

ABSTRACT
African swine fever virus (ASFV) codes for a putative histone-like protein (pA104R) with extensive sequence homology to bacterial proteins that are implicated in genome replication and packaging. Functional characterization of purified recombinant pA104R revealed that it binds to single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA) over a wide range of temperatures, pH values, and salt concentrations and in an ATP-independent manner, with an estimated binding site size of about 14 to 16 nucleotides. Using site-directed mutagenesis, the arginine located in pA104R's DNA-binding domain, at position 69, was found to be relevant for efficient DNA-binding activity. Together, pA104R and ASFV topoisomerase II (pP1192R) display DNA-supercoiling activity, although none of the proteins by themselves do, indicating that the two cooperate in this process. In ASFV-infected cells, A104R transcripts were detected from 2 h postinfection (hpi) onward, reaching a maximum concentration around 16 hpi. pA104R was detected from 12 hpi onward, localizing with viral DNA replication sites and being found exclusively in the Triton-insoluble fraction. Small interfering RNA (siRNA) knockdown experiments revealed that pA104R plays a critical role in viral DNA replication and gene expression, with transfected cells showing lower viral progeny numbers (up to a reduction of 82.0%), lower copy numbers of viral genomes (−78.3%), and reduced transcription of a late viral gene (−47.6%). Taken together, our results strongly suggest that pA104R participates in the modulation of viral DNA topology, probably being involved in viral DNA replication, transcription, and packaging, emphasizing that ASFV mutants lacking the A104R gene could be used as a strategy to develop a vaccine against ASFV.
IMPORTANCE Recently reintroduced in Europe, African swine fever virus (ASFV) causes a fatal disease in domestic pigs, causing high economic losses in affected countries, as no vaccine or treatment is currently available. Remarkably, ASFV is the only known mammalian virus that putatively codes for a histone-like protein (pA104R) that shares extensive sequence homology with bacterial histone-like proteins. In this study, we characterized the DNA-binding properties of pA104R, analyzed the functional importance of two conserved residues, and showed that pA104R and ASFV topoisomerase II cooperate and display DNA-supercoiling activity. Moreover, pA104R is expressed during the late phase of infection and accumulates in viral DNA replication sites, and its downregulation revealed that pA104R is required for viral DNA replication and transcription. These results suggest that pA104R participates in the modulation of viral DNA topology and genome packaging, indicating that A104R deletion mutants may be a good strategy for vaccine development against ASFV.
KEYWORDS: African swine fever virus, DNA binding, histone-like protein, pA104R, viral replication, viral transcription
INTRODUCTION
African swine fever virus (ASFV) causes a highly lethal disease that is considered one of the most threatening diseases for pig husbandry, with no vaccine or antiviral treatment available yet. Originally endemic in sub-Saharan Africa, African swine fever disease was introduced in the Transcaucasian countries (Georgia, Armenia, and Azerbaijan) and in the Russian Federation (2007), where it is maintained (1). In the course of the last few years, new outbreaks of ASFV infection have occurred in Ukraine (2012, 2014, and 2015); Belarus (2013); and Lithuania, Estonia, Latvia, and Poland (2014 and 2015) (2).
ASFV infects domestic and wild suids, as well as soft ticks of the genus Ornithodoros, and can be transmitted either directly by contact with infected animals or indirectly via tick bites and through virus-contaminated feed or fomites (3). While warthogs and bushpigs are asymptomatic carriers of the disease, acting as reservoirs of infection, the clinical signs in domestic pigs and wild boars may vary from acute to chronic forms, depending on the virulence of the ASFV strain and on the immunological status of the host (3, 4).
ASFV is the only member of the family Asfarviridae sharing some functional and structural similarities with other large eukaryotic viruses (e.g., poxviruses and iridoviruses), all belonging to the nucleocytoplasmic large DNA virus (NCLDV) clade (5). Its double-stranded DNA genome ranges from 170 to 190 kbp and includes between 151 and 167 open reading frames (ORFs) (6), including ORF A104R. This gene codes for a putative histone-like protein (pA104R), highly conserved among all ASFV isolates, that shares a sequence identity of 25 to 30% with two families of bacterial histone-like proteins (HU and IHF) and is the only histone-like protein encoded by a eukaryotic virus (7, 8). In addition, pA104R possesses the signature of histone-like proteins at residues 57 to 76 of its sequence (PROSITE PS00045), including 8 amino acid residues responsible for DNA interaction and conserved in bacterial histone-like proteins. In prokaryotes, these nucleoid-associated proteins are involved in DNA supercoiling and also play an important role in DNA replication, repair, recombination, and transcription (9–12) by interacting with topoisomerases (13–16). Besides their structural and sequence similarities to eukaryotic histones, these bacterial proteins are often termed histones due to their DNA-binding properties, low molecular weights, and high bending capacities (17–19).
In ASFV, it was demonstrated that pA104R binds to double-stranded DNA (dsDNA)- and single-stranded DNA (ssDNA)-cellulose columns (8), and although it was anticipated that pA104R might be involved in DNA-related processes or, alternatively, act as a transcription factor modulating viral gene expression (7, 8), its activity and biological role remain to be characterized. Therefore, in this study, we aimed to assess pA104R DNA-binding activity using electrophoretic mobility shift assay (EMSA); to understand the functional importance of two conserved residues, both located within the DNA-binding motif, by using site-directed mutagenesis; to verify whether pA104R has DNA-supercoiling activity; and to measure the mRNA and the protein expression levels of A104R, as well as the pA104R distribution pattern, using ASFV-infected Vero cells. Finally, RNA interference (RNAi) assays were performed to clarify the role of pA104R in infection.
RESULTS
pA104R forms distinct DNA-protein complexes in the presence of oligonucleotides with different lengths.
Although previous studies reported that the ASFV ORF A104R-encoded protein has significant sequence homology with bacterial histone-like proteins and pA104R binds to both single- and double-stranded calf thymus DNA-cellulose columns (7, 8), the DNA-binding activity of the protein remains poorly understood.
In order to evaluate pA104R DNA-binding affinity and the DNA binding site size, oligonucleotides with different lengths (from 10 to 50 nucleotides [nt]) were used in EMSA. Although no protein-DNA complexes were detected when the purified pA104R was incubated with very short oligonucleotides (10 nt) (data not shown), the viral protein was able to bind oligonucleotides with lengths of 20 to 50 nt (Fig. 1), revealing that the minimum DNA length required to elicit binding is between 11 and 20 nt. Additionally, two or three distinct protein-DNA complexes were identified when pA104R was incubated with ssDNA probes with a length of 40 nt or 50 nt, respectively (Fig. 1, arrows), whereas a single protein-DNA complex was detected when the reaction mixtures contained oligonucleotides of 20 or 30 nt (Fig. 1, asterisks), suggesting that the minimal DNA length required for pA104R binding is about 14 to 16 nt.
FIG 1.

pA104R binds to DNA fragments with different lengths and shows a binding site size of about 14 to 16 nt. EMSA was performed using 2.5 μM recombinant purified pA104R and 1 pmol of single-stranded oligonucleotides with increasing lengths (20, 30, 40, and 50 nt). After 30 min of incubation at room temperature, the reaction mixtures were subjected to native polyacrylamide gel electrophoresis, and biotin-labeled DNA was detected using a streptavidin-HRP antibody. The asterisks and arrows indicate bands corresponding to pA104R-DNA complexes. The unidentified DNA bands represent oligonucleotides annealed during incubation.
pA104R binds both ssDNA and dsDNA at a wide range of temperatures, pH, and salt concentrations and in an ATP-independent manner.
To further characterize pA104R DNA-binding activity, increasing concentrations of protein (0.05, 0.25, 0.5, 1.25, 2.5, 3.75, and 5 μM) were incubated with 1 pmol of 5′-biotin-labeled 30-nucleotide-long ssDNA or dsDNA for 30 min at room temperature (RT). EMSA showed that pA104R binds both ssDNA and dsDNA, at minimum concentrations of 1.25 μM (Fig. 2A) and 0.5 μM (Fig. 2B), respectively, with the intensity of the free-DNA band decreasing with increasing concentrations of pA104R. Indeed, for protein concentrations above 0.5 μM, no free dsDNA was present, in contrast to the large amounts of ssDNA found even at 5 μM concentration, proving that pA104R has higher binding affinity to dsDNA than to ssDNA. A supershift assay was also performed to confirm the specific binding of pA104R to DNA. The addition of pA104R antibody to the binding reaction mixture resulted in the presence of a supershifted band with lower mobility than when the antibody was not added (Fig. 2C). Since the serum produced against purified pA104R specifically detected this protein by Western blotting (Fig. 2D), the observed supershifted band corresponded to pA104R antibody bound to the protein-ssDNA and protein-dsDNA complexes.
FIG 2.

pA104R binds to dsDNA with higher affinity than to ssDNA. (A and B) Reaction mixtures containing increasing concentrations of recombinant purified ASFV pA104R and 1 pmol of biotin-labeled 30-nt ssDNA (A) or biotin-labeled 30-nt dsDNA (B) were incubated for 30 min at room temperature, followed by resolution on a native polyacrylamide gel and detection using a streptavidin-HRP antibody. (C) A supershift assay was performed by adding an anti-pA104R antibody to the binding mixture 5 min after the beginning of the reaction. (D) Specific recognition of the serum used was tested against the purified pA104R by Western blotting.
Although the formation of protein-DNA complexes was easily detected after a short incubation time (1 min), a larger number of complexes were formed with longer incubation times (Fig. 3A). EMSAs also revealed that the DNA-binding activity of pA104R was affected by the NaCl concentration and was higher at 0.1, 0.25, and 0.5 M; residual at 0 M and 1 M; and absent at 2 M (Fig. 3B). Furthermore, pA104R was able to bind DNA at a wide range of temperatures (4 to 37°C) (Fig. 3C) and pH values (Fig. 3D), with similar affinities and in an ATP-independent fashion (Fig. 3E). The pA104R binding site size remained constant under all experimental conditions.
FIG 3.

pA104R exhibits high DNA-binding affinity at a wide range of temperatures and pH values in an ATP-independent manner and is affected by ionic strength. Binding reaction mixtures containing 2.5 μM recombinant purified pA104R and 1 pmol of biotin-labeled 30-nt ssDNA were incubated for 30 min in a binding buffer. The incubation periods (A), NaCl concentrations (B), temperatures (C), pH values (D), and ATP concentrations (E) varied as indicated.
The arginine-69 residue is required for the DNA-binding activity of pA104R.
Although arginine at position 69 and proline at position 74 of pA104R, both belonging to the DNA-binding domain, are conserved in all ASFV isolates and in bacterial histone-like proteins (20), their roles in pA104R's DNA-binding activity are unknown. In order to evaluate if these residues are involved in protein-DNA interactions, three point mutants were generated by site-directed mutagenesis: pA104RR69A, pA104RR69K, and pA104RP74A.
Our results showed that replacement of the positively charged arginine at position 69 (Arg69) by a nonpolar amino acid (alanine) (pA104RR69A) severely reduces DNA-binding activity (Fig. 4B) in comparison to the wild-type (wt) protein (pA104Rwt) (Fig. 4A). Interestingly, when Arg69 was replaced by another positively charged amino acid (lysine) (pA104RR69K), the DNA-binding activity was not affected (Fig. 4C), suggesting that a positively charged residue at this position is critical for protein-DNA interactions. Finally, replacement of the proline at position 74 by an alanine residue (pA104RP74A) did not significantly change the ability of pA104R to bind DNA (Fig. 4D).
FIG 4.

Arg69 is needed for efficient pA104R DNA-binding activity, in contrast to the Pro74 residue. Wild-type pA104R (pA104Rwt) (A) and point mutants at arginine residue 69 (pA104RR69A and pA104RR69K) (B and C) or at proline residue 74 (pA104RP74A) (D) were added at increasing concentrations (1.25, 2.5, 3.75, and 5 μM) to 1 pmol of biotin-labeled 30-nt ssDNA and incubated for 30 min at room temperature.
pA104R cooperates with ASFV topoisomerase II (pP1192R) to modulate DNA supercoiling.
Considering that in bacteria, histone-like proteins and topoisomerases work together in spatial genome organization (13–15, 21), and knowing that ASFV also codes for a type II DNA topoisomerase (pP1192R), we aimed to investigate whether this phenomenon is maintained in ASFV. For this purpose, increasing concentrations of pA104R (0.01, 0.05, 0.1, 0.25, and 0.5 μM) were incubated with 150 ng of relaxed pBR322 DNA, in the presence or absence of ASFV topoisomerase II (0.01 μM), for 30 min at 37°C in a reaction volume of 20 μl. When pA104R was incubated alone with the relaxed plasmid, no supercoiling activity was detected. However, supercoiled DNA molecules were observed when pA104R, at high concentrations (0.25 and 0.5 μM), was added to a mixture of pP1192R and relaxed pBR322 DNA (Fig. 5). A similar, yet less pronounced, result was obtained when pA104R was added prior to the addition of pP1192R (data not shown).
FIG 5.

pA104R has DNA-supercoiling activity in the presence of ASFV topoisomerase II (pP1192R). pA104R and/or ASFV topoisomerase II was incubated at the indicated concentrations with 150 ng of relaxed pBR322 DNA for 30 min at 37°C and subjected to agarose gel electrophoresis. ASFV topoisomerase II was added to the incubation mixtures 10 min prior to pA104R addition.
The A104R gene encodes a late protein that localizes with viral DNA replication sites.
Although low levels of A104R transcripts were detected from 2 h postinfection (hpi) onward, the transcription pattern of the gene closely resembles the transcriptional dynamics of the ASFV B646L late gene compared to the viral CP204L early gene. Indeed, the A104R gene is mainly transcribed from 8 hpi onward, showing a maximum peak at 16 hpi, and is transcribed at much lower levels than the above-mentioned genes encoding capsid proteins (Fig. 6A). The immunoblot analysis revealed that pA104R is expressed in ASFV-infected Vero cells from 12 hpi onward, reaching a maximum concentration peak at 18 to 20 hpi (Fig. 6B). pA104R was not detected in infected cells exposed to cytosine arabinoside (AraC), an inhibitor of viral DNA replication and consequently of late-phase transcription, further supporting the notion that pA104R is a late viral protein (Fig. 6B).
FIG 6.
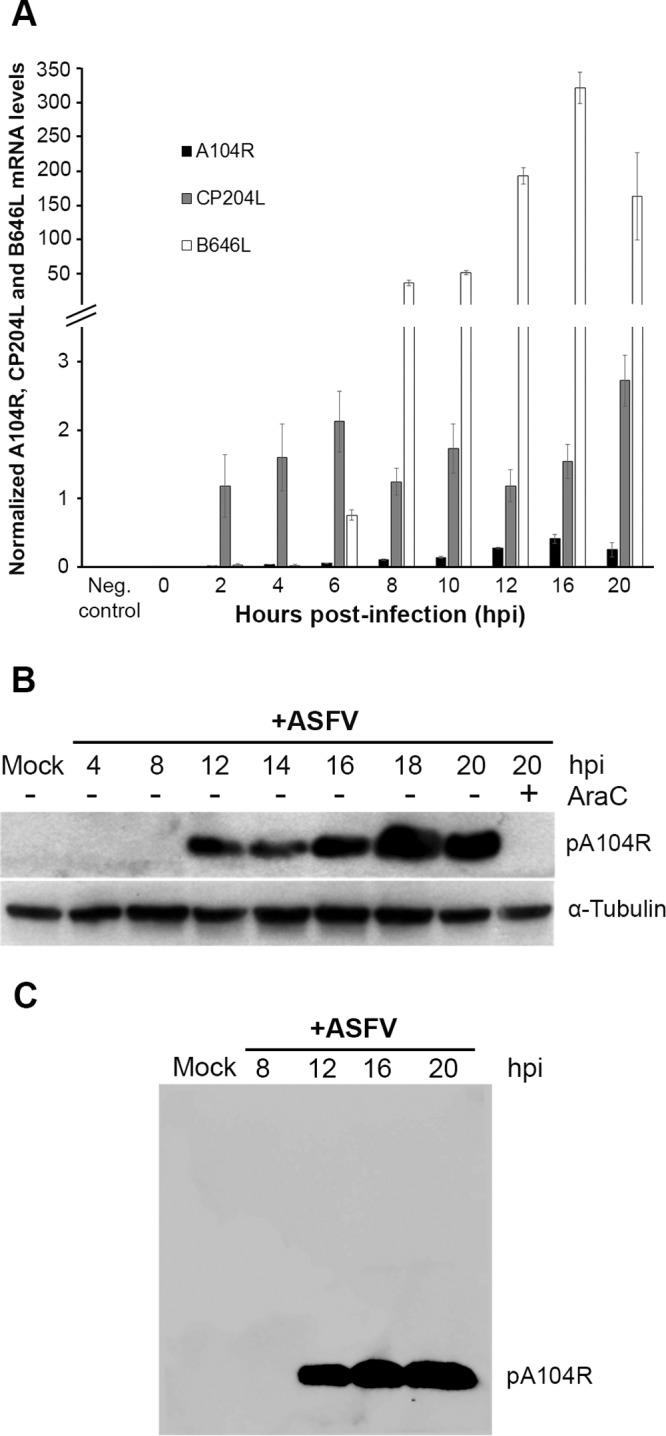
The A104R gene, encoding a late protein, is transcribed from 2 hpi. (A) A104R mRNA levels were measured by qRT-PCR at different time points after infection of Vero cells with the ASFV-Ba71V isolate (MOI = 1.5). Mock-infected cells were used as a control. CP204L (vp32) and B646L (vp72) mRNA levels were determined in parallel, as controls for early and late viral gene expression, respectively. The results are shown as averages ± standard errors (SE) between the number of molecules of each viral gene and the number of molecules of the cyclophilin A housekeeping gene. The results were obtained from two independent experiments run in duplicate. (B) Vero cells infected with the Ba71V isolate (MOI = 5) were harvested at the indicated time points. AraC (50 μg/ml) treatment was performed after the initial viral adsorption period (1 h), and cells collected at 20 hpi were lysed for the immunoblot assay. α-Tubulin was used as a loading control. (C) The specific recognition of the serum used was tested against whole infected-cell extracts by Western blotting.
Immunostaining studies revealed that pA104R shows a speckled nuclear distribution with nucleolar exclusion and accumulation in the cytoplasmic viral factories (Fig. 7A) from 12 hpi onward. The specificity of the serum used against whole infected-cell extracts is shown in Fig. 6C.
FIG 7.

pA104R localizes within cell nuclei and cytoplasmic viral factories and is bound to Triton X-100-insoluble components/structures. (A) Vero cells infected with the Ba71V isolate (MOI = 2) were fixed at 6, 12, and 16 hpi and analyzed by immunofluorescence for pA104R, ASFV, and DNA. In the merged images, pA104R, ASFV, and DAPI staining is shown in green, red, and blue, respectively. Representative images from at least two independent experiments are shown. (B) For the Triton X-100 fractionation procedure, Vero cells were resuspended in buffer containing 0.1% Tx and fractionated.
Taking into consideration the DNA-binding activity of pA104R and its subcellular localization in infected Vero cells, we aimed to evaluate if the viral protein is bound to the Triton X-100 (Tx)-insoluble elements (e.g., viral DNA, nuclear matrix, and cytoskeleton). The distribution of pA104R was investigated in Triton X-100 subfractions of whole infected-cell lysates harvested at 16 hpi. Remarkably, the polyclonal antibody raised against pA104R recognized only a 12-kDa band in the Triton X-100-insoluble fraction (Fig. 7B), indicating that pA104R is anchored to detergent-insoluble components/structures.
Knockdown of pA104R reduces viral infection.
To further explore the biological role of pA104R, Vero cells were transfected with two small interfering RNA (siRNA) sequences targeting A104R transcripts and then infected with the ASFV-Ba71V isolate. Taking into consideration that the siRNA-A104R_1 duplex did not reduce the A104R mRNA levels and that the siRNA-A104R_2 sequence leads to a reduction of around 27% at 16 hpi (Fig. 8), further siRNA studies were performed using this sequence.
FIG 8.

siRNA-A104R_2 reduces the mRNA levels of A104R by 27% at 16 hpi. Vero cells were transfected with siRNA-A104R_1 (50 nM) or siRNA-A104R_2 (50 nM) for 8 h and then infected with Ba71V (MOI = 0.1). A104R mRNA levels were quantified by qRT-PCR at 4, 8, and 16 hpi. Transfected noninfected cells were used as a negative control and nontransfected infected cells as a positive control. The results are shown as averages ± SE between the number of molecules of each viral gene and the number of molecules of the cyclophilin A housekeeping gene. The data were obtained from two independent experiments run in duplicate.
siRNA experiments revealed a lower number of viral progeny (−82.0%) (Fig. 9A), a lower number of viral genomes (−78.3%) (Fig. 9B), and reduced mRNA levels of a late ASFV gene that codes for the capsid protein p72 (B646L; −47.6%) (Fig. 9C) in A104R-depleted cells compared with the control cells (transfected with siRNA-GAPDH [glyceraldehyde-3-phosphate dehydrogenase]). No differences in mRNA levels of CP204L, an early viral gene, were found between the groups (Fig. 9C).
FIG 9.

A104R mRNA knockdown inhibits ASFV infection. (A) A reduction in viral yield (−82.0%) was detected in ASFV-infected Vero cells (MOI = 0.1) transfected with siRNA-A104R_2 (10 and 50 nM) in comparison to controls (nontransfected infected cells and GAPDH siRNA-transfected infected cells) at 72 hpi. The virus yield of each supernatant was calculated from the average of three independent experiments. The error bars represent standard errors of the mean values. (B) A104R-depleted cells showed a decreased number of ASFV genomes (−78.3%) compared to control cells at 72 hpi. The results were obtained from two independent experiments. (C) Decreased mRNA levels of B646L were observed in transfected infected cells (−47.6%, 16 hpi) compared with the nontransfected infected cells and GAPDH siRNA-transfected infected cells. CP204L mRNA levels were not altered. The results are shown as averages ± SE between the number of molecules of each viral gene and the number of molecules of the cyclophilin A housekeeping gene. The data were obtained from two independent experiments run in duplicate.
DISCUSSION
In this study, we found that purified recombinant pA104R binds to DNA, with higher affinity for dsDNA than for ssDNA, suggesting that at the cellular level, the protein is more capable of folding full-length ASFV genomes than intermediate single-stranded genomes. We also demonstrated that pA104R has a binding site size of around 14 to 16 nt and a minimal binding length of 11 to 20 nt, similar to those reported for other viral DNA-binding proteins (22, 23). The results obtained show that pA104R DNA-binding activity is stable at a wide range of temperatures (4°C to 37°C) and pH values (4 to 11), possibly to support ASFV replication in the soft tick vector (Ornithodoros spp.) and in swine. Indeed, ASFV replicates in the midgut epithelial cells of ticks at low pH values (24) and enters swine macrophages by a low-pH-dependent endocytic pathway (25). pA104R also binds DNA in an ATP-independent manner, suggesting that viral DNA packaging is a highly dynamic process, and a prompt DNA compaction may be critical to generate a high number of ASFV particles and to maintain a successful infection. pA104R-DNA interactions were not detected under high-ionic-strength conditions (≥2 M NaCl), indicating that pA104R-binding activity involves ion pair formation, as reported for other viral DNA-binding proteins (26–28).
Site-directed mutagenesis studies revealed that, as reported for other viral DNA-binding proteins (29–31), Arg69 is essential for the efficient DNA-binding activity of pA104R, probably due to its positive charge, since its replacement by the nonpolar amino acid alanine leads to reduction of pA104R activity (approximately 3-fold decrease) and its replacement by another positively charged amino acid (lysine) did not change the pA104R DNA-binding properties. In this study, a Pro74 residue could be replaced by a lysine without affecting the formation of pA104R-DNA complexes, indicating that van der Waals forces between Pro74 and nitrogenous bases are not essential for pA104R-DNA interactions, in contrast to previous findings for bacterial histone-like proteins (20, 32) and to in silico studies predicting that Pro74 is critical for pA104R's DNA-binding activity (7, 8).
Furthermore, we showed that pA104R has the ability to supercoil DNA in the presence of the recently characterized ASFV topoisomerase II (33–36), as reported for some bacterial histone-like proteins (13–16, 18, 37–39) and for some viral proteins involved in genome packaging (40–43). The results of the supercoiling assay suggest that pA104R may participate in ASFV genome packaging, a rate-limiting step for virus maturation. Indeed, ASFV has to package its large DNA genome (170 to 190 kbp) into particles that have a diameter of only about 80 nm (44), a very challenging event, since viral particles without DNA have been shown to be frequently released from infected cells by budding (45). Although the mechanism by which ASFV packages its genome into a preassembled capsid remains unknown, this study points to the putative role of pA104R in promoting a stable, organized, and compact nucleoid, which is further supported by the observation that pA104R is localized over the central nucleoid structure (8, 45).
In vitro results from cell cultures revealed that the A104R gene is mainly transcribed during the late phase of infection, showing higher mRNA levels at 16 hpi, although in much smaller amounts than two ASFV structural genes (CP204L and B646L), indicating that pA104R is more likely to be involved in fundamental viral processes (e.g., DNA replication and transcription) than in capsid formation. At the protein level, pA104R was detected from 12 hpi onward, and its expression was completely abrogated by AraC, confirming that pA104R is a late ASFV protein. The recruitment of pA104R to viral DNA replication sites (cytoplasmic factories) corroborates the results obtained with ASFV-infected macrophages (8) and strengthens the idea that the viral protein may participate in ASFV genome packaging. In parallel, its nuclear localization reinforces the concept that pA104R may also participate in the viral DNA replication that occurs inside this cellular compartment (46) and/or in the heterochromatization of the cell genome (47). The latter phenomenon could facilitate viral infection by silencing genes required to mount an immune response. Additionally, the nucleocytoplasmic distribution of pA104R is not mediated by the CRM1-dependent export pathway (data not shown). Finally, the partial depletion of A104R transcripts (−27% at 16 hpi) reduces viral progeny release (−82.0%), decreases the viral genome copy number (−78.3%), and represses the transcription of a late viral gene (B646; 47.6%) without interfering with the transcription of the early viral gene CP204L.
Taken together, our results demonstrate that pA104R plays a key role in ASFV infection, probably by modulating the topological state of viral DNA and DNA-dependent events (e.g., DNA replication and transcription), as well as in viral genome packaging, suggesting that an ASFV mutant lacking ORF A104R may be a good strategy for the development of a vaccine against ASF. Indeed, an ASFV A104R-defective mutant would be expected to invade host cells and to express a number of immediate-early gene products, providing antigens that could induce a protective immune response in pigs and producing noninfectious progeny that undergo only one round of replication (e.g., aberrant/immature ASFV particles without DNA). Taking this into consideration, this study also highlights the importance of the establishment of a new complementing cell line that expresses pA104R in order to isolate and propagate an ASFV mutant lacking ORF A104R obtained by homologous recombination.
MATERIALS AND METHODS
Cloning, expression, and purification of recombinant A104Rwt, A104RR69A, A104RR69K, and A104RP74A.
The complete ORF A104R, without the stop codon, was PCR amplified from Ba71V genomic DNA, using the A104R_Fw and A104R_Rev primers (Table 1), which contain NdeI and XhoI restriction endonuclease sites at their 5′ ends. The resulting PCR product was purified using the High Pure Viral Nucleic Acid kit (Roche) and then cloned in the vector pET24a (Novagen). The three single point mutants (R69A, P74A, and R69K) were generated using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies), following the manufacturer's instructions.
TABLE 1.
Primers used in the present study
| Target | Primer name | Sense sequence (5′–3′) | Orientation |
|---|---|---|---|
| ASFV-A104Ra | A104R_Fw | GGAATTCCATATGATGTCGACAAAAAAAAAGCCCACAATTA | Forward |
| ASFV-A104Ra | A104R_Rev | TCCGCTCGAGATTTAACATATCATGAACAGGTTTCAATGC | Reverse |
| ASFV-A104Ra | A104R_RT_Fwe | AGCGGCAGATACCCAGTTAA | Forward |
| ASFV-A104Ra | A104R_RT_Fw | ACCCGGAATCAAGTTCACCG | Forward |
| ASFV-A104Ra | A104R_RT_Rev | CGGCTTTATGTTCAGGCTTGG | Reverse |
| Cyclophilin A | Cyclo_Fw | AGACAAGGTTCCAAAGACAGCAG | Forward |
| Cyclophilin A | Cyclo_Rev | AGACTGAGTGGTTGGATGGCA | Reverse |
| Cyclophilin A | Cyclo_Fwe | TGCCATCCAACCACTCAGTCT | Forward |
| ASFV-B646La | VP72_Fw | ACGGCGCCCTCTAAAGGT | Forward |
| ASFV-B646La | VP72_Rev | CATGGTCAGCTTCAAACGTTTC | Reverse |
| ASFV-CP204La | VP32_Fw | TGCACATCCTCCTTTGAAACAT | Forward |
| ASFV-CP204La | VP32_Rev | TCTTTTGTGCAAGCATATACAGCTT | Reverse |
The primers were designed based on the full genome sequence of the ASFV-Ba71V isolate (GenBank/EBML accession number ASU18466).
For expression of the recombinant proteins, the Escherichia coli strain BL21(DE3)-pLysS (Novagen) was transformed with either a pET24a/A104R_6×His wild-type or single-point-mutated A104R version and grown in LB medium (10 g tryptone, 5 g Select yeast extract, 5 g NaCl, pH 7.2) supplemented with kanamycin (30 μg/ml) plus chloramphenicol (34 μg/ml) at 37°C with shaking at 200 rpm until the optical density at 600 nm (OD600) reached 0.1 to 0.2, measured with a Nanodrop 2000 (Thermo Scientific). Induction of protein expression was carried out by adding isopropyl-β-d-1-thiogalactopyranoside (IPTG) at a final concentration of 1 mM. Five hours after induction, bacterial cells were harvested by centrifugation (10,000 × g for 10 min; 4°C) and washed with sterile water. The pellet was resuspended in binding buffer (20 mM sodium phosphate, 500 mM NaCl, and 20 mM imidazole, pH 7.4). and the cells were lysed by addition of a lysis solution (0.2 mg/ml lysozyme, 20 μg/ml DNase, and 1 mM phenylmethylsulfonyl fluoride [PMSF]) and sonicated 5 times for 5 min each time on ice (5 cycles; 70% amplitude). Then, the lysates were centrifuged at 3,000 × g for 15 min, and the pellets were discarded. The extracts were then filtered through a 0.45-μm syringe filter (Rotilabo; Carl Roth) and incubated with Ni Sepharose 6 Fast Flow slurry (GE Healthcare) for 1 h, according to the manufacturer's instructions. The mixture was loaded onto a PD-10 column (GE Healthcare) and washed with binding buffer solution (20 mM sodium phosphate, 500 mM NaCl, pH 7.4) containing increasing concentrations of imidazole (40, 60, and 80 mM), and the recombinant protein was eluted with elution buffer (20 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole, pH 7.4). The fractions were collected in low-binding tubes and analyzed by SDS-PAGE, and the recombinant pA104R, purified under native conditions, was stored at −80°C until further use.
EMSA.
Biotin-labeled ssDNA molecules with different lengths (10, 20, 30, 40, and 50 nt) were synthetized by Stab Vida, Lda (Lisbon, Portugal), and the double-stranded oligonucleotides were obtained by hybridization with complementary unlabeled molecules as described by Loregian et al. (22). Briefly, equal amounts of complementary strands were mixed in annealing buffer (10 mM Tris, pH 7.8, 50 mM NaCl, and 1 mM EDTA), denatured at 95°C for 5 min, and annealed by gradual cooling to RT. The oligonucleotides (1 pmol) were then incubated with either purified wild-type pA104R or pA104R point mutants (pA104RR69A, pA104RR69K, or pA104RP74A), at different concentrations, in an EMSA buffer (20 μl) containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 2 mM EDTA, 5% (vol/vol) glycerol for 30 min at RT. After the addition of 5× loading buffer, the reaction products were subjected to SDS-PAGE electrophoresis (8 to 16%) (Bio-Rad) in a 0.5× Tris-borate-EDTA (TBE) buffer (Sigma-Aldrich) and electrophoretically transferred onto a positively charged nylon membrane (Amersham Hybond-N+; GE Healthcare). The DNA was cross-linked to the membrane with a UVC 500 linker UV chamber (700 mJ/cm2; 15 min; Hoefer). Then, the membranes were blocked overnight at 4°C with phosphate-buffered saline (PBS) plus 0.1% (vol/vol) Tween 20 (PBST) (Sigma-Aldrich) containing 5% (wt/vol) bovine serum albumin (BSA) (Sigma-Aldrich), followed by incubation with horseradish peroxidase (HRP)-conjugated streptavidin antibody (RPN1231; GE Healthcare) for 1 h at RT. After five wash steps with PBST (10 min each), chemiluminescence detection was performed with a Clarity Western ECL Substrate detection kit (Bio-Rad) according to the manufacturer's instructions, using Amersham hyperfilms (GE Healthcare).
Supercoiling assay.
The DNA-supercoiling activity of pA104R was assayed by monitoring the conversion of relaxed pBR322 DNA (TopoGen) to its supercoiled form, both in the presence and absence of ASFV DNA type II topoisomerase (pP1192R), purified as described by Coelho et al. (33). In order to perform the assay, relaxed pBR322 plasmid (150 ng) was incubated with increasing amounts of pA104R and, where indicated, in the presence of pP1192R (0.01 μM) at 37°C, using a reaction buffer (50 mM Tris, pH 7.5, 75 mM NaCl, 6 mM MgCl2, 1 mM dithiothreitol [DTT], 2 mM ATP) previously optimized for pP1192R activity assays (33) and confirmed to be suitable for pA104R-binding activity, in a final volume of 20 μl. Reactions were stopped by the addition of 5 μl of a stop solution containing 5% (vol/vol) SDS, 0.003% (wt/vol) bromophenol blue, and 25% (vol/vol) glycerol. The reaction mixtures were subjected to electrophoresis in 1% agarose gels (0.5× TBE), and the DNA was stained with ethidium bromide (0.5 μg/ml) and imaged under UV light.
Cells and viruses.
Vero E6 cells (kidney epithelial cells from the African green monkey, Chlorocebus aethiops) were obtained from the European Cell Culture Collection (ECACC) and maintained in Dulbecco modified Eagle's minimal essential medium (DMEM) supplemented with l-GlutaMAX, 10% heat-inactivated fetal bovine serum (FBS), 1× nonessential amino acids, and 2 mM l-glutamine (all from Gibco, Life Technologies). All cell cultures were grown at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
The Vero cell-adapted ASFV-Ba71V isolate was propagated as described previously (48), and infections were carried out at the indicated multiplicities of infection (MOI). At the end of the adsorption period (1 h), the inoculum was removed and the cells were washed twice with serum-free medium. Viral titers were determined by 50% tissue culture infectious dose (TCID50) titration using Vero E6 cells and the Spearman-Kärber method (49).
qRT-PCR.
Total RNA was extracted from ASFV-infected Vero cells (MOI = 1.5) at different time points of infection, using an RNeasy minikit and an RNase-free DNase set (both from Qiagen) and following the manufacturer's protocols. First-strand cDNA was synthesized from 2 μg total RNA using the Superscript II First Strand Synthesis System (Invitrogen) and analyzed in duplicate by quantitative reverse transcription-PCR (qRT-PCR). The real-time PCR mixtures contained 1 μl (1:20) of template cDNA, 2.5 μl of forward and reverse primers (at 50 nM) (Table 1), 12.5 μl of Maxima SYBR green/ROX qPCR master mix (Thermo Scientific), and sterile water to a final volume of 25 μl per tube. The thermal-cycling conditions were initial denaturation at 95°C for 10 min and 40 cycles of 95°C for 10 s and 60°C for 60 s, followed by storage at 4°C for further use. qRT-PCR analysis was performed in a 7300 real-time PCR system (Applied Biosystems), and the mRNA levels of viral genes (A104R, CP204L, and B646L genes) and of the reference gene (cyclophilin A gene) were quantified using the standard curves of different plasmids (pGEM-Teasy_A104R, pGEM-Teasy_CP204L, pGEM-Teasy_B646L, and pGEM-Teasy_Cyclophilin A). Only data from qRT-PCRs showing an amplification efficiency of ≥0.92 and an R2 value of ≥0.98 were considered in the analysis.
Quantification of ASFV genomes by qPCR.
Viral DNA was extracted from Ba71V-infected Vero cells (MOI = 0.1) transfected with siRNA targeting A104R or GAPDH (control group), at 72 hpi, using a High Pure Viral Nucleic Acid kit (Roche). The number of viral genomes was determined by quantitative PCR as described by King et al. (50).
Antibodies.
pA104R labeling was performed with a mouse polyclonal antiserum raised against pA104R using the purified recombinant protein (1:100). Young male mice (BALB/c; 4 to 6 weeks old) were injected subcutaneously with 100 μg of pA104R, purified as described above and combined with Freund's complete adjuvant. Following the primary injection, two booster injections were administered at 2-week intervals. Total blood specimens were collected 10 days after the final booster injection, and the sera were aliquoted and stored at −20°C until they were used for immunoblotting and immunofluorescence studies. The specificity of the polyclonal antiserum was tested against purified recombinant ASFV-pA104R and whole infected-cell extracts. The immunostaining of ASFV-infected cells was performed by incubation with an in-house anti-ASFV polyclonal antibody (1:100) produced in swine. Two secondary fluorescence-conjugated antibodies were used as follows: anti-mouse fluorescein isothiocyanate (FITC) (1:300; sc-2099; Santa Cruz Biotechnology) and anti-swine Texas Red (1:500; ab6775; Abcam).
For immunoblot analysis, three primary antibodies (anti-pA104R, 1:100; anti-α-tubulin, 1:1,250 [number 2125; Cell Signaling Technology]; anti-β-actin, 1:200 [SC-69879; Santa Cruz Biotechnology) and two HRP-conjugated secondary antibodies (anti-rabbit IgG, 1:20,000 [4010-05], and anti-mouse IgG, 1:32,500 [1010-05; both from Southern Biotech]) were used. All antibody dilutions were performed in blocking solution and incubated according to the manufacturers' recommendations.
Protein extraction and Western blotting.
Vero cells grown in 6-well plates (5.6 × 104 cells/cm2) were infected with the ASFV-Ba71V isolate (MOI = 5) and, when indicated, exposed to 50 μg/ml AraC (Sigma-Aldrich) after the adsorption period (1 h). Before sampling, mock-infected, infected, and AraC-treated infected cells were washed twice with PBS and lysed in ice-cold modified RIPA buffer (25 mM Tris, pH 8.2, 150 mM NaCl, 0.5% [vol/vol] NP-40, 0.5% [wt/vol] sodium deoxycholate, 0.1% [wt/vol] SDS) supplemented with protease inhibitor cocktail (Complete, Mini, EDTA free; Roche) and phosphatase inhibitor cocktail (PhosStop; Roche). Clarified whole-cell lysates harvested at 4, 8, 12, 14, 16, 18, and 20 hpi were further subjected to SDS-PAGE, using 8 to 16% (wt/vol) polyacrylamide separating gels, and transferred to a 0.2-μm-pore-diameter nitrocellulose membrane (Whatman; Schleider & Schuell) by electroblotting.
For Tx solubility analysis, after the wash step with PBS, a buffer containing 50 mM HEPES (pH 7.6), 100 mM NaCl, 2 mM EDTA, 250 mM sucrose, 0.1% Tx supplemented with protease (Complete, Mini, EDTA free; Roche) and phosphatase (PhosStop, Roche) inhibitors was added to the Vero cells. ASFV-infected and mock-infected cells were collected at 16 hpi, and after centrifugation (10,000 × g for 10 min at 4°C), the pellet containing Tx-insoluble proteins was lysed in RIPA buffer. Then, both Tx-soluble (supernatant) and Tx-insoluble fractions were analyzed in SDS-polyacrylamide (8 to 16% [wt/vol]) gel electrophoresis.
The blot membranes were blocked with PBST containing 5% (wt/vol) BSA (Sigma-Aldrich) for 1 h at RT and further incubated with specific primary antibodies (1 h at RT). Then, the secondary antibodies conjugated with HRP were also incubated for 1 h at RT. All the antibody incubations were followed by three 10-min wash steps with PBST, and protein detection was performed using a chemiluminescence detection kit (Pierce ECL Western blotting substrate; Thermo Scientific) on Amersham Hyperfilm ECL (GE Healthcare). α-Tubulin and β-actin were used as loading controls.
Immunofluorescence studies.
At the indicated time points, ASFV-infected Vero cells (MOI = 2) growing on glass coverslips in 24-well plates (5.0 × 104 cells/cm2) were fixed with 3.7% (wt/vol) paraformaldehyde in HPEM buffer {25 mM HEPES, 60 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], 10 mM EGTA, 1 mM MgCl2} for 10 min and permeabilized with PBS–Triton X-100 (0.2%, [vol/vol]) for 5 min at RT. The cells were then washed with PBS, blocked with PBST and BSA (1% [wt/vol]) for 30 min, and incubated with primary antibodies for 1 h. All procedures were performed at RT, and all antibody incubations were performed in a dark humidified chamber to prevent fluorochrome bleaching. Vectashield mounting medium with DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratories) was used to visualize the nucleus and the viral factories. Fluorescence images were acquired using a Leica DMIRE2 epifluorescence microscope equipped with a 40× objective and analyzed with ImageJ open-source software (version IJ 1.48g; National Institutes of Health, Bethesda, MD, USA) and Adobe Photoshop CS5 software (Adobe Systems, Inc.).
ASFV-A104R downregulation by siRNA.
Two siRNAs targeting A104R transcripts (Table 2) and a siRNA duplex targeting GAPDH (siRNA-GAPDH) (Silencer GAPDH siRNA Control; number 4605) were purchased from Ambion (Applied Biosystems). siRNA-A104R was designed based on the full genome of the ASFV-Ba71V isolate (GenBank/EBML accession number U18466.2). The transfection mixtures were prepared by adding siRNAs to Opti-MEM medium (Gibco, Life Technologies) and to HiPerFect transfection reagent (Qiagen), following the manufacturer's instructions. Before addition to the cell cultures, the mixtures were homogenized by pipetting and incubated for 30 min at RT to allow the formation of transfection complexes. Vero cells grown in 24-well plates (2.0 × 104 cells/cm2) were transfected with siRNAs at 10 and 50 nM. After 8 h, the transfection complexes were removed and replaced with fresh DMEM supplemented with 10% FBS before infection (MOI = 0.1). Infection was allowed to proceed for 72 h. The efficacies of the siRNAs were evaluated by qRT-PCR, comparing the A104R mRNA levels between mock-transfected cells and cultures transfected with siRNA-A104R_1 or siRNA-A104R_2. The antiviral effects of siRNA-A104R_2 were evaluated by quantifying the ASFV genomes and the cytopathic effect (CPE) and by virus yields at 72 hpi. In addition, the mRNA levels of an early and a late viral gene (CP204L and B646L genes) were determined by qRT-PCR and compared with those of nontransfected infected cells at 8 and 16 hpi.
TABLE 2.
Sequences of siRNAs used to knock down expression of pA104R in ASFV-infected Vero cells
| Targeta | Primer designation | Sequence (5′–3′) |
|---|---|---|
| ASFV-A104R | siRNA-A104R_1 | ACAUAAAGCCGUAAAGAUAUUUU |
| ASFV-A104R | siRNA-A104R_2 | GCAGAUACCCAGUUAAAUAUUUU |
The primers were designed based on the full genome sequence of the ASFV-Ba71V isolate (GenBank/EBML accession number ASU18466).
ACKNOWLEDGMENTS
This work was supported by the Fundação para a Ciência e a Tecnologia (CIISA-UID/CVT/00276/2013) and by the European Union's Seventh Framework Programme (FP7/2007-2013, 311931, ASFORCE). G.F. and F.B.F. were supported by doctoral scholarships from the Fundação para a Ciência e a Tecnologia (SFRH/BD/89426/2012, SFRH/BD/104261/2014).
REFERENCES
- 1.Sánchez-Vizcaíno JM, Mur L, Martínez-López B. 2013. African swine fever (ASF): five years around Europe. Vet Microbiol 165:45–50. doi: 10.1016/j.vetmic.2012.11.030. [DOI] [PubMed] [Google Scholar]
- 2.Gallardo MC, de la Reoyo A, Fernández-Pinero TJ, Iglesias I, Muñoz MJ, Arias ML. 2015. African swine fever: a global view of the current challenge. Porcine Health Manag 1:21. doi: 10.1186/s40813-015-0013-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costard S, Mur L, Lubroth J, Sanchez-Vizcaino JM, Pfeiffer DU. 2013. Epidemiology of African swine fever virus. Virus Res 173:191–197. doi: 10.1016/j.virusres.2012.10.030. [DOI] [PubMed] [Google Scholar]
- 4.Blome S, Gabriel C, Beer M. 2013. Pathogenesis of African swine fever in domestic pigs and European wild boar. Virus Res 173:122–130. doi: 10.1016/j.virusres.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 5.King AMQ, Adams MJ, Carsten EB, Lefkowitz EJ. 2012. Virus taxonomy: classification and nomenclature of viruses. Ninth Report of the International Committee on Taxonomy of Viruses, p 153–162. Elsevier Inc., Philadelphia, PA. [Google Scholar]
- 6.Dixon LK, Chapman DAG, Netherton CL, Upton C. 2013. African swine fever virus replication and genomics. Virus Res 173:3–14. doi: 10.1016/j.virusres.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 7.Neilan JG, Lu Z, Kutish GF, Sussman MD, Roberts PC, Yozawa T, Rock DL. 1993. An African swine fever virus gene with similarity to bacterial DNA binding proteins, bacterial integration host factors, and the Bacillus phage SPO1 transcription factor, TF1. Nucleic Acids Res 21:1496. doi: 10.1093/nar/21.6.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borca MV, Irusta PM, Kutish GF, Carillo C, Afonso CL, Burrage AT, Neilan JG, Rock DL. 1996. A structural DNA binding protein of African swine fever virus with similarity to bacterial histone-like proteins. Arch Virol 141:301–313. doi: 10.1007/BF01718401. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto M, Imhoff B, Ali MM, Kow YW. 2003. HU protein of Escherichia coli has a role in the repair of closely opposed lesions in DNA. J Biol Chem 278:28501–28507. doi: 10.1074/jbc.M303970200. [DOI] [PubMed] [Google Scholar]
- 10.Kar S, Edgar R, Adhya S. 2005. Nucleoid remodeling by an altered HU protein: reorganization of the transcription program. Proc Natl Acad Sci U S A 102:16397–16402. doi: 10.1073/pnas.0508032102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oberto J, Nabti S, Jooste V, Mignot H, Rouviere-Yaniv J. 2009. The HU regulon is composed of genes responding to anaerobiosis, acid stress, high osmolarity and SOS induction. PLoS One 4:e4367. doi: 10.1371/journal.pone.0004367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koli P, Sudan S, Fitzgerald D, Adhya S, Kar S. 2011. Conversion of commensal Escherichia coli K-12 to an invasive form via expression of a mutant histone-like protein. mBio 2:e00182-11. doi: 10.1128/mBio.00182-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bensaid A, Almeida A, Drlica K, Rouviere-Yaniv J. 1996. Cross-talk between topoisomerase I and HU in Escherichia coli. J Mol Biol 256:292–300. doi: 10.1006/jmbi.1996.0086. [DOI] [PubMed] [Google Scholar]
- 14.Malik M, Bensaid A, Rouviere-Yaniv J, Drlica K. 1996. Histone-like protein HU and bacterial DNA topology: suppression of an HU deficiency by gyrase mutations. J Mol Biol 256:66–76. doi: 10.1006/jmbi.1996.0068. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh S, Mallick B, Nagaraja V. 2014. Direct regulation of topoisomerase activity by a nucleoid-associated protein. Nucleic Acids Res 42:11156–11165. doi: 10.1093/nar/gku804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang JC. 2002. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 17.Rouvière-Yaniv J, Gros F. 1975. Characterization of a novel, low-molecular-weight DNA-binding protein from Escherichia coli. Proc Natl Acad Sci U S A 72:3428–3432. doi: 10.1073/pnas.72.9.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luijsterburg MS, Noom MC, Wuite GJL, Dame RT. 2006. The architectural role of nucleoid-associated proteins in the organization of bacterial chromatin: a molecular perspective. J Struct Biol 156:262–272. doi: 10.1016/j.jsb.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Grove A. 2011. Functional evolution of bacterial histone-like HU proteins. Curr Issues Mol Biol 13:1–12. [PubMed] [Google Scholar]
- 20.Luscombe NM, Thornton JM. 2002. Protein-DNA interactions: amino acid conservation and the effects of mutations on binding specificity. J Mol Biol 320:991–1009. doi: 10.1016/S0022-2836(02)00571-5. [DOI] [PubMed] [Google Scholar]
- 21.Macvanin M, Adhya S. 2012. Architectural organization in E. coli nucleoid. Biochim Biophys Acta 1819:830–835. doi: 10.1016/j.bbagrm.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loregian A, Sinigalia E, Mercorelli B, Palù G, Coen DM. 2007. Binding parameters and thermodynamics of the interaction of the human cytomegalovirus DNA polymerase accessory protein, UL44, with DNA: implications for the processivity mechanism. Nucleic Acids Res 35:4779–4791. doi: 10.1093/nar/gkm506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rochester SC, Traktman P. 1998. Characterization of the single-stranded DNA binding protein encoded by the vaccinia virus I3 gene. J Virol 72:2917–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sojka D, Franta Z, Horn M, Caffrey CR, Mareš M, Kopáček P. 2013. New insights into the machinery of blood digestion by ticks. Trends Parasitol 29:276–285. doi: 10.1016/j.pt.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Alonso C, Galindo I, Cuesta-Geijo MA, Cabezas M, Hernaez B, Muñoz-Moreno R. 2013. African swine fever virus-cell interactions: from virus entry to cell survival. Virus Res 173:42–57. doi: 10.1016/j.virusres.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komazin-Meredith G, Santos WL, Filman DJ, Hogle JM, Verdine GL, Coen DM. 2008. The positively charged surface of herpes simplex virus UL42 mediates DNA binding. J Biol Chem 283:6154–6161. doi: 10.1074/jbc.M708691200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang C, Komazin-Meredith G, Tian W, Coen DM, Hwang CB. 2009. Mutations that increase DNA binding by the processivity factor of herpes simplex virus affect virus production and DNA replication fidelity. J Virol 83:7573–7580. doi: 10.1128/JVI.00193-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Wu A, Wang YE, Quanquin N, Li C, Wang J, Chen H-W, Liu S, Liu P, Zhang H, Qin FX-F, Jiang T, Cheng G. 2015. Functional genomics reveals linkers critical for influenza polymerase. J Virol 90:2938–2947. doi: 10.1128/JVI.02400-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang PJ, Miller G. 2004. Autoregulation of DNA binding and protein stability of Kaposi's sarcoma-associated herpesvirus ORF50 protein. J Virol 78:10657–10673. doi: 10.1128/JVI.78.19.10657-10673.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thain A, Webster K, Emery D, Clarke AR, Gaston K. 1997. DNA binding and bending by the human papillomavirus type 16 E2 protein. Recognition of an extended binding site. J Biol Chem 272:8236–8242. [DOI] [PubMed] [Google Scholar]
- 31.Paquet F, Delalande O, Goffinont S, Culard F, Loth K, Asseline U, Castaing B, Landon C. 2014. Model of a DNA-protein complex of the architectural monomeric protein MC1 from Euryarchaea. PLoS One 9:e88809. doi: 10.1371/journal.pone.0088809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luscombe NM, Laskowski R, Thornton JM. 2001. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res 29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coelho J, Ferreira F, Martins C, Leitão A. 2016. Functional characterization and inhibition of the type II DNA topoisomerase coded by African swine fever virus. Virology 493:209–216. doi: 10.1016/j.virol.2016.03.023. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Beato R, Freije JM, Lopez-Otin C, Blasco R, Vinuela E, Salas ML. 1992. A gene homologous to topoisomerase II in African swine fever virus. Virology 188:938–947. doi: 10.1016/0042-6822(92)90558-7. [DOI] [PubMed] [Google Scholar]
- 35.Coelho J, Martins C, Ferreira F, Leitão A. 2015. African swine fever virus ORF P1192R codes for a functional type II DNA topoisomerase. Virology 474:82–93. doi: 10.1016/j.virol.2014.10.034. [DOI] [PubMed] [Google Scholar]
- 36.Freitas FB, Frouco G, Martins C, Leitão A, Ferreira F. 2016. In vitro inhibition of African swine fever virus-topoisomerase II disrupts viral replication. Antiviral Res 134:34–41. doi: 10.1016/j.antiviral.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 37.Kar S, Choi EJ, Guo F, Dimitriadis EK, Kotova SL, Adhya S. 2006. Right-handed DNA supercoiling by an octameric form of histone-like protein HU: modulation of cellular transcription. J Biol Chem 281:40144–40153. doi: 10.1074/jbc.M605576200. [DOI] [PubMed] [Google Scholar]
- 38.Swinger KK, Rice PA. 2004. IHF and HU: flexible architects of bent DNA. Curr Opin Struct Biol 14:28–35. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 39.Rimsky S, Travers A. 2011. Pervasive regulation of nucleoid structure and function by nucleoid-associated proteins. Curr Opin Microbiol 14:136–141. doi: 10.1016/j.mib.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Bogner E, Radsak K, Stinski MF. 1998. The gene product of human cytomegalovirus open reading frame UL56 binds the pac motif and has specific nuclease activity. J Virol 72:2259–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borst EM, Kleine-Albers J, Gabaev I, Babic M, Wagner K, Binz A, Degenhardt I, Kalesse M, Jonjic S, Bauerfeind R, Messerle M. 2013. The human cytomegalovirus UL51 protein is essential for viral genome cleavage-packaging and interacts with the terminase subunits pUL56 and pUL89. J Virol 87:1720–1732. doi: 10.1128/JVI.01955-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ostapchuk P, Yang J, Auffarth E, Hearing P. 2005. Functional interaction of the adenovirus IVa2 protein with adenovirus type 5 packaging sequences. J Virol 79:2831–2838. doi: 10.1128/JVI.79.5.2831-2838.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thoma C, Borst E, Messerle M, Rieger M, Hwang JS, Bogner E. 2006. Identification of the interaction domain of the small terminase subunit pUL89 with the large subunit pUL56 of human cytomegalovirus. Biochemistry 45:8855–8863. doi: 10.1021/bi0600796. [DOI] [PubMed] [Google Scholar]
- 44.Andrés G, Simón-Mateo C, Viñuela E. 1997. Assembly of African swine fever virus: role of polyprotein pp220. J Virol 71:2331–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salas ML, Andrés G. 2013. African swine fever virus morphogenesis. Virus Res 173:29–41. doi: 10.1016/j.virusres.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 46.Simões M, Martins C, Ferreira F. 2015. Early intranuclear replication of African swine fever virus genome modifies the landscape of the host cell nucleus. Virus Res 210:1–7. doi: 10.1016/j.virusres.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 47.Simões M, Rino J, Pinheiro I, Martins C, Ferreira F. 2015. Alterations of nuclear architecture and epigenetic signatures during African swine fever virus infection. Viruses 7:4978–4996. doi: 10.3390/v7092858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carrascosa AL, Bustos MJ, de Leon P. 2011. Methods for growing and titrating African swine fever virus: field and laboratory samples. Curr Protoc Cell Biol Chapter 53:Unit 26.14. doi: 10.1002/0471143030.cb2614s53. [DOI] [PubMed] [Google Scholar]
- 49.Kärber G. 1931. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn-Schmiedebergs Arch Exp Pathol Pharmakol 162:480–483. doi: 10.1007/BF01863914. [DOI] [Google Scholar]
- 50.King DP, Reid SM, Hutchings GH, Grierson SS, Wilkinson PJ, Dixon LK, Bastos ADS, Drew TW. 2003. Development of a TaqMan PCR assay with internal amplification control for the detection of African swine fever virus. J Virol Methods 107:53–61. doi: 10.1016/S0166-0934(02)00189-1. [DOI] [PubMed] [Google Scholar]