

ABSTRACT
Vibrio cholerae is the causative agent of the severe diarrheal disease cholera. V. cholerae thrives within the human host, where it replicates to high numbers, but it also persists within the aquatic environments of ocean and brackish water. To survive within these nutritionally diverse environments, V. cholerae must encode the necessary tools to acquire the essential nutrient iron in all forms it may encounter. A prior study of systems involved in iron transport in V. cholerae revealed the existence of vciB, which, while unable to directly transport iron, stimulates the transport of iron through ferrous (Fe2+) iron transport systems. We demonstrate here a role for VciB in V. cholerae in which VciB stimulates the reduction of Fe3+ to Fe2+, which can be subsequently transported into the cell with the ferrous iron transporter Feo. Iron reduction is independent of functional iron transport but is associated with the electron transport chain. Comparative analysis of VciB orthologs suggests a similar role for other proteins in the VciB family. Our data indicate that VciB is a dimer located in the inner membrane with three transmembrane segments and a large periplasmic loop. Directed mutagenesis of the protein reveals two highly conserved histidine residues required for function. Taken together, our results support a model whereby VciB reduces ferric iron using energy from the electron transport chain.
IMPORTANCE Vibrio cholerae is a prolific human pathogen and environmental organism. The acquisition of essential nutrients such as iron is critical for replication, and V. cholerae encodes a number of mechanisms to use iron from diverse environments. Here, we describe the V. cholerae protein VciB that increases the reduction of oxidized ferric iron (Fe3+) to the ferrous form (Fe2+), thus promoting iron acquisition through ferrous iron transporters. Analysis of VciB orthologs in Burkholderia and Aeromonas spp. suggest that they have a similar activity, allowing a functional assignment for this previously uncharacterized protein family. This study builds upon our understanding of proteins known to mediate iron reduction in bacteria.
KEYWORDS: Vibrio cholerae, iron acquisition, iron reductase
INTRODUCTION
Vibrio cholerae is the etiological agent of the severe diarrheal disease cholera. Not only a highly successful human pathogen, V. cholerae also thrives in its environmental reservoirs, ocean and brackish waters, where it associates with various flora and fauna (1). To survive in these diverse environments, V. cholerae uses a vast array of tools to acquire necessary nutrition. Iron is an essential nutrient with roles in respiration (2, 3), nucleotide reduction (4), and oxidative stress tolerance (5). In oxidizing environments, iron can be found in the ferric state (Fe3+), which has low solubility and forms insoluble complexes (6), while the more readily soluble ferrous iron (Fe2+) predominates in reducing environments. Within the human, iron is tightly complexed to carrier molecules or proteins (7), greatly restricting its availability to V. cholerae during infection. V. cholerae is found in all of these types of environmental conditions and thus has evolved systems to scavenge iron in a variety of forms (8). Iron not complexed to a carrier molecule can be acquired through the Feo ferrous iron transporter and the Fbp ferric iron transporter (9). V. cholerae also produces the siderophore vibriobactin (10–13), which is secreted into the environment and binds ferric iron. Iron bound to vibriobactin is transported into the cell via outer membrane receptors and inner membrane transport systems (14–16). Similar transport systems allow the use of iron from ferrichrome (17), enterobactin (18, 19), heme (20–23), and other small iron chelates (24).
In studies of iron transporters in V. cholerae, Mey et al. (25) identified a gene, vciB (VC0283), that played an accessory role in iron acquisition. VciB itself did not actively transport iron, but it stimulated iron transport when coexpressed with ferrous iron specific transporters, such as Feo in Escherichia coli and the unrelated ABC-type transporter Sit in Shigella flexneri. In contrast, VciB failed to stimulate iron transport through the ferric iron transporters V. cholerae Fbp or Haemophilus influenzae Hit. The ability of VciB to stimulate iron transport by the structurally dissimilar Feo and Sit systems led Mey et al. (25) to propose a model whereby VciB increased the reduction of ferric iron to ferrous iron, thus allowing for subsequent import through ferrous iron specific transporters.
We show here that VciB supports iron acquisition by increasing the ferric iron reductase activity in the cell, thus increasing the amount of the ferrous iron substrate for the Feo system. Orthologous proteins from Aeromonas hydrophila and Burkholderia species have similar functions, suggesting a shared role for this family of proteins. Further, we determined that VciB is a dimer and confirmed its predicted topology as an inner membrane protein with a large periplasmic loop. Mutagenesis identified two histidine residues essential for VciB function. These histidine mutants exert a dominant-negative effect on the wild-type chromosomal copy, indicating that VciB functions as a dimer.
RESULTS
VciB stimulates the growth of V. cholerae when Feo is the sole iron acquisition system.
Previous studies in E. coli indicated VciB stimulated iron acquisition by ferrous iron transport systems. However, no phenotypes in V. cholerae were identified for a vciB mutant in growth in vitro or in the infant mouse colonization model (25), likely due to the large number of iron transport systems in V. cholerae. Subsequently, the identification of all apparent iron transporters that support growth in laboratory conditions has allowed generation of isogenic V. cholerae iron transport mutants in which none or only one of these high-affinity iron acquisition systems was present, facilitating functional characterization of each individual system (26). The iron transport systems that support growth under laboratory conditions are Feo (ferrous iron transporter) (9), Fbp (ferric iron transporter) (9), Vct (transport of iron bound to catechol siderophores and an unidentified iron ligand) (18, 19), and Vib (synthesis of the siderophore vibriobactin) (10–13). To assess the role of VciB in V. cholerae, vciB was mutated in strains carrying only one of these functional iron transport systems and the growth of these individual strains was assessed using a colony size assay, which is a sensitive and reproducible measure of iron use (9, 10) (Fig. 1A). Consistent with the previous observation with ferric iron transporters (25), strains with functional Fbp (vFbp) or vibriobactin (vVib) were unaffected by the presence of vciB. The strain dependent upon the Vct system for growth (vVct) was also unaffected by the presence of vciB, suggesting that the uncharacterized iron ligand for this system is not free ferrous iron. The strain dependent upon the Feo system for iron acquisition was significantly impacted by the presence of vciB. Diameters of colonies formed by the Feo+ VciB+ strain were significantly larger than the Feo+ strain lacking VciB function. Although VciB stimulated ferrous iron transporters, including V. cholerae Feo in E. coli (25), this is the first evidence of a role for vciB in V. cholerae iron acquisition.
FIG 1.

VciB stimulates the growth of V. cholerae when Feo is the sole iron acquisition system. A colony size assay was used to assess the growth of the indicated strains. (A) Diameters of colonies formed by the wild-type (O395), vFeo (EPV104), vFbp (EPV115), vVct (EPV103), and vVib (EPV126) strains and a null strain (EPV102), along with vciB mutant derivatives in each background (EPV123, EPV137, EPV133, EPV136, and EPV131, respectively), were measured. The asterisk indicates significance as determined by one-way analysis of variance (ANOVA [Sidak]; P < 0.0001). (B) The effects of plasmid-encoded VciB on growth of Feo+ strains with mutations in vciA or vciB were determined by colony size assays as described above. (i) EPV104, which has wild-type chromosomal vciAB (Feo+ VciAB+), (ii) EPV153, which has a mutation in chromosomal vciB (Feo+ VciB−), and (iii) EPV154, which has a mutation in vciA that is polar on vciB (Feo+ VciAB−), were transformed with empty vector and plasmid-encoded vciB. Asterisks indicate significance as determined by multiple t tests (P < 0.0001). (C) Comparison of growth of wild-type (O395), null (EPV102), Feo+ VciB+ (EPV104), and Feo+ VciB− (EPV123) strains on unsupplemented LB agar and on L agar with 4 mM sodium ascorbate. Asterisks indicate significance as determined by a two-way ANOVA multiple-comparison test (Sidak; P < 0.0001). For all experiments, at least 10 well-isolated colonies were measured using a reticle after 24 h of incubation at 37°C. The data are means and standard deviations and are representative of biological replicates.
vciB is located in a two-gene operon downstream of vciA, which encodes a putative outer membrane protein. Function of VciB in E. coli does not require vciA; however, this could be due to the presence of an E. coli gene(s) that substitutes for the absence of vciA. To test the role of vciA in V. cholerae, vciA was deleted in the vFeo background. Deletion of the vciA coding region alone resulted in a similar growth defect to the deletion of vciB (Fig. 1B). However, the addition of vciB alone on a plasmid was sufficient to restore colony growth, indicating that the growth defect of the vciA mutation is due to a polar effect on vciB expression and demonstrates, as was shown in E. coli, that VciA is not required for VciB function in V. cholerae. Expression of vciB from the plasmid pVCvciB stimulated growth of the parent vFeo strain beyond that of the chromosomally encoded copy, suggesting that increasing the expression of vciB has a direct correlation to increased growth.
VciB is required for robust growth of the Feo+ strain in ferrous-iron limited conditions.
VciB was proposed to function as a ferric iron reductase due to its ability to stimulate iron acquisition through a variety of ferrous iron transporters, including the V. cholerae, E. coli, and S. flexneri Feo systems and the S. flexneri Sit ABC transporter, but not through the ferric transporters Fbp and Hit (25). If the role of VciB is to reduce ferric iron to ferrous iron, then vciB should have a maximal effect under conditions where ferrous iron is limiting. Conversely, the requirement for VciB should be reduced under conditions which favor ferrous iron. To assess whether increasing ferrous iron would stimulate growth in the absence of VciB, the reducing agent ascorbate was added to the medium to reduce ferric iron to its ferrous form (Fig. 1C). The presence of supplementary ascorbate strongly stimulates growth of both the Feo+ VciB+ and Feo+ VciB− strains. In the absence of ascorbate, the presence of vciB caused an increase in colony size, whereas in the presence of ascorbate, this effect was markedly reduced. This shows that VciB has a maximal effect under conditions where ferric iron is predominant. Under reducing conditions, where ferrous iron is expected to be more prevalent, the requirement for vciB is reduced. This is consistent with the proposed role of iron reduction by VciB to make ferric iron available for ferrous iron transporters (25).
Orthologs of vciB perform a similar function.
VciB belongs to a family of proteins with undetermined function (COG3295). Orthologs of V. cholerae VciB are found in a number of genera other than Vibrio, including Burkholderia and Aeromonas. Members of these genera are clinically important, and select Burkholderia species cause severe disease with high mortality rates (27), while Aeromonas species are opportunistic pathogens that cause gastroenteritis and necrotizing fasciitis (28). To assess whether orthologs found in these organisms share a similar function with V. cholerae VciB, we tested their ability to complement the vciB mutation in the Feo+ VciB− strain. The vciB homologs were amplified from Burkholderia mallei, Burkholderia thailandensis, and Aeromonas hydrophila genomic DNA and cloned into pUC18 (see Fig. S1 in the supplemental material). These plasmids were transformed into the V. cholerae Feo+ VciB− strain and function assessed through the colony size assay. All vciB homologs stimulated growth in this strain, although there was variation in the degree of stimulation, with A. hydrophila VciB, which is more closely related to V. cholerae than are the Burkholderia proteins, having the highest activity (Fig. 2). The ability of these orthologs to stimulate the growth of V. cholerae also depended upon a functional Feo system (Fig. 2). These data suggest that VciB homologs share a similar function in reducing iron. Interestingly, while A. hydrophila strains have a VciA ortholog upstream of the VciB ortholog (25), Burkholderia species do not appear to have VciA (29), suggesting a separate or obsolete function.
FIG 2.
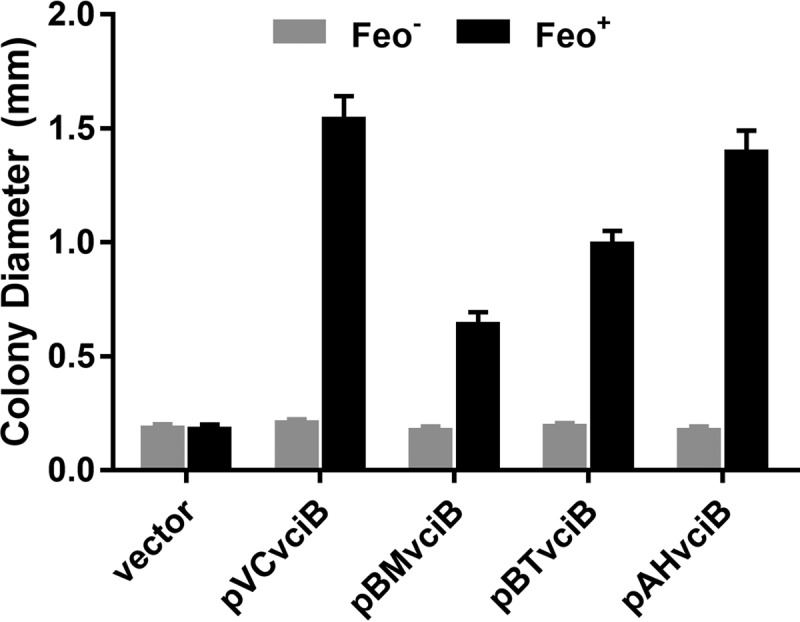
VciB orthologs share function. Orthologs cloned from B. mallei (pBMvciB), B. thailandensis (pBTvciB), and A. hydrophila (pAHvciB) were tested alongside empty vector (pUC18) and pVCvciB in the colony size assay using Feo+ VciB− (EPV153) and Feo− VciB− (EPV156) strain backgrounds. Ten well-isolated colonies were measured using a reticle after 24 h of incubation at 37°C; the data are means and standard deviations and are representative of biological replicates.
VciB confers increased ferric iron reductase activity to V. cholerae.
To test whether VciB supports the reduction of free ferric iron, the whole-cell ferric iron reduction assay described by Small and O'Brian (30) was adapted for use in V. cholerae. This assay takes advantage of the colorimetric reagent FerroZine, which binds specifically to ferrous iron, and the formation of a Fe2+-FerroZine complex results in a color change that can be quantified by measuring absorbance at 562 nm (31). Overexpression of the putative reductase should produce an excess of ferrous iron that can diffuse out of the cell where it is trapped by FerroZine in the supernatant. In addition, FerroZine is small enough to cross the outer membrane via porins, and it may bind ferrous iron at the site of reduction in the periplasm. Using the FerroZine assay, we tested whether a strain (Feo+ VciB−) with plasmid-encoded VciB (pVCvciB) would stimulate iron reduction relative to a strain carrying the empty vector (Fig. 3). A significant increase in the amount of reduced iron was observed in cultures of cells with pVCvciB, while only a low level was observed in cultures harboring the empty vector.
FIG 3.

VciB confers increased ferric iron reductase activity independently of Feo. Strains with (EPV153) or without (EPV156) feo carrying either empty vector or pVCvciB were assessed for the capacity to reduce iron over time using a whole-cell iron reductase assay. The A562 is proportional to the amount of Fe2+-FerroZine species. The data are means and standard deviations from three biological replicates.
Reductase activity is independent of Feo.
Because the growth assays for VciB activity required the presence of a functional ferrous iron transporter (25), it was not known whether VciB required Feo for its activity. Therefore, Feo+ and Feo− strains with pVCvciB were compared for reductase activity in the FerroZine assay (Fig. 3). The Feo− strain displayed a similar capacity to reduce iron, indicating that VciB functions independently of the Feo ferrous iron transporter (Fig. 3). These data indicate that VciB-mediated iron reduction is independent of iron transport, and thus a direct interaction between the iron transport system proteins and VciB is not necessary for VciB activity.
Deletion of nqr impacts iron reduction kinetics.
While our data suggest that VciB plays a role in iron reduction, it remains unclear whether VciB itself is the reductase and directly passes electrons to Fe3+ or if VciB is an intermediate in the process. VciB lacks homology to known reductases and the mechanism by which VciB increases ferrous iron is unknown. Many of the characterized bacterial reductases involved in iron acquisition are soluble and use cofactors (32). The Bradyrhizobium japonicum FrcB reductase is a membrane-associated cytochrome b-type protein that requires heme for reductase activity (30). For ferric iron reductases that allow iron to serve as the terminal electron acceptor, such as those studied in Shewanella oneidensis (33–35), Geobacter sulfurreducens (36, 37), and E. coli (34), electrons from the oxidation of NADH are passed through the electron transport chain to iron. Attempts to identify heme or other cofactors in samples of purified VciB have not been successful. Therefore, mutation of genes which serve a primary role in NADH oxidation was used to determine whether electron transport plays a role in VciB-mediated reduction of iron. V. cholerae uses either the Na+-translocating NADH:quinone oxidoreductase (Na+-NQR) or NADH dehydrogenase (NDH-2) to oxidize NADH and initiate the passage of electrons through electron transport chain constituents. We deleted the nqr locus, which encodes the Na+-NQR and tested the ability of pVCvciB to stimulate iron reduction in the absence of nqr. If VciB requires electrons from the electron transport chain, then the deletion of nqr should diminish or abolish iron reductase activity. Although the reduction of iron was observed, the kinetics of iron reduction was altered (Fig. 4). The nqr deletion strain displayed a reduced rate of iron reduction, suggesting that changes to the electron transport chain affect VciB function, but iron reduction does not depend solely on the Na+-NQR.
FIG 4.

VciB function requires the proton motive force. (A) Iron reduction by wild-type O395 and the Δnqr mutant (EPV162) harboring empty vector (pUC18) or pVCvciB was tested. Asterisks indicate significant differences between the wild type and the Δnqr mutant at the time point as determined by a two-way ANOVA multiple-comparison test (Sidak; P < 0.05). The data are means and standard deviations from three biological replicates. (B) Iron reduction was tested in EPV153 carrying either empty vector (pUC18) or pVCvciB. At 20 min, either a mock treatment with DMSO or CCCP (at a final concentration of 100 μM) was added to the assay. Asterisks indicate significant differences between the mock and CCCP treatments as determined by two-way ANOVA multiple-comparison test (Sidak; P < 0.05). The data indicate means and standard deviations from three biological replicates.
To test the involvement of the electron transport chain and the proton motive force, the proton-ionophore CCCP was introduced into the whole-cell iron reduction assay. CCCP neutralizes the proton motive force by allowing protons to pass freely through the inner membrane and interferes with the electron transport chain. Assays were carried out as described above and, after 20 min of incubation, CCCP or a mock treatment (dimethyl sulfoxide [DMSO]) was introduced into the reaction. The addition of CCCP immediately halted further color change in both the VciB and the vector control samples, indicating that an intact electron transport chain is essential for function of VciB and supports a role for VciB at the inner membrane.
VciB-mediated iron reduction does not require NapC.
In E. coli, a component of the nitrate reductase complex (NapC) directs electrons from the electron transport chain to iron (34). V. cholerae encodes a NapC ortholog with 72% identity to E. coli NapC based on BLAST (29). V. cholerae napC is expressed when the bacteria are grown aerobically in rich medium (38), conditions where vciB is also expressed. To determine whether V. cholerae NapC is required for VciB function, a napC mutant and parent strain were compared in their ability to stimulate iron reduction while harboring empty vector or pVCvciB (Fig. 5). The reduction of iron by both parent and napC mutant strains bearing pVCvciB in this assay was identical, indicating that NapC is not required for VciB to function. Interestingly, the napC mutant and parent strains bearing empty vector were markedly different at times 40 min and onward, with the napC mutant evolving less reduced iron over the course of this experiment. Together, these data suggest that while NapC does not contribute to VciB activity, it does contribute to the basal level of iron reduction in V. cholerae that is independent of VciB.
FIG 5.

NapC is not required for VciB-mediated iron reduction. V. cholerae ΔvciB (EPV164) and ΔvciB ΔnapC (EPV165) mutants were compared in a whole-cell iron reductase assay. Iron reduction was measured in cells harboring empty vector (pUC18) or pVCvciB. The data are means and standard deviations from three biological replicates. Asterisks indicate significant differences between strains carrying empty vector at the indicated time points as determined by two-way ANOVA multiple-comparison test (Sidak; P < 0.05).
Subcellular localization and membrane topology of VciB.
VciB is predicted to be located in the inner membrane with three transmembrane regions with a large periplasmic region and two short cytoplasmic segments (Fig. 6D) (39). To first confirm the subcellular localization of the VciB protein, an N-terminal V5 epitope tag was introduced into VciB encoded by the low-copy-number plasmid vector pWKS30 (pV5VciB). The function of this protein was confirmed through complementation of the growth defect of the Feo+ VciB− strain (Fig. 6A). Subcellular localization of V5-VciB was determined through the separation of the cytoplasmic, inner membrane, and outer membrane fractions by ultracentrifugation and subsequent SDS-PAGE and immunoblotting. V5-VciB was located in the inner membrane fraction but not in the outer membrane or cytoplasmic fraction (Fig. 6B), confirming the predicted inner membrane localization.
FIG 6.

VciB is an inner membrane protein with topology consistent with predictions. (A) The V5 epitope-tagged vciB plasmid construct and point mutant derivatives were assessed for the ability to complement the colony formation defect of the Feo+ VciB− strain (EPV153) in a colony size assay. All constructs had the C41S mutation, in addition to the mutations indicated. (B) Subcellular fractionation of bacterial cells using ultracentrifugation. Total lysate (total), cytoplasmic (cyto), inner membrane (inner), and outer membrane (outer) fractions are marked. (Top) Western blot using anti-V5 antibody. (Bottom) Coomassie blue-stained SDS-PAGE gel. (C) Representative Western blots of residues probed using SCAM. Lanes: –, negative control (no treatment); +, positive control (MalPEG treated); N, samples pretreated with NEM; M, samples pretreated with MTSES. (D) Supported topology model with V5 epitope tag (blue), Cys mutants (orange circles), and point mutations introduced in conserved residues (green squares). Transmembrane regions were predicted using TMHMM (39). The topology map was generated using the TeXtopo package in the LaTeX program (55).
To determine the membrane topology of V5-VciB, the substituted cysteine accessibility method (SCAM) described in Butler et al. (40) was performed. For this approach, single cysteines were introduced into the protein by mutagenesis of selected amino acid residues, and the accessibility of the individual Cys residues was determined by labeling them with PEGylated maleimide (MalPEG) in the presence or absence of reagents that block binding in the periplasm or block binding in either the periplasm or the cytoplasm. To make constructs with a single cysteine, the protein must lack Cys residues. Wild-type VciB has one cysteine (Cys41), but the protein retained full activity when the cysteine was mutagenized to serine (C41S) (Fig. 6A). Subsequent mutants constructed in the vciB C41S background were tested for function, and only those that retained function were tested for localization of the Cys (Fig. 6A).
The locations of the introduced Cys residues were determined by measuring their accessibility to PEGylation, which causes a size shift in immunoblotting when it binds Cys (Fig. 6C; also see Fig. S2 in the supplemental material). The size shift was determined in the presence or absence of NEM and MTSES, maleimide reagents that covalently bind Cys residues and block subsequent MalPEG binding, preventing a shift in the size of the protein. NEM is membrane permeable and blocks residues in both the cytoplasm and the periplasm, whereas the membrane-impermeable MTSES only blocks residues that are in the periplasm. Cys residues that were PEGylated only in the absence of NEM or MTSES are located in the periplasm (e.g., G225C) (Fig. 6C), where both reagents blocked MalPEG, and residues that were blocked by NEM but not by MTSES (e.g., S18C) (Fig. 6C) are in the cytoplasm, which is inaccessible to MTSES. Residues buried within the inner membrane, including the single cysteine in the wild-type VciB protein (C41) (Fig. 6C), were not blocked by either NEM or MTSES. The results of the SCAM shown in Fig. 6C are summarized in Table 1. The locations of residues determined by SCAM are consistent with the predicted model for VciB membrane topology shown in Fig. 6D. VciB possesses three transmembrane-spanning regions with a large periplasmic loop. With this topology model, we next assessed the role of specific amino acid residues in the function of VciB.
TABLE 1.
SCAM labeling by NEM, MTSES, and MalPEG
| Residue | Blocking resulta |
Interpretation (localization) | |
|---|---|---|---|
| NEM | MTSES | ||
| Ser18 | Blocked | Unblocked | Cytoplasm |
| Ile34 | Blocked | Unblocked | Cytoplasm |
| Cys41 | Unblocked | Unblocked | Membrane |
| Ser60 | Partial blocking | Partial blocking | Periplasm (partially exposed) |
| Ser73 | Blocked | Blocked | Periplasm |
| Ser75 | Blocked | Blocked | Periplasm |
| Ile106 | Blocked | Blocked | Periplasm |
| Ala110 | Blocked | Blocked | Periplasm |
| Leu114 | Blocked | Blocked | Periplasm |
| Ser139 | Blocked | Blocked | Periplasm |
| Ala160 | Blocked | Blocked | Periplasm |
| Thr171 | Blocked | Blocked | Periplasm |
| Leu181 | Unblocked | Unblocked | Membrane |
| Ser203 | Blocked | Unblocked | Cytoplasm |
| Ser215 | Unblocked | Unblocked | Membrane |
| Gly225 | Blocked | Blocked | Periplasm |
“Blocked” indicates that treatment prevented MalPEG binding and that retarded gel migration was not observed. “Unblocked” indicates that treatment did not prevent MalPEG binding and that retarded gel migration was observed. “Partial blocking” indicates that treatment did not completely block the residue and that both protein species were observed.
Histidines 38 and 166 are essential for VciB function.
To gain more insight into the function of VciB, conserved residues were mutated, and the effect of individual mutations on the activity of the protein was determined. The most conserved residues were identified through ClustalO (41) alignment of the first 100 reference proteins identified by BLAST (29), excluding Vibrionaceae, and those targeted through site-directed mutagenesis were H38, H61, H166, W33, W127, W197, Y142, and D180. Their positions are indicated on the topological map (Fig. 6D). The residues were mutagenized as indicated in the N-terminally V5-tagged VciB plasmid and tested for protein expression. Although H38N, H61N, H166N, W33Y, W127Y, W197Y, and D180N mutants were expressed at or near wild-type levels, Y142W expression or stability was poor, and it was not analyzed further (Fig. 7A). We then tested these mutants for their ability to complement the vciB mutation in the colony size assay. The H38N and H166N mutants failed to complement the growth phenotype (Fig. 7B). To ensure that the H38N and H166N mutations did not result in protein mislocalization, we confirmed inner membrane localization of both mutant species (Fig. 7C).
FIG 7.
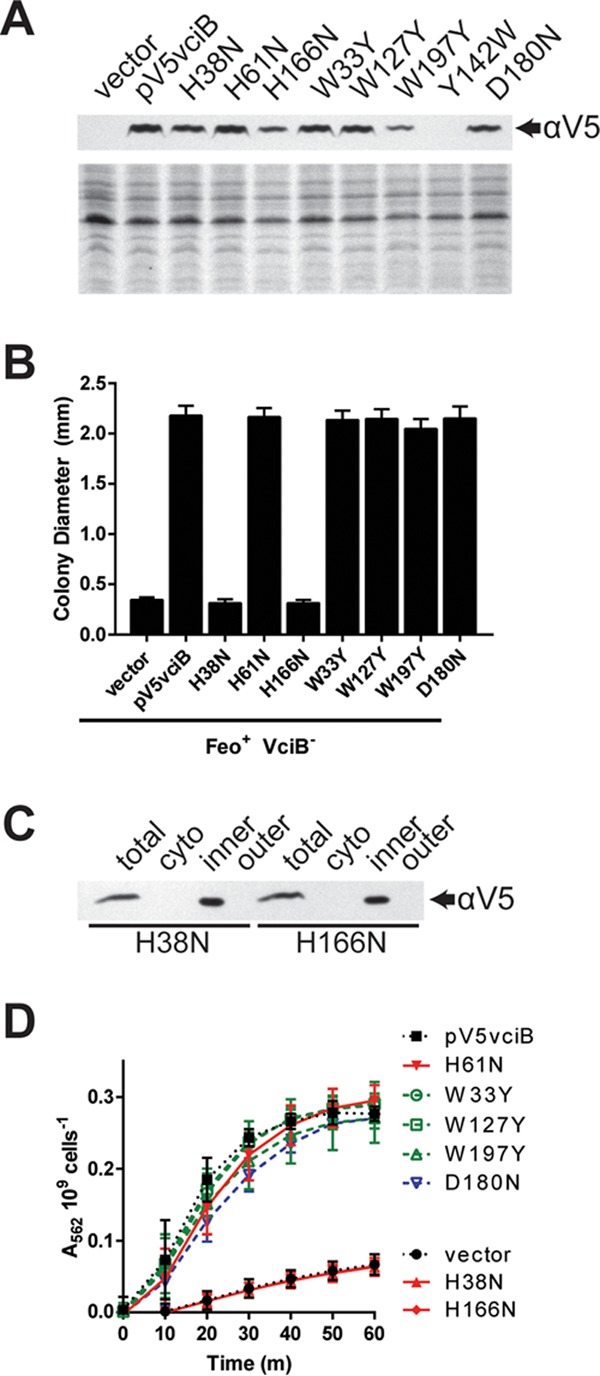
Identification of residues critical for VciB function. (A) (Top) Total cell lysates from EPV153 (Feo+ VciB−) bearing the indicated plasmids were probed for the V5 epitope. (Bottom) Coomassie blue-stained SDS-PAGE gel. (B) Point mutants generated in the V5-tagged VciB background were tested for the ability to complement the colony formation defect of the Feo+ VciB− strain (EPV153). Ten well-isolated colonies were measured using a reticle after 24 h of incubation at 37°C. The data are means and standard deviations and are representative of biological replicates. (C) Subcellular fractionation of cells with either V5-VciB H38N or V5-VciB H166N was used to determine localization. The total lysate (total), cytoplasmic (cyto), inner membrane (inner), and outer membrane (outer) fractions are marked. (D) Point mutants were tested for the capacity to reduce iron in the whole-cell iron reduction assay. H61N, W33Y, W127Y, W197Y, and D180N point mutants do not differ significantly from the wild-type pV5vciB sample; H38N and H166N mutants do not differ from the vector control. Significance was determined using a two-way ANOVA multiple-comparison test (Sidak).
The mutants also were tested using the whole-cell reductase assay to measure their capacity to reduce ferric iron. Although many of the tested residues are highly conserved and the mutants displayed no obvious defects in complementation of the growth phenotype, the whole-cell reductase assay might reveal subtle differences among these mutant protein species. All point mutant proteins capable of complementing the growth phenotype of the Feo+ VciB− strain also mediated efficient iron reduction in this assay (Fig. 7D). Cells harboring VciB with the mutation H38N or H166N, which did not complement the mutant growth phenotype, had only background levels of iron reduction. These data indicate essential roles for His38 and His166 in the function of VciB.
VciB functions as a dimer.
Given that VciB was shown to confer increased iron use in E. coli and Shigella (25), as well as in V. cholerae, it must either function independently to reduce iron or interact with highly conserved proteins, such as those of the electron transport chain. Since neither the Feo system (Fig. 2) nor NapC (Fig. 5) is necessary to observe VciB activity, we used chemical cross-linking with formaldehyde to probe for potential interacting partners. Bacterial cells expressing V5-VciB were treated with formaldehyde, and the proteins were analyzed by SDS-PAGE separation and immunoblotting. The predicted molecular mass of V5-VciB is 27.2 kDa; however, VciB is highly basic (pI = 10.66), and V5-VciB (pI = 10.32) migrates at approximately 23 kDa. A protein species with migration at roughly twice the size of the V5-VciB protein (46 kDa) was observed after formaldehyde treatment (Fig. 8A), suggesting that VciB may form a homodimeric protein species. To determine whether the mutated residues in the point mutants isolated in our mutagenesis (H38N and H166N) were necessary for this interaction, we assessed whether the large protein species was formed with these mutants. Indeed, the larger species could be detected for both H38N and H166N mutants, suggesting that the nonfunction of these mutants is due to another aspect of the protein chemistry. Although the level of the higher-molecular-weight protein species appears to be reduced with H38N, formaldehyde cross-linking is not quantitative.
FIG 8.
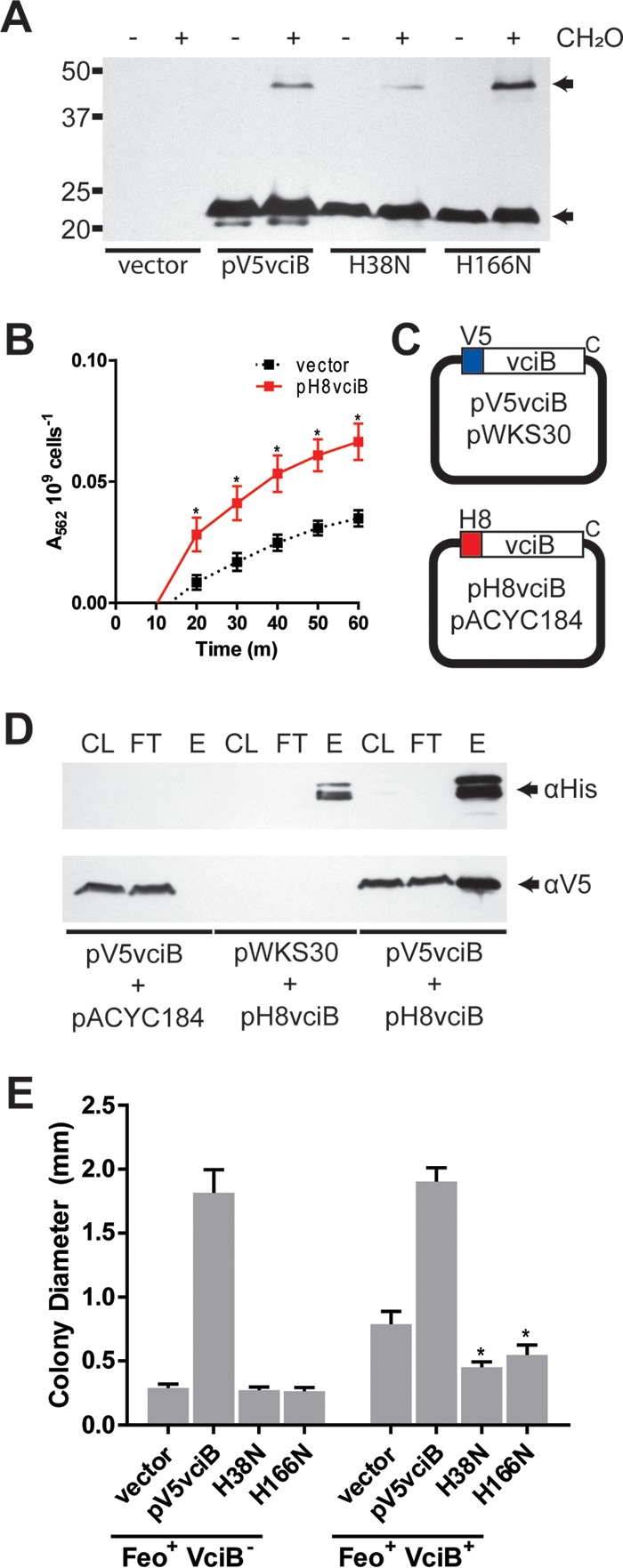
VciB forms a higher-order structure. (A) Formaldehyde-treated cells were lysed and subjected to SDS-PAGE separation and immunoblotting with an anti-V5 antibody. Monomeric and higher-molecular-weight V5-reactive proteins were detected as indicated (arrows). (B) An N-terminally 8×His epitope-tagged VciB was introduced into pACYC184; function was tested using a whole-cell iron reductase activity. The data are means and standard deviations from three biological replicates. Asterisks indicate significant differences between the vector and pH8vciB samples at respective time points as determined by two-way ANOVA multiple comparisons (Sidak; P < 0.05). (C) Plasmids used for coaffinity purification. Compatible plasmids were used to N-terminally tag VciB with the V5 tag and 8×His tag separately. (D) Clarified lysate (CL), affinity purification flowthrough (FT), and elution (E) fractions from cells carrying the indicated plasmids were separated using SDS-PAGE. All lysates were incubated with Ni-NTA–agarose, and the fractions were probed using anti-His and anti-V5 antibodies as indicated. (E) Colony sizes were determined for strains EPV153 (Feo+ VciB−) and EPV104 (Feo+ VciB+) bearing plasmids as noted. Asterisks indicate significant difference between EPV104 with plasmids harboring the H38N and H166N point mutants and EPV104 with empty vector as determined by two-way ANOVA multiple comparisons (Holm-Sidak; P < 0.0001).
While this larger protein species may represent an interaction between VciB and an unidentified protein, the size suggested it could be a VciB dimer. To determine whether VciB interacts with itself, a construct encoding VciB with an 8×His epitope on the N terminus of VciB was cloned into the pWKS30-compatible plasmid pACYC184. The function of pH8VciB was confirmed by using the whole-cell iron reduction assay (Fig. 8B). Iron reduction was weak relative to previously observed activity in other plasmid systems (Fig. 3 and 7), but significantly different from empty vector. pH8vciB and pV5vciB were transformed into the vciB deletion mutant to assess for potential interactions between the two protein species (Fig. 8C). If VciB interacts with itself, then the V5-VciB should copurify upon affinity purification of the His-tagged H8vciB. Consistent with VciB-VciB interaction, we found V5-VciB in elution fractions when His-tagged VciB was purified using Ni-nitrilotriacetic acid (NTA) under detergent conditions (Fig. 8D). Surprisingly, this interaction was maintained in the absence of formaldehyde treatment, suggesting that the interaction is sufficiently strong that cross-linking is not required to maintain the association during purification. The data from the formaldehyde cross-linking and copurification together suggest dimerization by VciB.
To assess whether or not dimerization is necessary for VciB to function, plasmids encoding the point mutations H38N and H166N were introduced into the strain EPV104 (Feo+ VciB+), which retains the chromosomal vciB locus. Given the higher copy number of the plasmid-encoded VciB, we reasoned that the point mutant species could saturate association with the wild-type VciB and result in a dominant-negative effect. Indeed, the introduction of either H38N or H166N into EPV104 resulted in a reduction in colony size (Fig. 8E). Although this reduction was not equivalent to the poor growth of the vciB mutant EPV153 (Feo+ VciB−), it is likely that the formation of the wild-type VciB homodimer was not fully prevented, resulting in low levels of activity and weak stimulation. Altogether, these data confirm homodimerization by VciB and indicate that dimerization is likely required for the function of VciB in mediating iron reduction.
DISCUSSION
Iron acquisition is critical to the survival of most bacteria. The maintenance of numerous V. cholerae iron acquisition genes supports the importance of iron for V. cholerae and is consistent with a broad variety of iron sources available to this organism. As V. cholerae transitions among ocean and brackish water environments and into the human host through the course of its life cycle, it encounters vastly differing oxygen, pH, and nutrient conditions. These conditions can drastically affect the availability and form(s) of iron, and V. cholerae has evolved a number of iron transport systems to take advantage of the available iron sources. In addition to high-affinity transport systems, ferric iron reductases represent a mechanism for increasing the availability of soluble ferrous iron. This could be important in oxidizing environments, such as alkaline ocean waters or at the surface of the intestinal epithelium. Reductases may also allow the use of small ferric iron chelates or siderophores that would not otherwise be accessible iron sources. The fact that vciB is in an operon with vciA, an iron-regulated gene with homology to siderophore receptors, suggests that VciB might originally have been acquired as part of a siderophore-mediated iron acquisition system. If VciA transported an iron siderophore complex into the periplasm and there was no cognate inner membrane transporter, VciB's role could be to release the iron for subsequent transport through the Feo system. An analogous system has been reported in Pseudomonas aeruginosa, where the inner membrane protein FoxP allows use of siderophores as iron sources in the absence of the specific inner membrane transporters for those siderophores (42). However, no ligand has been identified for VciA, no vciA homolog is present in some species that have vciB, and VciB can increase iron acquisition in the absence of VciA.
Ferric reductases have been identified in a number of bacterial species. Both B. japonicum (30) and Listeria monocytogenes (43, 44) use reductases to increase the amount of ferrous iron that can be transported for use by the bacterial cell. The Bradyrhizobium reductase FrcB resembles proteins of the cytochrome b superfamily and mediates iron reduction through two heme prosthetic groups (30). Small and O'Brian (30) were able to purify FrcB to demonstrate heme association and iron-dependent oxidation of the heme groups in vitro. This iron reduction process likely facilitates ferrous iron transport through the B. japonicum Feo system (45). In the Gram-positive pathogen L. monocytogenes, an unidentified surface-associated protein is involved in the reduction of iron and is predicted to aid in iron acquisition of the intracellular pathogen, which lacks apparent siderophores for iron scavenging (44). NapC from E. coli, a tetraheme cytochrome c protein, represents an additional family of proteins capable of iron reduction (34). Fre, another protein identified in E. coli, is able to reduce ferric iron through a reduced flavin intermediate (46). VciB and its orthologs lack homology to FrcB, NapC, and Fre, and no proteins similar to VciB are identified from the sequenced L. monocytogenes as determined by BLAST (29), suggesting that VciB represents a previously unrecognized class of proteins involved in bacterial iron acquisition.
VciB is a cytoplasmic membrane protein, and the SCAM analysis is consistent with the protein having three transmembrane domains and a large periplasmic loop. The protein is found as a dimer, and the dimer can be isolated even in the absence of chemical cross-linking, suggesting that there is a tight interaction between the VciB monomers. The dimer is likely the active form of the protein, since expression of an inactive mutant form of the protein reduces the activity of the wild type (Fig. 8E). Thus, the mutant appears to act as a dominant negative by forming inactive heterodimers with the wild-type VciB.
The mechanism by which VciB mediates iron reduction has yet to be determined. It is likely that it is a reductase, since it can function in different species (25), and it does not appear to interact with any protein other than itself. However, the reductase activity has only been observed in whole cells, and it is not possible to attribute the enzyme activity to VciB until the activity of the purified protein can be shown. Reduction of ferric iron appears to occur in the periplasm, since the ferrous iron is available to the Feo transport system, and there is no growth stimulation in the absence of Feo, indicating that the ferrous iron must be transported from the periplasm to the cytoplasm by the Feo system. Further, the ferrous iron produced when VciB is expressed is accessible to the ferrous chelator FerroZine when it is present in the medium. Although Feo is required to see the stimulation of growth by VciB, Feo is not required for VciB activity, since there was no difference in the level of ferric iron reduction when the assay was performed in the presence or absence of Feo.
VciB is a member of a family of proteins of unknown function, COG3295. This is a family of integral membrane proteins with putative PepSY_TM-like domains (47). Studies on the PepSY family of proteins suggest that these domains serve regulatory roles for the protein itself or for other associated proteins (48). A common amino acid sequence shared among family members (hyd-D-hyd-X-X-G, where hyd indicates a hydrophobic amino acid) is not completely conserved in the V. cholerae VciB sequence (SDLAAV, residues 179 to 184). Although mutations (D180N and L181C) in the conserved motif do not impair function of VciB in our assays (Fig. 6 and 7), it remains unclear what purpose, if any, these conserved regions serve for this family of proteins. Additional mutagenesis targeting conserved residues in other parts of the protein did identify critical histidine residues. His38 and His166 were both required for growth stimulation and ferric reductase activity. His38 localizes within the membrane, and His166 is in the periplasmic loop of the protein. Histidine residues may be required for binding cations (49), binding a cofactor (30), or aiding in electron transfer to ferric iron in concert with tyrosine (50). Most of the known iron reductases use heme to reduce the ferric iron (30, 37, 51). In preliminary studies, heme has not yet been found to be part of the active VciB complex. A potential source of the electrons for reduction is the electron transport chain, and deletion of the Na+-NQR complex (nqr) alters iron reduction kinetics (Fig. 4). However, V. cholerae encodes alternative methods for NADH oxidization by other proteins such as NDH-2 (52), and other carbon substrates can be oxidized to provide energy to the electron transport chain. This may explain why iron reduction, while impaired, is not abolished in the absence of nqr. However, the addition of the proton-ionophore CCCP abolishes activity immediately in our assay. Thus, we propose a model (Fig. 9) in which VciB is able to acquire electrons from the electron transport chain (ETC), originating from either the Na+-NQR, NDH-2, or alternative oxidation pathway within the ETC and pass them to ferric iron as the terminal electron acceptor. Reduced iron can subsequently be transported through the Feo system.
FIG 9.
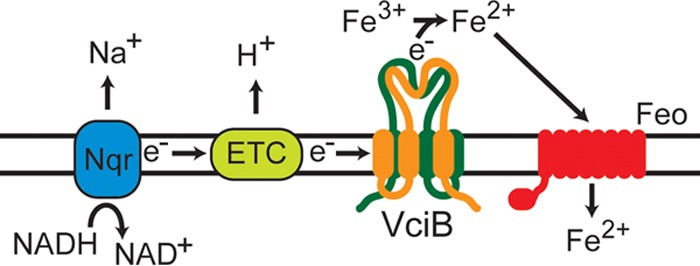
Proposed model for VciB function. Functional VciB forms a dimeric structure in the inner membrane. Electrons from the electron transport chain, either from the Na+-NQR (shown) or oxidation of other energy sources by other electron transport chain (ETC) constituents, are passed from VciB to Fe3+ to result in reduction to Fe2+. It is not clear at this time if VciB directly mediates iron reduction.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The bacterial strains and plasmids used in this study are listed in Table 2. Strains were maintained at −80°C in tryptic soy broth with 20% glycerol. Strains were routinely grown in Luria-Bertani (LB) broth (1% [wt/vol] tryptone, 0.5% [wt/vol] yeast extract, 1% [wt/vol] sodium chloride) or on LB agar (1.5% [wt/vol] agar) at 37°C. For experiments comparing wild-type V. cholerae O395 and the Δnqr strain, a modified LB medium was used (1% [wt/vol] tryptone, 0.5% [wt/vol] yeast extract, 1% [wt/vol] sodium chloride, 0.4% [wt/vol] sucrose, and 100 mM Tris [pH 7.5]). Hemin (5 μM) supplementation was used for the routine growth of strains. Antibiotics were used as follows for V. cholerae strains: 75 μg/ml streptomycin, 50 μg/ml kanamycin, 100 μg/ml ampicillin, 5 μg/ml chloramphenicol, and 5 μg/ml gentamicin. For E. coli strains, 50 μg/ml kanamycin, 100 μg/ml ampicillin, and 25 μg/ml chloramphenicol were used.
TABLE 2.
Strains and plasmids
| Strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| V. cholerae | ||
| O395 | Classical biotype | 56 |
| EPV102 (null) | O395 ΔvibB fbpA::cam vctP::Gentr feoABC::kan ΔviuA | 26 |
| EPV131 (null vciB::kan) | O395 ΔvibB fbpA::cam vctP::Gentr feoABC::kan ΔviuA vciB::kan | This study |
| EPV156 (null ΔvciB) | O395 ΔvibB fbpA::cam vctP::Gentr feoABC::kan ΔviuA ΔvciB | This study |
| EPV104 (vFeo) | O395 ΔvibB fbpA::cam vctP::Gentr viuA | 26 |
| EPV123 (vFeo vciB::kan) | O395 ΔvibB fbpA::cam vctP::Gentr viuA vciB::kan | This study |
| EPV153 (vFeo ΔvciB) | O395 ΔvibB fbpA::cam vctP::Gentr viuA ΔvciB | This study |
| EPV154 (vFeo ΔvciA) | O395 ΔvibB fbpA::cam vctP::Gentr viuA ΔvciA | This study |
| EPV115 (vFbp) | O395 ΔvibB ΔviuA vctP::Gentr feoABC::kan | 26 |
| EPV137 (vFbp vciB::kan) | O395 ΔvibB ΔviuA vctP::Gentr feoABC::kan vciB::kan | This study |
| EPV126 (vVib) | O395 feoABC::kan vctP::Gentr fbpA::cam | 26 |
| EPV136 (vVib vciB::kan) | O395 feoABC::kan vctP::Gentr fbpA::cam vciB::kan | This study |
| EPV103 (vVct) | O395 feoB::tmp ΔvibB fbpA::cam ΔviuA | 26 |
| EPV133 (vVct vciB::kan) | O395 feoB::tmp ΔvibB fbpA::cam ΔviuA vciB::kan | This study |
| EPV162 | O395 Δnqr | This study |
| EPV164 | O395 ΔvciB | This study |
| EPV165 | O395 ΔvciB ΔnapC | This study |
| E. coli | ||
| DH5α | Cloning strain | 57 |
| DH5α λpir | Cloning strain | 57 |
| SM10 λpir | Conjugation strain | 58 |
| Other strains | ||
| B. mallei ATCC 23344 | Source of genomic DNA | 59 |
| B. thailandensis ATCC 700388 | Burkholderia thailandensis strain used for isolating genomic DNA | 60 |
| A. hydrophila BG-2 | Aeromonas hydrophila isolate used for isolating genomic DNA | Texas Department of Health |
| Plasmids | ||
| pUC18 | Cloning vector | 61 |
| pWKS30 | Cloning vector | 62 |
| pACYC184 | Cloning vector | 63 |
| pCVD442N | Suicide vector pCVD442 with a NotI adapter inserted in the SacI site | 9 |
| pAMS23 | pHM5 carrying vciB::kan | 25 |
| pSΔvciΒ | pCVD442N carrying sequences for the deletion of the vciB locus | This study |
| pSΔvciΑ | pCVD442N carrying sequences for the deletion of the vciA locus | This study |
| pSΔnqr | pCVD442N carrying sequences for the deletion of the nqr operon | This study |
| pSΔnapC | pCVD442N carrying sequences for the deletion of napC | This study |
| pVCvciB | V. cholerae vciB cloned into pUC18 | This study |
| pBMvciB | B. mallei vciB cloned into pUC18 | This study |
| pBTvciB | B. thailandensis vciB cloned into pUC18 | This study |
| pAHvciB | A. hydrophila vciB cloned into pUC18 | This study |
| pV5vciB | V. cholerae vciB fused to an N-terminal V5 epitope tag cloned into pWKS30 | This study |
| pH8vciB | V. cholerae vciB fused to an N-terminal 8×His epitope tag cloned into pACYC184 | This study |
| pV5vciBC41S | Derivative of pV5vciB in which vciB Cys41 is mutagenized to Ser | This study |
| pV5vciB*S18C | Derivative of pV5vciBC41S with a single cysteine substitution at S18 | This study |
| pV5vciB*I34C | Derivative of pV5vciBC41S with a single cysteine substitution at N31 | This study |
| pV5vciB*S60C | Derivative of pV5vciBC41S with a single cysteine substitution at S60 | This study |
| pV5vciB*S73C | Derivative of pV5vciBC41S with a single cysteine substitution at S73 | This study |
| pV5vciB*S75C | Derivative of pV5vciBC41S with a single cysteine substitution at S89 | This study |
| pV5vciB*I106C | Derivative of pV5vciBC41S with a single cysteine substitution at I106 | This study |
| pV5vciB*A110C | Derivative of pV5vciBC41S with a single cysteine substitution at L114 | This study |
| pV5vciB*S139C | Derivative of pV5vciBC41S with a single cysteine substitution at S139 | This study |
| pV5vciB*A160C | Derivative of pV5vciBC41S with a single cysteine substitution at A160 | This study |
| pV5vciB*T171C | Derivative of pV5vciBC41S with a single cysteine substitution at T171 | This study |
| pV5vciB*L181C | Derivative of pV5vciBC41S with a single cysteine substitution at L181 | This study |
| pV5vciB*S203C | Derivative of pV5vciBC41S with a single cysteine substitution at S203 | This study |
| pV5vciB*S215C | Derivative of pV5vciBC41S with a single cysteine substitution at S215 | This study |
| pV5vciB*G225C | Derivative of pV5vciBC41S with a single cysteine substitution at G225 | This study |
| pV5vciBH38N | Derivative of pV5vciB with an H38N substitution | This study |
| pV5vciBH61N | Derivative of pV5vciB with an H61N substitution | This study |
| pV5vciBH166N | Derivative of pV5vciB with an H166N substitution | This study |
| pV5vciBW33Y | Derivative of pV5vciB with a W33Y substitution | This study |
| pV5vciBW127Y | Derivative of pV5vciB with a W127Y substitution | This study |
| pV5vciBW197Y | Derivative of pV5vciB with a W197Y substitution | This study |
| pV5vciBY142W | Derivative of pV5vciB with a Y142W substitution | This study |
| pV5vciBD180N | Derivative of pV5vciB with a D180N substitution | This study |
Gentr, gentamicin resistance.
Colony size assays.
Colony size assays, which are more reproducible and more sensitive than broth cultures for measuring effects of iron reduction on growth, were performed as described previously (9, 26). Overnight cultures of strains grown in LB medium supplemented with heme and appropriate antibiotics were diluted and plated onto LB agar plates with supplements as indicated. Plates were incubated at 37°C for 24 h, and the diameters of at least 10 well-isolated colonies within a field were measured using a reticle. When included, sodium ascorbate was prepared immediately before use and added at the final concentrations indicated.
Primers and plasmid construction.
The sequences of primers used in this study are listed in Table S1 in the supplemental material. All inserts and mutations were confirmed through DNA sequencing at the University of Texas Institute for Cellular and Molecular Biology DNA core sequencing facility. To generate deletion constructs, flanking regions were amplified from genomic DNA by PCR using the primers indicated below. The PCR products were joined using splice overlap and cloned into the SmaI site of the suicide vector pCVD442N.
To generate the following deletion constructs, flanking regions were amplified from genomic DNA by PCR using the indicated primer pairs: (i) pSΔvciB, primers VciB.2659.for-VciB.3658.rev and VciB.4337.SO.for-VciB.5336.rev; (ii) pSΔvciA, primers VciA.596.for-VciA.1596.rev and VciA.3639.SO.for-VciA.4638.rev; (iii) pSΔnqr, primers Nqr.1045.for-Nqr.2250.SO.rev and Nqr.7328.for-Nqr.8338.rev (the nqr deletion generated by this plasmid has endpoints identical to that described by Barquera et al. [53]); and (iv) pSΔnapC, primers NapC.369.for-NapC.1268.SO.rev and NapC.3731.for-NapC.4630.rev.
To generate the following expression constructs for V. cholerae vciB and orthologous genes, upstream and coding regions of each respective gene were amplified by PCR with the indicated primer pairs: (i) pVCvciB (V. cholerae), primers VciB.3337.EcoRI and VciB.4336.rev.XbaI (the PCR product was digested with EcoRI/XbaI and cloned into EcoRI/XbaI-cut pUC18 to generate pVCvciB); (ii) pBTvciB (Burkholderia thailandensis), primers BTvciB.EcoRI.for and BTvciB.rev (the PCR product was digested with EcoRI and cloned into EcoRI/SmaI-digested pUC18 to generate pBTvciB); (iii) pBMvciB (Burkholderia mallei), primers BMvciB.EcoRI.1578.for and BMvciB.2524.rev (the PCR product was digested with EcoRI and cloned into EcoRI/SmaI-digested pUC18 to generate pBMvciB); and (iv) pAHvciB (Aeromonas hydrophila), primers AHvciB.EcoRI.for and AHvciB.rev (the PCR product was digested with EcoRI and cloned into EcoRI/SmaI-digested pUC18 to generate pAHvciB).
To generate pV5vciB and mutant derivatives, V. cholerae vciB was amplified from O395 genomic DNA using the primers VciB.V5.for.EcoRI and VciB.4336.rev.XbaI, which introduced an N-terminal V5 epitope tag. The resultant product was ligated into EcoRI/XbaI-digested pWKS30. The primers VciB.V5fix.for.EcoRV and VciB.V5fix.rev.EcoRV were used to remove additional plasmid sequence to allow vciB transcription from the pWKS30 lacZ promoter. The PCR product was digested with EcoRV and self-ligated to generate pV5vciB. Site-directed mutant derivatives of pV5vciB were generated using the QuikChange (Agilent Technologies, Inc.) protocol or back-to-back primer amplification protocol (54).
To generate pH8vciB, V. cholerae vciB was amplified from O395 genomic DNA using the primers pET16b.vciB.NdeI.for and pET16b.vciB.XhoI.rev. The PCR product was digested using NdeI and XhoI and ligated into the respective sites in pET16b. The vciB locus and 8×His N-terminal tag sequence were amplified using the primers H8.for and VciB.4336.rev and cloned into the EcoRV restriction site in pACYC184. The 8×His-VciB construct interrupts and runs antiparallel to the tetR gene carried by pACYC184.
Strain construction.
Deletion of vciB, vciA, nqr, or napC or disruption of vciB was carried out using the plasmid pSΔvciB, pSΔvciA, pSΔnqr, pSΔnapC, or pAMS23, respectively. Conjugation using SM10 λpir and sucrose selection on LBA with 10% sucrose was performed as described previously (23).
Whole-cell iron reductase activity assay.
The reduction assay was modified from Small et al. (30) as follows. Strains bearing the indicated plasmids grown on LB agar plates were inoculated into LB medium with appropriate antibiotics and grown overnight. The cells were pelleted, washed once in phosphate-buffered saline (PBS), and then resuspended in assay buffer containing 100 mM Tris (pH 7.5), 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 0.2 mM FeCl3, 0.2% succinate, and 0.25 mM FerroZine [3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-p,p′-disulfonic acid monosodium salt hydrate; Sigma] at a density of 4 × 109 to 7 × 109 bacteria as determined from the optical density (A650). Samples were incubated at 37°C with aeration, and aliquots were removed at 10-min intervals. Aliquots were centrifuged to pellet bacterial cells, and the supernatants were collected in a 96-well plate. The absorbance at 562 nm, indicative of the ferrous-FerroZine species (31), was measured, and values were normalized against the bacterial cell count determined initially. For studies with the proton ionophore CCCP (carbonyl cyanide 3-chlorophenylhydrazone), either CCCP (100 μM final concentration) dissolved in DMSO or DMSO alone was added to the assay suspension at the 20-min time point; the reaction was monitored as described above.
Subcellular localization of V5-VciB and mutant derivatives.
Analyses of V5-VciB and mutant derivatives were performed in a vciB deletion background unless otherwise noted. Overnight cultures of strains bearing the plasmid-encoded V5 epitope-tagged wild-type VciB or V5 epitope-tagged point mutant derivatives of VciB were diluted 1:200 into fresh LB media with ampicillin and heme. Cultures were grown to mid-logarithmic growth phase, and cells were harvested by centrifugation. Cell pellets were resuspended in 10 mM Na2HPO4–5 mM MgSO4 (pH 7) with SigmaFAST protease inhibitor cocktail, lysed by sonication, and centrifuged at 12,000 × g for 30 min at 4°C. A fraction of the whole-cell lysate was removed for subsequent SDS-PAGE and immunoblotting. The remaining supernatant was centrifuged at 135,000 × g for 45 min at 4°C to separate the membrane fraction, and the supernatant containing the cytoplasmic fraction was collected. The pellet was then resuspended in double-distilled H2O (ddH2O) and centrifuged at 135,000 × g for 45 min at 4°C. The pellet was resuspended in 1% (wt/vol) sodium lauroyl sarcosinate in ddH2O, followed by incubation for 2 h at 4°C to solubilize the inner membrane. The sample was centrifuged at 135,000 × g for 45 min at 4°C. The supernatant containing the inner membrane fraction was collected. The pellet was washed once with 1% sodium lauroyl sarcosinate in ddH2O, and the pellet containing the outer membrane fraction was resuspended in ddH2O. Samples were stored at −20°C.
SCAM.
The protocol for the substituted-cysteine accessibility method (SCAM) was adapted from Butler et al. (40). A cysteine-less variant of V5-VciB was generated through site-directed mutagenesis, and function was verified through the ability of the plasmid to complement the chromosomal vciB deletion in colony size assays. Site-directed mutagenesis was used to change selected amino acid residues to cysteine residues in the cysteine-less background. The function for each substitution was verified as described above. For labeling and discrimination of amino acid localization relative to the inner membrane, overnight cultures were diluted and grown to mid-logarithmic phase. A total of 2.1 × 109 cells were pelleted and washed once in PBS. Pellets were resuspended in 210 μl of PBS containing 1 mM phenylmethylsulfonyl fluoride and SigmaFAST protease inhibitor cocktail. Cell suspensions were split into four separate treatments. One 50-μl aliquot was treated for 1 h at room temperature with 5 mM N-ethylmaleimide (NEM; Sigma), a membrane-permeable reagent that forms a covalent bond with exposed cysteine residues in both the periplasm and the cytoplasm. One 50-μl aliquot was treated for 1 h at room temperature with 5 μM sodium (2-sulfonatoethyl)methanethiosulfonate (MTSES; Cayman Chemical), an inner-membrane-impermeable reagent that reacts with exposed cysteine residues only in the periplasm. Two 50-μl aliquots were left untreated in the initial labeling but were incubated at room temperature with rotation alongside the NEM- and MTSES-treated samples.
After the initial treatment, the cells were pelleted, washed once in PBS, and resuspended in 50 μl of lysis buffer (15 mM Tris-HCl [pH 7.4], 1% SDS, 6 M urea). The NEM- and MTSES-treated samples were incubated with 5 mM methoxypolyethylene glycol maleimide (molecular weight, 5,000; MalPEG), as well as one of the untreated samples for a positive labeling control. The remaining aliquot was left untreated as a negative control. After 1 h of incubation at room temperature, 50 μl of 2× AB buffer (6.84 mM Na2HPO4, 3.16 mM NaH2PO4, 50 mM Tris-HCl [pH 6.8], 6 M urea, 1% β-mercaptoethanol, 3% SDS, 10% [vol/vol] glycerol, 0.1% [wt/vol] bromophenol blue) was added to all samples, and the samples were boiled for 7 min prior to SDS-PAGE separation and immunoblotting. A topology map (Fig. 6D) was generated using the TeXtopo package in the LaTeX program (55).
Cross-linking of V5-tagged VciB in vivo.
Formaldehyde concentrations ranging from 1.5 to 2% (vol/vol) were used to cross-link proteins in whole cells. Overnight cultures of strains harboring plasmids were diluted 1:200 into fresh media containing antibiotics. At mid-logarithmic phase (A650 = 0.4 to 0.7), the cells were pelleted, washed once in PBS, and resuspended in PBS containing formaldehyde. The cell suspension was incubated with rotation at room temperature for 10 to 15 min. The cells were then pelleted and resuspended in PBS containing 1.25 M glycine. The cells were next pelleted and resuspended in sample buffer for analysis by SDS-PAGE and immunoblotting.
Affinity purification of H8-tagged VciB.
Overnight cultures of cells with indicated plasmids were diluted into fresh medium with appropriate antibiotics and grown to mid-logarithmic phase. Cells were pelleted and lysed by resuspension in modified radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 0.1% SDS, 0.75% deoxycholate, 1% NP-40, and 10 mM imidazole). The suspension was incubated on ice for 10 min, sonicated, and centrifuged at 12,000 × g for 20 min. The clarified lysate was incubated with Ni-NTA–agarose (G-Biosciences) with rotation for 90 min at 4°C; the sample was then pelleted and washed repeatedly with modified RIPA buffer. Proteins were eluted by boiling in SDS sample buffer (2×) for 10 min. Elution fractions were separated by SDS-PAGE, and proteins were detected by immunoblotting.
Supplementary Material
ACKNOWLEDGMENTS
We thank Christopher Hatcher and Alfredo Torres for B. thailandensis and Burkholderia mallei genomic DNA. We are indebted to Elizabeth Wyckoff and Alexandra Mey for scientific discussions and critical review of the manuscript. We are grateful to Marvin Whiteley, Bryan Davies, Ian Molineux, and Emily Que, as well as to members of the Payne laboratory, for helpful discussions.
This study was funded by the National Institutes of Health, National Institute of Allergy and Infectious Diseases, under grant AI091957.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00874-16.
REFERENCES
- 1.Islam MS, Drasar BS, Sack RB. 1994. The aquatic flora and fauna as reservoirs of Vibrio cholerae: a review. J Diarrhoeal Dis Res 12:87–96. [PubMed] [Google Scholar]
- 2.Mayfield JA, Dehner CA, DuBois JL. 2011. Recent advances in bacterial heme protein biochemistry. Curr Opin Chem Biol 15:260–266. doi: 10.1016/j.cbpa.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lill R. 2009. Function and biogenesis of iron-sulphur proteins. Nature 460:831–838. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 4.Thelander L, Reichard P. 1979. Reduction of ribonucleotides. Annu Rev Biochem 48:133–158. doi: 10.1146/annurev.bi.48.070179.001025. [DOI] [PubMed] [Google Scholar]
- 5.Gregory EM, Yost FJ, Fridovich I. 1973. Superoxide dismutases of Escherichia coli: intracellular localization and functions. J Bacteriol 115:987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hem JD. 1972. Chemical factors that influence the availability of iron and manganese in aqueous systems. Geol Soc Am Spec Pap 140:17–24. [Google Scholar]
- 7.Becker KW, Skaar EP. 2014. Metal limitation and toxicity at the interface between host and pathogen. FEMS Microbiol Rev 38:1235–1249. doi: 10.1111/1574-6976.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Payne SM, Mey AR, Wyckoff EE. 2016. Vibrio iron transport: evolutionary adaptation to life in multiple environments. Microbiol Mol Biol Rev 80:69–90. doi: 10.1128/MMBR.00046-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wyckoff EE, Mey AR, Leimbach A, Fisher CF, Payne SM. 2006. Characterization of ferric and ferrous iron transport systems in Vibrio cholerae. J Bacteriol 188:6515–6523. doi: 10.1128/JB.00626-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keating TA, Marshall CG, Walsh CT. 2000. Vibriobactin biosynthesis in Vibrio cholerae: VibH is an amide synthase homologous to nonribosomal peptide synthetase condensation domains. Biochemistry (Mosc) 39:15513–15521. doi: 10.1021/bi001651a. [DOI] [PubMed] [Google Scholar]
- 11.Keating TA, Marshall CG, Walsh CT. 2000. Reconstitution and characterization of the Vibrio cholerae vibriobactin synthetase from VibB, VibE, VibF, and VibH. Biochemistry (Mosc) 39:15522–15530. doi: 10.1021/bi0016523. [DOI] [PubMed] [Google Scholar]
- 12.Marshall CG, Burkart MD, Keating TA, Walsh CT. 2001. Heterocycle formation in vibriobactin biosynthesis: alternative substrate utilization and identification of a condensed intermediate. Biochemistry (Mosc) 40:10655–10663. doi: 10.1021/bi010937s. [DOI] [PubMed] [Google Scholar]
- 13.Marshall CG, Hillson NJ, Walsh CT. 2002. Catalytic mapping of the vibriobactin biosynthetic enzyme VibF. Biochemistry (Mosc) 41:244–250. doi: 10.1021/bi011852u. [DOI] [PubMed] [Google Scholar]
- 14.Butterton JR, Stoebner JA, Payne SM, Calderwood SB. 1992. Cloning, sequencing, and transcriptional regulation of viuA, the gene encoding the ferric vibriobactin receptor of Vibrio cholerae. J Bacteriol 174:3729–3738. doi: 10.1128/jb.174.11.3729-3738.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoebner JA, Butterton JR, Calderwood SB, Payne SM. 1992. Identification of the vibriobactin receptor of Vibrio cholerae. J Bacteriol 174:3270–3274. doi: 10.1128/jb.174.10.3270-3274.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wyckoff EE, Valle AM, Smith SL, Payne SM. 1999. A multifunctional ATP-binding cassette transporter system from Vibrio cholerae transports vibriobactin and enterobactin. J Bacteriol 181:7588–7596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rogers MB, Sexton JA, DeCastro GJ, Calderwood SB. 2000. Identification of an operon required for ferrichrome iron utilization in Vibrio cholerae. J Bacteriol 182:2350–2353. doi: 10.1128/JB.182.8.2350-2353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wyckoff EE, Payne SM. 2011. The Vibrio cholerae VctPDGC system transports catechol siderophores and a siderophore-free iron ligand. Mol Microbiol 81:1446–1458. doi: 10.1111/j.1365-2958.2011.07775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mey AR, Wyckoff EE, Oglesby AG, Rab E, Taylor RK, Payne SM. 2002. Identification of the Vibrio cholerae enterobactin receptors VctA and IrgA: IrgA is not required for virulence. Infect Immun 70:3419–3426. doi: 10.1128/IAI.70.7.3419-3426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henderson DP, Payne SM. 1994. Vibrio cholerae iron transport systems: roles of heme and siderophore iron transport in virulence and identification of a gene associated with multiple iron transport systems. Infect Immun 62:5120–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henderson DP, Payne SM. 1993. Cloning and characterization of the Vibrio cholerae genes encoding the utilization of iron from haemin and haemoglobin. Mol Microbiol 7:461–469. doi: 10.1111/j.1365-2958.1993.tb01137.x. [DOI] [PubMed] [Google Scholar]
- 22.Henderson DP, Payne SM. 1994. Characterization of the Vibrio cholerae outer membrane heme transport protein HutA: sequence of the gene, regulation of expression, and homology to the family of TonB-dependent proteins. J Bacteriol 176:3269–3277. doi: 10.1128/jb.176.11.3269-3277.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mey AR, Payne SM. 2001. Haem utilization in Vibrio cholerae involves multiple TonB-dependent haem receptors. Mol Microbiol 42:835–849. doi: 10.1046/j.1365-2958.2001.02683.x. [DOI] [PubMed] [Google Scholar]
- 24.Wyckoff EE, Allred BE, Raymond KN, Payne SM. 2015. Catechol siderophore transport by Vibrio cholerae. J Bacteriol 197:2840–2859. doi: 10.1128/JB.00417-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mey AR, Wyckoff EE, Hoover LA, Fisher CR, Payne SM. 2008. Vibrio cholerae VciB promotes iron uptake via ferrous iron transporters. J Bacteriol 190:5953–5962. doi: 10.1128/JB.00569-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng ED, Wyckoff EE, Mey AR, Fisher CR, Payne SM. 2015. Nonredundant roles of iron acquisition systems in Vibrio cholerae. Infect Immun 84:511–523. doi: 10.1128/IAI.01301-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatcher CL, Muruato LA, Torres AG. 2015. Recent advances in Burkholderia mallei and B. pseudomallei research. Curr Trop Med Rep 2:62–69. doi: 10.1007/s40475-015-0042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janda JM, Abbott SL. 2010. The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin Microbiol Rev 23:35–73. doi: 10.1128/CMR.00039-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 30.Small SK, O'Brian MR. 2011. The Bradyrhizobium japonicum frcB gene encodes a diheme ferric reductase. J Bacteriol 193:4088–4094. doi: 10.1128/JB.05064-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stookey LL. 1970. Ferrozine: a new spectrophotometric reagent for iron. Anal Chem 42:779–781. doi: 10.1021/ac60289a016. [DOI] [Google Scholar]
- 32.Schröder I, Johnson E, De Vries S. 2003. Microbial ferric iron reductases. FEMS Microbiol Rev 27:427–447. doi: 10.1016/S0168-6445(03)00043-3. [DOI] [PubMed] [Google Scholar]
- 33.Ruebush SS, Brantley SL, Tien M. 2006. Reduction of soluble and insoluble iron forms by membrane fractions of Shewanella oneidensis grown under aerobic and anaerobic conditions. Appl Environ Microbiol 72:2925–2935. doi: 10.1128/AEM.72.4.2925-2935.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gescher JS, Cordova CD, Spormann AM. 2008. Dissimilatory iron reduction in Escherichia coli: identification of CymA of Shewanella oneidensis and NapC of E. coli as ferric reductases. Mol Microbiol 68:706–719. doi: 10.1111/j.1365-2958.2008.06183.x. [DOI] [PubMed] [Google Scholar]
- 35.Schuetz B, Schicklberger M, Kuermann J, Spormann AM, Gescher J. 2009. Periplasmic electron transfer via the c-type cytochromes MtrA and FccA of Shewanella oneidensis MR-1. Appl Environ Microbiol 75:7789–7796. doi: 10.1128/AEM.01834-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaspard S, Vazquez F, Holliger C. 1998. Localization and solubilization of the iron(III) reductase of Geobacter sulfurreducens. Appl Environ Microbiol 64:3188–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lloyd JR, Leang C, Hodges Myerson AL, Coppi MV, Cuifo S, Methe B, Sandler SJ, Lovley DR. 2003. Biochemical and genetic characterization of PpcA, a periplasmic c-type cytochrome in Geobacter sulfurreducens. Biochem J 369:153–161. doi: 10.1042/bj20020597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mey AR, Wyckoff EE, Kanukurthy V, Fisher CR, Payne SM. 2005. Iron and Fur regulation in Vibrio cholerae and the role of Fur in virulence. Infect Immun 73:8167–8178. doi: 10.1128/IAI.73.12.8167-8178.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 40.Butler EK, Davis RM, Bari V, Nicholson PA, Ruiz N. 2013. Structure-function analysis of MurJ reveals a solvent-exposed cavity containing residues essential for peptidoglycan biogenesis in Escherichia coli. J Bacteriol 195:4639–4649. doi: 10.1128/JB.00731-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuív PÓ Keogh D, Clarke P, O'Connell M. 2007. FoxB of Pseudomonas aeruginosa functions in the utilization of the xenosiderophores ferrichrome, ferrioxamine B, and schizokinen: evidence for transport redundancy at the inner membrane. J Bacteriol 189:284–287. doi: 10.1128/JB.01142-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deneer HG, Boychuk I. 1993. Reduction of ferric iron by Listeria monocytogenes and other species of Listeria. Can J Microbiol 39:480–485. doi: 10.1139/m93-068. [DOI] [PubMed] [Google Scholar]
- 44.Deneer HG, Healey V, Boychuk I. 1995. Reduction of exogenous ferric iron by a surface-associated ferric reductase of Listeria spp. Microbiol Read Engl 141(Pt 8):1985–1992. doi: 10.1099/13500872-141-8-1985. [DOI] [PubMed] [Google Scholar]
- 45.Sankari S, O'Brian MR. 2016. The Bradyrhizobium japonicum ferrous iron transporter FeoAB is required for ferric iron utilization in free living aerobic cells and for symbiosis. J Biol Chem 291:15653–15662. doi: 10.1074/jbc.M116.734129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coves J, Fontecave M. 1993. Reduction and mobilization of iron by a NAD(P)H:flavin oxidoreductase from Escherichia coli. Eur J Biochem 211:635–641. doi: 10.1111/j.1432-1033.1993.tb17591.x. [DOI] [PubMed] [Google Scholar]
- 47.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A. 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeats C, Rawlings ND, Bateman A. 2004. The PepSY domain: a regulator of peptidase activity in the microbial environment? Trends Biochem Sci 29:169–172. doi: 10.1016/j.tibs.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 49.Barnes MR. (ed). 2007. Bioinformatics for geneticists: a bioinformatics primer for the analysis of genetic data, 2nd ed. Wiley, Chichester, England. [Google Scholar]
- 50.Pagba CV, McCaslin TG, Chi S-H, Perry JW, Barry BA. 2016. Proton-coupled electron transfer and a tyrosine-histidine pair in a photosystem II-inspired β-hairpin maquette: kinetics on the picosecond time scale. J Phys Chem B 120:1259–1272. doi: 10.1021/acs.jpcb.6b00560. [DOI] [PubMed] [Google Scholar]
- 51.Myers CR, Myers JM. 1997. Cloning and sequence of cymA, a gene encoding a tetraheme cytochrome c required for reduction of iron(III), fumarate, and nitrate by Shewanella putrefaciens MR-1. J Bacteriol 179:1143–1152. doi: 10.1128/jb.179.4.1143-1152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Minato Y, Fassio SR, Reddekopp RL, Häse CC. 2014. Inhibition of the sodium-translocating NADH-ubiquinone oxidoreductase [Na+-NQR] decreases cholera toxin production in Vibrio cholerae O1 at the late exponential growth phase. Microb Pathog 66:36–39. doi: 10.1016/j.micpath.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barquera B, Hellwig P, Zhou W, Morgan JE, Häse CC, Gosink KK, Nilges M, Bruesehoff PJ, Roth A, Lancaster CRD, Gennis RB. 2002. Purification and characterization of the recombinant Na+-translocating NADH:quinone oxidoreductase from Vibrio cholerae. Biochemistry (Mosc) 41:3781–3789. doi: 10.1021/bi011873o. [DOI] [PubMed] [Google Scholar]
- 54.Kunkel TA. 1985. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A 82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beitz E. 2000. T(E)Xtopo: shaded membrane protein topology plots in LAT(E)X2ε. Bioinforma Oxf Engl 16:1050–1051. doi: 10.1093/bioinformatics/16.11.1050. [DOI] [PubMed] [Google Scholar]
- 56.Mekalanos JJ, Swartz DJ, Pearson GD, Harford N, Groyne F, de Wilde M. 1983. Cholera toxin genes: nucleotide sequence, deletion analysis and vaccine development. Nature 306:551–557. doi: 10.1038/306551a0. [DOI] [PubMed] [Google Scholar]
- 57.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 58.Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires ToxR. J Bacteriol 170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nierman WC, DeShazer D, Kim HS, Tettelin H, Nelson KE, Feldblyum T, Ulrich RL, Ronning CM, Brinkac LM, Daugherty SC, Davidsen TD, Deboy RT, Dimitrov G, Dodson RJ, Durkin AS, Gwinn ML, Haft DH, Khouri H, Kolonay JF, Madupu R, Mohammoud Y, Nelson WC, Radune D, Romero CM, Sarria S, Selengut J, Shamblin C, Sullivan SA, White O, Yu Y, Zafar N, Zhou L, Fraser CM. 2004. Structural flexibility in the Burkholderia mallei genome. Proc Natl Acad Sci U S A 101:14246–14251. doi: 10.1073/pnas.0403306101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brett PJ, DeShazer D, Woods DE. 1998. Burkholderia thailandensis sp. nov., a Burkholderia pseudomallei-like species. Int J Syst Bacteriol 48(Pt 1):317–320. doi: 10.1099/00207713-48-1-317. [DOI] [PubMed] [Google Scholar]
- 61.Norrander J, Kempe T, Messing J. 1983. Construction of improved M13 vectors using oligodeoxynucleotide-directed mutagenesis. Gene 26:101–106. doi: 10.1016/0378-1119(83)90040-9. [DOI] [PubMed] [Google Scholar]
- 62.Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing, and gene expression in Escherichia coli. Gene 100:195–199. doi: 10.1016/0378-1119(91)90366-J. [DOI] [PubMed] [Google Scholar]
- 63.Chang AC, Cohen SN. 1978. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol 134:1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.