

ABSTRACT
Upon herpes simplex virus 1 (HSV-1) infection, the CD98 heavy chain (CD98hc) is redistributed around the nuclear membrane (NM), where it promotes viral de-envelopment during the nuclear egress of nucleocapsids. In this study, we attempted to identify the factor(s) involved in CD98hc accumulation and demonstrated the following: (i) the null mutation of HSV-1 UL34 caused specific dispersion throughout the cytoplasm of CD98hc and the HSV-1 de-envelopment regulators, glycoproteins B and H (gB and gH); (ii) as observed with CD98hc, gB, and gH, wild-type HSV-1 infection caused redistribution of the endoplasmic reticulum (ER) markers calnexin and ERp57 around the NM, whereas the UL34-null mutation caused cytoplasmic dispersion of these markers; (iii) the ER markers colocalized efficiently with CD98hc, gB, and gH in the presence and absence of UL34 in HSV-1-infected cells; (iv) at the ultrastructural level, wild-type HSV-1 infection caused ER compression around the NM, whereas the UL34-null mutation caused cytoplasmic dispersion of the ER; and (v) the UL34-null mutation significantly decreased the colocalization efficiency of lamin protein markers of the NM with CD98hc and gB. Collectively, these results indicate that HSV-1 infection causes redistribution of the ER around the NM, with resulting accumulation of ER-associated CD98hc, gB, and gH around the NM and that UL34 is required for ER redistribution, as well as for efficient recruitment to the NM of the ER-associated de-envelopment factors. Our study suggests that HSV-1 induces remodeling of the global ER architecture for recruitment of regulators mediating viral nuclear egress to the NM.
IMPORTANCE The ER is an important cellular organelle that exists as a complex network extending throughout the cytoplasm. Although viruses often remodel the ER to facilitate viral replication, information on the effects of herpesvirus infections on ER morphological integrity is limited. Here, we showed that HSV-1 infection led to compression of the global ER architecture around the NM, resulting in accumulation of ER-associated regulators associated with nuclear egress of HSV-1 nucleocapsids. We also identified HSV-1 UL34 as a viral factor that mediated ER remodeling. Furthermore, we demonstrated that UL34 was required for efficient targeting of these regulators to the NM. To our knowledge, this is the first report showing that a herpesvirus remodels ER global architecture. Our study also provides insight into the mechanism by which the regulators for HSV-1 nuclear egress are recruited to the NM, where this viral event occurs.
KEYWORDS: ER, herpes simplex virus, organelle structure
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is classified in the subfamily Alphaherpesvirinae of the family Herpesviridae. It is one of the best-studied members of the family and is an important human pathogen, causing a variety of disease states, including mucocutaneous diseases, keratitis, skin diseases, and encephalitis (1). After entry of HSV-1 into the host cell, the nucleocapsid is transported to a nuclear pore, enabling the entry of the viral genome into the nucleus and initiation of viral gene expression (1). Viral DNA replication, capsid assembly, and packaging of replicated viral genomes into capsids take place in the nucleus (1). The nascent progeny nucleocapsids of HSV-1, like those of other herpesviruses, then traverse the inner nuclear membrane (INM) and outer nuclear membrane (ONM) by a unique nuclear egress mechanism designated vesicle-mediated nucleocytoplasmic transport (2, 3). In this nucleocytoplasmic transport system, progeny nucleocapsids acquire a primary envelope by budding through the INM into the perinuclear space between the INM and the ONM (primary envelopment), and the enveloped nucleocapsids then fuse with the ONM and are released into the cytoplasm (de-envelopment) (2, 3). A heterodimeric complex of HSV-1 proteins UL31 and UL34, which are conserved in all subfamilies of herpesviruses, is critical for primary HSV-1 envelopment and has been designated the nuclear egress complex (NEC) (1–8). UL34 is a type II integral membrane protein that is targeted to both the INM and the ONM (9, 10). UL31 is a soluble component that is held in close apposition to the inner and outer surfaces of the INM and ONM, respectively, through its interaction with UL34 (4). Therefore, the topology of the NEC indicates that it should be displayed on the inner surfaces of primary enveloped virions. The NEC has been reported to play multiple roles in primary envelopment, including deformation of the INM (6–8, 11); recruitment of nucleocapsids to the INM by binding to a capsid protein (2, 3, 12); and recruitment of cellular factors, such as members of the protein kinase C (PKC) family, which have been thought to dissolve the nuclear lamina by phosphorylation of lamin proteins in order to facilitate access of nucleocapsids to the INM (2, 3, 13, 14). In addition to the core components UL31 and UL34, the NEC appears to contain the HSV-1 serine/threonine protein kinase Us3, the major HSV-1 structural protein UL47, the HSV-1 regulatory protein ICP22, and the cellular protein p32 (10, 15–17). Consistent with this observation, Us3, UL47, and p32 were shown to be components of the primary enveloped virion (10, 16, 17); UL47 and ICP22 were found to be required for efficient primary envelopment of HSV-1 (15, 16); and p32 and Us3 affected the phosphorylation status of the nuclear lamina (18–20), which might result in its disintegration, leading to efficient access of nucleocapsids to the INM, as described above.
Several viral and cellular regulatory proteins involved in the HSV-1 de-envelopment step have been identified. Mutation(s) or depletion(s) that abrogates either the expression or catalytic activity of HSV-1 Us3; the expression of both HSV-1 envelope glycoproteins B (gB) and H (gH); the phosphorylation of UL31; both the Us3 phosphorylation of gB and gH expression; and the expression of the cellular proteins p32, CD98 heavy chain (CD98hc), and β1 integrin have been reported to induce membranous structures that contain primary enveloped virions, which are invaginations of the INM into the nucleoplasm, and to induce the aberrant accumulation of primary enveloped virions in the perinuclear space and within the induced invagination structures (17, 21–24). Aberrant accumulation of virions within membranous invagination structures has been thought to reflect an imbalance between the rate of virion delivery into the perinuclear space and the rate of egress from this space: the rate of virion egress from the perinuclear space may have decreased, while the rate of egress from the nucleoplasm may have not changed or not decreased to the same degree. Therefore, Us3, gB, gH, UL31, p32, CD98hc, and β1 integrin were suggested to be the regulators for HSV-1 de-envelopment (17, 21–24). Curiously, in uninfected cells, p32, CD98hc, and β1 integrin are localized predominantly in the cytoplasm and/or at the cell surface, and HSV-1 infection leads to redistribution of these cellular proteins to the region around the NM (17, 24). The accumulation of p32 around the NM requires its HSV-1 binding partner, UL47 (17), whereas the mechanisms for the redistribution of CD98hc and β1 integrin remain unknown.
CD98hc is a type II membrane glycoprotein that acts as an amino acid transporter on the cell surface by associating through its extracellular domain with one of several light chains (25). It also regulates integrin signaling, which is involved in cell adhesion and migration, by associating with β1 and β3 integrins through its cytoplasmic and transmembrane domains (26–29). As observed with HSV-1 de-envelopment, CD98hc was found to regulate membrane fusion mediated by several enveloped RNA viruses and to require β1 integrin for this activity (30–33). These findings suggest that CD98hc and β1 integrin may be common regulators for membrane fusion mediated by these enveloped viruses. In this study, we attempted to identify the factor(s) involved in the accumulation of CD98hc around the NM of HSV-1-infected cells. Specifically, since CD98hc was reported to interact with multiple HSV-1 and cellular proteins, including gB, gH, UL31, UL34, Us3, and β1 integrin (24), we investigated whether these protein interactors with CD98hc were involved in the regulation of CD98hc localization in HSV-1-infected cells.
RESULTS
Characterization of recombinant viruses generated in this study.
To examine the effect of UL34 on the localization of CD98hc in HSV-1-infected cells, we constructed the UL34-null mutant virus YK722(ΔUL34) and its repaired virus, YK723(ΔUL34-repair) (Fig. 1). As expected, HEp-2 cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) expressed UL34, but cells infected with YK722(ΔUL34) did not (Fig. 2A). In agreement with a previous report (34), the level of accumulation of UL31 in HEp-2 cells infected with YK722(ΔUL34) was slightly lower than that in cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) (Fig. 2A). In contrast, HEp-2 cells infected with YK722(ΔUL34) at a multiplicity of infection (MOI) of 5 for 24 h accumulated the immediate-early gene products ICP0 and ICP27, the early gene products ICP8 and UL12, and the late gene products and gB at levels similar to those infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) (Fig. 2A). These results indicate that that YK722(ΔUL34) mostly retained the wild-type expression levels of the viral proteins examined in these studies. Viral production of YK722(ΔUL34) in HEp-2 cells at MOIs of 5 and 0.01 was remarkably impaired compared to that of wild-type HSV-1(F) (Fig. 2B). UL31, the binding partner of UL34, appeared as a distinct, smooth line localized to the nuclear rim in HEp-2 cells infected with wild-type HSV-1(F), whereas it was localized to and diffusely distributed throughout the nucleus and was colocalized with a nuclear viral protein, UL12 (35), in cells infected with YK722(ΔUL34) (Fig. 2C). Wild-type-virus replication and UL31 localization were restored in cells infected with YK723(ΔUL34-repair) (Fig. 2B and C). These features of YK722(ΔUL34) were in agreement with previous reports characterizing other UL34 deletion mutants (4).
FIG 1.

Schematic diagrams of the genome structure of wild-type HSV-1(F) and the relevant domains of the recombinant viruses generated in this study. 1, wild-type HSV-1(F) genome; 2, domain of the UL33 to UL35 genes; 3, domain of the UL34 gene; 4 and 5, recombinant viruses with mutations in the UL34 gene.
FIG 2.

Characterization of UL34-Vero cells and recombinant viruses generated in this study. (A) (Left) Vero and UL34-Vero cells mock infected or infected with wild-type HSV-1(F) or YK722(ΔUL34) at an MOI of 5 for 18 h were analyzed by immunoblotting with the indicated antibodies. (Right) HEp-2 cells mock infected or infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) at an MOI of 5 for 24 h were analyzed by immunoblotting with the indicated antibodies. (B) HEp-2 cells were infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) at an MOI of 5 (left) or 0.01 (right). Total virus from the cell culture supernatants and infected cells was harvested and assayed on UL34-Vero cells. (C) HEp-2 cells were mock infected or infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) at an MOI of 5 for 24 h and then fixed, permeabilized, stained with the indicated antibodies, and examined by confocal microscopy. Scale bars, 10 μm.
HSV-1 and cellular protein interactions with CD98hc affected the localization of CD98hc in HSV-1-infected cells.
To examine the effects of the HSV-1 protein interactors with CD98hc, including gB, gH, Us3, UL34, and UL31, on the localization of CD98hc in HSV-1-infected cells, HEp-2 cells were mock infected or infected with wild-type HSV-1(F), YK701(ΔgB), YK713(ΔgH), YK720(ΔUL31), R7041(ΔUs3), YK722(ΔUL34), or YK723(ΔUL34-repair) at an MOI of 5 (Fig. 3, 4, and 5; see Fig. 7 and 8). In addition, to examine the effects of cellular interaction between CD98hc and β1 integrin, sh-β1 integrin-HEp-2 cells (in which the expression of β1 integrin is reduced by the expression of short hairpin RNA [shRNA] against β1 integrin compared to that in sh-Luc-HEp-2 cells, which express control shRNA against firefly luciferase [Fig. 6A]) (24) and sh-Luc-HEp-2 cells were mock infected or infected with wild-type HSV-1(F) at an MOI of 5. These infected cells were fixed at 24 h after infection, and localization of CD98hc, gB, gH, UL31, UL34, cadherin, lamin A/C, and/or lamin B1 was examined by immunofluorescence. Cadherin was shown to be a marker of the plasma membrane, and lamin A/C and lamin B1 were shown to be markers of the NM (36, 37).
FIG 3.
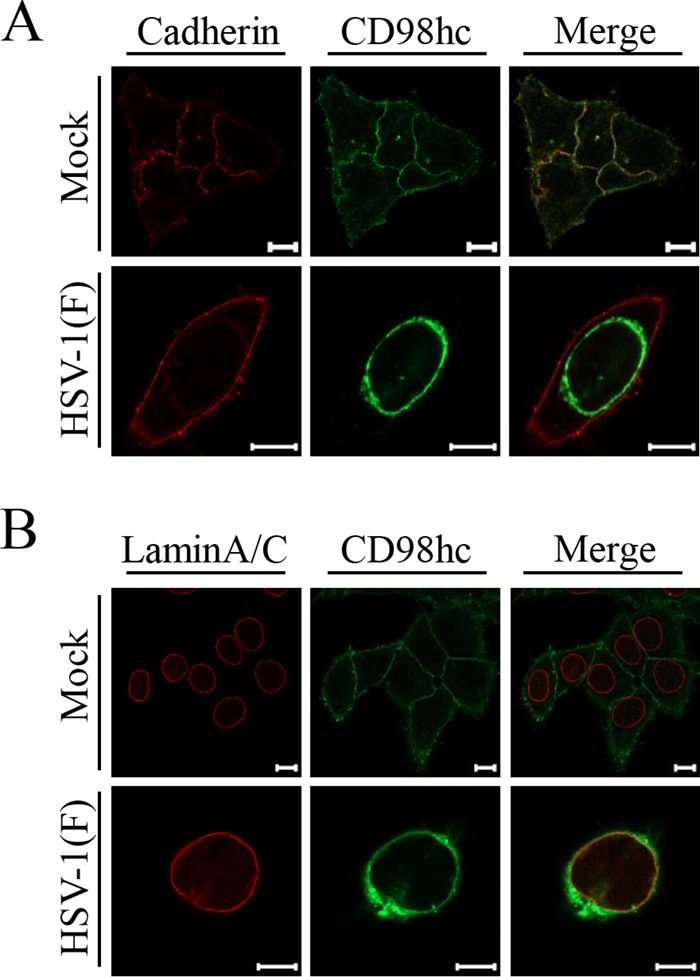
Localization of CD98hc in HSV-1-infected cells. HEp-2 cells were mock infected or infected with wild-type HSV-1(F) at an MOI of 5 for 24 h and then fixed, permeabilized, stained with anti-CD98hc antibody in combination with anti-cadherin (A) or anti-lamin A/C (B), and examined by confocal microscopy. Scale bars, 10 μm.
FIG 4.

Effects of mutations in gH, gB, or UL31 on localization of CD98hc in HSV-1-infected cells. HEp-2 cells were mock infected or infected with wild-type HSV-1(F) (A to D), YK713(ΔgH) (A), YK701(ΔgB) (B), R7040(ΔUs3) (C), or YK720(ΔUL31) (D) at an MOI of 5 for 24 h and then fixed, permeabilized, stained with the indicated antibodies, and examined by confocal microscopy. Scale bars, 10 μm.
FIG 5.
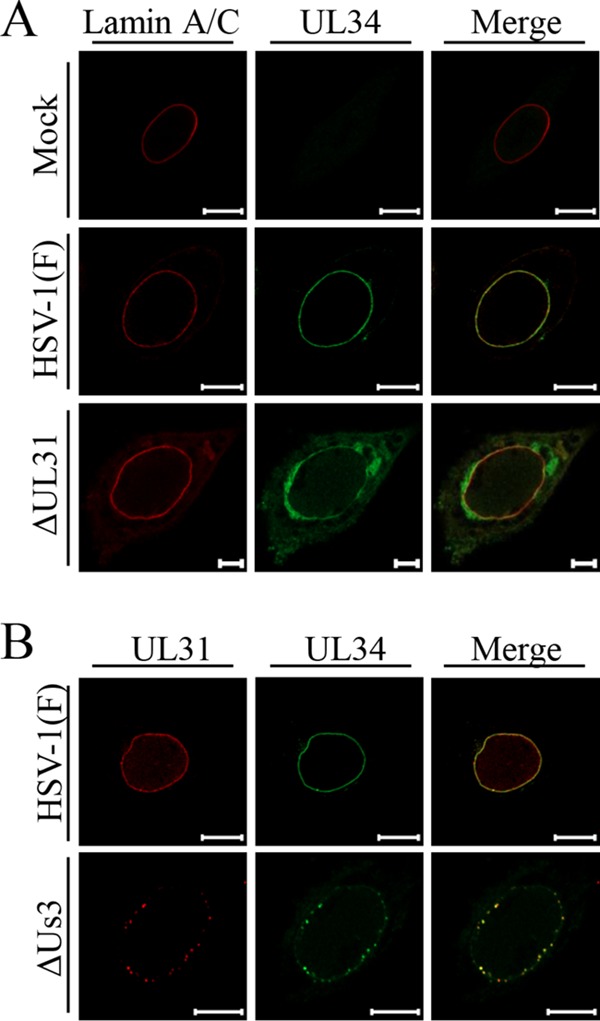
Effects of mutations in UL31 or Us3 on localization of UL34 and/or UL31 in HSV-1-infected cells. HEp-2 cells were mock infected (A) or infected with wild-type HSV-1(F) (A and B), YK720(ΔUL31) (A), or R7040(ΔUs3) (B) at an MOI of 5 for 24 h and then fixed, permeabilized, stained with the indicated antibodies, and examined by confocal microscopy. Scale bars, 10 μm.
FIG 7.
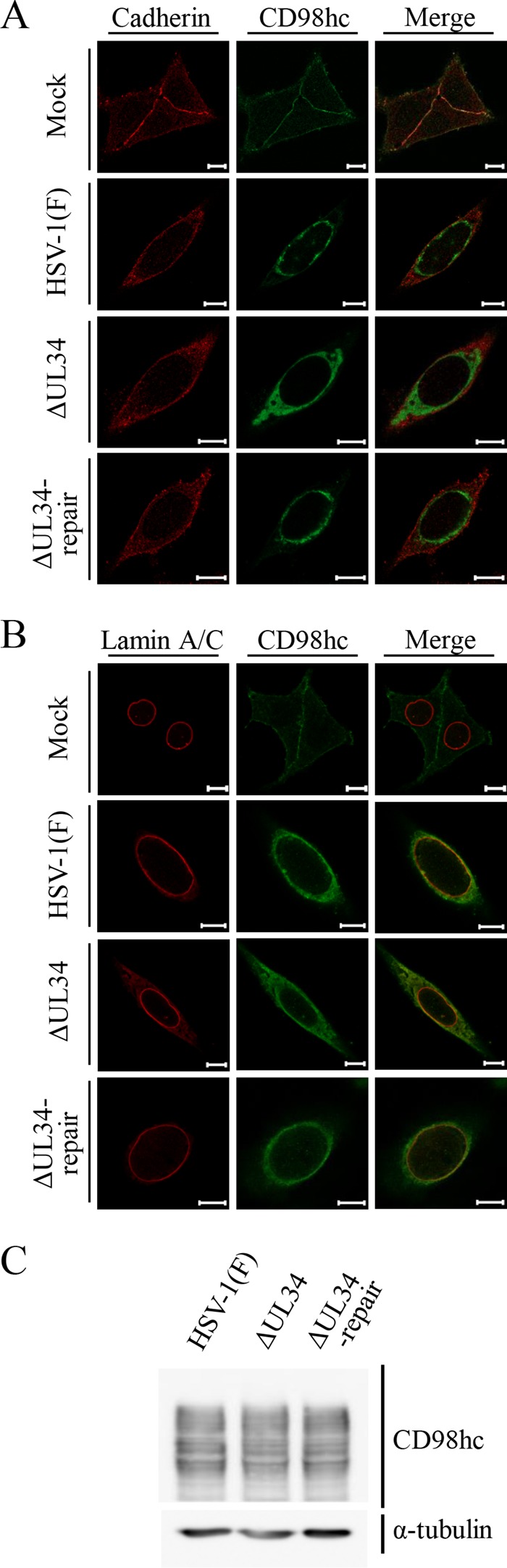
Effects of mutation in UL34 on localization and accumulation of CD98hc in HSV-1-infected cells. (A and B) HEp-2 cells were mock infected or infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) at an MOI of 5 for 24 h. The cells were fixed, permeabilized, stained with anti-CD98hc antibody in combination with anti-cadherin (A) or anti-lamin A/C (B) antibody, and examined by confocal microscopy. Scale bars, 10 μm. (C) The cells were also analyzed by immunoblotting with the indicated antibodies.
FIG 8.

Effects of mutation in UL34 on localization of gB (A) and gH (B) and on colocalization of CD98hc with gB (C) or gH (D) in HSV-1-infected cells. HEp-2 cells were mock infected or infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) at an MOI of 5 for 24 h and then fixed, permeabilized, stained with the indicated antibodies, and examined by confocal microscopy. Scale bars, 10 μm.
FIG 6.
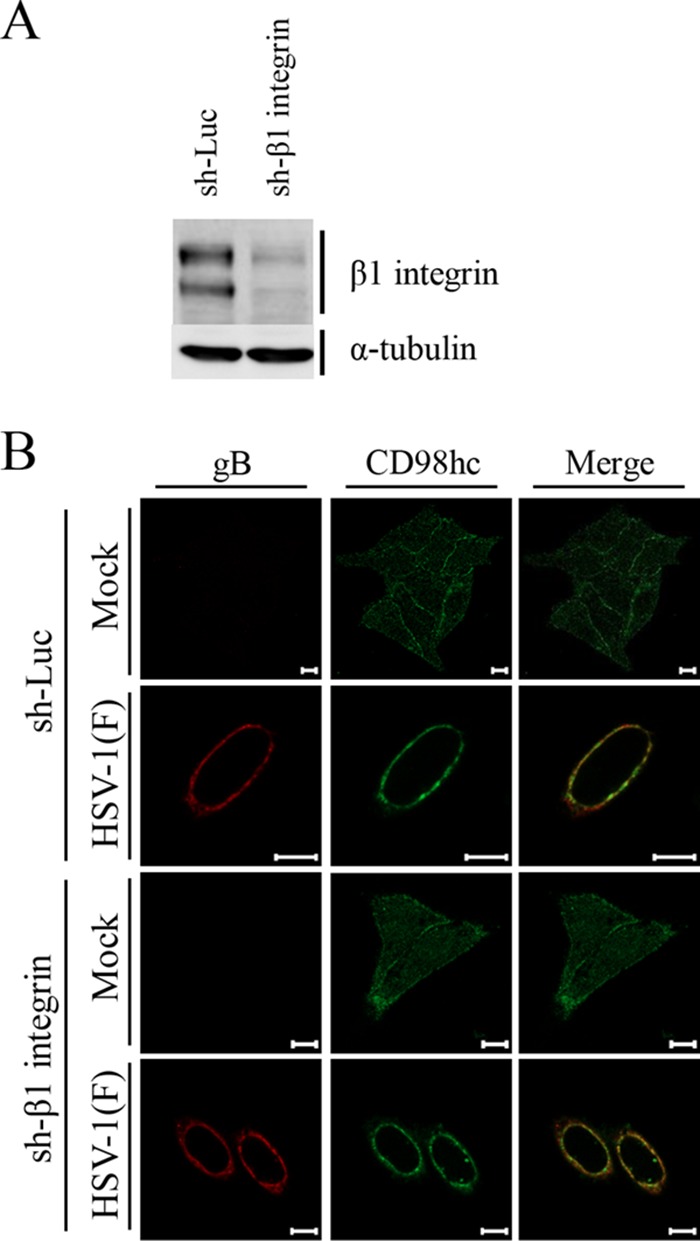
Effects of β1 integrin knockdown on localization of CD98hc in HSV-1 infected cells. (A) Expression of β1 integrin in sh-Luc-HEp-2 and sh-β1 integrin-HEp-2 cells was analyzed by immunoblotting with the indicated antibodies. (B) sh-Luc-HEp-2 and sh-β1 integrin-HEp-2 cells were mock infected or infected with wild-type HSV-1(F) at an MOI of 5 for 24 h and then fixed, permeabilized, stained with the indicated antibodies, and examined by confocal microscopy. Scale bars, 10 μm.
As reported previously (24), CD98hc was detected predominantly at the plasma membrane and indeed colocalized with cadherin in mock-infected cells (Fig. 3A). In contrast, CD98hc was predominantly detected around the nuclear rim and colocalized partially with lamin A/C (Fig. 3B) and more completely with gB and gH (Fig. 4) in wild-type-HSV-1(F)-infected cells. Similarly, in cells infected with YK713(ΔgH), YK701(ΔgB), R7041(ΔUs3), or YK720(ΔUL31), CD98hc was detected around the nuclear rim and colocalized with gB or gH (Fig. 4). We verified that, in agreement with previous reports (4, 38), the UL31- and Us3-null mutations caused dislocation of UL34 in HSV-1-infected cells: in cells infected with YK720(ΔUL31) or R7041(ΔUs3), UL34 was detected both at the nuclear rim and in the perinuclear region in the cytoplasm (Fig. 5A) or as punctate structures at the nuclear rim (Fig. 5B), respectively. Furthermore, wild-type HSV-1(F) infection caused the accumulation of CD98hc around the nuclear rim in sh-β1 integrin-HEp-2 cells, as observed in sh-Luc-HEp-2 cells (Fig. 6B).
In contrast, the distribution of CD98hc in cells infected with YK722(ΔUL34) was apparently different from its distribution in cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) (Fig. 7). Thus, CD98hc was more diffusely distributed throughout the cytoplasm in cells infected with YK722(ΔUL34) than in cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) (Fig. 7 and 8). Similarly, in cells infected with YK722(ΔUL34), gB and gH were also more diffusely distributed throughout the cytoplasm than in cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) (Fig. 8A and B) and were colocalized with CD98hc in these infected cells (Fig. 8C and D). The level of accumulation of CD98hc in cells infected with YK722(ΔUL34) was similar to that in cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) (Fig. 7C).
Taken together, the results of the interactions of the HSV-1 proteins gB, gH, Us3, UL34, and UL31 with CD98hc for the localization of CD98hc in HSV-1-infected cells indicate that UL34 was required for the appropriate localization of CD98hc, gB, and gH in HSV-1-infected cells, but gB, gH, Us3, UL31, and β1 integrin were not.
Effect of UL34 on the distribution of the ER in HSV-1-infected cells.
gB and gH have been reported to be predominantly colocalized with NM and ER markers in wild-type HSV-1-infected cells (21, 39, 40). As shown in Fig. 6 and 7, the localizations of CD98hc, gB, and gH were simultaneously changed in similar ways by the absence of UL34 in HSV-1infected cells. These observations led us to hypothesize that, similarly to gB and gH in HSV-1-infected cells, CD98hc is also associated with the endoplasmic reticulum (ER) and that UL34 is involved in the regulation of the ER global architecture and thereby regulates the localization of ER-associated proteins. To test this hypothesis, HEp-2 cells mock infected or infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) were fixed, and the localization of the ER markers calnexin and ERp57 (41, 42) was analyzed by immunofluorescence. As reported previously (41, 42), calnexin and ERp57 were distributed throughout the cytoplasm in mock-infected cells (Fig. 9). In contrast, these ER marker proteins were concentrated around the nuclear rim in cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) and were partially colocalized with lamin A/C or lamin B1 (Fig. 9). In cells infected with YK722(ΔUL34), the ER marker proteins were more diffusely distributed in the cytoplasm than in cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) (Fig. 9). The distributions of calnexin and ERp57 in cells infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) were similar to those of gB, gH, and CD98hc. Indeed, these ER marker proteins mostly colocalized with gB, gH, and CD98hc in the infected cells (Fig. 10).
FIG 9.
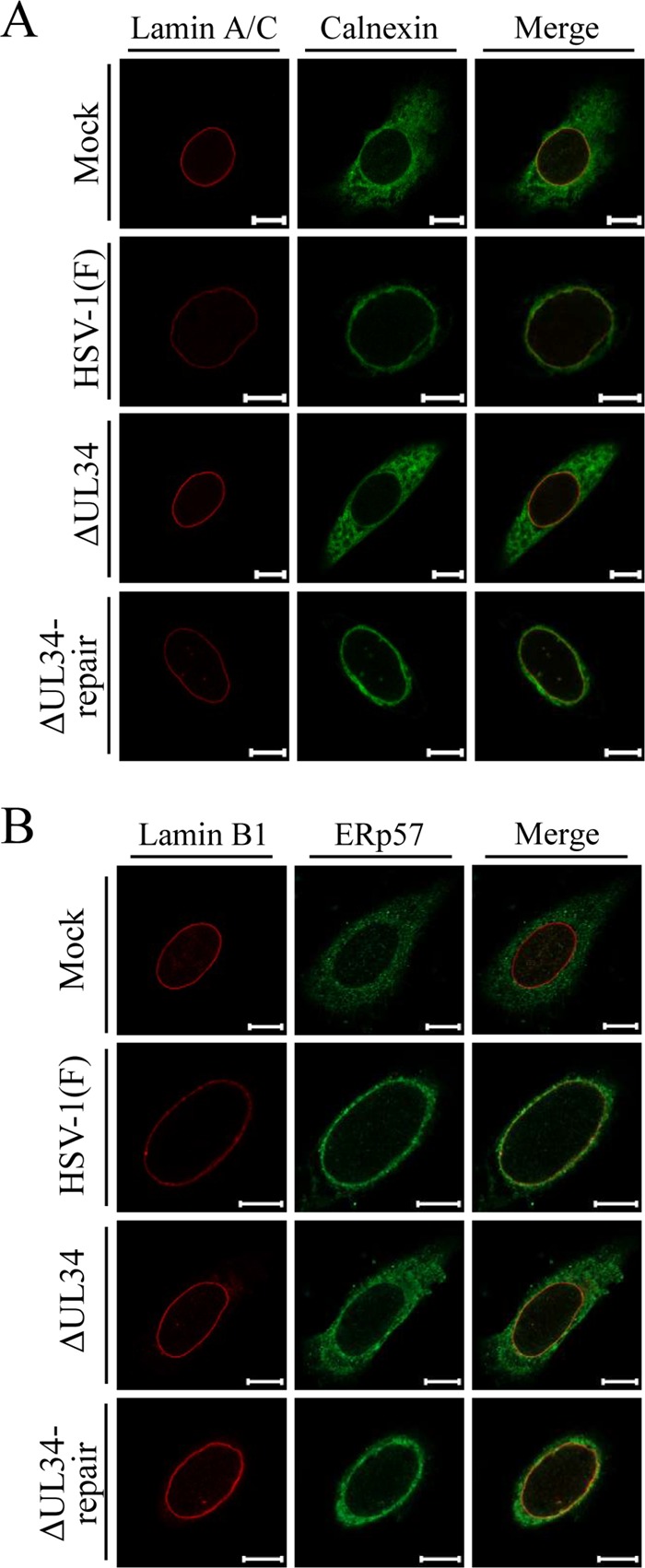
Effect of mutation in UL34 on localization of the ER markers calnexin (A) and ERp57 (B) in HSV-1-infected cells. HEp-2 cells were mock infected or infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) at an MOI of 5 for 24 h and then fixed, permeabilized, stained with the indicated antibodies, and examined by confocal microscopy. Scale bars, 10 μm.
FIG 10.
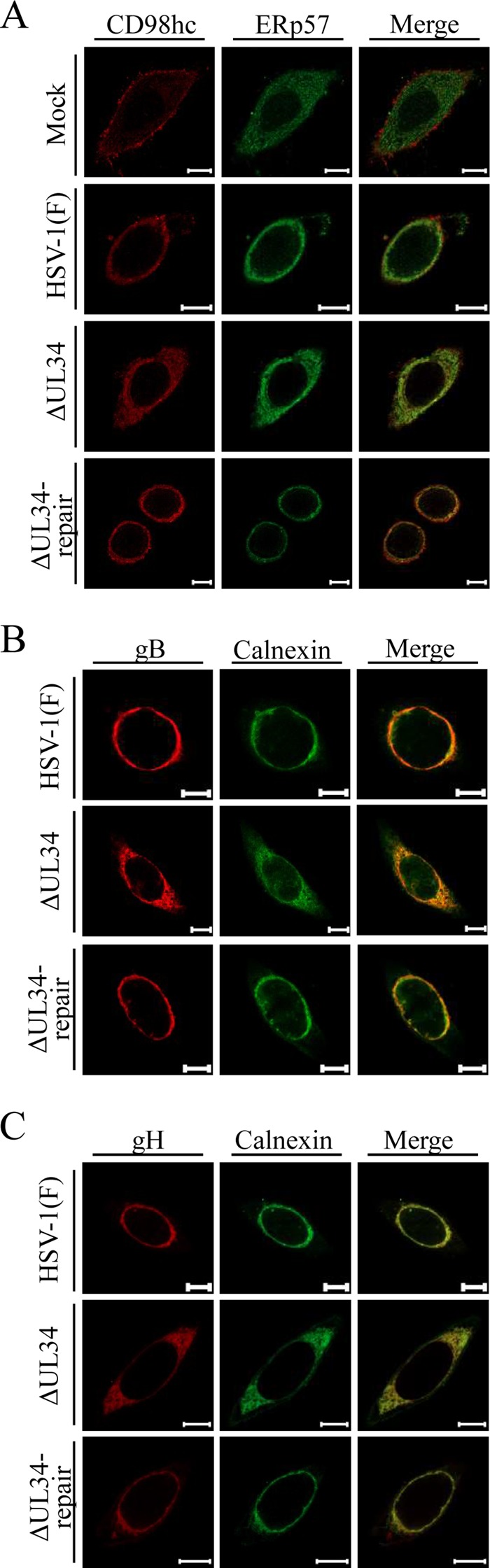
Effect of mutation in UL34 on colocalization of the ER markers ERp57 (A) and calnexin (B and C) with each of the de-envelopment factors CD98hc (A), gB (B), and gH (C) in HSV-1-infected cells. HEp-2 cells were mock infected or infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) at an MOI of 5 for 24 h and then fixed, permeabilized, stained with the indicated antibodies, and examined by confocal microscopy. Scale bars, 10 μm.
To investigate the effect of UL34 on the distribution of the ER in HSV-1-infected cells at the ultrastructural level, HEp-2 cells infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) were analyzed by electron microscopy. At the ultrastructural level, the ER membranes were distributed throughout the cytoplasm in mock-infected cells (Fig. 11), as described previously (43). In agreement with the immunofluorescence analysis of the ER marker proteins (Fig. 9 and 10), the ER membranes were concentrated around the nuclear rim in cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) and were more diffusely distributed throughout the cytoplasm in cells infected with YK722(ΔUL34) than in cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) (Fig. 11).
FIG 11.

Effect of mutation in UL34 on the distribution of the ER in HSV-1-infected cells at the ultrastructural level. HEp-2 cells were mock infected (A) or infected with wild-type HSV-1(F) (B), YK722(ΔUL34) (C), or YK723(ΔUL34-repair) (D) at an MOI of 30 for 24 h; embedded; sectioned; stained; and examined by transmission electron microscopy. The arrowheads indicate the ER. Nu, nucleus; Cy, cytoplasm. Scale bars, 1 μm.
Collectively, these results suggest that UL34 was required for proper global architecture of the ER in HSV-1-infected cells. gB, gH, and CD98hc appeared to be tightly associated with the ER in HSV-1-infected cells, and therefore, their localization was closely linked to the distribution of the ER.
Effect of ectopic expression of UL34 and/or UL31 on the distribution of the ER.
To examine whether UL34 by itself can regulate the distribution of the ER marker and the localization of CD98hc, HEp-2 cells were transfected with a UL34 expression plasmid or an empty expression plasmid, and the localizations of UL34 and calnexin or CD98hc were analyzed by immunofluorescence. In agreement with a previous report (4), transiently expressed UL34 was mainly detected as aggregates in the cytoplasm (Fig. 12A and C). As shown in Fig. 12A, the pattern of localization of calnexin in cells transfected with the empty plasmid was apparently different from that in cells transiently expressing UL34. Specifically, a fraction of calnexin was recruited to the UL34 aggregates and colocalized with UL34 (Fig. 12A). In contrast, ectopic expression of UL34 had no effect on the pattern of localization of CD98hc (Fig. 12C). These results indicate that UL34 has the ability to regulate the distribution of the ER marker in the absence of any other HSV-1 protein, whereas it was not sufficient to regulate the localization of CD98hc without HSV-1 infection.
FIG 12.

Localization of calnexin and CD98hc in cells ectopically expressing UL34 and/or UL31 or in HSV-1-infected cells. (A to D) HEp-2 cells were transfected with the empty plasmid alone (A to D), the UL34 expression plasmid alone (A and C), or the UL34 and UL31 expression plasmids (B and D) for 48 h and then fixed, permeabilized, stained with anti-calnexin (A and B) or anti-CD98hc (C and D) antibody in combination with the indicated antibodies, and examined by confocal microscopy. (E) HEp-2 cells were mock infected or infected with wild-type HSV-1(F) at an MOI of 5 for 24 h and then fixed, permeabilized, stained with anti-UL34 antibody and anti-calnexin antibody, and examined by confocal microscopy. Scale bars, 10 μm.
The ectopic coexpression of UL34 and UL31 reportedly targets these viral proteins to the NM (4). Therefore, we also examined the effects of the ectopic coexpression of UL34 and UL31 on the localization of the ER marker calnexin and on the localization of CD98hc. In agreement with previous reports (4), the ectopic coexpression of UL34 and UL31 led to the localization of these viral proteins predominantly at the nuclear rim (Fig. 12B and D). In cells expressing both UL34 and UL31, calnexin was recruited around the nuclear rim and was partially colocalized with these viral proteins at the nuclear rim (Fig. 12B), as was also observed in wild-type HSV-1(F)-infected cells (Fig. 12E). In contrast, the coexpression of UL34 and UL31 had no effect on the localization of CD98hc (Fig. 12D). These results suggest that the NEC was sufficient for the recruitment of the ER to the region around the nuclear rim.
Effects of UL34 on recruitment of CD98hc, of gB, and, in addition, of gD to the NM.
Finally, we examined whether UL34 was involved in the recruitment to the NM of the ER-associated viral and cellular regulators of HSV-1 nuclear egress, which include CD98hc and gB. We also investigated the involvement of UL34 in recruitment of gD to the NM, since UL34 was reported to promote gD localization at the NM (44). To this end, HEp-2 cells infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) were fixed, and immunofluorescence analysis of the localization of the NM marker lamin A/C or lamin B1, CD98hc, gB, and gD was performed. Colocalization between each of the NM markers and CD98hc, gB, or gD was quantified. In agreement with a previous report (44), the efficiency of colocalization between gD and lamin B1 in cells infected with YK722(ΔUL34) was significantly lower than that in cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) (Fig. 13A), confirming that UL34 promoted gD localization at the NM. Similar results were also obtained for efficiencies of colocalization between CD98hc and lamin A/C, and gB and lamin B1 (Fig. 13B and C). The differences in intensity levels of the fluorescence emissions of gB, gD, CD98hc, lamin B1, and lamin A/C between cells infected with YK722(ΔUL34) and cells infected with wild-type HSV-1(F) or YK723(ΔUL34-repair) were either not significant (gB, gD, lamin B1, and lamin A/C) or did not decrease (CD98hc) (Fig. 14), eliminating the possibility that the effects of the UL34-null mutation on the colocalization efficiencies of gB, gD, and CD98hc and on the colocalization efficiencies of the NM markers were not due to the decreased expression of these cellular and viral proteins in infected cells. The results indicate that UL34 is required for efficient accumulation of gD, gB, and CD98hc at the NM.
FIG 13.
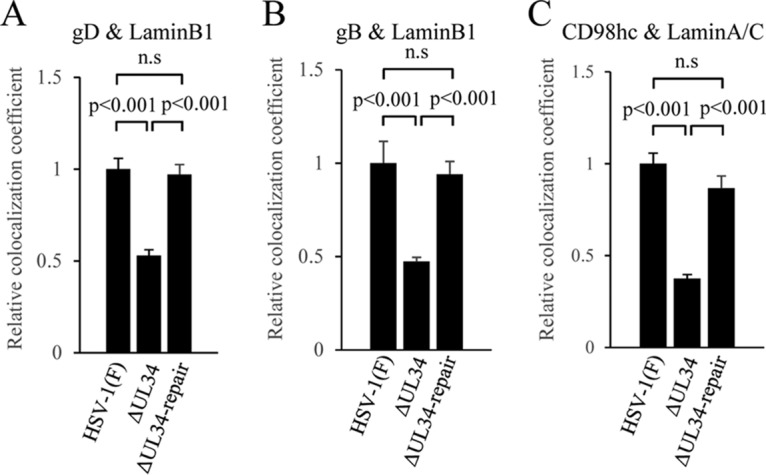
Effects of mutation in UL34 on colocalization of the NM markers with gD, gB, or CD98hc in HSV-1-infected cells. HEp-2 cells were infected with wild-type HSV-1(F), YK722(ΔUL34), or YK723(ΔUL34-repair) at an MOI of 5 for 24 h and then fixed; permeabilized; stained with anti-lamin B1 (A and B) or anti-lamin A/C (C) in combination with anti-gD (A), anti-gB (B), or anti-CD98hc antibody (C); and examined by confocal microscopy. Colocalization between gD and lamin B1 (A), gB and lamin B1 (B), or CD98hc and lamin A/C (C) was quantified using Manders' colocalization coefficient. Each value is the mean and standard error (n = 40) and is expressed relative to the mean value of wild-type HSV-1(F)-infected HEp-2 cells, which was normalized to 1. Statistical analysis was performed by one-way analysis of variance with the Tukey test; n.s., not significant.
FIG 14.
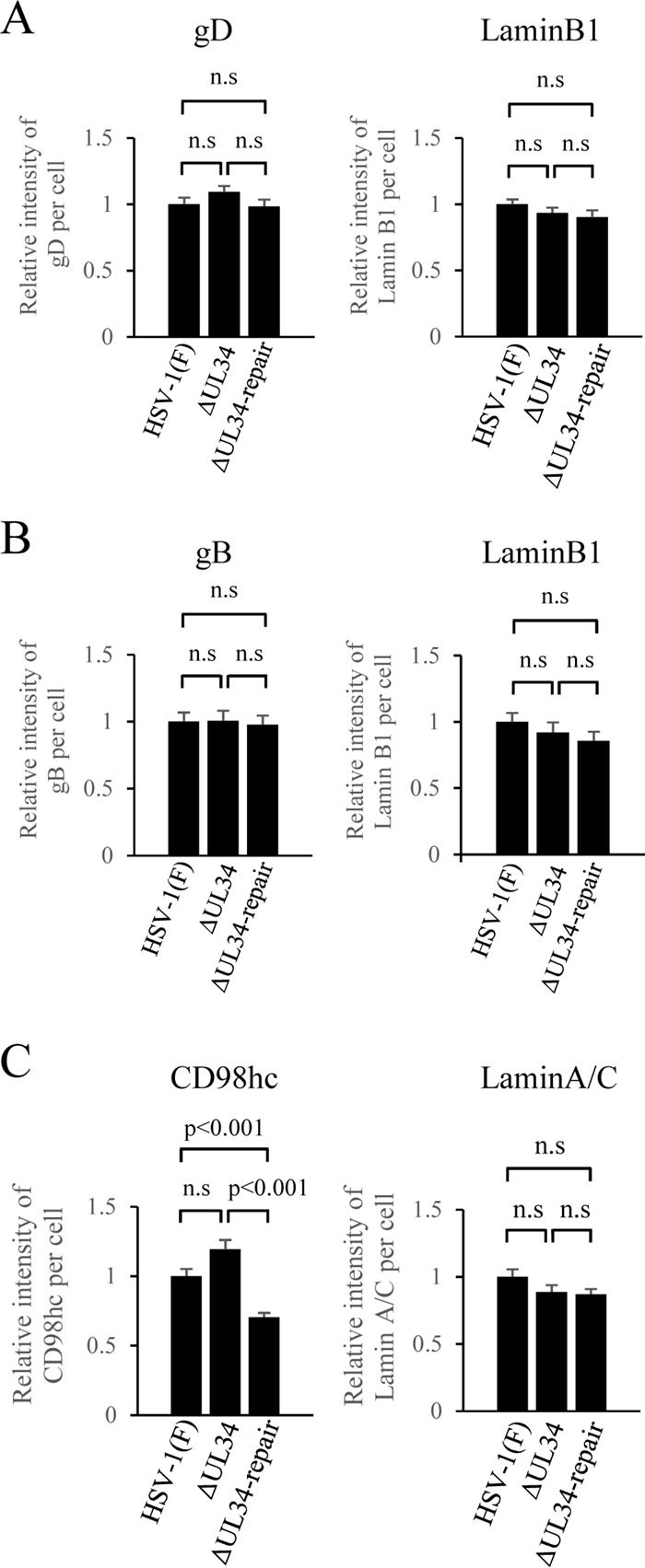
Effect of mutation in UL34 on the total intensity of fluorescence of gD (A), gB (B), or CD98hc (C) in HSV-1-infected cells detected by confocal microscopy. Total intensities per cell in the infected HEp-2 cells analyzed in Fig. 13 were obtained using ZEN software (Zeiss). Each value is the mean and standard error (n = 40) and is expressed relative to the mean value of wild-type HSV-1(F)-infected HEp-2 cells, which was normalized to 1. Statistical analysis was performed by one-way analysis of variance with the Tukey test; n.s., not significant.
DISCUSSION
Viruses frequently induce remodeling of the membranes of host cellular organelles, which appears to facilitate viral replication, especially replication of viral genomes, and the morphogenesis of virions (45–50). Indeed, RNA and DNA viruses that replicate their genomes in the cytoplasm utilize the remodeled cellular membranes to create compartments, which are designated replication factories (45–50). These compartments appear to function as physical support for the coordinated accumulation of the viral and cellular components required for efficient viral replication and/or as a barrier that prevents exposure of viral nucleic acids to the host's immune system (45–51). In contrast, information on the remodeling of the membranes of cellular organelles mediated by HSV-1 infection has been limited. Thus, it has been reported that HSV-1 infection modifies the Golgi apparatus and the trans-Golgi network (TGN) (52), i.e., whereas these organelles are localized in the juxtanuclear cytoplasmic domain in normal cells, they are dispersed throughout the cytoplasm in infected cells (52). However, in the case of HSV-1 infection, the biological significance of the modification of organelle membranes by HSV-1 remains largely unknown.
The ER is the largest host cellular organelle and forms a complex network that extends from the NM to the plasma membrane (43). This membranous organelle plays a central role in the synthesis, modification, and transport of secretory and membrane proteins and is the site of the biosynthesis, processing, and transport of lipids (53). Interestingly, the ER is most often hijacked by a variety of viruses for the creation of viral replication factories. Therefore, viruses that use the ER in this way cause substantial alterations in its morphology (45–47, 50). However, information on the effects of herpesvirus infections on the morphological integrity of the ER is scarce. In this study, we showed that HSV-1 infection dramatically altered the global architecture of the ER and that UL34 was required for the HSV-1-induced alteration of the ER. To our knowledge, this is the first report to show that a herpesvirus remodels the morphology of the ER and to identify the herpesvirus protein responsible and required for ER remodeling. Since the mechanism by which the spatial integrity of the ER in normal cells is regulated largely remains to be elucidated, we suggest that UL34 could be used as a probe for investigating the cellular mechanism involved in regulating the spatial integrity of the ER.
Newly synthesized membrane proteins are sorted from the ER into various membranous compartments and to the plasma membrane. The membrane of the ER is continuous with the ONM and INM, but not with either the plasma membrane or the membranes of other cytoplasmic organelles, such as the Golgi apparatus, TGN, and endosomes (43). Therefore, the mechanisms by which membrane proteins target the plasma membrane and the cytoplasmic membranous organelles are distinct from those by which membrane proteins specifically accumulate at the NM. The membrane proteins targeted to the plasma membrane and the cytoplasmic membranous organelles are in general sorted from the ER by the vesicular trafficking system (54). In contrast, recent live-imaging and mathematical analyses support the diffusion and retention model for specific targeting of membrane proteins to the NM (55, 56). Thus, newly synthesized membrane proteins targeted to the NM are inserted into the membranes of the ER and are thought to move laterally from the ER to the ONM and/or the INM by free diffusion (57, 58). It appears that enrichment of the INM proteins in the INM occurs by interaction with nuclear binding partners, such as the lamin proteins or chromatin, which would be required to retain INM proteins at the INM (55, 56). Enrichment of the ONM proteins in the ONM is driven by interaction with INM proteins in the perinuclear space (59). In agreement with this model, it has been reported that HSV-1 gD interacts with the HSV-1 NEC, and the efficient retention of HSV-1 gD at the INM requires the NEC in HSV-1-infected cells (44). At present, although it remains to be elucidated whether our study findings that the UL34-mediated ER compression around the NM and the UL34-mediated recruitment of the de-envelopment regulators to the NM are related, it is conceivable that the compression of the ER around the NM could physically promote the diffusion of membrane proteins from the ER toward the NM, thereby leading to their accumulation at the NM in HSV-1-infected cells. Moreover, the HSV-1 NEC was shown to localize at both the INM and ONM (10), and therefore, it is possible that, for the cellular membrane protein CD98hc, which has been shown to interact with UL34 and UL31 (24), its retention at the NM might be further promoted by its interaction with the HSV-1 NEC.
In this and previous studies (24), the level of CD98hc localization at the plasma membrane in HSV-1-infected cells was shown to be remarkably reduced compared to that in mock-infected cells. These observations suggest that HSV-1 infection changed the extent of CD98hc retention at the plasma membrane, which is determined by the balance between the rate of its delivery to the plasma membrane and the rate of its internalization into the cytoplasm (60). Conceivably, HSV-1 infection inhibits the transport of this cellular membrane protein to the plasma membrane and/or enhances its endocytosis. Notably, whereas we showed that the ectopic coexpression of UL34 and UL31 caused redistribution of the ER marker calnexin around the NM, which was also observed with markers of the ER in HSV-1-infected cells, the ectopic coexpression of UL34 and UL31 had no effect on the retention of CD98hc at the plasma membrane and did not cause it to accumulate around the NM. This result was in contrast to the effect of UL34 on CD98hc accumulation around the NM in HSV-1-infected cells. These observations suggest that HSV-1 infection induces an HSV-1 regulator(s) other than UL34 and UL31 or a cellular regulator(s) that inhibits the retention of CD98hc at the plasma membrane, causing the accumulation of CD98hc at the ER in infected cells, and then the UL34 and UL31 proteins somehow induce the compression of the ER around the INM, resulting in the accumulation of CD98hc around the NM.
Many mechanisms have evolved in a variety of viruses, including the herpesviruses, that restrict the retention of viral and cellular proteins at the plasma membrane, especially proteins such as the major histocompatibility complex (MHC) molecules, which target infected cells for the host immune response (61–64). In fact, various herpesvirus proteins, such as human cytomegalovirus Us2 and Us11 and the open reading frame 66 (ORF66) protein kinase encoded by varicella-zoster virus, have been reported to inhibit the transport of MHC molecules to the cell surface through the vesicular trafficking system (61, 64). HSV-1 Us3 was shown to inhibit the expression of gB, which is known to be an inducer of the host immune response (65), at the cell surface by enhancing its endocytosis (66, 67). Further studies that should be of interest and are under way in our laboratory include both identifying the HSV-1 and/or cellular protein(s) involved in inhibiting the retention of CD98hc at the plasma membrane and clarifying the role of such a protein(s) in inhibiting the retention of CD98hc at the plasma membrane.
MATERIALS AND METHODS
Cells and viruses.
Vero, rabbit skin, and HEp-2 cells were described previously (68, 69). Flp-In-CV-1/α27-gB, UL31-CV1, and Vero-gH cells expressing gB, UL31, and gH, respectively, were described previously (39, 70, 71). sh-Luc-HEp-2 and sh-β1 integrin-HEp-2 cells expressing shRNA against firefly luciferase and shRNA against human β1 integrin, respectively, were described previously (24). The HSV-1 wild-type strain HSV-1(F), gB-null mutant virus YK701(ΔgB), UL31-null mutant virus YK720(ΔUL31), Us3-null mutant virus R7041(ΔUs3), and gH-null mutant virus YK713(ΔgH) were described previously (15, 39, 70, 72). R7041(ΔUs3) was kindly provided by Bernard Roizman. For experiments with YK720(ΔUL31), wild-type HSV-1(F) and YK720(ΔUL31) viruses were propagated and assayed in UL31-CV1 cells. For experiments with YK701(ΔgB), wild-type HSV-1(F) and YK701(ΔgB) viruses were propagated and assayed in Flp-In-CV-1/α27-gB cells. For experiments with YK713(ΔgH), wild-type HSV-1(F) and YK713(ΔgH) viruses were propagated and assayed in Vero-gH cells.
Plasmids.
The plasmids pcDNA3.1-UL34 and pcDNA3.1-UL31, which were used for expression of HSV-1 UL34 and UL31, respectively, were generated by cloning fragments containing these ORFs, which had been amplified by PCR from the HSV-1(F) genome, into pcDNA3.1(−) (Invitrogen). The transfer plasmid pBS-UL34-rep, used to generate the recombinant virus YK723(ΔUL34 repair) (Fig. 1), in which the UL34 deletion in YK722(ΔUL34) was repaired, was constructed by cloning the PCR-amplified domain containing the UL34 ORF (a 513-bp upstream sequence flanking the UL34 start codon and a 500-bp downstream sequence flanking the UL34 stop codon) from the HSV-1(F) genome into pBluescript II KS(+) (Stratagene). The UL34 gene sequence, optimized for expression in human cells, was synthesized by GenScript and cloned into pMxs-puro to construct pMxs-UL34o, which was used for generating cell lines stably expressing UL34. The optimized U34 sequence was as follows: 5′-ATGGCTGGGCTGGGGAAACCTTACACTGGACATCCTGGGGACGCCTTTGAGGGGCTGGTGCAGAGAATTAGACTGATTGTGCCTTCAACCCTGAGAGGCGGCGACGGCGAGGCTGGCCCTTACAGCCCAAGCTCCCTGCCATCCCGGTGCGCCTTCCAGTTTCACGGCCACGACGGCTCTGATGAGAGCTTCCCCATCGAATACGTGCTGCGGCTGATGAACGATTGGGCCGAGGTGCCCTGTAACCCTTATCTGCGGATCCAGAATACAGGCGTGAGCGTGCTGTTTCAGGGCTTCTTTCACAGACCTCACAACGCTCCAGGCGGCGCTATCACCCCCGAGAGAACAAATGTGATCCTGGGCTCTACCGAGACCACAGGCCTGAGCCTGGGCGACCTGGATACAATCAAGGGCCGGCTGGGCCTGGACGCTAGACCCATGATGGCCTCCATGTGGATCTCTTGCTTCGTGAGAATGCCCAGGGTGCAGCTGGCCTTCCGGTTTATGGGCCCTGAGGATGCCGGCAGAACCCGGAGAATCCTGTGCAGGGCTGCTGAGCAGGCTATCACCAGGCGGAGAAGGACACGGAGATCCCGGGAGGCCTATGGCGCTGAGGCTGGCCTGGGCGTGGCTGGCACCGGCTTCAGGGCTAGAGGCGACGGCTTTGGCCCACTGCCCCTGCTGACACAGGGCCCATCTAGACCTTGGCACCAGGCCCTGAGGGGCCTGAAGCACCTGAGAATCGGCCCTCCTGCCCTGGTGCTGGCCGCAGGACTGGTGCTGGGAGCCGCCATTTGGTGGGTCGTCGGGGCAGGGGCAAGACTGTGA-3′.
Establishment of Vero cells stably expressing UL34.
Plat-GP cells were cotransfected with pMDG and pMxs-UL34o, and the supernatants were harvested. Vero cells were transduced with the supernatants and selected with puromycin (5 μg/ml), as described previously (73). Resistant cells were cloned from a single colony and designated UL34-Vero cells. Expression of UL34 protein in UL34-Vero cells was confirmed by immunoblotting (Fig. 2A).
Mutagenesis of viral genomes in E. coli and generation of recombinant HSV-1.
A UL34-null mutant virus, YK722(ΔUL34) (Fig. 1), in which the UL34 gene was disrupted by replacing UL34 codons 2 to 228 with a foreign gene cassette containing an I-SceI site and a kanamycin resistance gene, was generated by Red-mediated mutagenesis using Escherichia coli GS1783, carrying pYEbac102 (68), a full-length infectious HSV-1(F) clone, as described previously (74), except that the primers 5′-GTTTACGCGGGCACGCACGCTCCCATCGCGGGCGCCATGGAGGATGACGACGATAAGTAGGG-3′ and 5′-CCGCAGGGCCTGGTGCCACGGGCGGGAGGGCCCTTGGGTTCAACCAATTAACCAATTCTGATTAG-3′ were used, and Vero-UL34 cells were transfected with pYEbac102 carrying the UL34-null mutation, with the use of Lipofectamine 2000. The replacement of UL34 codons 2 to 228 with a foreign gene was previously used to generate an HSV-1 UL34-null mutant (5). The recombinant virus strain YK723(ΔUL34-repair), in which the UL34-null mutation in YK722 was repaired, was generated by cotransfection of rabbit skin cells with pYEbac102, carrying the UL34-null mutation, and pBS-UL34-rep, as described previously (15). Viruses were isolated from plaques and purified 2 additional times on Vero cells. Restoration of the original UL34 sequence was confirmed by sequencing. In experiments with YK722(ΔUL34), wild-type HSV-1(F), YK722(ΔUL34), and YK723(ΔUL34-repair), viruses were propagated and assayed in Vero-UL34 cells.
Antibodies.
Commercial mouse monoclonal antibodies to gB (H1817; Virusys), gH (52-S; American Type Culture Collection), gD (BL6; Santa Cruz Biotechnology), ICP0 (5H7; Santa Cruz Biotechnology), ICP27 (8.F.137B; Abcam), ICP8 (10A3; Chemicon), lamin A/C (636; Santa Cruz Biotechnology), ERp57 (MaP.ERp57; Santa Cruz Biotechnology), pancadherin (CH-19; Sigma), and α-tubulin (DM1A; Sigma); commercial rabbit polyclonal antibodies to CD98hc (H-300; Santa Cruz Biotechnology), calnexin (c4731; Sigma), and lamin B1 (ab16048-100; Abcam); and commercial goat polyclonal antibody to β1 integrin (N20; Santa Cruz Biotechnology) were used in this study. The rabbit polyclonal antibodies to UL12, UL34, and UL31, and mouse polyclonal antibody to UL31 were described previously (16, 75). Rabbit polyclonal antibody to UL31 was used for immunoblotting, and mouse polyclonal antibody to UL31 was used for immunofluorescence assay. Chicken polyclonal antibody to UL34 (a generous gift from R. Roller) was described previously (4) and was used for immunofluorescence assay with mouse polyclonal antibody to UL31 or rabbit polyclonal antibody to calnexin.
Immunoblotting and immunofluorescence assay.
Immunoblotting was performed as described previously (76). Immunofluorescence assays were performed as described previously (24), except that samples were examined with an LSM5 or LSM800 laser scanning confocal microscope (Zeiss).
Measurement of colocalization in infected cells.
Images acquired by the LSM800 microscope (Zeiss) were analyzed with the colocalization function in ZEN2.1 software (Zeiss). Regions of the image that did not contain a visual signal were selected and used to threshold the image. Manders' colocalization coefficient (M) was calculated using the following equation, as described previously (77–79): M = ΣiXi.colocalized/ΣiXi, where Xi is equal to the intensity of marker X at a pixel and Xi.colocalized is the intensity of the pixels where the intensity of the other marker is greater than the threshold value. An M value of 1.0 indicates 100% colocalization, and 0 indicates 0% colocalization.
Electroporation.
The indicated expression plasmids were electroporated into HEp-2 cells with the use of a Super Electroporator NEPA21 type 2 (NEPA Gene), according to the manufacturer's instructions.
ACKNOWLEDGMENTS
We thank Tomoko Ando and Yoshie Asakura for their excellent technical assistance and Bernard Roizman and Richard Roller for providing the Us3-null mutant virus R7040 and the chicken polyclonal antibody to UL34, respectively.
This study was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS); Grants-in-Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan (16H06433, 16H06429, and 16K21723); a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) from the Japan Agency for Medical Research and Development (AMED); and grants from the Takeda Science Foundation and the Mitsubishi Foundation.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott-Williams &Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 3.Mettenleiter TC, Muller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol 15:170–178. doi: 10.1111/cmi.12044. [DOI] [PubMed] [Google Scholar]
- 4.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D. 2000. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J Virol 74:117–129. doi: 10.1128/JVI.74.1.117-129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bigalke JM, Heuser T, Nicastro D, Heldwein EE. 2014. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5:4131. doi: 10.1038/ncomms5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bigalke JM, Heldwein EE. 2015. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J 34:2921–2936. doi: 10.15252/embj.201592359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, Klupp BG, Siebert CA, Vasishtan D, Bauerlein FJ, Cheleski J, Werner S, Guttmann P, Rehbein S, Henzler K, Demmerle J, Adler B, Koszinowski U, Schermelleh L, Schneider G, Enquist LW, Plitzko JM, Mettenleiter TC, Grunewald K. 2015. Structural basis of vesicle formation at the inner nuclear membrane. Cell 163:1692–1701. doi: 10.1016/j.cell.2015.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiba C, Daikoku T, Goshima F, Takakuwa H, Yamauchi Y, Koiwai O, Nishiyama Y. 2000. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J Gen Virol 81:2397–2405. doi: 10.1099/0022-1317-81-10-2397. [DOI] [PubMed] [Google Scholar]
- 10.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci U S A 104:7241–7246. doi: 10.1073/pnas.0701757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang K, Baines JD. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc Natl Acad Sci U S A 108:14276–14281. doi: 10.1073/pnas.1108564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854–857. doi: 10.1126/science.1071506. [DOI] [PubMed] [Google Scholar]
- 14.Park R, Baines JD. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol 80:494–504. doi: 10.1128/JVI.80.1.494-504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maruzuru Y, Shindo K, Liu Z, Oyama M, Kozuka-Hata H, Arii J, Kato A, Kawaguchi Y. 2014. Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J Virol 88:7445–7454. doi: 10.1128/JVI.01057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y. 2014. Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88:4657–4667. doi: 10.1128/JVI.00137-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z, Kato A, Oyama M, Kozuka-Hata H, Arii J, Kawaguchi Y. 2015. Role of host cell p32 in herpes simplex virus 1 de-envelopment during viral nuclear egress. J Virol 89:8982–8998. doi: 10.1128/JVI.01220-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Yang Y, Wu S, Pan S, Zhou C, Ma Y, Ru Y, Dong S, He B, Zhang C, Cao Y. 2014. p32 is a novel target for viral protein ICP34.5 of herpes simplex virus type 1 and facilitates viral nuclear egress. J Biol Chem 289:35795–35805. doi: 10.1074/jbc.M114.603845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mou F, Forest T, Baines JD. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J Virol 81:6459–6470. doi: 10.1128/JVI.00380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mou F, Wills EG, Park R, Baines JD. 2008. Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus U(L)34-encoded protein to the inner nuclear membrane. J Virol 82:8094–8104. doi: 10.1128/JVI.00874-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci U S A 104:10187–10192. doi: 10.1073/pnas.0703790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mou F, Wills E, Baines JD. 2009. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J Virol 83:5181–5191. doi: 10.1128/JVI.00090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J Virol 83:3115–3126. doi: 10.1128/JVI.01462-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirohata Y, Arii J, Liu Z, Shindo K, Oyama M, Kozuka-Hata H, Sagara H, Kato A, Kawaguchi Y. 2015. Herpes simplex virus 1 recruits CD98 heavy chain and beta1 integrin to the nuclear membrane for viral de-envelopment. J Virol 89:7799–7812. doi: 10.1128/JVI.00741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, Kanai Y. 2004. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch 447:532–542. doi: 10.1007/s00424-003-1086-z. [DOI] [PubMed] [Google Scholar]
- 26.Fenczik CA, Sethi T, Ramos JW, Hughes PE, Ginsberg MH. 1997. Complementation of dominant suppression implicates CD98 in integrin activation. Nature 390:81–85. doi: 10.1038/36349. [DOI] [PubMed] [Google Scholar]
- 27.Zent R, Fenczik CA, Calderwood DA, Liu S, Dellos M, Ginsberg MH. 2000. Class- and splice variant-specific association of CD98 with integrin beta cytoplasmic domains. J Biol Chem 275:5059–5064. doi: 10.1074/jbc.275.7.5059. [DOI] [PubMed] [Google Scholar]
- 28.Feral CC, Nishiya N, Fenczik CA, Stuhlmann H, Slepak M, Ginsberg MH. 2005. CD98hc (SLC3A2) mediates integrin signaling. Proc Natl Acad Sci U S A 102:355–360. doi: 10.1073/pnas.0404852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prager GW, Feral CC, Kim C, Han J, Ginsberg MH. 2007. CD98hc (SLC3A2) interaction with the integrin beta subunit cytoplasmic domain mediates adhesive signaling. J Biol Chem 282:24477–24484. doi: 10.1074/jbc.M702877200. [DOI] [PubMed] [Google Scholar]
- 30.Ito Y, Komada H, Kusagawa S, Tsurudome M, Matsumura H, Kawano M, Ohta H, Nishio M. 1992. Fusion regulation proteins on the cell surface: isolation and characterization of monoclonal antibodies which enhance giant polykaryocyte formation in Newcastle disease virus-infected cell lines of human origin. J Virol 66:5999–6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohta H, Tsurudome M, Matsumura H, Koga Y, Morikawa S, Kawano M, Kusugawa S, Komada H, Nishio M, Ito Y. 1994. Molecular and biological characterization of fusion regulatory proteins (FRPs): anti-FRP mAbs induced HIV-mediated cell fusion via an integrin system. EMBO J 13:2044–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohgimoto S, Tabata N, Suga S, Nishio M, Ohta H, Tsurudome M, Komada H, Kawano M, Watanabe N, Ito Y. 1995. Molecular characterization of fusion regulatory protein-1 (FRP-1) that induces multinucleated giant cell formation of monocytes and HIV gp160-mediated cell fusion. FRP-1 and 4F2/CD98 are identical molecules. J Immunol 155:3585–3592. [PubMed] [Google Scholar]
- 33.Okamoto K, Tsurudome M, Ohgimoto S, Kawano M, Nishio M, Komada H, Ito M, Sakakura Y, Ito Y. 1997. An anti-fusion regulatory protein-1 monoclonal antibody suppresses human parainfluenza virus type 2-induced cell fusion. J Gen Virol 78:83–89. doi: 10.1099/0022-1317-78-1-83. [DOI] [PubMed] [Google Scholar]
- 34.Ye GJ, Roizman B. 2000. The essential protein encoded by the UL31 gene of herpes simplex virus 1 depends for its stability on the presence of UL34 protein. Proc Natl Acad Sci U S A 97:11002–11007. doi: 10.1073/pnas.97.20.11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banks LM, Halliburton IW, Purifoy DJ, Killington RA, Powell KL. 1985. Studies on the herpes simplex virus alkaline nuclease: detection of type-common and type-specific epitopes on the enzyme. J Gen Virol 66:1–14. doi: 10.1099/0022-1317-66-1-1. [DOI] [PubMed] [Google Scholar]
- 36.Leckband D, Prakasam A. 2006. Mechanism and dynamics of cadherin adhesion. Annu Rev Biomed Eng 8:259–287. doi: 10.1146/annurev.bioeng.8.061505.095753. [DOI] [PubMed] [Google Scholar]
- 37.Stuurman N, Heins S, Aebi U. 1998. Nuclear lamins: their structure, assembly, and interactions. J Struct Biol 122:42–66. doi: 10.1006/jsbi.1998.3987. [DOI] [PubMed] [Google Scholar]
- 38.Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J Virol 78:399–412. doi: 10.1128/JVI.78.1.399-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirohata Y, Kato A, Oyama M, Kozuka-Hata H, Koyanagi N, Arii J, Kawaguchi Y. 2015. Interactome analysis of herpes simplex virus 1 envelope glycoprotein H. Microbiol Immunol 59:331–337. doi: 10.1111/1348-0421.12255. [DOI] [PubMed] [Google Scholar]
- 40.Imai T, Sagou K, Arii J, Kawaguchi Y. 2010. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J Virol 84:153–162. doi: 10.1128/JVI.01447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wada I, Rindress D, Cameron PH, Ou WJ, Doherty JJ II, Louvard D, Bell AW, Dignard D, Thomas DY, Bergeron JJ. 1991. SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem 266:19599–19610. [PubMed] [Google Scholar]
- 42.Ellgaard L, Ruddock LW. 2005. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep 6:28–32. doi: 10.1038/sj.embor.7400311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.English AR, Voeltz GK. 2013. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb Perspect Biol 5:a013227. doi: 10.1101/cshperspect.a013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wills E, Mou F, Baines JD. 2009. The U(L)31 and U(L)34 gene products of herpes simplex virus 1 are required for optimal localization of viral glycoproteins D and M to the inner nuclear membranes of infected cells. J Virol 83:4800–4809. doi: 10.1128/JVI.02431-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tolonen N, Doglio L, Schleich S, Krijnse Locker J. 2001. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol Biol Cell 12:2031–2046. doi: 10.1091/mbc.12.7.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 81:11861–11869. doi: 10.1128/JVI.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suhy DA, Giddings TH Jr, Kirkegaard K. 2000. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol 74:8953–8965. doi: 10.1128/JVI.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Romero-Brey I, Bartenschlager R. 2016. Endoplasmic reticulum: the favorite intracellular niche for viral replication and assembly. Viruses 8:E160. doi: 10.3390/v8060160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chan YK, Gack MU. 2016. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol 14:360–373. doi: 10.1038/nrmicro.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Campadelli G, Brandimarti R, Di Lazzaro C, Ward PL, Roizman B, Torrisi MR. 1993. Fragmentation and dispersal of Golgi proteins and redistribution of glycoproteins and glycolipids processed through the Golgi apparatus after infection with herpes simplex virus 1. Proc Natl Acad Sci U S A 90:2798–2802. doi: 10.1073/pnas.90.7.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwarz DS, Blower MD. 2016. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci 73:79–94. doi: 10.1007/s00018-015-2052-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borgese N. 2016. Getting membrane proteins on and off the shuttle bus between the endoplasmic reticulum and the Golgi complex. J Cell Sci 129:1537–1545. doi: 10.1242/jcs.183335. [DOI] [PubMed] [Google Scholar]
- 55.Ungricht R, Klann M, Horvath P, Kutay U. 2015. Diffusion and retention are major determinants of protein targeting to the inner nuclear membrane. J Cell Biol 209:687–703. doi: 10.1083/jcb.201409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boni A, Politi AZ, Strnad P, Xiang W, Hossain MJ, Ellenberg J. 2015. Live imaging and modeling of inner nuclear membrane targeting reveals its molecular requirements in mammalian cells. J Cell Biol 209:705–720. doi: 10.1083/jcb.201409133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mattaj IW. 2004. Sorting out the nuclear envelope from the endoplasmic reticulum. Nat Rev Mol Cell Biol 5:65–69. doi: 10.1038/nrm1263. [DOI] [PubMed] [Google Scholar]
- 58.Lusk CP, Blobel G, King MC. 2007. Highway to the inner nuclear membrane: rules for the road. Nat Rev Mol Cell Biol 8:414–420. doi: 10.1038/nrm2165. [DOI] [PubMed] [Google Scholar]
- 59.Padmakumar VC, Libotte T, Lu W, Zaim H, Abraham S, Noegel AA, Gotzmann J, Foisner R, Karakesisoglou I. 2005. The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J Cell Sci 118:3419–3430. doi: 10.1242/jcs.02471. [DOI] [PubMed] [Google Scholar]
- 60.Glick BS, Nakano A. 2009. Membrane traffic within the Golgi apparatus. Annu Rev Cell Dev Biol 25:113–132. doi: 10.1146/annurev.cellbio.24.110707.175421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansen TH, Bouvier M. 2009. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol 9:503–513. doi: 10.1038/nri2575. [DOI] [PubMed] [Google Scholar]
- 62.Tomazin R, Boname J, Hegde NR, Lewinsohn DM, Altschuler Y, Jones TR, Cresswell P, Nelson JA, Riddell SR, Johnson DC. 1999. Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat Med 5:1039–1043. doi: 10.1038/12478. [DOI] [PubMed] [Google Scholar]
- 63.Chaudhry A, Verghese DA, Das SR, Jameel S, George A, Bal V, Mayor S, Rath S. 2009. HIV-1 Nef promotes endocytosis of cell surface MHC class II molecules via a constitutive pathway. J Immunol 183:2415–2424. doi: 10.4049/jimmunol.0804014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abendroth A, Lin I, Slobedman B, Ploegh H, Arvin AM. 2001. Varicella-zoster virus retains major histocompatibility complex class I proteins in the Golgi compartment of infected cells. J Virol 75:4878–4888. doi: 10.1128/JVI.75.10.4878-4888.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bishop GA, Glorioso JC, Schwartz SA. 1983. Relationship between expression of herpes simplex virus glycoproteins and susceptibility of target cells to human natural killer activity. J Exp Med 157:1544–1561. doi: 10.1084/jem.157.5.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Imai T, Arii J, Minowa A, Kakimoto A, Koyanagi N, Kato A, Kawaguchi Y. 2011. Role of the herpes simplex virus 1 Us3 kinase phosphorylation site and endocytosis motifs in the intracellular transport and neurovirulence of envelope glycoprotein B. J Virol 85:5003–5015. doi: 10.1128/JVI.02314-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J Virol 83:250–261. doi: 10.1128/JVI.01451-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J Virol 82:5198–5211. doi: 10.1128/JVI.02681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, Spear PG, Lanier LL, Arase H. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944. doi: 10.1016/j.cell.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liang L, Tanaka M, Kawaguchi Y, Baines JD. 2004. Cell lines that support replication of a novel herpes simplex virus 1 UL31 deletion mutant can properly target UL34 protein to the nuclear rim in the absence of UL31. Virology 329:68–76. doi: 10.1016/j.virol.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 72.Purves FC, Spector D, Roizman B. 1991. The herpes simplex virus 1 protein kinase encoded by the US3 gene mediates posttranslational modification of the phosphoprotein encoded by the UL34 gene. J Virol 65:5757–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arii J, Uema M, Morimoto T, Sagara H, Akashi H, Ono E, Arase H, Kawaguchi Y. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor alpha. J Virol 83:4520–4527. doi: 10.1128/JVI.02601-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fujii H, Mugitani M, Koyanagi N, Liu Z, Tsuda S, Arii J, Kato A, Kawaguchi Y. 2014. Role of the nuclease activities encoded by herpes simplex virus 1 UL12 in viral replication and neurovirulence. J Virol 88:2359–2364. doi: 10.1128/JVI.03621-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol 71:7328–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khandelwal P, Ruiz WG, Balestreire-Hawryluk E, Weisz OA, Goldenring JR, Apodaca G. 2008. Rab11a-dependent exocytosis of discoidal/fusiform vesicles in bladder umbrella cells. Proc Natl Acad Sci U S A 105:15773–15778. doi: 10.1073/pnas.0805636105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fichter KM, Flajolet M, Greengard P, Vu TQ. 2010. Kinetics of G-protein-coupled receptor endosomal trafficking pathways revealed by single quantum dots. Proc Natl Acad Sci U S A 107:18658–18663. doi: 10.1073/pnas.1013763107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dunn KW, Kamocka MM, McDonald JH. 2011. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]