region overlaps. EMBO J 4:3315–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernhardt TG, Roof WD, Young R. 2000. Genetic evidence that the bacteriophage ϕX174 lysis protein inhibits cell wall synthesis. Proc Natl Acad Sci U S A 97:4297–4302. doi: 10.1073/pnas.97.8.4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernhardt TG, Wang IN, Struck DK, Young R. 2001. A protein antibiotic in the phage Qβ virion: diversity in lysis targets. Science 292:2326–2329. doi: 10.1126/science.1058289. [DOI] [PubMed] [Google Scholar]
- 6.Bernhardt TG, Struck DK, Young R. 2001. The lysis protein E of ϕX174 is a specific inhibitor of the MraY-catalyzed step in peptidoglycan synthesis. J Biol Chem 276:6093–6097. doi: 10.1074/jbc.M007638200. [DOI] [PubMed] [Google Scholar]
- 7.Roof WD, Horne SM, Young KD, Young R. 1994. slyD, a host gene required for ϕX174 lysis, is related to the FK506-binding protein family of peptidyl-prolyl cis-trans-isomerases. J Biol Chem 269:2902–2910. [PubMed] [Google Scholar]
- 8.Bernhardt TG, Wang IN, Struck DK, Young R. 2002. Breaking free: “protein antibiotics” and phage lysis. Res Microbiol 153:493–501. doi: 10.1016/S0923-2508(02)01330-X. [DOI] [PubMed] [Google Scholar]
- 9.Maratea D, Young K, Young R. 1985. Deletion and fusion analysis of the ϕX174 lysis gene E. Gene 40:39–46. doi: 10.1016/0378-1119(85)90022-8. [DOI] [PubMed] [Google Scholar]
- 10.Höltje JV, van Duin J. 1984. MS2 phage induced lysis of E. coli depends upon the activity of the bacterial autolysins, p 195–199. In Nombela C. (ed), Microbial cell wall synthesis and autolysis. Elsevier Science Publishers, New York, NY. [Google Scholar]
- 11.Goessens WHF, Driessen AJM, Wilschut J, van Duin J. 1988. A synthetic peptide corresponding to the C-terminal 25 residues of phage MS2-coded lysis protein dissipates the proton-motive force in Escherichia coli membrane vesicles by generating hydrophilic pores. EMBO J 7:867–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walderich B, Höltje JV. 1989. Specific localization of the lysis protein of bacteriophage MS2 in membrane adhesion sites of Escherichia coli. J Bacteriol 171:3331–3336. doi: 10.1128/jb.171.6.3331-3336.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walderich B, Ursinus-Wosner A, van Duin J, Höltje JV. 1988. Induction of the autolytic system of Escherichia coli by specific insertion of bacteriophage MS2 lysis protein into the bacterial cell envelope. J Bacteriol 170:5027–5033. doi: 10.1128/jb.170.11.5027-5033.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McIntosh BK. 2008. Bacteriophage MS2 L protein: genetic and biochemical characterization. Ph.D. dissertation. Texas A&M University, College Station, TX. [Google Scholar]
- 15.Lopilato J, Bortner S, Beckwith J. 1986. Mutations in a new chromosomal gene of Escherichia coli K-12, pcnB, reduce plasmid copy number of pBR322 and its derivatives. Mol Gen Genet 205:285–290. doi: 10.1007/BF00430440. [DOI] [PubMed] [Google Scholar]
- 16.Bardwell JC, Tilly K, Craig E, King J, Zylicz M, Georgopoulos C. 1986. The nucleotide sequence of the Escherichia coli K12 dnaJ+ gene. A gene that encodes a heat shock protein. J Biol Chem 261:1782–1785. [PubMed] [Google Scholar]
- 17.Höltje JV, Fiedler W, Rotering H, Walderich B, van Duin J. 1988. Lysis induction of Escherichia coli by the cloned lysis protein of the phage MS2 depends on the presence of osmoregulatory membrane-derived oligosaccharides. J Biol Chem 263:3539–3541. [PubMed] [Google Scholar]
- 18.Kellenberger E. 1990. The ‘Bayer bridges’ confronted with results from improved electron microscopy methods. Mol Microbiol 4:697–705. doi: 10.1111/j.1365-2958.1990.tb00640.x. [DOI] [PubMed] [Google Scholar]
- 19.Lubitz W, Halfmann G, Plapp R. 1984. Lysis of Escherichia coli after infection with ϕX174 depends on the regulation of the cellular autolytic system. J Gen Microbiol 130:1079–1087. [DOI] [PubMed] [Google Scholar]
- 20.Shi YY, Hong XG, Wang CC. 2005. The C-terminal (331–376) sequence of Escherichia coli DnaJ is essential for dimerization and chaperone activity: a small angle X-ray scattering study in solution. J Biol Chem 280:22761–22768. doi: 10.1074/jbc.M503643200. [DOI] [PubMed] [Google Scholar]
- 21.Puvirajesinghe TM, Elantak L, Lignon S, Franche N, Ilbert M, Ansaldi M. 2012. DnaJ (Hsp40 protein) binding to folded substrate impacts KplE1 prophage excision efficiency. J Biol Chem 287:14169–14177. doi: 10.1074/jbc.M111.331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt BF, Berkhout B, Overbeek GP, van Strien A, van Duin J. 1987. Determination of the RNA secondary structure that regulates lysis gene expression in bacteriophage MS2. J Mol Biol 195:505–516. doi: 10.1016/0022-2836(87)90179-3. [DOI] [PubMed] [Google Scholar]
- 23.Berkhout B. 1986. Translational control mechanisms in RNA bacteriophage MS2. Ph.D. dissertation Leiden University, Leiden, The Netherlands. [Google Scholar]
- 24.Adhin MR, van Duin J. 1990. Scanning model of translational reinitiation in Eubacteria. J Mol Biol 213:811–818. doi: 10.1016/S0022-2836(05)80265-7. [DOI] [PubMed] [Google Scholar]
- 25.Smith DL, Chang CY, Young R. 1998. The λ holin accumulates beyond the lethal triggering concentration under hyper-expression conditions. Gene Expr 7:39–52. [PMC free article] [PubMed] [Google Scholar]
- 26.Miller JH. 1972. Experiments in molecular genetics, p 201–205. Cold Spring Harbor Laboratory, Cold Spring Harbor NY. [Google Scholar]
- 27.Miller JH. 1972. Experiments in molecular genetics, p 431 Cold Spring Harbor Laboratory, Cold Spring Harbor NY. [Google Scholar]
- 28.Berry J, Savva C, Holzenburg A, Young R. 2010. The lambda spanin components Rz and Rz1 undergo tertiary and quaternary rearrangements upon complex formation. Protein Sci 19:1967–1977. doi: 10.1002/pro.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moussa SH, Kuznetsov V, Tran TA, Sacchettini JC, Young R. 2012. Protein determinants of phage T4 lysis inhibition. Protein Sci 21:571–582. doi: 10.1002/pro.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guyer MS, Reed RR, Steitz JA, Low KB. 1981. Identification of a sex-factor-affinity site in E. coli as gamma delta. Cold Spring Harb Symp Quant Biol 45:135–140. [DOI] [PubMed] [Google Scholar]
- 31.Bernhardt TG, de Boer PA. 2003. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol 48:1171–1182. doi: 10.1046/j.1365-2958.2003.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.