

ABSTRACT
Human cytomegalovirus (HCMV) genome encapsidation requires several essential viral proteins, among them pUL56, pUL89, and the recently described pUL51, which constitute the viral terminase. To gain insight into terminase complex assembly, we investigated interactions between the individual subunits. For analysis in the viral context, HCMV bacterial artificial chromosomes carrying deletions in the open reading frames encoding the terminase proteins were used. These experiments were complemented by transient-transfection assays with plasmids expressing the terminase components. We found that if one terminase protein was missing, the levels of the other terminase proteins were markedly diminished, which could be overcome by proteasome inhibition or providing the missing subunit in trans. These data imply that sequestration of the individual subunits within the terminase complex protects them from proteasomal turnover. The finding that efficient interactions among the terminase proteins occurred only when all three were present together is reminiscent of a folding-upon-binding principle leading to cooperative stability. Furthermore, whereas pUL56 was translocated into the nucleus on its own, correct nuclear localization of pUL51 and pUL89 again required all three terminase constituents. Altogether, these features point to a model of the HCMV terminase as a multiprotein complex in which the three players regulate each other concerning stability, subcellular localization, and assembly into the functional tripartite holoenzyme.
IMPORTANCE HCMV is a major risk factor in immunocompromised individuals, and congenital CMV infection is the leading viral cause for long-term sequelae, including deafness and mental retardation. The current treatment of CMV disease is based on drugs sharing the same mechanism, namely, inhibiting viral DNA replication, and often results in adverse side effects and the appearance of resistant virus strains. Recently, the HCMV terminase has emerged as an auspicious target for novel antiviral drugs. A new drug candidate inhibiting the HCMV terminase, Letermovir, displayed excellent potency in clinical trials; however, its precise mode of action is not understood yet. Here, we describe the mutual dependence of the HCMV terminase constituents for their assembly into a functional terminase complex. Besides providing new basic insights into terminase formation, these results will be valuable when studying the mechanism of action for drugs targeting the HCMV terminase and developing additional substances interfering with viral genome encapsidation.
KEYWORDS: complex assembly, cytomegalovirus, nuclear import, protein interactions, protein stability, terminase
INTRODUCTION
Human cytomegalovirus (HCMV) has the largest genome of all mammalian DNA viruses, and packaging of its double-stranded DNA into preformed capsids has similarities with that of tailed bacteriophages. In phages, viral proteins called terminases interact with both the viral DNA and prohead to accomplish packaging (1, 2). For HCMV, several lines of evidence indicate that the viral terminase comprises the proteins pUL56 and pUL89, with pUL56 binding to the capsid portal protein pUL104 as well as to the packaging signals on the concatemeric viral DNA (3, 4) and pUL89 mediating cleavage after exactly one genome copy has been encapsidated (5). Analyses of the pUL89 structure revealed similarities between herpesvirus and bacteriophage terminases and showed that the endonuclease domain is contained within the pUL89 C terminus (6–8). Modeling of the herpesviral pUL56 orthologs disclosed structural similarity to human topoisomerase I, suggesting a role in processing of the viral DNA as proposed by Visalli et al. (9). ATPase activity has been reported for pUL56 (10, 11), which thus may provide the energy required to package the HCMV genome against electrostatic repulsions and entropic penalty, as the DNA is translocated into the capsids. The presence of so-called Walker A and B motifs (12) in the amino acid sequence of pUL89 (13, 14) suggests that this subunit can bind ATP as well and contribute to ATP hydrolysis; however, this remains to be investigated. In general, terminases belong to the most powerful molecular machines in nature, even exceeding the strength of—for instance—the myosin and kinesin motors (15, 16).
Besides the portal and the terminase proteins pUL56 and pUL89, several other essential viral proteins were shown to be necessary for HCMV genome encapsidation. These essential proteins are pUL52, whose specific role is not defined yet (17), as well as pUL77 and pUL93, which are capsid constituents presumably building the capsid vertex-specific complex (18–21). Moreover, we recently identified pUL51 as a third component of the HCMV terminase, as it forms a complex with pUL56 and pUL89 in infected cells and was found to be crucial for viral genome cleavage-packaging (22).
HCMV infection is a considerable risk factor for immunocompromised patients such as transplant recipients (23). All currently available medications to fight acute HCMV infection and disease are associated with adverse effects and target the same viral process, namely, replication of the viral DNA genome. Upon prolonged application of the antiviral drugs, resistant HCMV strains can emerge, and due to the shared mode of action, cross-resistance against different drugs is not uncommon (24). It is therefore desirable to develop new antiviral substances that interfere with other processes of the viral life cycle, and targeting viral DNA encapsidation is considered a particularly attractive approach (25–27). One of the most promising new drug candidates, Letermovir (MK-8228; AIC246), which demonstrated excellent efficacy and safety in phase 2 clinical trials in kidney transplant patients and in hematopoietic stem cell recipients (28, 29) and recently met the primary endpoint in a phase III clinical study (30) (ClinicalTrials registration no. NCT02137772) prevents cleavage of the viral DNA concatemers and formation of mature virions, apparently by targeting the terminase subunit pUL56 (31–34). Importantly, Letermovir is able to inhibit HCMV variants that are resistant to other antiviral agents (35–37), and compared to the approved HCMV drugs, its toxicity is negligible. The favorable properties of Letermovir and the novel target structure have sparked new interest in the functioning of the HCMV terminase complex, particularly as the precise molecular mechanism of inhibition is not yet known.
Investigation into the interplay among the terminase subunits—preferentially in the context of infection—is a prerequisite to understand the mode of action of drugs blocking genome encapsidation. However, studies with CMVs carrying mutations in the genes encoding the terminase subunits are scarce (14, 22). For UL56 and UL89, little more is known than that disruption of these open reading frames (ORFs) blocks the viral life cycle (38, 39). This is in part owed to the experience that establishing complementing cells for HCMV mutants is challenging (40). Consequently, most data concerning the terminase subunits were obtained with transient-transfection assays or in vitro experiments with lysates of cells infected by wild-type virus. Nevertheless, many questions regarding the assembly of the terminase complex are open, and the molecular mechanisms governing terminase function remain poorly understood. This applies particularly to the role of the pUL51 subunit, which is the smallest protein of the terminase constituents. We have recently described the phenotype of an HCMV mutant in which a destabilizing domain (ddFKBP [destabilizing domain of FK506 binding protein]) was added to pUL51 (22, 41). The resulting fusion protein is unstable and hence degraded, thus resembling the phenotype of a knockout mutant, whereas upon addition of a small synthetic ligand (shield-1), the protein is stabilized and can exert its biological function. Taking advantage of this technique, we could assign an essential role to pUL51 in cleavage-packaging of the HCMV genome (22). However, it has not yet been determined whether pUL51 interacts directly with either pUL56 or pUL89 or with both of these terminase subunits, and therefore, its exact role within the terminase complex remains undefined. A recent publication reported on the consequences of the interplay of the terminase subunits for subcellular distribution employing transient expression of the HCMV terminase proteins, yet direct protein-protein interactions were not addressed (42). Moreover, previous studies that were based on yeast two-hybrid analyses and transient expression of isolated proteins in mammalian cells proposed a plethora of viral interaction partners and gave rise to the hypothesis that pUL51 (and its orthologs in other herpesviruses) may serve as hub proteins linking viral DNA cleavage to encapsidation and nuclear egress of DNA-filled capsids (43–45). Still, most of the proposed interactions await confirmation in infected cells.
Here, we report on the construction of HCMV genomes lacking large parts of either the UL56 or UL89 ORF and on the consequences of the deletions for (i) expression of the remaining terminase subunits, (ii) their subcellular localization, and (iii) their interaction. An HCMV bacterial artificial chromosome (BAC) in which the UL51 ORF is disrupted was also included in the analysis (41). This study became possible through application of an adenovirus particle-mediated transfection protocol (adenofection), which allows efficient transfer of HCMV BACs into permissive cells and investigation of viral proteins of BAC-transfected cells (46), thereby circumventing the need for generating complementing systems for the mutants. These experiments were complemented by transient-transfection assays of cells with plasmids expressing the terminase proteins. Compared to cells transfected with the parental HCMV genome, the levels of the remaining terminase subunits were markedly diminished in cells harboring the genomes that lacked one of the terminase ORFs. The observation that the protein levels could be rescued upon inhibition of the proteasome suggests that formation of the terminase complex protects the individual subunits from degradation. For translocation of pUL51 and pUL89 into the cell nucleus, the presence of all three subunits was required, whereas for pUL56, nuclear import occurred by default. Furthermore, we demonstrated direct interactions among the individual terminase constituents, which occurred efficiently only in the presence of all three subunits. Altogether, our data point to mutual interplay between the terminase proteins as a prerequisite for assembly and nuclear localization of the tripartite holoenzyme.
RESULTS
Construction of HCMV BAC genomes with deletions in the ORFs encoding the terminase subunits.
We recently showed that pUL51 is associated with pUL56 and pUL89 in HCMV-infected cells, indicating that pUL51 is a third component of the HCMV terminase (22). To investigate how the terminase complex is formed, in particular which interactions are still possible when one terminase subunit is missing, we generated HCMV BAC genomes in which either the UL51, UL56, or UL89 ORF is disrupted (Fig. 1). The pHG-ΔUL51 genome described before (22, 41) carries a small deletion within the UL51 ORF (Fig. 1A, construct 1). To construct the UL56 null genome (Fig. 1A, construct 2), the first 1,928 nucleotides (out of 2,553) of the UL56 ORF were deleted, and for the UL89 knockout genome (Fig. 1A, construct 3), most of exon 2 (1,049 nucleotides out of 1,137) was removed, including the sequences encoding the proposed interaction domain for pUL56 (6, 47). Care was taken to retain putative regulatory sequences of neighboring essential ORFs. Due to the lack of a specific pUL51 antibody suitable for immunoblotting, we used the HCMV BAC pHG-UL51-SF/HA (22) as the basis for constructing pHG-ΔUL56 and pHG-ΔUL89, which comprises an epitope-tagged pUL51 version (Strep/FLAG/HA [Strep stands for Strep-tag, and HA stands for hemagglutinin] tag) together with its putative promoter sequences inserted at an ectopic position of the UL51 null genome (Fig. 1A, construct 4). In previous studies, we demonstrated that ectopic insertion of N-terminally tagged pUL51 versions gives rise to recombinant virus exhibiting growth properties comparable to those of the parental HCMV strain (22, 41). Likewise, the replacement of nonessential viral genes in the US region of the HCMV genome by the BAC vector and enhanced green fluorescent protein (EGFP) sequences did not interfere with viral growth in cell culture (48, 49). Successful mutagenesis was confirmed by restriction analysis (Fig. 1B) and sequencing of the relevant parts within the BAC genomes. Upon transfection of permissive cells, the mutant HCMV BACs were found to be noninfectious (Fig. 1C), confirming that each of the terminase proteins is essential for production of viral progeny.
FIG 1.
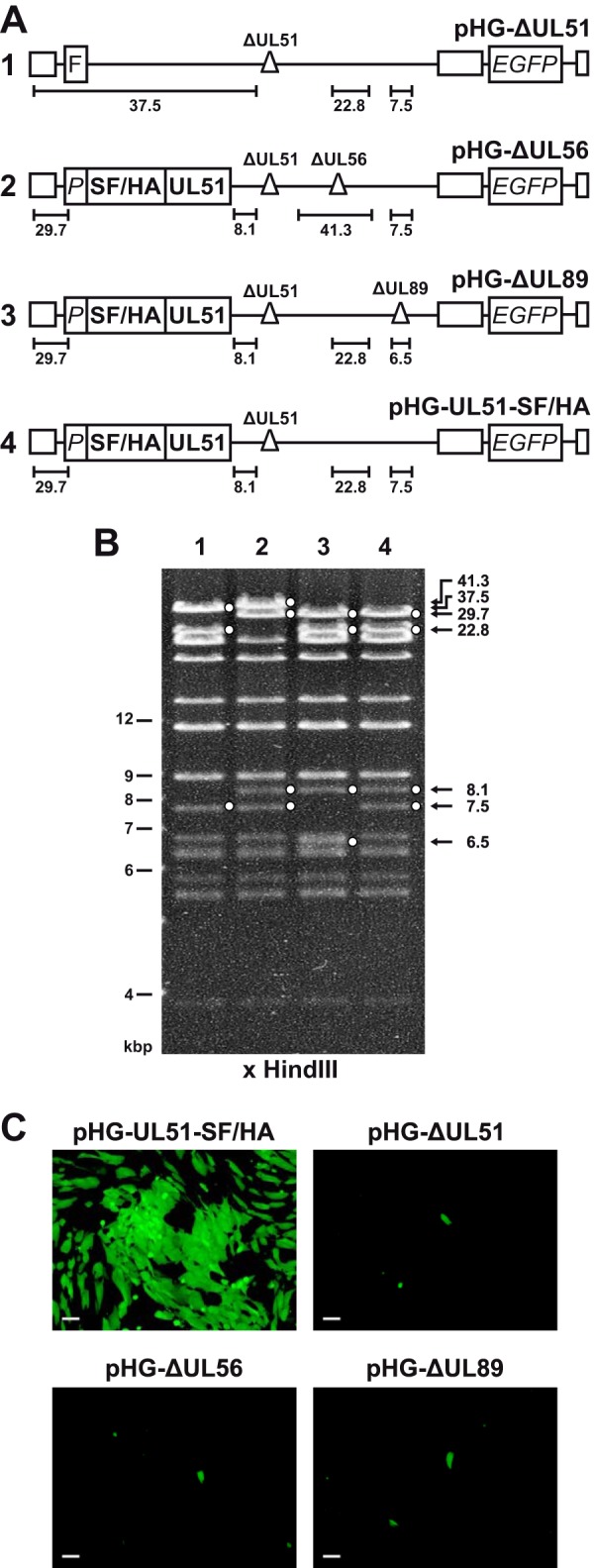
HCMV BAC genomes used in this study. (A) Schematic representation of the generated BACs containing the following elements: FLP recombination target (FRT [F]) site replacing the nonessential ORFs UL1 to UL10; UL51 promoter region (P); sequences encoding Strep-tag II, FLAG, and HA epitope tags (SF/HA); deletions (Δ) disrupting the indicated ORFs; ORF encoding the enhanced green fluorescent protein (EGFP). The terminal and internal repeat regions are indicated by open boxes. HindIII DNA fragments characteristic of the respective viral genomes are indicated by black lines. The illustration is not drawn to scale. (B) Restriction analysis of the recombinant HCMV genomes. BAC DNA was cut with HindIII, separated by agarose gel electrophoresis, and stained with ethidium bromide. Relevant bands characterizing the HCMV genomes are indicated by solid white circles. The lanes are numbered according to constructs 1 to 4 in the schematic drawings shown in panel A. (C) Ability of recombinant HCMV BAC genomes to generate infectious progeny virus. RPE-1 cells were adenofected with the indicated BACs. On day 4 posttransfection, cells were harvested and added to a monolayer of HFF cells, and 10 days later, viral spread was monitored by UV light microscopy. Bars, 100 μm.
The absence of one terminase component affects the protein levels of the others.
Utilizing the HCMV deletion genomes, we first studied expression of the remaining terminase proteins in the absence of the third component. Since no complementing cell lines are available to reconstitute the corresponding virus mutants, we applied the recently described adenovirus-mediated transfection protocol (adenofection) which allows direct investigation of BAC-transfected cells. During previous analyses, we have shown that upon adenofection, viral gene expression occurs with kinetics similar to that of infected cells, that the majority of adenofected cells successfully enters the late phase of the HCMV infection cycle, and that adenofection is well suited to study the consequences of deletion of various essential genes from the viral genome (18, 46). Human telomerase reverse transcriptase-immortalized-RPE-1 cells (RPE-1 cells) were adenofected with the recombinant HCMV BAC genomes lacking either UL51, UL56, or UL89 or with the parental BAC pHG-UL51-SF/HA. Whole-cell lysates were prepared on day 4 posttransfection and analyzed by immunoblotting. Figure 2A (left blots) shows that in the absence of one terminase subunit, the levels of the others were reduced, whereas the levels of the viral control proteins pUL52 (expressed with late kinetics) and pUL44 (an early viral protein) remained unchanged. The disruption of UL56 had the greatest impact, as the levels of both pUL51 and pUL89 were markedly diminished (to about 5% for pUL51 and to 30% for pUL89 [Fig. 2A, right graphs]). pUL51 was more strongly affected by the lack of pUL89 than pUL56 was (reduction of UL51 protein level to 30% [on average] and of the pUL56 level to 50%), and the absence of pUL51 led to decreases in the amounts of pUL56 and pUL89 to approximately 35% and 55%, respectively. Serial dilutions of the samples prior to quantification gave the same results (not shown). In contrast, the lack of pUL52, another protein involved in genome cleavage-packaging, which is not part of the terminase complex (22), did not influence pUL56 or pUL89 protein levels, as demonstrated following transfection of cells with an HCMV BAC deleted for the UL52 ORF (pHG-ΔUL52 [17] [Fig. 2B]). This result is in agreement with our previous finding that in the absence of pUL52, neither the nuclear targeting of pUL51, pUL56, and pUL89 nor the interaction between pUL56 and pUL89 is disturbed (46). Thus, our data point to a specific role of each individual terminase subunit for maintaining adequate protein levels of the other two. To assess whether the reduced protein amounts might result from changes at the transcriptional level, we analyzed the UL51, UL56, and UL89 transcript amounts in RPE-1 cells adenofected with the HCMV null genomes (Fig. 2C). This demonstrated that the absence of one of the terminase proteins does not affect the RNA levels of the remaining two proteins.
FIG 2.

Reduced amounts of the terminase subunits in cells transfected with HCMV BAC genomes in which either UL51, UL56, UL89, or UL52 is disrupted. (A, left) RPE-1 cells were adenofected with the indicated BACs or were mock transfected. On day 4 posttransfection, cells were harvested, whole-cell lysates were prepared, and immunoblot analysis was performed with the antibodies indicated. The HCMV late protein pUL52 and the early protein pUL44 served as controls for both transfection efficiency and viral gene expression, and GAPDH was used as a loading control. The asterisks denote unspecific reactivity with a cellular protein. (Right) Immunoblot signals of three independent experiments (means ± standard deviations [SD] [error bars]) were quantified with ImageJ software using membranes exposed for a few seconds only. For normalization, UL51, UL56, and UL89 protein levels of cells transfected with the parental BAC pHG-UL51-SF/HA were set at 100%. (B) RPE-1 cells were adenofected with the BAC genome pHG-ΔUL52 carrying a deletion within the UL52 ORF or with the parental BAC pHG and analyzed by immunoblotting as in panel A with the antibodies indicated (HCMV major capsid [late] protein [MCP]). Quantification of signals was done as described above for panel A. (C) RPE-1 cells adenofected with the indicated HCMV BACs were used for preparation of total RNA on day 4 posttransfection. UL51, UL56, UL89, and UL52 transcript levels were determined by quantitative RT-PCR, relative RNA levels were calculated using UL52 as the internal control and normalized to the values for cells transfected with the parental BAC pHG-UL51-SF/HA. Data are representative of two independent experiments. (D and E) Interactions between the terminase subunits in RPE-1 cells adenofected with the indicated BACs. pUL51-SF/HA was pulled down using Strep-Tactin Sepharose (D), or pUL56 and pUL89 were immunoprecipitated (IP) with specific antibodies from cell lysates prepared on day 4 posttransfection (E). Eluted proteins were analyzed by immunoblotting using antibodies directed against the HA tag (for pUL51), pUL56, or pUL89. IgG heavy chains (HC) and light chains (LC) served as controls.
We next investigated interactions between two of the terminase subunits when the third one is missing. To this end, pUL51, pUL56, or pUL89 was pulled down from lysates of RPE-1 cells adenofected with the above-mentioned HCMV knockout genomes, and associated terminase subunits were detected by immunoblotting (Fig. 2D and E). In the absence of pUL89, pUL51 and pUL56 could still interact with each other (Fig. 2D, lane 3, and Fig. 2E, lane 3), albeit considerably larger amounts of the respective proteins copurified when all three subunits were present (Fig. 2D, lane 4, and Fig. 2E, lane 4). This hints at a role of pUL89 in mediating efficient pUL51-pUL56 interaction. Overall however, the interpretation of these interaction experiments was impeded by the diminished protein levels as shown in Fig. 2A. For instance, in the absence of pUL56, the abundance of pUL51 was strongly decreased (Fig. 2A, lane 2), and thus, very little pUL51 could be pulled down from pHG-ΔUL56-adenofected cells, and no pUL89 was found to be associated with pUL51 (Fig. 2D, lane 2). Still, one cannot deduce that pUL89 no longer bound to pUL51, especially because the total amount of pUL89 available for interaction in this setting was strongly diminished (Fig. 2A, lane 2). Similar limitations apply to the reciprocal pUL89 immunoprecipitation (IP) experiment depicted in Fig. 2E, lane 6, and thus, a conclusion about the pUL51-pUL89 interaction in the absence of pUL56 cannot be drawn. Conversely, upon UL51 deletion, reasonable amounts of pUL56 and pUL89 were detected (Fig. 2A, lane 1), yet binding of pUL89 to pUL56 following pUL56 IP was essentially abolished (Fig. 2E, lane 1), whereas ample amounts of pUL89 were associated with pUL56 in the presence of pUL51 (Fig. 2E, lane 4). This suggests that pUL51 is needed for stable interactions between pUL56 and pUL89.
Efficient formation of the HCMV terminase complex is achieved only in the presence of all three subunits.
Due to the limitations described above, we decided to examine terminase assembly further by applying transient transfection of HCMV-permissive RPE-1 cells using plasmids expressing either pUL51 (fused to a Strep-FLAG-HA epitope tag), pUL56, or pUL89 (Fig. 3A). The expression plasmids were transfected alone or in different combinations, and whole-cell lysates were analyzed by immunoblotting. Overall, in these experimental settings, fewer differences in the protein levels of the terminase components were observed (Fig. 3B), suggesting that virus-specific pathways contribute to the decreased levels of the terminase constituents seen in BAC-transfected cells. For instance, HCMV upregulates both the level and activity of proteasomes, which was found to promote viral gene expression (50). Nevertheless, upon transfection of the pcDNA constructs, somewhat decreased amounts of pUL56 were also seen when expressed alone (Fig. 3B, lane 2) or together with pUL89 (Fig. 3B, lane 6). To check for interactions among the transiently expressed terminase proteins, we first pulled down pUL51-SF/HA and tested for copurifying pUL56 or pUL89 by immunoblotting. As is obvious from Fig. 3C, pUL51 was pulled down from the cell lysates shown in Fig. 3B with comparable efficiencies, yet considerably less pUL56 or pUL89 was associated with pUL51 when the respective third subunit was missing (compare lanes 4 and 5 to lane 7). This indicates that pUL51 can directly bind pUL56 and pUL89, yet robust interactions are established only when all three subunits are present together. To confirm this finding and to further evaluate the role of pUL51 in mediating binding of pUL56 to pUL89, reciprocal coimmunoprecipitation (co-IP) experiments were performed by pulling down either pUL89 or pUL56. In agreement with previous findings (22) and probably due to overlap of the binding sites of the pUL89 monoclonal antibody (MAb) and the interaction domain for pUL56, IP with the pUL89-specific MAb yielded very little pUL56 (data not shown), and therefore, the outcome was inconclusive. IP utilizing the pUL56 MAb led to efficient coprecipitation of all terminase proteins after expressing the three subunits together (Fig. 3D, lane 7) and verified the requirement of pUL89 for strong interaction between pUL51 and pUL56 (Fig. 3D, compare lane 4 to lane 7). However, the reduced amounts of pUL56 in lysates of cells expressing pUL56 alone or in combination with pUL89 (compare lanes 2 and 6 in Fig. 3B) resulted in small amounts of pUL56 being immunoprecipitated from these cells compared to cells transfected with all three pcDNA constructs (Fig. 3D, compare lanes 2 and 6 to lane 7). Therefore, we adjusted the immunoprecipitated pUL56 to equal levels by diluting the respective samples. Figure 3E shows that substantially less pUL89 was bound to pUL56 when pUL51 was absent (compare lane 3 to lane 5 and compare lane 4 to lane 6), confirming an important role of pUL51 in establishing robust pUL56-pUL89 interaction. Taken together, each terminase protein is able to interact directly with the other two, although efficient interactions between the individual subunits occur only when all three components are available simultaneously.
FIG 3.

Efficient interactions require all three terminase proteins. (A) Schematic representations of the expression plasmids used in this study. The major immediate early promoter of HCMV (CMV), sequences for Strep-tag II, FLAG, and HA epitope tags (SF/HA) and polyadenylation signal sequence (pA) are indicated. (B) RPE-1 cells were transfected with the expression plasmids depicted in panel A, either alone or in the indicated combinations, or were mock transfected. On day 2 posttransfection, whole-cell lysates were prepared and analyzed by immunoblotting using antibodies directed against the HA tag (for pUL51), against pUL56, pUL89, or GAPDH (which served as a loading control). The asterisks denote unspecific reactivity with a cellular protein. (C) The epitope-tagged pUL51 was pulled down using Strep-Tactin beads, and pUL56 or pUL89 bound to it was assessed by immunoblotting. (D) Following immunoprecipitation (IP) of pUL56, coprecipitated pUL51 or pUL89 was detected by immunoblotting. IgG HC, immunoglobulin heavy chain. (E) The samples displayed in panel D, lanes 6 and 7, were diluted to adjust immunoprecipitated pUL56 to comparable levels, and interacting pUL51 and pUL89 was analyzed by immunoblotting.
The terminase proteins protect each other from proteasomal turnover.
As is seen in Fig. 3B, pUL56 abundance seemed to depend on the presence of pUL51, which might be explained by degradation of pUL56 when pUL51 is missing. To test this, RPE-1 cells transfected with the pUL56 expression plasmid were incubated with the proteasome inhibitor MG132 prior to harvesting. UL56 protein amounts were only moderately increased upon 5 h of proteasome inhibition (Fig. 4A, lanes 1 and 2), yet following 24 h of MG132 treatment, they were similar to those found after coexpression of pUL56 with pUL51 (Fig. 4A, lanes 3 and 4). Expression of pUL89 in addition to pUL51 did not further increase pUL56 levels (Fig. 4A, lane 5). This finding points to a role of pUL51 in stabilizing pUL56. In order to substantiate this hypothesis, the pUL56 expression plasmid was cotransfected with increasing amounts of the pUL51 construct. As shown in Fig. 4B (top panel), increasing pUL51 amounts resulted in a dose-dependent accumulation of pUL56, indicating that pUL51 expression is able to rescue UL56 protein levels. Interestingly, pUL56 amounts did not increase further upon addition of MG132 to cells coexpressing pUL51 and pUL56, indicating that pUL51 is sufficient to prevent proteasome-mediated turnover of pUL56 (Fig. 4B, bottom panel, compare lane 2 to lane 4).
FIG 4.

Mutual protection of the terminase subunits from proteasomal turnover. (A) RPE-1 cells were transfected with equal amounts of pcDNA-UL56 and treated with MG132 (5 μM) for the time periods indicated before harvesting (lanes 1 to 3), or cotransfected with pcDNA-UL56 and pcDNA-UL51-SF/HA (lane 4), or cotransfected with all three expression constructs (lane 5), or mock transfected (lane 6). Total cell lysates were prepared on day 2 posttransfection and analyzed by immunoblotting. (B, top) Stabilization of pUL56 levels by coexpression of pUL51. RPE-1 cells were cotransfected with a constant amount of pcDNA-UL56 and increasing amounts of pcDNA-UL51-SF/HA or were mock transfected and then analyzed by immunoblotting. (Bottom) Stabilization of pUL56 in the presence of both pUL51 and MG132. RPE-1 cells were transfected with the indicated expression plasmids and cultivated with MG132 (+) or without MG132 (−) for 24 h before harvesting. (C and D) Rescue of the terminase protein levels in BAC-transfected cells through proteasome inhibition (C) or by expressing the missing terminase constituent (D). (C) Adenofected RPE-1 cells were treated with MG132 (5 μM) for 24 h before harvesting (+) or were treated with solvent (dimethyl sulfoxide [DMSO]) only (−). Expression of the indicated proteins was examined by immunoblotting on day 4 posttransfection. (D) RPE-1 cells were transfected with the parental pHG-UL51-SF/HA BAC or the deletion BAC genomes indicated, with (+) or without (−) the pcDNA constructs encoding either the UL51, UL56, or UL89 protein. Analysis was done as described above for panel C. Please note that due to the high levels of pUL51, pUL56, and pUL89 in cells that received the expression plasmids (lanes 2, 5, and 8), the corresponding proteins expressed by the parental BAC pHG-UL51-SF/HA (lanes 3, 6, and 9) were detected only after overexposure of the blots (not shown).
To examine whether an analogous mechanism accounts for the diminished levels of the terminase subunits in BAC-transfected cells (Fig. 2A), we adenofected RPE-1 cells with the deletion BAC genomes or the parental BAC, followed by incubation with MG132 before preparing total cell lysates (Fig. 4C). Indeed, proteasome inhibition restored the levels of pUL56 and pUL89 expressed from pHG-ΔUL51 to those obtained after transfection of the parental BAC pHG-UL51-SF/HA (Fig. 4C, lanes 1 to 4), and the same was found for pUL51 and pUL89 levels in the absence of pUL56 (Fig. 4C, lanes 5 to 8), as well as for pUL51 and pUL56 in cells adenofected with pHG-ΔUL89 (Fig. 4C, lanes 9 to 12). Treatment of the BAC-transfected cells with the proteasome inhibitor epoxomicin had the same effect (data not shown). We therefore concluded that each terminase subunit is needed to protect the others from proteasomal turnover, presumably by mutual stabilization through sequestration upon complex formation. Attempts to coimmunoprecipitate the terminase proteins from these cell lysates were not successful, inasmuch as almost no protein could be precipitated with the respective antibodies (data not shown), indicating that the proteins were no longer accessible to the antibodies. A likely explanation for this finding is that upon MG132 treatment, the proteins became insoluble. In fact, protein aggregation upon proteasome inhibition occurs quite frequently (51).
If the hypothesis that the HCMV terminase subunits stabilize each other holds true, the abundance of the terminase proteins in cells transfected with the deletion BACs should be rescued when the missing subunit is provided in trans. As depicted in Fig. 4D, in RPE-1 cells adenofected with the HCMV BAC pHG-ΔUL51 and the expression plasmid encoding pUL51, the levels of pUL56 and pUL89 were restored to the levels obtained after transfection of the parental BAC pHG-UL51-SF/HA (Fig. 4D, lanes 1 to 3), and the same was found for pUL51 and pUL56 levels following expression of pUL89 in pHG-ΔUL89-transfected cells (Fig. 4D, lanes 7 to 9). Similarly, pUL56 increased the abundance of pUL51 and pUL89 in pHG-ΔUL56-transfected cells—almost reaching the levels achieved after transfection of the parental BAC pHG-UL51-SF/HA (Fig. 4D, lanes 4 to 6). These results are all the more remarkable, as cotransfection of the expression plasmids led to smaller amounts of other viral proteins, such as pUL44 and pUL52 (Fig. 4D). Furthermore, a control plasmid expressing the unrelated Kaposi's sarcoma-associated herpesvirus (KSHV) ORF52 protein did not elicit elevated levels of the terminase components (data not shown). We therefore concluded that the proteasome-dependent turnover of the HCMV terminase subunits can be overcome by adding the missing component, which implies that the specific interplay among the terminase proteins results in their stabilization.
Nuclear targeting of pUL51 and pUL89 depends on the presence of the other terminase subunits.
We finally asked how the subcellular distribution of the individual terminase proteins is influenced by the other subunits. Previous studies have demonstrated that all three terminase proteins are found within the nuclei of HCMV-infected fibroblasts (22) or of HEK-293T cells cotransfected with plasmids encoding the UL51, UL56, and UL89 proteins (42). First, we adenofected RPE-1 cells with the HCMV BAC null genomes or the parental BAC, and analyzed them by confocal laser scanning microscopy. In line with the reduced levels of the terminase proteins when one component was lacking (Fig. 2A), the signals acquired upon transfection of the deletion BACs were faint and could be visualized only by using microscope settings that resulted in overexposure of the control cells (RPE-1 adenofected with the parental pHG-UL51-SF/HA BAC). Accordingly, strong signals were seen for pUL51, pUL56, and pUL89 (Fig. 5A, top left panel), yet the homogenous nuclear staining concealed their enrichment in viral replication compartments. pUL56 was detected in the nucleus when pUL51 or pUL89 was missing (Fig. 5A, right panels), and pUL89 was found in the cytoplasm in the absence of pUL51 or pUL56 (Fig. 5A, top right panel and bottom left panel). pUL51 was both nuclear and cytoplasmic in pHG-ΔUL56-transfected cells (Fig. 5A, bottom left panel), and when expressed from pHG-ΔUL89, weak nuclear staining was seen (Fig. 5A, bottom right panel).
FIG 5.

Dependence of the subcellular localization of pUL51 and pUL89 on the presence of the other terminase subunits. (A) RPE-1 cells adenofected with the indicated BAC genomes were probed 4 days later with antibodies directed against the HA tag (for pUL51), pUL56, or pUL89 and were analyzed by confocal laser scanning microscopy. (B) HeLa cells were transfected with expression plasmids encoding pUL51, pUL56, or pUL89, either alone or in the given combinations, together with pEGFP-C1 to mark the transfected cells. After 2 days, cells were analyzed as described above for panel A. In panels A and B, contours of the nuclei are marked by white dashed lines. The numbers below each panel represent the proportion of cells exhibiting the localization pattern shown in the respective micrograph (e.g., 19 cells exhibiting the localization pattern/20 total cells). Images displaying the same antibody staining were taken with identical microscope settings. Bars, 10 μm.
To complement these results and to assess the localization of the terminase subunits when expressed in isolation, we also analyzed cells transiently transfected with the expression plasmids. As expected, when pUL51, pUL56, and pUL89 were present simultaneously, all three were found in the nucleus (Fig. 5B, top left panel). In contrast, when expressed alone, pUL51 was equally distributed between the cytoplasm and the nucleus, whereas pUL56 was confined to the nucleus, and pUL89 was predominantly localized to the cytoplasm (Fig. 5B, top right panel). Notably, pairwise expression of the terminase components resulted in the same subcellular distribution of the proteins as observed after expressing the subunits separately (Fig. 5B, bottom panels). Taking all these results together, we concluded that correct nuclear localization of both pUL51 and pUL89 requires the concurrent presence of all three terminase subunits. Conversely, pUL56 is located in the nucleus when expressed alone or in the absence of either pUL51 or pUL89, which is in accordance with the presence of a nuclear localization signal in the UL56 protein (52, 53).
DISCUSSION
In this work, we investigated HCMV terminase complex formation and studied for the first time consequences of UL56 and UL89 deletions in the context of the HCMV genome, as well as direct interactions between the terminase subunits. We found that upon deletion of either the UL51, UL56, or UL89 ORF from the HCMV genome, the protein levels of the remaining terminase components were markedly reduced, which could be overcome by proteasome inhibition or by providing the missing subunit in trans. Moreover, analysis of cells transiently transfected with expression plasmids indicated that efficient interactions among the terminase proteins require all three subunits together. Similarly, the nuclear targeting of pUL51 and pUL89 depended on the presence of the other terminase constituents. To sum up, these data indicate that each of the terminase subunits affects the others with regard to their stability, correct subcellular localization, and assembly into a functional terminase complex.
In a previous study employing a conditional UL51 viral mutant, we reported that in infected cells, pUL56 and pUL89 became virtually undetected by immunofluorescence analysis following knockdown of pUL51 (22). By applying more sensitive microscope settings, we now saw that in cells adenofected with the UL51 null genome, pUL56 was nuclear, whereas pUL89 was restricted to the cytoplasm. In general, pUL89, which does not contain a nuclear localization signal (NLS) according to in silico analyses (53), was cytoplasmic when one of the other terminase components was missing. pUL51, for which no classical NLS was predicted (53), was distributed throughout the cell in the absence of pUL56 or pUL89. This can be explained by the small size of pUL51, allowing diffusion through the nuclear pore complex. Conversely, pUL56 was nuclear when pUL51 or pUL89 was lacking. The pUL51 and pUL89 distribution patterns are comparable to those described by Wang et al. using transient transfection of HEK-293T cells or baculovirus-mediated expression in insect cells (42). In contrast to their data, we found that pUL56 is imported into the nucleus when expressed in the absence of pUL51 and/or pUL89, which was somehow expected, as pUL56 comprises a functional nuclear localization signal (52). As already discussed by Wang et al. (42), discrepancies in subcellular localization of pUL56 may result from the use of different cell types in their study and the study of Giesen and colleagues (52), and the same explanation presumably also accounts for the difference in our data. Nevertheless, Wang et al. (42) also stated that the presence of pUL51 does not direct pUL89 to the nucleus and that nuclear localization of each terminase component is greatest upon coexpression of all three proteins. This is in line with our data, which imply that the pairwise presence of only two of the HCMV terminase subunits is not sufficient to translocate pUL51 or pUL89 to the nucleus and that all three subunits are required together for nuclear localization of the HCMV terminase complex.
A major result of this study is that in cells adenofected with HCMV BAC genomes lacking either the UL51, UL56, or UL89 ORF, the amounts of the remaining terminase subunits were diminished. This finding for the HCMV terminase components is novel, as this aspect has not systematically been addressed for other herpesviruses. Although it was reported that herpes simplex virus 1 (HSV-1) pUL28 and pUL33 (the orthologs of HCMV pUL56 and pUL51, respectively) can stabilize each other (54–56), virus mutants with deletions in the ORFs encoding the three terminase constituents were not compared side by side as we did here. We now show a mutual dependence of all three terminase subunits to preserve appropriate protein levels of each component. This is best explained by sequestration of the individual subunits within the terminase complex, thereby withdrawing them from turnover by the proteasome. Indeed, we found that proteasome inhibition restored the levels of the terminase proteins to the same extent as the addition of the missing subunit in trans did. This is reminiscent of a mechanism termed cooperative stability, which describes the protection from degradation through loading of a given protein into a larger complex (57). This process ensures that monomeric subunits are removed before they might interfere with the proper function of the final multiprotein complex. In this context, one can easily envisage that the isolated terminase components can exert deleterious effects on the viral life cycle by improperly acting on viral DNA, such as, for instance, occupation of virus genomes with nonfunctional terminase components, non-sequence-specific or premature genome cleavage, futile ATP hydrolysis, or by acting on other essential viral and cellular pathways. In addition, pUL51, pUL56, and pUL89 are produced with identical kinetics in HCMV-infected cells (58), in agreement with the observation that the synthesis of monomers of a multiprotein entity subjected to the cooperative stability principle is often synchronized, in order to guarantee the simultaneous presence of all subunits in appropriate amounts when the biological function of the complex is required (59). Overall, our data argue in favor of a model of the HCMV terminase as a multiprotein complex in which the individual subunits stabilize each other upon assembly. Such a setting was also proposed for other CMV protein complexes, namely, for the murine CMV (MCMV) US22 protein family (60) and for the HCMV major and small capsid proteins (18), as well as for the cellular anaphase-promoting complex, a multiprotein assembly targeted by HCMV (61).
Contrary to the results obtained here with the UL51 null genome, our previous data using the HCMV-ddFKBP-UL51 mutant did not reveal reduced levels of pUL56 or pUL89 when pUL51 was knocked down. In the former experiments, the UL51 protein was first synthesized and subsequently destabilized and degraded, with some of it still persisting (22). This suggests that the transient presence of pUL51 together with its residual amounts were sufficient to increase pUL56 and pUL89 levels. Furthermore, the results obtained in this study indicate that the effect of pUL51 on enhancing pUL56 amounts did not require the steady interaction of the two proteins in transiently transfected cells, as this was achieved only after coexpression of pUL89. It therefore seems that a weak or temporary interaction of pUL51 with pUL56 is already sufficient for pUL56 stabilization.
Another question we addressed here for the first time was which direct interactions can take place between individual terminase subunits and whether they are influenced by the presence of the third one. In the viral context, this analysis was impaired by the markedly decreased levels of the terminase proteins. Using this approach, the only interaction seen was between pUL51 and pUL56 when pUL89 was lacking. Therefore, this aspect was investigated by means of transient-transfection assays. In this setting, the amounts of the terminase proteins were less affected by the lack of one component. A reason for this could be divergent regulation upon isolated expression compared to the viral context, and additionally, proteasomal degradation rates can be reduced at high protein concentrations (57). The transient-transfection experiments demonstrated that terminase complex formation occurs independently of HCMV DNA and procapsids; nevertheless, it is conceivable that terminase assembly might be further enhanced in infected cells when the portal protein and viral genomes are present. In addition, direct binding of pUL51 to both pUL56 and pUL89, as well as of pUL56 to pUL89, was disclosed. The latter has been reported before, based on glutathione S-transferase (GST) pulldown and IP assays with lysates of infected cells (10, 47). Importantly, on top of that, our data now indicate a conjoint cooperation among the HCMV terminase proteins for holoenzyme formation, because the pairwise expressed terminase constituents did not interact efficiently until the respective third subunit was present. This observation suggests that conformational changes induced through mutual interaction of all three terminase components are a prerequisite for stable complex formation. Such a mechanism, termed folding upon binding, is a well-known principle governing protein complex assembly (59), and is characterized by an increase in the strength of the interactions within the complex. Moreover, successful complex assembly then protects the individual players from degradation. Conversely, unassembled subunits often exhibit aberrant conformations and are thus recognized by the cellular quality control machinery. This is in agreement with our results seen for the terminase proteins in cells adenofected with the deletion BAC genomes. Accordingly, one may ascribe chaperone-like functions to the terminase components. In general, molecular chaperones are defined as proteins that interact with other proteins to stabilize them, to aid them in adopting their functionally active conformation, and to ensure their proper subcellular localization (62, 63). In macromolecular complexes, the individual subunits themselves being part of the complex can take over these functions, without eventually dissociating from the successfully folded and assembled proteins (57, 59). A role for pUL51 and its herpesviral orthologs in promoting folding and assembly of the terminase subunits, rather than a role in the enzymatic function of the holoenzyme, has been proposed (42, 56, 64). Our data support this assumption, as pUL51 is obviously needed to maintain appropriate pUL56 and pUL89 levels and to enhance the pUL56-pUL89 interaction. However, this property is not restricted to pUL51, but also applies to the other terminase subunits.
The capability of terminase subunits to undergo conformational changes is supported by structural information available for the nuclease domains of HSV-1 pUL15 and HCMV pUL89 (6, 7), which suggests an induced fit mechanism with regard to DNA binding (8). Moreover, conformational changes are a universal prerequisite for the generation of force in molecular nanomotors (2, 65, 66). Structural analyses of the isolated pUL56 and pUL89 terminase subunits have been reported (5–7, 67); however, in view of the folding-upon-binding principle that presumably also applies to the HCMV terminase, it remains an important goal to determine the structure of the complex as a whole and to elucidate conformational changes of the individual subunits. Further conformational alterations likely also occur upon binding to the viral genome and the capsid portal.
In sum, we provide evidence of the mutual dependence of the HCMV terminase subunits with regard to protein levels, subcellular localization, and complex assembly, which altogether might protect from detrimental effects of incomplete terminase assemblies on viral or cellular pathways critical for the HCMV life cycle. This information about the interplay among the terminase proteins is key to gaining knowledge about the mode of action of promising new antiviral drugs targeting viral DNA encapsidation and will help to develop additional antiviral substances to fight CMV disease.
MATERIALS AND METHODS
Cell culture.
Human foreskin fibroblasts (HFF) and hTERT-RPE-1 cells (human telomerase reverse transcriptase-immortalized-RPE-1 cells) (Clontech, Palo Alto, CA) were propagated as previously described (46, 48), and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM GlutaMAX-I; Gibco), supplemented with 10% fetal calf serum (FCS).
Plasmids.
The expression vector pcDNA-UL51-SF/HA was constructed by PCR amplification of the epitope-tagged UL51 ORF by the use of primers UL51-SF/HA.for (for stands for forward) (5′-CGCGAATTCCCACCATGGATTATAAAGATGATG-3′) and UL51-SF/HA.rev (rev stands for reverse) (5′-ATAGCGGCCGCTTATTTACCCGGCGCCGACT-3′) with plasmid pOri6K-UL51-SF (22) as the template. The resulting PCR product was cloned into plasmid pcDNA3.1(+) (Invitrogen) via the NotI and EcoRI sites. To generate the pcDNA-ORF52-HA vector, the Kaposi's sarcoma-associated herpesvirus (KSHV) ORF52 was amplified from a KSHV bacterial artificial chromosome (BAC) (kindly provided by Sandra Koch, Hannover Medical School) using primers ORF52-HA.for (5′-CCACTAGTCCAGTGTGGTGGATGTACCCATACGACGTCCCAGACTACGCTGCCGCGCCCAGGGGCAGACCCAAAAAG-3′) and ORF52-HA.rev (5′-CGGGCCCTCTAGACTCGAGCTCAGTCATCAACCCCCGCCCGT-3′) and cloned between the EcoRI and NotI sites of pcDNA3.1(+) by Gibson Assembly according to the manufacturer's instruction (New England BioLabs). The integrity of the plasmids was verified by restriction analysis and sequencing. pEGFP-C1 was obtained from Clontech. pcDNA-UL56 was constructed by PCR amplification of ORF UL56 using the primers 5′UL56EcoRI (5′-ATGCGAATTCATGGAGATGAATTTGTTACAGAAACTATGCGTAGTGTGTTCGA-3′) and 3′UL56NotI (5′-CTCGAGCGGCCGCTTAACGCAGACTACCAGGCACCAGATCCTGG-ATT-3′) and BAC pHG (17) as the template. The resulting PCR product was inserted via In-Fusion cloning (Clontech) into pcDNA3.1(+) using the EcoRI and NotI sites. pcDNA-UL89 was constructed likewise by PCR amplification of human codon-optimized ORF UL89 (GeneArt, Germany) using primers 5_UL89codopt (codopt stands for codon optimized) (5′-GGCCGCATTTGCGAATTCGCCACCATGCTGAGAGGCGATAGCGCCGCC-3′) and 3′UL89codopt (5′-GGGCCCTCTAGCTCGAGTCATCAGGACACCCGGAACCG-3′) followed by In-Fusion cloning into pcDNA3.1(+) via the EcoRI and XhoI sites.
HCMV bacterial artificial chromosomes.
Recombinant HCMV BACs generated in this study are based on the BAC-cloned AD169 strain (48). HCMV BACs pHG-ΔUL56 and pHG-ΔUL89, which carry a deletion in either the 5′ region of the UL56 ORF or in exon 2 of the UL89 ORF, were constructed by en passant mutagenesis (68). To this end, a tetracycline resistance cassette was PCR amplified from pCP16 (69) with primer pairs UL56-ko.for (ko stands for knockout) (5′-GTCCTTGACGTGGGGTAGTACGCCCGCGTTGTCGCAGGCAGACTCGACTCGCTCACGTAGGGATAACAGGGTAATGATGTGCTTAAAAACTTACTCA-3′) and UL56-ko.rev (5′-AGACCGGACGACAGCGTCTCGTACGTGAGCGAGTCGAGTCTGCCTGCGACAACGCGGGCGTGATTCCCTTTGTCAACAGCAAT-3′) or UL89-ko.for (5′-ATATCGTCACACAGGTAGGTGGCCATGATGTCACGTAGAATCAAGACCTAGGAGCGGGTTAGGGATTGGCTTTAGGGATAACAGGGTAATGATGTGCTTAAAAACTTACT-3′) and UL89-ko.rev (5′-AGCATCCGAGGACAAAACTTCCACTTGCTGGGTAAGCCAATCCCTAACCCGCTCCTAGGTCTTGATTCTACGTGATTCCCTTTGTCAACAGCA-3′). The resulting PCR products were recombined with HCMV BAC pHG-UL51-SF/HA (equivalent to pHD-UL51-SF [22] except that it also carries an enhanced green fluorescent protein [EGFP] gene under the control of the CMV major immediate early promoter) by red-α, -β, -γ-mediated recombination in Escherichia coli strain GS1783 (68) (a kind gift of Gregory Smith, Northwestern University, Chicago, IL). The integrity of the generated BACs was verified by restriction analysis and sequencing. pHG-ΔUL52 lacking the HCMV UL52 ORF was described elsewhere (17), and construction of pHG-ΔUL51 in which the UL51 ORF is disrupted was reported before (22).
Transfection of HeLa and RPE-1 cells.
Transient transfection of HeLa cells was done by using the jetPEI reagent according to the manufacturer's instructions (PolyPlus). Briefly, 1.2 × 106 cells were seeded in 10-cm dishes 24 h prior to transfection with plasmids (0.8 pmol; 1.6 pmol in the case of pcDNA-UL56). When cells were transfected with one or two of the expression plasmids, the amount of total DNA was adjusted with herring sperm DNA (Gibco BRL). For mock transfection, herring sperm DNA only was used. The DNA and the jetPEI reagent (29.4 μl) were diluted separately in a final volume of 250 μl of 150 mM NaCl. Following 1-min incubation at room temperature (rt), the DNA solutions were mixed with the diluted jetPEI reagent and incubated for another 25 min at rt before adding the transfection mixes to the cells. After 4 h, the cells were washed twice with phosphate-buffered saline (PBS) and further incubated in complete medium for 2 days. To transfect hTERT-RPE-1 cells with HCMV BACs, 1.5 × 106 cells were seeded into 10-cm dishes 24 h prior to transfection with BAC DNA (3 μg), applying the adenovirus particle-mediated gene delivery protocol as described recently (46), and analyzed on day 4 posttransfection. For cotransfection of hTERT-RPE-1 cells with HCMV BACs and expression plasmids, 2 × 105 cells were seeded in 6-well plates 24 h prior to adenofection with one of the BAC constructs (0.5 μg) plus either pcDNA-UL51-SF/HA, pcDNA-UL56, pcDNA-UL89, or pcDNA-ORF52-HA (10 ng each).
Immunoblotting and immunofluorescence microscopy.
For analysis of whole-cell lysates by immunoblotting, cells were harvested by trypsinization and lysed in Roti-Load 1 buffer (Roth, Karlsruhe, Germany) followed by boiling at 99°C for 5 min. Cell lysates or immunoprecipitated material was subjected to SDS-PAGE, and proteins were transferred to nitrocellulose membranes. Antibody dilutions were 1:1,000 for the rabbit anti-HA MAb (clone C29F4; Cell Signaling), 1:100 for anti-pUL56 and anti-pUL89 mouse hybridoma supernatants (CapRI, Rijeka, Croatia) (22), 1:1,000 for the mouse anti-pUL44 MAb (kindly provided by Bodo Plachter, University of Mainz, Mainz, Germany), 1:200 for the anti-pUL52 mouse hybridoma supernatant (22), 1:2,000 for the rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) MAb (clone 14C10; Cell Signaling), and 1:100 for the anti-major capsid protein (anti-MCP) mouse hybridoma supernatant (a kind gift of Klaus Radsak, University of Marburg, Marburg, Germany). Horseradish peroxidase-coupled secondary antibodies were goat anti-mouse IgG (1:3,500) (catalog no. 32230; Pierce) and goat anti-rabbit IgG (1:2,000) (catalog no. 32260; Pierce). Signals were visualized by chemiluminescence using the SuperSignal West Femto maximum sensitivity substrate (catalog no. 34096; Thermo Scientific) and a LAS-3000 imager (Fujifilm). The acquired pictures were further processed with Adobe Photoshop CS4 version 11.0. Signals were quantified with ImageJ version 1.47v.
Immunofluorescence microscopy of transfected cells was performed on day 2 (transiently transfected HeLa cells) or day 4 posttransfection (BAC-adenofected RPE-1 cells) as reported elsewhere (17). Primary antibody dilutions were as follows: 1:500 for rabbit anti-HA MAb and 1:10 for anti-pUL56 and anti-pUL89 mouse hybridoma supernatants. Secondary antibodies used were Alexa Fluor 568-labeled goat anti-mouse IgG (catalog no. A11031; Invitrogen), and Alexa Fluor 568-labeled goat anti-rabbit IgG (catalog no. A11036; Invitrogen), each at 1:500 dilution. Images were taken with a LSM 510 Meta confocal laser scanning microscope (Zeiss) and further processed using AxioVision (version 4.9.1.0) and Adobe Photoshop CS4.
Affinity purification and immunoprecipitation.
RPE-1 cells transiently transfected with expression plasmids or adenofected with HCMV BACs were harvested 2 or 4 days posttransfection. The cells were lysed in 50 mM Tris-HCl (pH 8.0)–300 mM KCl–0.5 mM EDTA–0.5% NP-40–1 mM dithiothreitol in the presence of protease inhibitors for 15 min on ice. Insoluble material was removed by centrifugation for 15 min at 16,100 × g and 4°C, and the supernatants were incubated with 60 μl of Strep-Tactin Sepharose (catalog no. 2-1201-002; IBA) in a rotating wheel for 3 h at 4°C. Beads were washed five times with lysis buffer, and bound proteins were recovered by adding Roti-Load 1 and boiling for 5 min at 99°C. For coimmunoprecipitation, cells were lysed in IP buffer (50 mM Tris-HCl [pH 7.4]–300 mM KCl–5 mM EDTA–0.5% NP-40) containing protease inhibitors, and insoluble material was pelleted as described above. After preclearance of the cell lysates with 40 μl of protein G Sepharose (catalog no. 17-0618-02; GE Healthcare) for 1 h at 4°C, the samples were supplemented with 3 μg of either the pUL56 or pUL89 MAb purified by protein G affinity chromatography as described previously (70) plus 40 μl of protein G Sepharose, followed by incubation in a rotating wheel for 2 h at 4°C. The beads were washed five times with lysis buffer, and proteins were eluted as mentioned above.
RNA extraction and quantitative RT-PCR.
Total RNA was extracted from BAC-adenofected RPE-1 cells 4 days posttransfection using the RNeasy Plus minikit (Qiagen). One microgram of total RNA was reverse transcribed into cDNA in a total volume of 20 μl using the QuantiTect reverse transcription kit according to the manufacturer's protocol (Qiagen). The quantitative reverse transcription-PCR (qRT-PCR) was performed using 2 μl of cDNA product in a total reaction mix of 20 μl containing 10 μl of Brilliant III Ultra-Fast SYBR green Low ROX QPCR master mix as well as 400 nM forward and reverse gene-specific primers on the Stratagene Mx3000P QPCR system as described by the manufacturer (Agilent Technologies). Primers used were as follows: UL51-qPCR.for (qPCR stands for quantitative PCR) (5′-CGCCAATTGTACCCATACGACG-3′) and UL51-qPCR.rev (5′-GTTTTCCTCCTCTCCGTCGTCA-3′), UL56-qPCR.for (5′-TTCTGTACCGCAGCCAATACCA-3′) and UL56-qPCR.rev (5′-GGCAGACTCTTGTGGATCAGGT-3′), UL89-qPCR.for (5′-CTGGCCCAGAATACCACCAAGA-3′) and UL89-qPCR.rev (5′-GCACACGTAAGAGACCACGTTG-3′), and UL52-qPCR.for (5′-GTGAAACTGGCCATCTGTCACG-3′) and UL52-qPCR.rev (5′-TGAAGAGTGTGATGCAGAGCCA-3′). Amplification conditions were as follows: denaturation at 95°C for 3 min, followed by 40 cycles, with 1 cycle consisting of 95°C for 5 s and 60°C for 20 s. Three technical replicates were performed, and a control reaction mixture lacking reverse transcriptase as well as nontemplate controls were included for each sample. Data were analyzed using the qPCR software MxPro version 3.0 (Agilent Technologies). Relative transcript levels of UL51, UL56, and UL89 were calculated according to the 2−ΔΔCT quantification method (71) using UL52 transcripts as an internal control, and were normalized to the values of cells transfected with the parental BAC pHG-UL51-SF/HA.
ACKNOWLEDGMENTS
We are grateful to Bodo Plachter (University of Mainz, Mainz, Germany) and Klaus Radsak (University of Marburg, Marburg, Germany) for generously sharing antibodies and to Gregory Smith (Northwestern University, Chicago, IL, USA) for providing the E. coli strain GS1783. Wiebke Jäger contributed to construction of a pUL51 expression plasmid, and Jennifer Kleine-Albers was involved in BAC mutagenesis.
This work was funded by Deutsche Forschungsgemeinschaft (DFG; individual grant BO4196/1-2 to Eva Maria Borst) and in part by Deutsches Zentrum für Infektionsforschung (DZIF; grant IICH07.0802 to Martin Messerle).
S.N., E.M.B., T.G., P.L., and M.M. conceived and designed experiments. S.N., E.M.B., L.S., T.G., and K.W. performed experiments. S.N., E.M.B., K.W., L.S., and M.M. analyzed data. S.N., E.M.B. and M.M. wrote the manuscript.
REFERENCES
- 1.Casjens SR. 2011. The DNA-packaging nanomotor of tailed bacteriophages. Nat Rev Microbiol 9:647–657. doi: 10.1038/nrmicro2632. [DOI] [PubMed] [Google Scholar]
- 2.Feiss M, Rao VB. 2012. The bacteriophage DNA packaging machine. Adv Exp Med Biol 726:489–509. doi: 10.1007/978-1-4614-0980-9_22. [DOI] [PubMed] [Google Scholar]
- 3.Dittmer A, Drach JC, Townsend LB, Fischer A, Bogner E. 2005. Interaction of the putative human cytomegalovirus portal protein pUL104 with the large terminase subunit pUL56 and its inhibition by benzimidazole-d-ribonucleosides. J Virol 79:14660–14667. doi: 10.1128/JVI.79.23.14660-14667.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bogner E, Radsak K, Stinski MF. 1998. The gene product of human cytomegalovirus open reading frame UL56 binds the pac motif and has specific nuclease activity. J Virol 72:2259–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheffczik H, Savva CG, Holzenburg A, Kolesnikova L, Bogner E. 2002. The terminase subunits pUL56 and pUL89 of human cytomegalovirus are DNA-metabolizing proteins with toroidal structure. Nucleic Acids Res 30:1695–1703. doi: 10.1093/nar/30.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Couvreux A, Hantz S, Marquant R, Champier G, Alain S, Morellet N, Bouaziz S. 2010. Insight into the structure of the pUL89 C-terminal domain of the human cytomegalovirus terminase complex. Proteins 78:1520–1530. doi: 10.1002/prot.22669. [DOI] [PubMed] [Google Scholar]
- 7.Nadal M, Mas PJ, Blanco AG, Arnan C, Sola M, Hart DJ, Coll M. 2010. Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc Natl Acad Sci U S A 107:16078–16083. doi: 10.1073/pnas.1007144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selvarajan Sigamani S, Zhao H, Kamau YN, Baines JD, Tang L. 2013. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J Virol 87:7140–7148. doi: 10.1128/JVI.00311-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Visalli MA, House BL, Lahrman FJ, Visalli RJ. 2015. Intermolecular complementation between two varicella-zoster virus pORF30 terminase domains essential for DNA encapsidation. J Virol 89:10010–10022. doi: 10.1128/JVI.01313-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwang JS, Bogner E. 2002. ATPase activity of the terminase subunit pUL56 of human cytomegalovirus. J Biol Chem 277:6943–6948. doi: 10.1074/jbc.M108984200. [DOI] [PubMed] [Google Scholar]
- 11.Scholz B, Rechter S, Drach JC, Townsend LB, Bogner E. 2003. Identification of the ATP-binding site in the terminase subunit pUL56 of human cytomegalovirus. Nucleic Acids Res 31:1426–1433. doi: 10.1093/nar/gkg229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker JE, Saraste M, Runswick MJ, Gay NJ. 1982. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J 1:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Champier G, Hantz S, Couvreux A, Stuppfler S, Mazeron MC, Bouaziz S, Denis F, Alain S. 2007. New functional domains of human cytomegalovirus pUL89 predicted by sequence analysis and three-dimensional modelling of the catalytic site DEXDc. Antivir Ther 12:217–232. [PubMed] [Google Scholar]
- 14.Wang JB, McVoy MA. 2008. Mutagenesis of the murine cytomegalovirus M56 terminase gene. J Gen Virol 89:2864–2868. doi: 10.1099/vir.0.2008/003137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun S, Rao VB, Rossmann MG. 2010. Genome packaging in viruses. Curr Opin Struct Biol 20:114–120. doi: 10.1016/j.sbi.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petrov AS, Harvey SC. 2008. Packaging double-helical DNA into viral capsids: structures, forces, and energetics. Biophys J 95:497–502. doi: 10.1529/biophysj.108.131797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borst EM, Wagner K, Binz A, Sodeik B, Messerle M. 2008. The essential human cytomegalovirus gene UL52 is required for cleavage-packaging of the viral genome. J Virol 82:2065–2078. doi: 10.1128/JVI.01967-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borst EM, Bauerfeind R, Binz A, Stephan TM, Neuber S, Wagner K, Steinbruck L, Sodeik B, Lenac RT, Jonjic S, Messerle M. 2016. The essential human cytomegalovirus proteins pUL77 and pUL93 are structural components necessary for viral genome encapsidation. J Virol 90:5860–5875. doi: 10.1128/JVI.00384-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Köppen-Rung P, Dittmer A, Bogner E. 2016. Intracellular distribution of capsid-associated pUL77 of human cytomegalovirus and interactions with packaging proteins and pUL93. J Virol 90:5876–5885. doi: 10.1128/JVI.00351-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeRussy BM, Tandon R. 2015. Human cytomegalovirus pUL93 is required for viral genome cleavage and packaging. J Virol 89:12221–12225. doi: 10.1128/JVI.02382-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeRussy BM, Boland MT, Tandon R. 2016. Human cytomegalovirus pUL93 links nucleocapsid maturation and nuclear egress. J Virol 90:7109–7117. doi: 10.1128/JVI.00728-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borst EM, Kleine-Albers J, Gabaev I, Babic M, Wagner K, Binz A, Degenhardt I, Kalesse M, Jonjic S, Bauerfeind R, Messerle M. 2013. The human cytomegalovirus UL51 protein is essential for viral genome cleavage-packaging and interacts with the terminase subunits pUL56 and pUL89. J Virol 87:1720–1732. doi: 10.1128/JVI.01955-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mocarski ES, Shenk T, Griffiths PD, Pass RF. 2013. Cytomegaloviruses, p 1960–2014. In Knipe DM, Howley PM (ed), Fields virology, 6th ed Lippincott, Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 24.Hakki M, Chou S. 2011. The biology of cytomegalovirus drug resistance. Curr Opin Infect Dis 24:605–611. doi: 10.1097/QCO.0b013e32834cfb58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bogner E. 2002. Human cytomegalovirus terminase as a target for antiviral chemotherapy. Rev Med Virol 12:115–127. doi: 10.1002/rmv.344. [DOI] [PubMed] [Google Scholar]
- 26.Baines JD. 2011. Herpes simplex virus capsid assembly and DNA packaging: a present and future antiviral drug target. Trends Microbiol 19:606–613. doi: 10.1016/j.tim.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Pi F, Zhao Z, Chelikani V, Yoder K, Kvaratskhelia M, Guo P. 2016. Development of potent antiviral drugs inspired by viral hexameric DNA-packaging motors with revolving mechanism. J Virol 90:8036–8046. doi: 10.1128/JVI.00508-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoelben S, Arns W, Renders L, Hummel J, Muhlfeld A, Stangl M, Fischereder M, Gwinner W, Suwelack B, Witzke O, Durr M, Beelen DW, Michel D, Lischka P, Zimmermann H, Rubsamen-Schaeff H, Budde K. 2014. Preemptive treatment of cytomegalovirus infection in kidney transplant recipients with letermovir: results of a Phase 2a study. Transpl Int 27:77–86. doi: 10.1111/tri.12225. [DOI] [PubMed] [Google Scholar]
- 29.Chemaly RF, Ullmann AJ, Stoelben S, Richard MP, Bornhauser M, Groth C, Einsele H, Silverman M, Mullane KM, Brown J, Nowak H, Kolling K, Stobernack HP, Lischka P, Zimmermann H, Rubsamen-Schaeff H, Champlin RE, Ehninger G. 2014. Letermovir for cytomegalovirus prophylaxis in hematopoietic-cell transplantation. N Engl J Med 370:1781–1789. doi: 10.1056/NEJMoa1309533. [DOI] [PubMed] [Google Scholar]
- 30.Merck Sharp & Dohme. 19 October 2016. Merck announces pivotal phase 3 study of Letermovir, an investigational antiviral medicine for prevention of cytomegalovirus (CMV) infection in high-risk bone marrow transplant patients, met primary endpoint. Merck Sharp & Dohme, Kenilworth, NJ: http://www.mercknewsroom.com/news-release/corporate-news/merck-announces-pivotal-phase-3-study-letermovir-investigational-antivir. [Google Scholar]
- 31.Goldner T, Hewlett G, Ettischer N, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. 2011. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J Virol 85:10884–10893. doi: 10.1128/JVI.05265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldner T, Hempel C, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. 2014. Geno- and phenotypic characterization of human cytomegalovirus mutants selected in vitro after letermovir (AIC246) exposure. Antimicrob Agents Chemother 58:610–613. doi: 10.1128/AAC.01794-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chou S. 2015. Rapid in vitro evolution of human cytomegalovirus UL56 mutations that confer Letermovir resistance. Antimicrob Agents Chemother 59:6588–6593. doi: 10.1128/AAC.01623-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lischka P, Michel D, Zimmermann H. 2016. Characterization of cytomegalovirus breakthrough events in a phase 2 prophylaxis trial of Letermovir (AIC246, MK 8228). J Infect Dis 213:23–30. doi: 10.1093/infdis/jiv352. [DOI] [PubMed] [Google Scholar]
- 35.Lischka P, Hewlett G, Wunberg T, Baumeister J, Paulsen D, Goldner T, Ruebsamen-Schaeff H, Zimmermann H. 2010. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob Agents Chemother 54:1290–1297. doi: 10.1128/AAC.01596-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaul DR, Stoelben S, Cober E, Ojo T, Sandusky E, Lischka P, Zimmermann H, Rubsamen-Schaeff H. 2011. First report of successful treatment of multidrug-resistant cytomegalovirus disease with the novel anti-CMV compound AIC246. Am J Transplant 11:1079–1084. doi: 10.1111/j.1600-6143.2011.03530.x. [DOI] [PubMed] [Google Scholar]
- 37.Marschall M, Stamminger T, Urban A, Wildum S, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. 2012. In vitro evaluation of the activities of the novel anticytomegalovirus compound AIC246 (letermovir) against herpesviruses and other human pathogenic viruses. Antimicrob Agents Chemother 56:1135–1137. doi: 10.1128/AAC.05908-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A 100:12396–12401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruzsics Z, Borst EM, Bosse JB, Brune W, Messerle M. 2013. Manipulating CMV genomes by BAC mutagenesis: strategies and applications, p 38–58. In Reddehase MJ. (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol 1 Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 41.Glass M, Busche A, Wagner K, Messerle M, Borst EM. 2009. Conditional and reversible disruption of essential herpesvirus proteins. Nat Methods 6:577–579. doi: 10.1038/nmeth.1346. [DOI] [PubMed] [Google Scholar]
- 42.Wang JB, Zhu Y, McVoy MA, Parris DS. 2012. Changes in subcellular localization reveal interactions between human cytomegalovirus terminase subunits. Virol J 9:315. doi: 10.1186/1743-422X-9-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uetz P, Dong YA, Zeretzke C, Atzler C, Baiker A, Berger B, Rajagopala SV, Roupelieva M, Rose D, Fossum E, Haas J. 2006. Herpesviral protein networks and their interaction with the human proteome. Science 311:239–242. doi: 10.1126/science.1116804. [DOI] [PubMed] [Google Scholar]
- 44.Fossum E, Friedel CC, Rajagopala SV, Titz B, Baiker A, Schmidt T, Kraus T, Stellberger T, Rutenberg C, Suthram S, Bandyopadhyay S, Rose D, von Brunn A, Uhlmann M, Zeretzke C, Dong YA, Boulet H, Koegl M, Bailer SM, Koszinowski U, Ideker T, Uetz P, Zimmer R, Haas J. 2009. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog 5:e1000570. doi: 10.1371/journal.ppat.1000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vizoso Pinto MG, Pothineni VR, Haase R, Woidy M, Lotz-Havla AS, Gersting SW, Muntau AC, Haas J, Sommer M, Arvin AM, Baiker A. 2011. Varicella zoster virus ORF25 gene product: an essential hub protein linking encapsidation proteins and the nuclear egress complex. J Proteome Res 10:5374–5382. doi: 10.1021/pr200628s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elbasani E, Gabaev I, Steinbrück L, Messerle M, Borst EM. 2014. Analysis of essential viral gene functions after highly efficient adenofection of cells with cloned human cytomegalovirus genomes. Viruses 6:354–370. doi: 10.3390/v6010354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thoma C, Borst E, Messerle M, Rieger M, Hwang JS, Bogner E. 2006. Identification of the interaction domain of the small terminase subunit pUL89 with the large subunit pUL56 of human cytomegalovirus. Biochemistry 45:8855–8863. doi: 10.1021/bi0600796. [DOI] [PubMed] [Google Scholar]
- 48.Borst EM, Hahn G, Koszinowski UH, Messerle M. 1999. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: a new approach for construction of HCMV mutants. J Virol 73:8320–8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borst EM, Messerle M. 2000. Development of a cytomegalovirus vector for somatic gene therapy. Bone Marrow Transplant 25(Suppl 2):S80–S82. [DOI] [PubMed] [Google Scholar]
- 50.Tran K, Mahr JA, Spector DH. 2010. Proteasome subunits relocalize during human cytomegalovirus infection, and proteasome activity is necessary for efficient viral gene transcription. J Virol 84:3079–3093. doi: 10.1128/JVI.02236-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salomons FA, Menendez-Benito V, Bottcher C, McCray BA, Taylor JP, Dantuma NP. 2009. Selective accumulation of aggregation-prone proteasome substrates in response to proteotoxic stress. Mol Cell Biol 29:1774–1785. doi: 10.1128/MCB.01485-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giesen K, Radsak K, Bogner E. 2000. The potential terminase subunit of human cytomegalovirus, pUL56, is translocated into the nucleus by its own nuclear localization signal and interacts with importin alpha. J Gen Virol 81:2231–2244. doi: 10.1099/0022-1317-81-9-2231. [DOI] [PubMed] [Google Scholar]
- 53.Sankhala RS, Lokareddy RK, Cingolani G. 2016. Divergent evolution of nuclear localization signal sequences in herpesvirus terminase subunits. J Biol Chem 291:11420–11433. doi: 10.1074/jbc.M116.724393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang K, Baines JD. 2006. The putative terminase subunit of herpes simplex virus 1 encoded by UL28 is necessary and sufficient to mediate interaction between pUL15 and pUL33. J Virol 80:5733–5739. doi: 10.1128/JVI.00125-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacobson JG, Yang K, Baines JD, Homa FL. 2006. Linker insertion mutations in the herpes simplex virus type 1 UL28 gene: effects on UL28 interaction with UL15 and UL33 and identification of a second-site mutation in the UL15 gene that suppresses a lethal UL28 mutation. J Virol 80:12312–12323. doi: 10.1128/JVI.01766-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang K, Poon AP, Roizman B, Baines JD. 2008. Temperature-sensitive mutations in the putative herpes simplex virus type 1 terminase subunits pUL15 and pUL33 preclude viral DNA cleavage/packaging and interaction with pUL28 at the nonpermissive temperature. J Virol 82:487–494. doi: 10.1128/JVI.01875-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Asher G, Reuven N, Shaul Y. 2006. 20S proteasomes and protein degradation “by default”. Bioessays 28:844–849. doi: 10.1002/bies.20447. [DOI] [PubMed] [Google Scholar]
- 58.Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang EC, Aicheler R, Murrell I, Wilkinson GW, Lehner PJ, Gygi SP. 2014. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157:1460–1472. doi: 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chari A, Fischer U. 2010. Cellular strategies for the assembly of molecular machines. Trends Biochem Sci 35:676–683. doi: 10.1016/j.tibs.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 60.Bolin LL, Hanson LK, Slater JS, Kerry JA, Campbell AE. 2010. Murine cytomegalovirus US22 protein pM140 protects its binding partner, pM141, from proteasome-dependent but ubiquitin-independent degradation. J Virol 84:2164–2168. doi: 10.1128/JVI.01739-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clark E, Spector DH. 2015. Studies on the contribution of human cytomegalovirus UL21a and UL97 to viral growth and inactivation of the anaphase-promoting complex/cyclosome (APC/C) E3 ubiquitin ligase reveal a unique cellular mechanism for downmodulation of the APC/C subunits APC1, APC4, and APC5 J Virol 89:6928–6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Balchin D, Hayer-Hartl M, Hartl FU. 2016. In vivo aspects of protein folding and quality control. Science 353:aac4354. doi: 10.1126/science.aac4354. [DOI] [PubMed] [Google Scholar]
- 63.Hartl FU. 1996. Molecular chaperones in cellular protein folding. Nature 381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 64.Visalli RJ, Knepper J, Goshorn B, Vanover K, Burnside DM, Irven K, McGauley R, Visalli M. 2009. Characterization of the varicella-zoster virus ORF25 gene product: pORF25 interacts with multiple DNA encapsidation proteins. Virus Res 144:58–64. doi: 10.1016/j.virusres.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo P, Lee TJ. 2007. Viral nanomotors for packaging of dsDNA and dsRNA. Mol Microbiol 64:886–903. doi: 10.1111/j.1365-2958.2007.05706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guo P, Noji H, Yengo CM, Zhao Z, Grainge I. 2016. Biological nanomotors with a revolution, linear, or rotation motion mechanism. Microbiol Mol Biol Rev 80:161–186. doi: 10.1128/MMBR.00056-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Savva CG, Holzenburg A, Bogner E. 2004. Insights into the structure of human cytomegalovirus large terminase subunit pUL56. FEBS Lett 563:135–140. doi: 10.1016/S0014-5793(04)00283-2. [DOI] [PubMed] [Google Scholar]
- 68.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 69.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 70.Houston A, Williams JM, Rovis TL, Shanley DK, O'Riordan RT, Kiely PA, Ball M, Barry OP, Kelly J, Fanning A, MacSharry J, Mandelboim O, Singer BB, Jonjic S, Moore T. 2016. Pregnancy-specific glycoprotein expression in normal gastrointestinal tract and in tumors detected with novel monoclonal antibodies. MAbs 8:491–500. doi: 10.1080/19420862.2015.1134410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]