

ABSTRACT
Recent studies have reported that host microRNAs (miRNAs) regulate infections by several types of viruses via various mechanisms and that inhibition of the miRNA processing factors enhances or prevents viral infection. However, it has not been clarified whether these effects of miRNAs extend to adenovirus (Ad) infection. Here we show that miR-27a and -b efficiently inhibit infection with an Ad via the downregulation of SNAP25 and TXN2, which are members of the SNARE proteins and the thioredoxin family, respectively. Approximately 80% reductions in Ad genomic copy number were found in cells transfected with miR-27a/b mimics, whereas there were approximately 2.5- to 5-fold larger copy numbers of the Ad genome following transfection with miR-27a/b inhibitors. Microarray gene expression analysis and in silico analysis demonstrated that SNAP25 and TXN2 are target genes of miR-27a/b. A reporter assay using plasmids containing the 3′ untranslated regions of the SNAP25 and TXN2 genes showed that miR-27a/b directly suppressed SNAP25 and TXN2 expression through posttranscriptional gene silencing. Knockdown of SNAP25 led to a significant inhibition of Ad entry into cells. Knockdown of TXN2 induced cell cycle arrest at G1 phase, leading to a reduction in Ad replication. In addition, overexpression of Ad-encoded small noncoding RNAs (VA-RNAs) restored the miR-27a/b-mediated reduction in infection level with a VA-RNA-lacking Ad mutant due to the VA-RNA-mediated inhibition of miR-27a/b expression. These results indicate that miR-27a and -b suppress SNAP25 and TXN2 expression via posttranscriptional gene silencing, leading to efficient suppression of Ad infection.
IMPORTANCE Adenovirus (Ad) is widely used as a platform for replication-incompetent Ad vectors (Adv) and replication-competent oncolytic Ad (OAd) in gene therapy and virotherapy. Regulation of Ad infection is highly important for efficient gene therapies using both Adv and OAd. In this study, we demonstrate that miR-27a and -b, which are widely expressed in host cells, suppress SNAP25 and TXN2 expression through posttranscriptional gene silencing. Suppression of SNAP25 and TXN2 expression leads to inhibition of Ad entry into cells and to cell cycle arrest, respectively, leading to efficient suppression of Ad infection. Our findings provide important clues to the improvement of gene therapies using both Adv and OAd.
KEYWORDS: SNAP25, TXN2, adenoviruses, miR-27, posttranscriptional gene silencing
INTRODUCTION
MicroRNAs (miRNAs) are small noncoding RNAs of approximately 22 nucleotides (nt) that regulate various biological phenomena via posttranscriptional gene silencing. miRNAs are initially transcribed as long pri-miRNAs, followed by processing by the RNase III enzyme Drosha, producing pre-miRNAs. The pre-miRNAs are then exported from the nucleus to the cytoplasm and processed by Dicer, producing miRNAs. Finally, the miRNAs are incorporated into the RNA-induced silencing complex (RISC), which inhibits translation of mRNAs possessing sequences complementary to the miRNAs (1).
Several previous reports demonstrated that host miRNAs inhibited viral infection via various mechanisms (2–5). Triboulet et al. showed that a host miR-17/92 cluster directly suppressed expression of the cellular p300/CREB-binding protein (CBP)-associated factor (PCAF), leading to inhibition of HIV-1 replication (3). Viral genomes as well as host genes are also targeted by host miRNAs. Otsuka et al. demonstrated that host miR-24 and miR-93 directly targeted the genome of vesicular stomatitis virus (VSV) via posttranscriptional gene silencing, leading to inhibition of VSV replication (4). Various types of viruses encode inhibitors of general or specific miRNAs in order to circumvent miRNA-mediated inhibition of virus infection (2, 6–12). Moreover, knockdown of cellular factors involved in miRNA-mediated posttranscriptional gene silencing, including Dicer, also reduces miRNA expression levels, leading to enhancement of infection with various types of viruses, including mouse cytomegalovirus (MCMV) and VSV (2, 4, 5). Thus, numerous miRNAs that regulate virus infection have been reported, but the mechanisms of miRNA-mediated regulation of virus infection have not been clarified completely. Clarification of the mechanisms of miRNA-mediated regulation of virus infection would lead to identification of not only novel genes involved in virus infection but also novel targets for the development of antiviral agents.
Adenoviruses (Ad), which are nonenveloped icosahedral viruses with double-stranded DNA genomes, often infect the gastrointestinal and respiratory tracts, causing a wide range of illnesses, such as colds, sore throat, and diarrhea (13). In addition, Ad, particularly those of Ad serotype 5, are widely used as a framework for replication-incompetent Ad vectors and oncolytic Ad (14). Infection with Ad is also considered to be regulated by certain miRNAs because Ad, like other viruses, possesses an miRNA inhibitor system. Ad expresses Ad-encoded small noncoding RNAs (VA-RNAs), which inhibit production of miRNAs (15–23). VA-RNAs were originally demonstrated to inhibit double-stranded RNA-dependent kinase (PKR), leading to efficient translation of viral proteins (24–27); more recently, however, VA-RNAs were reported to inhibit miRNA production (15–23). VA-RNAs inhibit Dicer expression by inhibiting nuclear export of Dicer mRNA, leading to suppression of miRNA production (15). In addition, VA-RNAs are exported from the nucleus by exportin-5 and processed by Dicer, producing VA-RNA-derived miRNAs (mivaRNAs) (17, 18, 21, 22). Competitive inhibition occurs between the processing of miRNA precursors and VA-RNAs via the steps described above (16–22). We also recently demonstrated that Ago2 knockdown led to significant promotion of Ad infection (28), suggesting that miRNA-mediated posttranscriptional silencing inhibits Ad infection; however, it remained to be revealed how and which miRNAs inhibit Ad infection. Elucidation of the mechanism of miRNA-mediated regulation of Ad infection may lead to the development of efficient therapies for Ad-related diseases as well as the development of novel Ad vectors and oncolytic Ad.
In this study, we focused on miR-27 as an miRNA crucial for the regulation of Ad infection, because miR-27 has been reported to be involved in infections with other viruses, including MCMV and herpesvirus saimiri (HVS). In addition, these viruses encode inhibitors of miR-27 (10–12). Ad also expresses miRNA inhibitors, or VA-RNAs, as described above. Qi et al. reported that various miRNAs, including miR-27, were downregulated following infection with viruses of Ad serotype 3 (29). In addition, miR-27 is expressed in a variety of cells and is involved in various cell activities (30–32). These findings led us to consider that miR-27 might regulate Ad infection. We demonstrate here that miR-27 dramatically inhibits Ad infection by suppressing the expression of the miR-27 target genes, encoding SNAP25 and TXN2.
RESULTS
miR-27a/b-mediated inhibition of Ad infection.
In order to examine whether miR-27a and -b are involved in Ad infection, HeLa cells, H1299 cells, human umbilical vein endothelial cells (HUVECs), and normal human lung fibroblasts (NHLFs) were transfected with miR-27a/b mimics or inhibitors, followed by infection with wild-type Ad (WT-Ad). We previously demonstrated that miR-27a and -b are expressed in HeLa cells (28). Detectable levels of miR-27a/b were also expressed in the other cells used in this study (Fig. 1A). The copy number of the WT-Ad genome was reduced more than 80% in HeLa cells transfected with miR-27a/b mimics (Fig. 1B). Even with 1 nM miR-27a/b mimics, we observed highly efficient inhibition of Ad infection. In contrast, an increase in the copy number of the WT-Ad genome was observed in HeLa cells transfected with miR-27a/b inhibitors, in a dose-dependent manner (Fig. 1C). Similar results were found with H1299 cells and two types of normal cells, HUVECs and NHLFs (Fig. 1D). Not only the WT-Ad genome copy numbers but also infectious unit (IFU) titers of the WT-Ad progeny were decreased and increased in HeLa cells transfected with miR-27a/b mimics and inhibitors, respectively (Fig. 1E). Overexpression of either precursor of miR-27a/b, pre-miR-27a or -b, also significantly decreased the genome copy numbers of WT-Ad (Fig. 1F). These results indicated that miR-27a/b negatively regulated Ad infection.
FIG 1.

Inhibition of Ad infection by miR-27a/b. (A) The copy numbers of miR-27a/b in HeLa cells, H1299 cells, HUVECs, and NHLFs were determined by quantitative RT-PCR analysis. (B to D) HeLa cells were transfected with miR-27a/b mimics (B) or inhibitors (C) at the indicated doses, or H1299 cells, HUVECs, and NHLFs were transfected with miR-27a/b mimics or inhibitors at 20 nM (D). The cells were then infected with WT-Ad at 100 VP/cell. After 24 h of incubation, the copy numbers of WT-Ad genomic DNA in the cells were determined by quantitative PCR analysis. (E) HeLa cells were transfected with miR-27a/b mimics or inhibitors at 20 nM, followed by infection with WT-Ad at 100 VP/cell. After 24 h of incubation, IFU titers of the WT-Ad progeny in the cells were determined by infectious titer assay. (F) HeLa cells were transfected with a control plasmid (pHM5-U6) or a pre-miR-27a/b-expressing plasmid (pHM5-U6-pre-miR-27a or -b), followed by infection with WT-Ad at 100 VP/cell. After 24 h of incubation, the copy numbers of WT-Ad genomic DNA in the cells were determined. The data are expressed as means and SD (n = 3 or 4). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Identification of miR-27a/b target genes involved in Ad infection.
In order to identify miR-27a/b target genes, we performed in silico analysis using a sequence-based miRNA target prediction program, TargetScan, and a microarray gene expression assay using RNA samples from HeLa cells transfected with each miR-27a/b mimic to search out miR-27a/b target genes. Among the top 50 genes listed by the in silico analysis using TargetScan (33), we searched for genes that were significantly downregulated in miR-27a/b mimic-transfected cells in the microarray analysis. These analyses yielded 24 genes as putative miR-27a/b target genes (Fig. 2A). To examine which of these 24 genes are actually targeted by miR-27a/b, the mRNA levels of the 24 putative target genes were evaluated by quantitative reverse transcription-PCR (RT-PCR) analysis following transfection with miR-27a/b mimics. The results showed that the mRNA levels of 11 of the 24 genes were significantly reduced by both of the miR-27a/b mimics (Fig. 2B). In order to examine the effects of these 11 genes on Ad infection, HeLa cells were transfected with small interfering RNAs (siRNAs) against mRNAs of the 11 genes, followed by infection with WT-Ad. We confirmed that transfection with the siRNAs induced significant knockdown of the target genes (Table 1). Significant reductions in the WT-Ad genomic copy number were found following transfection with siRNAs against GOLM1 (siGOLM1), SNAP25 (siSNAP25), and TXN2 (siTXN2) (Fig. 2C). Treatment with several siRNAs (e.g., siRNAs against TMUB1 and TMBIM6) led to significant increases in the WT-Ad genomic copy number (Fig. 2C). siRNAs targeting different regions of the same gene mRNAs (siSNAP25#2 and siTXN2#2) mediated significant decreases in the WT-Ad genomic copy number, whereas siGOLM1#2 had no significant effects on the WT-Ad genomic copy number in the cells (Fig. 2D). The WT-Ad genomic copy number was also reduced by knockdown of SNAP25 or TXN2 in HUVECs and NHLFs (Fig. 2E). Western blotting demonstrated significant knockdown of GOLM1, SNAP25, and TXN2 at the protein level following transfection with the respective siRNAs (Fig. 2F to H). On the other hand, cotransfection with SNAP25- or TXN2-expressing plasmids partly restored the miR-27a/b-mediated inhibition of Ad infection (Fig. 2I). These results suggested that the miR-27a/b-mediated suppression of SNAP25 and TXN2 expression led to the reduction in Ad infection.
FIG 2.

Identification of miR-27a/b target genes involved in Ad infection. (A) Work flow for the identification of miR-27a/b target genes. Microarray and in silico analyses were performed for the identification of miR-27a/b target genes. (B) HeLa cells were transfected with miR-27a/b mimics at 20 nM. After 48 h of incubation, expression levels of the putative target genes were determined by quantitative RT-PCR analysis. (C to E) HeLa cells (C and D), HUVECs (E), and NHLFs (E) were transfected with the indicated siRNAs at 50 nM, followed by infection with WT-Ad at 100 VP/cell. After 24 h of incubation, the copy numbers of WT-Ad genomic DNA in the cells were determined by quantitative PCR analysis. The data are expressed as means and SD (n = 3 or 4). (F to H) HeLa cells were transfected with the indicated siRNAs at 50 nM. After 48 h of incubation, GOLM1 (F), SNAP25 (G), and TXN2 (H) protein levels were determined by Western blotting. (I) HeLa cells were cotransfected with miR-27a/b mimics and a SNAP25- or TXN2-expressing plasmid (pSNAP25 or pTXN2), followed by infection with WT-Ad at 100 VP/cell. After 24 h of incubation, the copy numbers of WT-Ad genomic DNA in the cells were determined by quantitative PCR analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
TABLE 1.
siRNAs used in this study
| siRNA no. | Name | Target sequence (5′–3′) or product name | Company | Knockdown efficiencya |
|---|---|---|---|---|
| 1 | siGOLM1 | AACAAGCTGTACCAGGACGAA | Gene Design | 0.02 |
| 2 | siSNAP25 | GTTGGATGAGCAAGGCGAA | Gene Design | 0.37 |
| 3 | siSNAP25#2 | GGGTAACAAATGATGCCCG | Gene Design | 0.51 |
| 4 | siNR2F6 | CGGUGCUGGGCAUCGACAATT | Gene Design | 0.24 |
| 5 | siADORA2B | GUAUCUAGCUAAUAUGUAUUU | Gene Design | 0.50 |
| 6 | siTMUB1 | CCTCAATGATTCAGAGCAG | Gene Design | 0.09 |
| 7 | siPDK4 | GGACGTAAGAGATTCTCAT | Gene Design | 0.23 |
| 8 | siSLC7A11 | AAATGCCCAGATATGCATCGT | Gene Design | 0.59 |
| 9 | siTXN2#2 | GGATGGACCTGACTTTCAA | Gene Design | 0.04 |
| 10 | siSNAP23 | CGCATAACTAATGATGCCA | Gene Design | |
| 10 | siSTX4 | GCAACTCAATGCAGTCCGATT | Gene Design | |
| 10 | siANKRD40 | Hs_ANKRD40_1 FlexiTube siRNA | Qiagen | 0.13 |
| 11 | siGPAM | Hs_GPAM_6 FlexiTube siRNA | Qiagen | 0.15 |
| 12 | siTXN2 | Hs_TXN2_2 FlexiTube siRNA | Qiagen | 0.02 |
| 13 | siTMBIM6 | Hs_TEGT_6 FlexiTube siRNA | Qiagen | 0.01 |
| 14 | siPAPSS2 | Hs_PAPSS2_1 FlexiTube siRNA | Qiagen | 0.06 |
| 15 | siSFXN2 | Hs_SFXN2_6 FlexiTube siRNA | Qiagen | 0.17 |
| 16 | siGOLM1#2 | Hs_GOLPH2_5 FlexiTube siRNA | Qiagen | 0.09 |
Determined in HeLa cells following transfection with the indicated siRNA at 50 nM. mRNA levels in siControl-transfected cells were normalized to 1.
miR-27a/b-mediated suppression of SNAP25 and TXN2 expression via posttranscriptional gene silencing.
In order to examine whether miR-27a and -b directly regulate SNAP25 and TXN2 gene expression via posttranscriptional gene silencing, a reporter assay was performed using psiCHECK-2-SNAP25-3′UTR and -TXN2-3′UTR (Fig. 3A). miR-27a/b mimics significantly suppressed the expression levels of the reporter genes containing the 3′ UTR of the SNAP25 or TXN2 gene (Fig. 3B). The reporter gene expression was restored by introducing mutations in the predicted seed-matched sequence of the miR-27a/b binding site of each 3′ UTR (Fig. 3A and B). The protein levels of SNAP25 and TXN2 were also reduced by transfection with miR-27a/b mimics (Fig. 3C and D), similarly to those following transfection with siSNAP25 or siTXN2 (Fig. 2G and H). These results indicated that miR-27a/b directly suppressed SNAP25 and TXN2 gene expression via posttranscriptional gene silencing.
FIG 3.

Suppression of SNAP25 and TXN2 expression by miR-27a/b mimics. (A) Schematic diagrams of reporter gene expression cassettes containing wild-type and mutated 3′ UTRs of the SNAP25 and TXN2 genes downstream of the Renilla luciferase (RLuc) gene (psiCHECK-2-SNAP25-3′UTR, -SNAP25-3′UTRmut, -TXN2-3′UTR, and -TXN2-3′UTRmut). (B) HeLa cells were cotransfected with miR-27a/b mimics and a control plasmid (psiCHECK-2) or the indicated reporter plasmids. After 48 h of incubation, luciferase activities in the cells were determined. The data show RLuc activities normalized to firefly luciferase (FLuc) activities. The data are expressed as means and SD (n = 3 or 4). (C and D) HeLa cells were transfected with miR-27a/b mimics at 20 nM. After 48 h of incubation, SNAP25 (C) and TXN2 (D) protein levels were determined by Western blotting. ***, P < 0.001.
Inhibition of Ad entry in SNAP25-knockdown cells.
Next, we examined the roles of SNAP25 in Ad infection. SNAP25 is a component of the SNARE complex that mediates membrane fusion events, such as exocytosis and endocytosis (34). In Ad infection, Ad is internalized into cells by clathrin-dependent endocytosis (35). We hypothesized that miR-27a/b-mediated suppression of SNAP25 expression leads to inhibition of Ad entry into cells. A flow cytometric analysis demonstrated that the expression level of coxsackievirus-adenovirus receptor (CAR), a primary receptor of Ad, on the cell surface was not altered by SNAP25 knockdown (Fig. 4A). In order to examine the involvement of SNAP25 in Ad entry, the genome copy number of internalized WT-Ad was determined at an early time point after WT-Ad infection. Three hours after infection with WT-Ad, the copy number of the WT-Ad genome was reduced by approximately 50% and 30% in cells transfected with miR-27a/b mimics and siSNAP25, respectively (Fig. 4B). Similar results were found with normal cells (HUVECs) (Fig. 4C). In order to exclude the possibility that SNAP25 knockdown altered the replication levels of internalized Ad, SNAP25-knockdown HeLa cells were transduced with a replication-incompetent Ad vector expressing luciferase (Ad-Luc). The transduction efficiencies of Ad-Luc were significantly reduced by SNAP25 knockdown (Fig. 4D), suggesting that the internalization of Ad into cells was suppressed by SNAP25 knockdown. Next, in order to perform imaging analysis of Ad entry into cells, SNAP25-knockdown HeLa cells were infected with a Cy3-labeled Ad. The cells were also transfected with pGFP-Rab7, resulting in green fluorescent protein (GFP) labeling of Rab7 as a late endosomal marker. One hour after infection with the Cy3-labeled Ad, this Ad was efficiently internalized into cells and mainly localized to the late endosome in siControl-transfected cells (Fig. 4E). On the other hand, the amounts of Cy3-labeled Ad in SNAP25-knockdown cells were lower than those in control cells. Cy3-labeled Ad was not localized to the GFP-labeled late endosome 1 h after infection of SNAP25-knockdown cells (Fig. 4E). We next used a noninfectious ts1 mutant Ad (36) produced by virus amplification at a nonpermissive temperature (39°C). When the ts1 mutant Ad is grown at 39°C, a mutant Ad which is not able to escape from the endosomes to the cytoplasm after internalization into cells is produced (36). The copy number of the ts1 mutant Ad genome was also decreased by approximately 50% by SNAP25 knockdown (Fig. 4F). These results suggested that SNAP25 was involved in a viral entry step prior to endosomal escape. In order to further examine the involvement of SNAP25 in Ad entry into cells, HeLa cells were treated with a recombinant Clostridium botulinum BONT-E light chain (BONT-E LC), which cleaves the SNAP25 protein (37). A significant reduction of the Ad genomic copy number was found following treatment with BONT-E LC 24 h after infection (Fig. 4G). Next, we examined the effects of other SNARE proteins, SNAP23 and STX4, on Ad infection. Western blotting demonstrated a significant knockdown of SNAP23 and STX4 at the protein level following transfection with siRNAs (Fig. 4H and I). Treatment with siRNAs against SNAP23 (siSNAP23) and STX4 (siSTX4) led to significant reductions in the WT-Ad genomic copy number (Fig. 4J). These results indicated that knockdown of SNAP25 led to the inhibition of Ad infection at the step of viral entry into cells.
FIG 4.

Inhibition of Ad entry by knockdown of SNAP25. (A) HeLa cells were transfected with siSNAP25 at 50 nM. After 48 h of incubation, the cells were collected and analyzed by flow cytometry for the expression of coxsackievirus-adenovirus receptor (CAR). Open histograms represent isotype control staining. Filled histograms represent specific staining with an anti-CAR antibody. MFI, mean fluorescence intensity. (B and C) HeLa cells (B) and HUVECs (C) were transfected with miR-27a/b mimics at 20 nM or with siSNAP25 at 50 nM, followed by infection with WT-Ad at 100 VP/cell. After 3 h of incubation, the copy numbers of WT-Ad genomic DNA in the cells were determined by quantitative PCR analysis. (D) HeLa cells were transfected with siSNAP25 at 50 nM, followed by transduction with Ad-Luc at 100 VP/cell. After 12 h of incubation, luciferase activities in the cells were determined. (E) HeLa cells were cotransfected with a plasmid expressing a fusion protein of GFP and Rab7 (pGFP-Rab7) and with siSNAP25 at 50 nM, followed by infection with the Cy3-labeled Ad at 100 VP/cell. After 1 h of incubation, phase-contrast, GFP fluorescence (Rab7), and Cy3 fluorescence (Ad) photomicrographs of the cells were obtained. Representative images from multiple experiments are shown. Bar, 10 μm. (F) HeLa cells were transfected with siSNAP25 at 50 nM, followed by infection with a ts1 mutant Ad at 100 VP/cell. After 3 h of incubation, the copy numbers of WT-Ad genomic DNA in the cells were determined. (G) HeLa cells were pretreated with recombinant BONT-E LC at 50 nM for 12 h, followed by infection with WT-Ad at 100 VP/cell. After 24 h of incubation, the copy numbers of WT-Ad genomic DNA in the cells were determined. (H and I) HeLa cells were transfected with the indicated siRNAs at 50 nM. After 48 h of incubation, SNAP23 (H) and STX4 (I) protein levels were determined by Western blotting. (J) HeLa cells were transfected with the indicated siRNAs at 50 nM, followed by infection with WT-Ad at 100 VP/cell. After 24 h of incubation, the copy numbers of WT-Ad genomic DNA in the cells were determined. The data are expressed as means and SD (n = 3 or 4). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Reduction in Ad replication in TXN2-knockdown cells.
Next, we examined the involvement of TXN2 in Ad infection. The mitochondrial thioredoxin TXN2 is one of the redox-active proteins (38). A previous report demonstrated that TXN2-knockout cells showed a decreased cell proliferation rate (39). In the present study, we therefore hypothesized that miR-27a/b-mediated knockdown of TXN2 would result in cell cycle arrest, leading to a reduction in Ad infection levels. The proliferation of TXN2-knockdown cells was slower than that of control cells in this study (Fig. 5A). In order to examine the cell cycle properties of TXN2-knockdown cells, we utilized HeLa-FUCCI cells, which are HeLa cells stably expressing a fluorescent ubiquitination-based cell cycle indicator (FUCCI) (40). HeLa-FUCCI cells show red, yellow, and green fluorescence in their nuclei during the G0-G1, S, and G2-M phases, respectively. A significant increase in the proportion of cells in G0-G1 phase was observed following transfection with miR-27a/b mimics (Fig. 5B) and siTXN2 (Fig. 5C), suggesting that miR-27a/b-mediated knockdown of TXN2 induced G1 arrest. In order to examine the effects of this cell cycle arrest in Ad replication, HeLa cells were treated with a cyclin D kinase 4/6 (CDK4/6) inhibitor (PD0332991) that is known to induce G1 arrest (41), followed by infection with WT-Ad. Treatment with PD0332991 led to a >40% reduction in the Ad genome copy number (Fig. 5D). Ad entry into cells was not inhibited by TXN2 knockdown (Fig. 5E). These results suggested that miR-27a/b-mediated suppression of TXN2 expression induced cell cycle arrest, leading to a reduction in Ad replication levels.
FIG 5.

Reduction in Ad replication via cell cycle arrest by knockdown of TXN. (A) HeLa cells (2 × 105 cells/well) were transfected with siTXN2 at 50 nM. At the indicated days posttransfection, the cell numbers were counted. (B and C) HeLa-FUCCI cells were transfected with miR-27a/b mimics (B) and siTXN2 (C) at 20 nM and 50 nM, respectively. After 48 h of incubation, fluorescence photomicrographs of the cells were obtained. Cells in the G0-G1, S, and G2-M phases show red, yellow, and green fluorescence, respectively. Representative images from multiple experiments are shown. Percentages of cells in each phase are shown in the bar graphs. (D) HeLa cells were pretreated with PD0332991 at 1 μM, followed by infection with WT-Ad at 100 VP/cell. After 24 h of incubation, the copy numbers of WT-Ad genomic DNA in the cells were determined by quantitative PCR analysis. DMSO, dimethyl sulfoxide. (E) HeLa cells were transfected with siTXN2 at 50 nM, followed by infection with WT-Ad at 100 VP/cell. After 3 h of incubation, the copy numbers of WT-Ad genomic DNA in the cells were determined. The data are expressed as means and SD (n = 3 or 4). ***, P < 0.001; n.s., not significant.
Prevention of miR-27a/b-mediated inhibition of Ad infection by VA-RNAs.
VA-RNAs inhibit miRNA maturation by competitive inhibition of miRNA processing in several steps, including transport by Exportin-5, cleavage by Dicer, and incorporation into the RISC (16–22). In order to examine whether VA-RNAs inhibit host miR-27a/b expression, miR-27a/b expression levels were examined in HeLa cells following transfection with a VA-RNA-expressing plasmid (pAdVAntage). Transfection with pAdVAntage resulted in an approximately 50% reduction in host miR-27a/b expression (Fig. 6A). Taking into consideration that miR-27a/b significantly inhibited Ad infection (Fig. 1), these results suggested that Ad might partly circumvent the miR-27a/b-mediated inhibition of Ad infection via VA-RNA expression. In order to examine whether VA-RNAs have impacts on the miR-27a/b-mediated inhibition of Ad infection, HeLa cells were infected with Sub720, a VA-RNA-deleted Ad, following transfection with pre-miR-27a/b-expressing plasmids. A >50% reduction in the Sub720 genome copy number was found in HeLa cells transfected with pre-miR-27a/b-expressing plasmids compared to that in cells transfected with a control plasmid (Fig. 6B). Transfection with siSNAP25 or siTXN2 led to a significant decrease in the Sub720 genomic copy number (Fig. 6C). On the other hand, cotransfection with the VA-RNA-expressing plasmid pAdVAntage restored the miR-27a/b-mediated inhibition of Ad infection (Fig. 6D). These results indicate that VA-RNAs can suppress miR-27a/b expression, leading to efficient Ad infection.
FIG 6.
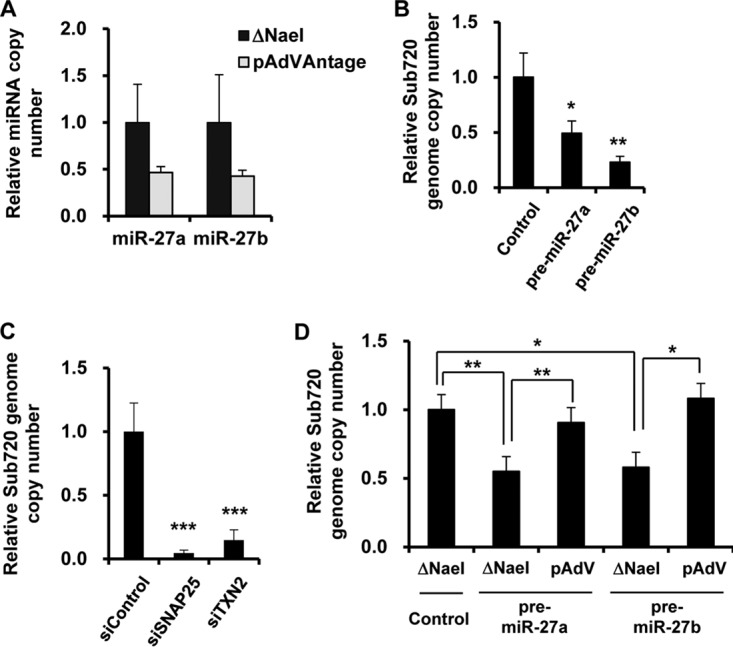
Restoration of miR-27a/b-mediated inhibition of Ad infection by overexpression of VA-RNAs. (A) HeLa cells were transfected with a control plasmid (ΔNaeI; pAdVAntage-ΔNaeI) or a VA-RNA-expressing plasmid (pAdVAntage). After 48 h of incubation, the copy numbers of miR-27a/b in the cells were determined by quantitative RT-PCR analysis. (B and C) HeLa cells were transfected with a pre-miR-27a- or -b-expressing plasmid (pHM5-U6-pre-miR-27a or -b) (B) or with siRNA at 50 nM (C), followed by infection with Sub720 at 100 VP/cell. After 24 h of incubation, the copy numbers of Sub720 genomic DNA were determined by quantitative PCR analysis. (D) HeLa cells were cotransfected with pHM5-U6-pre-miR-27a or -b and pAdVAntage, followed by infection with Sub720 at 100 VP/cell. After 24 h of incubation, the copy numbers of Sub720 genomic DNA in the cells were determined. The data are expressed as means and SD (n = 4). *, P < 0.05; **, P < 0.01.
DISCUSSION
For many viruses, the mechanism of infection is positively or negatively regulated by miRNA-mediated posttranscriptional silencing (4, 5, 42, 43); however, it has remained to be elucidated whether miRNAs regulate Ad infection. The aim of this study was to examine whether Ad infection is regulated by miRNA-mediated posttranscriptional silencing and to identify miRNAs and their target genes involved in Ad infection. Our previous studies demonstrated that Dicer-mediated cleavage of Ad-encoded small noncoding RNAs (VA-RNAs) negatively regulated Ad replication and that Dicer knockdown led to the promotion of Ad replication (28, 44). In addition, knockdown of Ago2, which is a major component of the RISC, also promoted Ad replication, although the level of Ad-replication enhancement induced by Ago2 knockdown was lower than that by Dicer knockdown. These results suggest that Ad infection is also regulated by miRNA-mediated posttranscriptional gene silencing. In this study, we demonstrated that miR-27a/b overexpression inhibited Ad infection (Fig. 1) and that miR-27a/b directly suppressed the expression of SNAP25 and TXN2 via posttranscriptional gene silencing (Fig. 2 and 3). The suppression of SNAP25 expression led to inhibition of Ad entry into cells (Fig. 4), while that of TXN2 inhibited Ad replication as a result of induction of G1 arrest (Fig. 5). These results indicate that miR-27a and -b suppress SNAP25 and TXN2 expression via posttranscriptional gene silencing, leading to efficient inhibition of Ad infection.
SNAP25 is one of the target SNARE (t-SNARE) proteins, which are located in the cellular membrane. t-SNARE proteins interact with vesicle SNARE (v-SNARE) proteins and subsequently mediate membrane fusion (45). Several previous reports demonstrated that the SNARE complex is involved in viral infection (46–48). Meier et al. reported that siRNA-mediated knockdown of the SNARE protein VAMP3 resulted in the inhibition of endocytic intracellular trafficking of a bunyavirus, Uukuniemi virus, indicating that SNARE protein-mediated membrane fusion is needed for the efficient entry of Uukuniemi virus into cells (49). Ad infection was also inhibited by knockdown of SNAP25 at the step of viral entry (Fig. 4). These findings suggest that SNARE proteins might be involved in endocytosis-dependent infections with various types of viruses and that functional inhibition of the SNARE proteins might result in the inhibition of viral infection.
The mitochondrion-specific thioredoxin TXN2 (also called Trx2) exhibits redox activities against oxidative stress, such as that from reactive oxygen species (ROS), and regulates mitochondrial apoptotic signaling. Following stimulation by various pathogens, including viral infections, excessive ROS are produced and accumulate in cells, leading to the induction of apoptosis or necrosis. In order to examine whether the reduction in Ad replication in TXN2-knockdown cells is attributable to Ad-induced ROS accumulation, we treated the TXN2-knockdown cells with an ROS inhibitor, N-acetyl-l-cysteine (NAC). Treatment with NAC did not reverse the reduction in Ad replication in TXN2-knockdown cells (data not shown), suggesting that this reduction was independent of ROS accumulation. TXN2 is also known to be involved in cell proliferation. Conrad et al. demonstrated that the growth of TXN2-knockout mouse embryonic fibroblasts (MEFs) was slower than that of wild-type MEFs (39). Consistent with their report, we found that TXN2 knockdown inhibited efficient cell growth due to cell cycle arrest (Fig. 5A to C). In addition, Ad did not replicate efficiently following cell cycle arrest (Fig. 5D).
As shown in Fig. 2C, knockdown of several genes, such as TMUB1 and TMBIM6, led to significant increases in Ad infection (Fig. 2C). TMUB1 (also known as HOPS) and TMBIM6 (also known as TEGT or BI-1) are genes associated with translational regulation and apoptosis, respectively (50, 51). It would be interesting to examine whether these genes are involved in the regulation of Ad infection.
Overexpression of VA-RNAs partly restored miR-27a/b-mediated inhibition of Ad infection (Fig. 6); however, VA-RNA-mediated antagonism of the miR-27-mediated inhibition of SNAP25 expression and Ad internalization is less likely to make a major contribution to VA-RNA-mediated upregulation of Ad infection. Although the miR-27a/b-mediated suppression of SNAP25 expression led to the inhibition of Ad infection at the step of viral entry (Fig. 4), it is difficult for VA-RNAs to enhance viral entry via suppression of SNAP25 expression, since VA-RNAs should not be expressed prior to viral internalization into cells. VA-RNAs might make a greater contribution to Ad infection as inhibitors of the miR-27a/b-mediated suppression of TXN2 expression, not SNAP25 expression, because knockdown of TXN2 led to a significant reduction in Ad replication, but not at a viral entry step (Fig. 5).
Previous studies demonstrated that miR-27a/b inhibited infections by two types of herpesviruses: MCMV and HVS (10–12). In order to counteract the miR-27a/b-mediated inhibition of MCMV and HVS infections, these viruses encode inhibitors of miR-27a/b. The MCMV-encoded inhibitor of miR-27a/b is the m169 transcript, which binds to host miR-27a/b in a sequence-dependent manner and induces the degradation of miR-27a/b (12). The degradation of miR-27a/b, in turn, leads to the efficient replication of MCMV (12); however, the mechanism of the miR-27a/b-mediated inhibition of MCMV replication remained to be clarified. Another herpesvirus, HVS, also encodes viral U-rich noncoding RNAs (HSURs), which are miR-27a/b-specific inhibitors (10, 11). In the present experiments, Ad infection was inhibited by miR-27a/b through the suppression of SNAP25 and TXN2 expression. In addition, the knockdown of SNAP25 and TXN2 suppressed infection by another type of herpesvirus, herpes simplex virus 1 (HSV-1) (M. Machitani, F. Sakurai, and H. Mizuguchi, unpublished data). These results suggest that miR-27a/b-mediated suppression of SNAP25 and TXN2 expression is involved in infection with herpesviruses, including MCMV and HVS.
Oncolytic viruses have great potential for cancer treatment, and various types of oncolytic virus have been used for oncolytic virotherapy in both basic studies and clinical trials (52, 53). In order to enhance the therapeutic efficacy of oncolytic virotherapy, various oncolytic viruses have been genetically engineered to express antitumor genes, including the genes expressing p53, interferons (IFNs), and granulocyte-macrophage colony-stimulating factor (GM-CSF) (52–54). As shown in Fig. 1, inhibition of miR-27a/b activity largely promoted Ad infection in the present study, suggesting that an oncolytic Ad expressing an inhibitor of miR-27a/b would show more efficient infection and superior oncolytic activities.
In summary, we have demonstrated that miR-27a and -b directly suppress the expression of SNAP25 and TXN2 via posttranscriptional silencing, resulting in the inhibition of Ad infection.
MATERIALS AND METHODS
Cells and reagents.
HeLa (a human epithelial carcinoma cell line; RCB0007) and HeLa-FUCCI (a HeLa cell transformant expressing FUCCI; RCB2812) (40) cells were obtained from the JCRB Cell Bank (Tokyo, Japan). The other cell lines were obtained from the American Type Culture Collection (ATCC) (Manassas, VA). HEK293 (a transformed embryonic kidney cell line), 293T (a transformed embryonic kidney cell line expressing the simian virus 40 [SV40] large T antigen), HeLa, and HeLa-FUCCI cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS), streptomycin (100 μg/ml), and penicillin (100 U/ml). H1299 (a non-small cell lung carcinoma cell line) cells were cultured in RPMI 1640 supplemented with 10% FBS, streptomycin (100 μg/ml), and penicillin (100 U/ml). Human umbilical vein endothelial cells (HUVECs) and normal human lung fibroblasts (NHLFs) were cultured in the medium recommended by the supplier (Lonza, Basel, Switzerland). PD0332991, a selective CDK4/6 inhibitor, was purchased from Sigma-Aldrich Japan (Tokyo, Japan). Recombinant C. botulinum BONT-E light chain (BONT-E LC) was purchased from R&D Systems (Minneapolis, MN). The antibodies used in this study were as follows: rabbit anti-GOLM1 (anti-GOLPH2) (EPR3606), anti-SNAP25 (EPR3274), anti-TXN2 (EPR15225), anti-SNAP23 (EPR8538), and anti-Syntaxin 4 (anti-STX4) (EPR15473) antibodies (all from Abcam, Cambridge, United Kingdom); a mouse anti-SNAP25 antibody (SP14) (Millipore, Bedford, MA); rabbit anti-TXN2 (D1C9L) (Cell Signaling Technology, Danvers, MA); mouse anti-β-actin (AC-15) (Sigma-Aldrich); and a rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) antibody (Trevigen, Gaithersburg, MD).
Viruses.
WT-Ad (Ad serotype 5) was obtained from ATCC and was propagated in HEK293 cells. A replication-incompetent Ad vector expressing luciferase (Ad-Luc) was previously constructed (55) and was propagated in HEK293 cells. Sub720 (28), a mutant Ad serotype 5 virus lacking the expression of both VA-RNA I and II, was propagated in 293T cells. A noninfectious temperature-sensitive (ts1) Ad mutant (36) (a kind gift from Andrew P. Byrnes, Food and Drug Administration, Bethesda, MD), which can be internalized into cells but cannot escape from endosomes, was propagated in HEK293 cells at 39°C as previously described (36). The adenoviruses were amplified and purified by two rounds of cesium chloride gradient ultracentrifugation, dialyzed, and stored at −80°C. The virus particle (VP) titers were determined by a spectrophotometric method (56).
Plasmids.
The pre-miR-27a- and -b-expressing plasmids, pHM5-U6-pre-miR-27a and -b, were constructed as follows. A BspMI fragment of pHM5-U6 (57) was ligated with oligonucleotides encoding pre-miR-27a, pre-miR-27a-S, and pre-miR-27a-AS, resulting in pHM5-U6-pre-miR-27a. pHM5-U6-pre-miR-27b was similarly constructed using the corresponding oligonucleotides. The sequences of the oligonucleotides are shown in Table 2. The miR-27a/b expression levels of these plasmids were confirmed by quantitative RT-PCR analysis (data not shown). A plasmid expressing a fusion protein of GFP and Rab7 (pGFP-Rab7) (58) was obtained from Addgene (plasmid 12605; Addgene, Cambridge, MA).
TABLE 2.
Oligonucleotides and primers used in this study
| Oligonucleotide no. | Name | Sequence (5′–3′)a |
|---|---|---|
| 1 | pre-miR-27a-S | CACCAGGGCTTAGCTGCTTGTGAGCAGGGTCCACACCAAGTCGTGTTCACAGTGGCTAAGTTCCGCTTTTT |
| 2 | pre-miR-27a-AS | GCATAAAAAGCGGAACTTAGCCACTGTGAACACGACTTGGTGTGGACCCTGCTCACAAGCAGCTAAGCCCT |
| 3 | pre-miR-27b-S | CACCAGAGCTTAGCTGATTGGTGAACAGTGATTGGTTTCCGCTTTGTTCACAGTGGCTAAGTTCTGCTTTTT |
| 4 | pre-miR-27b-AS | GCATAAAAAGCAGAACTTAGCCACTGTGAACAAAGCGGAAACCAATCACTGTTCACCAATCAGCTAAGCTCT |
| 5 | Ad5-F | GGGATCGTCTACCTCCTTTTGA |
| 6 | Ad5-R | GGGCAGCAGCGGATGAT |
| 7 | Ad5-probe | FAM-ACAGAAACCCGCGCTACCATACTGGAG-TAMRA |
| 8 | GAPDH-F | GGTGGTCTCCTCTGACTTCAACA |
| 9 | GAPDH-R | GTGGTCGTTGAGGGCAATG |
| 10 | GAPDH-probe | FAM-CACTCCTCCACCTTTGACGCTGGG-TAMRA |
| 11 | ts1-F | GAACCACCTACCCTTCACG |
| 12 | ts1-R | CCGCCAACATTACAGACTCG |
| 13 | RPS6KA5-F | CTCCTCACTGTCAAGCACGAG |
| 14 | RPS6KA5-R | GCCTTTTGAACGATTGTTGCCT |
| 15 | ANKRD40-F | GCGGCCTTAGGGGACATTC |
| 16 | ANKRD40-R | GTCCAGCCGTTGACCTCATT |
| 17 | GOLM1-F | TGGCCTGCATCATCGTCTTG |
| 18 | GOLM1-R | CCCTGGAACTCGTTCTTCTTCA |
| 19 | PRKX-F | GCAGGACTTTGACACGCTG |
| 20 | PRKX-R | GGCGAAGAAATGCTTGGCTG |
| 21 | GABRP-F | CTTGGCCTTCGTGTGTCTGAG |
| 22 | GABRP-R | CCGACCTCGACGTTGAACT |
| 23 | ATP6V1A-F | GGGTGCAGCCATGTATGAG |
| 24 | ATP6V1A-R | TGCGAAGTACAGGATCTCCAA |
| 25 | CSRP2-F | TGGGAGGACCGTGTACCAC |
| 26 | CSRP2-R | CCGTAGCCTTTTGGCCCATA |
| 27 | PDS5B-F | GATGTTCGCTTACTGGTAGCC |
| 28 | PDS5B-R | TCTAGCCCCTTCAACTGTCTT |
| 29 | GPAM-F | GATGTAAGCACACAAGTGAGGA |
| 30 | GPAM-R | TCCGACTCATTAGGCTTTCTTTC |
| 31 | SNAP25-F | ACCAGTTGGCTGATGAGTCG |
| 32 | SNAP25-R | CAAAGTCCTGATACCAGCATCTT |
| 33 | DCUN1D4-F | GCCGCCGCTGTCAATTTTC |
| 34 | DCUN1D4-R | AGGTTCAGCTTATTAAGGGTGTG |
| 35 | TXN2-F | CTGGTGGCCTGACTGTAACAC |
| 36 | TXN2-R | TGACCACTCGGTCTTGAAAGT |
| 37 | NR2F6-F | GAGCGGCAAGCATTACGGT |
| 38 | NR2F6-R | GGCAGGTGTAGCTGAGGTT |
| 39 | SEC62-F | CCAGCAGAAATGAGAGTAGGTG |
| 40 | SEC62-R | GAGTCAATGAAGCCCACATCA |
| 41 | CDH11-F | AGAGAGCCCAGTACACGTTGA |
| 42 | CDH11-R | TTGGCATGATAGGTCTCGTGC |
| 43 | ADORA2B-F | TGCACTGACTTCTACGGCTG |
| 44 | ADORA2B-R | GGTCCCCGTGACCAAACTT |
| 45 | TMUB1-F | GCAGCTACCGACAGCATGAG |
| 46 | TMUB1-R | ACCTGCTCTGAATCATTGAGGA |
| 47 | PDK4-F | GGAGCATTTCTCGCGCTACA |
| 48 | PDK4-R | ACAGGCAATTCTTGTCGCAAA |
| 49 | SLC7A11-F | TCTCCAAAGGAGGTTACCTGC |
| 50 | SLC7A11-R | AGACTCCCCTCAGTAAAGTGAC |
| 51 | TMBIM6-F | CATATAACCCCGTCAACGCAG |
| 52 | TMBIM6-R | GCAGCCGCCACAAACATAC |
| 53 | MBTD1-F | GGCATGGCTACCTGTGAGATG |
| 54 | MBTD1-R | GGCCAAAATGCTTGCCTTCT |
| 55 | PAPSS2-F | AGACGGAGAACCAGCAGAAAT |
| 56 | PAPSS2-R | CACACGGTACATCCTCGGAAC |
| 57 | SFXN2-F | GTCAATATCCCCATGATGCGAC |
| 58 | SFXN2-R | AGCTCTCCGGGAATGACCAA |
| 59 | TNRC18-F | TCTCTGTCGCTGAGTAACGTC |
| 60 | TNRC18-R | CCCCGTCACCATGAGGTTG |
| 61 | TXN2-3′UTR-F | CGACCTCGAGATCAGAGGATGGTGGTGCTG |
| 62 | TXN2-3′UTR-R | TTGCGGCCGCGGAGGCACCTTGAGACTTCC |
| 63 | SNAP25-3′UTRmut1-F | CCACAGTATTGTTCTTGTAAAAAGCTTACATTCCACAGAGTTACTGCCACGG |
| 64 | SNAP25-3′UTRmut1-R | CCGTGGCAGTAACTCTGTGGAATGTAAGCTTTTTACAAGAACAATACTGTGG |
| 65 | SNAP25-3′UTRmut2-F | GGAGAGAGCAATCTTGCGGATCCAACAGTGTGGATGTAAATTTTATAAGGC |
| 66 | SNAP25-3′UTRmut2-R | GCCTTATAAAATTTACATCCACACTGTTGGATCCGCAAGATTGCTCTCTCC |
| 67 | SNAP25-3′UTRmut3-F | GAATGAAATATAAAAGCTTAGATAAATATCATTATAGCATGTAATATTAAATTCCTCC |
| 68 | SNAP25-3′UTRmut3-R | GGAGGAATTTAATATTACATGCTATAATGATATTTATCTAAGCTTTTATATTTCATTC |
| 69 | TXN2-3′UTRmut1-F | CTCATTTTTTGTAGGTCACTAGTAAAGAACTTAGTCATCCCTTCCACCTC |
| 70 | TXN2-3′UTRmut1-R | GAGGTGGAAGGGATGACTAAGTTCTTTACTAGTGACCTACAAAAAATGAG |
| 71 | TXN2-3′UTRmut2-F | CATCCCTTCCACCTCCTAGTCTAGAGAACAGACCCTGGGTCC |
| 72 | TXN2-3′UTRmut2-R | GGACCCAGGGTCTGTTCTCTAGACTAGGAGGTGGAAGGGATG |
| 73 | TXN2-cloning-F | GGACCCAGGGTCTGTTCTCTAGACTAGGAGGTGGAAGGGATG |
| 74 | TXN2-cloning-R | GGACCCAGGGTCTGTTCTCTAGACTAGGAGGTGGAAGGGATG |
FAM, 6-carboxyfluorescein; TAMRA, 6-carboxytetramethylrhodamine.
The SNAP25- and TXN2-expressing plasmids, pSNAP25 and pTXN2, were constructed as follows. The fragment encoding the SNAP25 or TXN2 gene was synthesized (Greiner Bio-One, Tokyo, Japan) or amplified by a PCR using cDNA prepared from HeLa cells, respectively. These fragments were then cloned into p3XFLAG-CMV10 (Sigma-Aldrich), resulting in pSNAP25 and pTXN2. The sequences of the primers used in this study are given in Table 2.
pAdVAntage-ΔNaeI (28), a control plasmid lacking VA-RNA expression, was previously constructed using pAdVAntage (Promega, Madison, WI), which encodes VA-RNA I and II under the control of an endogenous RNA polymerase III promoter.
Transfection with siRNA, miRNA mimics, and inhibitors.
mirVana miRNA mimics and inhibitors were purchased from Life Technologies (Carlsbad, CA). Control siRNA was purchased from Qiagen (Allstars negative control siRNA; Qiagen, Hilden, Germany). Other siRNAs were obtained from Qiagen and Gene Design (Osaka, Japan). The siRNAs used in this study are shown in Table 1. Cells were transfected with the siRNAs, miRNA mimics, or inhibitors by use of Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions.
Determination of Ad genome copy number.
Total DNA, including Ad genomic DNA, was isolated from cells infected with Ad by use of a DNeasy blood and tissue kit (Qiagen). After isolation, the copy number of Ad genomic DNA was quantified using the StepOnePlus real-time PCR system (Life Technologies) as previously described (59). The sequences of the primers and probes used in this study are provided in Table 2. For determination of the Ad genome copy numbers internalized into cells, cells were treated with trypsin to remove the virus particles on the cell surface, followed by isolation of total DNA.
Infectious titer assay.
Following infection with WT-Ad, cells were recovered and subjected to 3 cycles of freezing and thawing. After centrifugation, the supernatants were added to HEK293 cells. After 48 h of incubation, the numbers of cells infected with Ad were analyzed using an Adeno-X rapid titer kit (Clontech, Mountain View, CA).
Quantitative RT-PCR analysis.
Total RNA was isolated from cells by use of Isogen (Nippon Gene, Tokyo, Japan). cDNA was synthesized using 500 ng of total RNA and a Superscript Vilo cDNA synthesis kit (Life Technologies). Quantitative RT-PCR analysis was performed using Fast SYBR green master mix (Life Technologies) and the StepOnePlus real-time PCR system (Life Technologies). The sequences of the primers used in this study are shown in Table 2.
miRNA expression levels were determined by quantitative RT-PCR analysis using a TaqMan microRNA reverse transcription kit, a TaqMan microRNA assay kit, and the StepOnePlus real-time PCR system (Life Technologies).
Microarray gene expression analysis of cells transfected with miR-27a/b mimics.
HeLa cells were transfected with miR-27a/b mimics at 20 nM. After 48 h of incubation, total RNA was isolated. Microarray gene expression analysis was performed by TaKaRa Bio (Shiga, Japan). Briefly, the quality of RNA samples was checked using an Agilent 2100 Bioanalyzer platform (Agilent Technologies, Santa Clara, CA). One hundred nanograms of each total RNA sample was amplified and labeled using an Agilent Low Input Quick Amp labeling kit (Agilent Technologies) following the manufacturer's protocol. The labeled samples were hybridized and washed using an Agilent gene expression hybridization kit and wash buffer kit (Agilent Technologies). Fluorescence signals of the hybridized Agilent microarrays were detected using an Agilent SureScan microarray scanner (Agilent Technologies). Agilent feature extraction software was used to read and process the microarray image files.
Prediction of miR-27a/b target genes.
miR-27a/b target genes and the putative binding sites in the 3′ untranslated region (3′ UTR) of each gene were predicted using TargetScan Human (33).
Reporter plasmids and reporter assay.
The reporter plasmids psiCHECK-2-SNAP25-3′UTR and -TXN2-3′UTR, which carry the sequences of the wild-type 3′ UTRs of the SNAP25 and TXN2 genes, respectively, downstream of the Renilla luciferase (RLuc) gene, were constructed as follows. A fragment containing the 3′ UTR of the SNAP25 gene was synthesized by Greiner Bio-One. The 3′ UTR of the TXN2 gene was amplified by a PCR using cDNA prepared from HeLa cells. The 3′ UTR sequences of the SNAP25 and TXN2 genes contain the miR-27a/b target sequences predicted by TargetScan. The fragments were ligated with an XhoI/NotI fragment of psiCHECK-2 (Promega, Madison, WI), resulting in psiCHECK-2-SNAP25-3′UTR and -TXN2-3′UTR. Next, the seed sequences of the predicted miR-27a/b target sites were mutated by use of a QuikChange site-directed mutagenesis kit (Agilent Technologies) and the primers SNAP25-3′UTRmut1-F/R, SNAP25-3′UTRmut2-F/R, or SNAP25-3′UTRmut3-F/R, resulting in psiCHECK-2-SNAP25-3′UTRmut. psiCHECK-2-TXN2-3′UTRmut was similarly constructed using the corresponding oligonucleotides. The sequences of the primers and oligonucleotides used in this study are provided in Table 2. HeLa cells were cotransfected with the reporter plasmids and miR-27a/b mimics. After 48 h of incubation, luciferase activities in the cells were determined using a dual-luciferase reporter assay system (Promega).
Western blotting.
Western blotting was performed as previously described (23). Briefly, whole-cell extracts were prepared and electrophoresed in 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels under reducing conditions, followed by electrotransfer to polyvinylidene difluoride (PVDF) membranes (Millipore). After blocking with 5% skim milk prepared in TBS-T (Tris-buffered saline with 0.1% Tween 20), the membranes were incubated with the primary antibodies, followed by incubation in the presence of horseradish peroxidase (HRP)-labeled anti-mouse or -rabbit IgG antibody (Cell Signaling Technology).
Determination of expression levels of CAR.
Expression levels of human CAR on the cell surface were measured by flow cytometry and quantified by use of FlowJo software (Tree Star, San Carlos, CA) as previously described (60).
Analysis of viral entry into cells by use of a fluorescence-labeled Ad.
Ad-Luc was labeled with Cy3 as previously described (61). Briefly, Ad-Luc was conjugated with Amersham Cy3 monoreactive dye (GE Healthcare, Tokyo, Japan) in sodium carbonate buffer. After 1 h of incubation at room temperature, the Cy3-labeled Ad was purified by cesium chloride gradient ultracentrifugation, dialyzed, and stored at −80°C. The VP titer was determined by a spectrophotometric method (56).
For the evaluation of viral entry into SNAP25-knockdown cells, HeLa cells were cotransfected with pGFP-Rab7 and siSNAP25 at 50 nM, followed by infection with the Cy3-labeled Ad at 100 VP/cell. After 1 h of incubation, phase-contrast, GFP fluorescence (Rab7), and Cy3 fluorescence (Ad) photomicrographs of the cells were obtained.
Statistical analysis.
Statistical significance was determined using Student's t test. Data are presented as means ± standard deviations (SD).
Accession number(s).
The microarray gene expression data have been submitted to the Gene Expression Omnibus (GEO) under accession number GSE85587.
ACKNOWLEDGMENTS
We thank Eri Shimaoka (Graduate School of Pharmaceutical Sciences, Osaka University, Osaka, Japan), Eri Hosoyamada (Graduate School of Pharmaceutical Sciences, Osaka University, Osaka, Japan), and Tomoko Yamaguchi (National Institutes of Biomedical Innovation, Health and Nutrition, Osaka, Japan) for their help.
This work was supported by grants-in-aid for scientific research (A and B) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan and by the Japan Agency for Medical Research and Development (AMED). M. Machitani and K. Wakabayashi are research fellows of the Japan Society for the Promotion of Science.
REFERENCES
- 1.Ambros V. 2004. The functions of animal microRNAs. Nature 431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 2.Skalsky RL, Cullen BR. 2010. Viruses, microRNAs, and host interactions. Annu Rev Microbiol 64:123–141. doi: 10.1146/annurev.micro.112408.134243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Triboulet R, Mari B, Lin YL, Chable-Bessia C, Bennasser Y, Lebrigand K, Cardinaud B, Maurin T, Barbry P, Baillat V, Reynes J, Corbeau P, Jeang KT, Benkirane M. 2007. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 315:1579–1582. doi: 10.1126/science.1136319. [DOI] [PubMed] [Google Scholar]
- 4.Otsuka M, Jing Q, Georgel P, New L, Chen J, Mols J, Kang YJ, Jiang Z, Du X, Cook R, Das SC, Pattnaik AK, Beutler B, Han J. 2007. Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity 27:123–134. doi: 10.1016/j.immuni.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 5.Ostermann E, Tuddenham L, Macquin C, Alsaleh G, Schreiber-Becker J, Tanguy M, Bahram S, Pfeffer S, Georgel P. 2012. Deregulation of type I IFN-dependent genes correlates with increased susceptibility to cytomegalovirus acute infection of dicer mutant mice. PLoS One 7:e43744. doi: 10.1371/journal.pone.0043744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Lu J, Han Y, Fan X, Ding SW. 2013. RNA interference functions as an antiviral immunity mechanism in mammals. Science 342:231–234. doi: 10.1126/science.1241911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maillard PV, Ciaudo C, Marchais A, Li Y, Jay F, Ding SW, Voinnet O. 2013. Antiviral RNA interference in mammalian cells. Science 342:235–238. doi: 10.1126/science.1241930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennasser Y, Le SY, Benkirane M, Jeang KT. 2005. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 22:607–619. doi: 10.1016/j.immuni.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 9.Sullivan CS, Ganem D. 2005. A virus-encoded inhibitor that blocks RNA interference in mammalian cells. J Virol 79:7371–7379. doi: 10.1128/JVI.79.12.7371-7379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cazalla D, Yario T, Steitz JA. 2010. Down-regulation of a host microRNA by a herpesvirus saimiri noncoding RNA. Science 328:1563–1566. doi: 10.1126/science.1187197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo YE, Riley KJ, Iwasaki A, Steitz JA. 2014. Alternative capture of noncoding RNAs or protein-coding genes by herpesviruses to alter host T cell function. Mol Cell 54:67–79. doi: 10.1016/j.molcel.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marcinowski L, Tanguy M, Krmpotic A, Radle B, Lisnic VJ, Tuddenham L, Chane-Woon-Ming B, Ruzsics Z, Erhard F, Benkartek C, Babic M, Zimmer R, Trgovcich J, Koszinowski UH, Jonjic S, Pfeffer S, Dolken L. 2012. Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog 8:e1002510. doi: 10.1371/journal.ppat.1002510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Echavarria M. 2008. Adenoviruses in immunocompromised hosts. Clin Microbiol Rev 21:704–715. doi: 10.1128/CMR.00052-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volpers C, Kochanek S. 2004. Adenoviral vectors for gene transfer and therapy. J Gene Med 6:S164–S171. doi: 10.1002/jgm.496. [DOI] [PubMed] [Google Scholar]
- 15.Bennasser Y, Chable-Bessia C, Triboulet R, Gibbings D, Gwizdek C, Dargemont C, Kremer EJ, Voinnet O, Benkirane M. 2011. Competition for XPO5 binding between Dicer mRNA, pre-miRNA and viral RNA regulates human Dicer levels. Nat Struct Mol Biol 18:323–327. doi: 10.1038/nsmb.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gwizdek C, Ossareh-Nazari B, Brownawell AM, Doglio A, Bertrand E, Macara IG, Dargemont C. 2003. Exportin-5 mediates nuclear export of minihelix-containing RNAs. J Biol Chem 278:5505–5508. doi: 10.1074/jbc.C200668200. [DOI] [PubMed] [Google Scholar]
- 17.Sano M, Kato Y, Taira K. 2006. Sequence-specific interference by small RNAs derived from adenovirus VAI RNA. FEBS Lett 580:1553–1564. doi: 10.1016/j.febslet.2006.01.085. [DOI] [PubMed] [Google Scholar]
- 18.Lu S, Cullen BR. 2004. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and microRNA biogenesis. J Virol 78:12868–12876. doi: 10.1128/JVI.78.23.12868-12876.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu N, Segerman B, Zhou X, Akusjarvi G. 2007. Adenovirus virus-associated RNAII-derived small RNAs are efficiently incorporated into the RNA-induced silencing complex and associate with polyribosomes. J Virol 81:10540–10549. doi: 10.1128/JVI.00885-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersson MG, Haasnoot PC, Xu N, Berenjian S, Berkhout B, Akusjarvi G. 2005. Suppression of RNA interference by adenovirus virus-associated RNA. J Virol 79:9556–9565. doi: 10.1128/JVI.79.15.9556-9565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aparicio O, Carnero E, Abad X, Razquin N, Guruceaga E, Segura V, Fortes P. 2010. Adenovirus VA RNA-derived miRNAs target cellular genes involved in cell growth, gene expression and DNA repair. Nucleic Acids Res 38:750–763. doi: 10.1093/nar/gkp1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aparicio O, Razquin N, Zaratiegui M, Narvaiza I, Fortes P. 2006. Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J Virol 80:1376–1384. doi: 10.1128/JVI.80.3.1376-1384.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Machitani M, Sakurai F, Katayama K, Tachibana M, Suzuki T, Matsui H, Yamaguchi T, Mizuguchi H. 2013. Improving adenovirus vector-mediated RNAi efficiency by lacking the expression of virus-associated RNAs. Virus Res 178:357–363. doi: 10.1016/j.virusres.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 24.O'Malley RP, Mariano TM, Siekierka J, Mathews MB. 1986. A mechanism for the control of protein synthesis by adenovirus VA RNAI. Cell 44:391–400. doi: 10.1016/0092-8674(86)90460-5. [DOI] [PubMed] [Google Scholar]
- 25.Kitajewski J, Schneider RJ, Safer B, Munemitsu SM, Samuel CE, Thimmappaya B, Shenk T. 1986. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 45:195–200. doi: 10.1016/0092-8674(86)90383-1. [DOI] [PubMed] [Google Scholar]
- 26.Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Machitani M, Yamaguchi T, Shimizu K, Sakurai F, Katayama K, Kawabata K, Mizuguchi H. 2011. Adenovirus vector-derived VA-RNA-mediated innate immune responses. Pharmaceutics 3:338–353. doi: 10.3390/pharmaceutics3030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Machitani M, Sakurai F, Wakabayashi K, Tomita K, Tachibana M, Mizuguchi H. 2016. Dicer functions as an antiviral system against human adenoviruses via cleavage of adenovirus-encoded noncoding RNA. Sci Rep 6:27598. doi: 10.1038/srep27598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi Y, Tu J, Cui L, Guo X, Shi Z, Li S, Shi W, Shan Y, Ge Y, Shan J, Wang H, Lu Z. 2010. High-throughput sequencing of microRNAs in adenovirus type 3 infected human laryngeal epithelial cells. J Biomed Biotechnol 2010:915980. doi: 10.1155/2010/915980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guttilla IK, White BA. 2009. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem 284:23204–23216. doi: 10.1074/jbc.M109.031427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin Q, Gao Z, Alarcon RM, Ye J, Yun Z. 2009. A role of miR-27 in the regulation of adipogenesis. FEBS J 276:2348–2358. doi: 10.1111/j.1742-4658.2009.06967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Q, Gallagher R, Ufret-Vincenty R, Li X, Olson EN, Wang S. 2011. Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23∼27∼24 clusters. Proc Natl Acad Sci U S A 108:8287–8292. doi: 10.1073/pnas.1105254108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedman RC, Farh KK, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. 1993. SNAP receptors implicated in vesicle targeting and fusion. Nature 362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 35.Meier O, Greber UF. 2004. Adenovirus endocytosis. J Gene Med 6(Suppl 1):S152–S163. doi: 10.1002/jgm.553. [DOI] [PubMed] [Google Scholar]
- 36.Tian J, Xu Z, Smith JS, Hofherr SE, Barry MA, Byrnes AP. 2009. Adenovirus activates complement by distinctly different mechanisms in vitro and in vivo: indirect complement activation by virions in vivo. J Virol 83:5648–5658. doi: 10.1128/JVI.00082-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Binz T, Blasi J, Yamasaki S, Baumeister A, Link E, Sudhof TC, Jahn R, Niemann H. 1994. Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J Biol Chem 269:1617–1620. [PubMed] [Google Scholar]
- 38.Tanaka T, Hosoi F, Yamaguchi-Iwai Y, Nakamura H, Masutani H, Ueda S, Nishiyama A, Takeda S, Wada H, Spyrou G, Yodoi J. 2002. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J 21:1695–1703. doi: 10.1093/emboj/21.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conrad M, Jakupoglu C, Moreno SG, Lippl S, Banjac A, Schneider M, Beck H, Hatzopoulos AK, Just U, Sinowatz F, Schmahl W, Chien KR, Wurst W, Bornkamm GW, Brielmeier M. 2004. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol Cell Biol 24:9414–9423. doi: 10.1128/MCB.24.21.9414-9423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, Imamura T, Ogawa M, Masai H, Miyawaki A. 2008. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- 41.Baughn LB, Di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R, Niesvizky R, Cho H, Ely S, Moore MA, Chen-Kiang S. 2006. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res 66:7661–7667. doi: 10.1158/0008-5472.CAN-06-1098. [DOI] [PubMed] [Google Scholar]
- 42.Sullivan CS, Ganem D. 2005. MicroRNAs and viral infection. Mol Cell 20:3–7. doi: 10.1016/j.molcel.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 43.Wilson JA, Zhang C, Huys A, Richardson CD. 2011. Human Ago2 is required for efficient microRNA 122 regulation of hepatitis C virus RNA accumulation and translation. J Virol 85:2342–2350. doi: 10.1128/JVI.02046-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Machitani M, Sakurai F, Wakabayashi K, Tachibana M, Fujiwara T, Mizuguchi H. 2017. Enhanced oncolytic activities of the telomerase-specific replication-competent adenovirus expressing short-hairpin RNA against Dicer. Mol Cancer Ther 16:251–259. doi: 10.1158/1535-7163.MCT-16-0383. [DOI] [PubMed] [Google Scholar]
- 45.Sudhof TC, Rothman JE. 2009. Membrane fusion: grappling with SNARE and SM proteins. Science 323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cepeda V, Fraile-Ramos A. 2011. A role for the SNARE protein syntaxin 3 in human cytomegalovirus morphogenesis. Cell Microbiol 13:846–858. doi: 10.1111/j.1462-5822.2011.01583.x. [DOI] [PubMed] [Google Scholar]
- 47.Liu ST, Sharon-Friling R, Ivanova P, Milne SB, Myers DS, Rabinowitz JD, Brown HA, Shenk T. 2011. Synaptic vesicle-like lipidome of human cytomegalovirus virions reveals a role for SNARE machinery in virion egress. Proc Natl Acad Sci U S A 108:12869–12874. doi: 10.1073/pnas.1109796108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nonnenmacher ME, Cintrat JC, Gillet D, Weber T. 2015. Syntaxin 5-dependent retrograde transport to the trans-Golgi network is required for adeno-associated virus transduction. J Virol 89:1673–1687. doi: 10.1128/JVI.02520-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meier R, Franceschini A, Horvath P, Tetard M, Mancini R, von Mering C, Helenius A, Lozach PY. 2014. Genome-wide small interfering RNA screens reveal VAMP3 as a novel host factor required for Uukuniemi virus late penetration. J Virol 88:8565–8578. doi: 10.1128/JVI.00388-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Della Fazia MA, Castelli M, Bartoli D, Pieroni S, Pettirossi V, Piobbico D, Viola-Magni M, Servillo G. 2005. HOPS: a novel cAMP-dependent shuttling protein involved in protein synthesis regulation. J Cell Sci 118:3185–3194. doi: 10.1242/jcs.02452. [DOI] [PubMed] [Google Scholar]
- 51.Grzmil M, Thelen P, Hemmerlein B, Schweyer S, Voigt S, Mury D, Burfeind P. 2003. Bax inhibitor-1 is overexpressed in prostate cancer and its specific down-regulation by RNA interference leads to cell death in human prostate carcinoma cells. Am J Pathol 163:543–552. doi: 10.1016/S0002-9440(10)63682-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Russell SJ, Peng KW, Bell JC. 2012. Oncolytic virotherapy. Nat Biotechnol 30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miest TS, Cattaneo R. 2014. New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol 12:23–34. doi: 10.1038/nrmicro3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim JH, Oh JY, Park BH, Lee DE, Kim JS, Park HE, Roh MS, Je JE, Yoon JH, Thorne SH, Kirn D, Hwang TH. 2006. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther 14:361–370. doi: 10.1016/j.ymthe.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 55.Mizuguchi H, Koizumi N, Hosono T, Utoguchi N, Watanabe Y, Kay MA, Hayakawa T. 2001. A simplified system for constructing recombinant adenoviral vectors containing heterologous peptides in the HI loop of their fiber knob. Gene Ther 8:730–735. doi: 10.1038/sj.gt.3301453. [DOI] [PubMed] [Google Scholar]
- 56.Mizuguchi H, Kay MA. 1998. Efficient construction of a recombinant adenovirus vector by an improved in vitro ligation method. Hum Gene Ther 9:2577–2583. [DOI] [PubMed] [Google Scholar]
- 57.Mizuguchi H, Funakoshi N, Hosono T, Sakurai F, Kawabata K, Yamaguchi T, Hayakawa T. 2007. Rapid construction of small interfering RNA-expressing adenoviral vectors on the basis of direct cloning of short hairpin RNA-coding DNAs. Hum Gene Ther 18:74–80. doi: 10.1089/hum.2006.129. [DOI] [PubMed] [Google Scholar]
- 58.Choudhury A, Dominguez M, Puri V, Sharma DK, Narita K, Wheatley CL, Marks DL, Pagano RE. 2002. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J Clin Invest 109:1541–1550. doi: 10.1172/JCI0215420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Machitani M, Sakurai F, Wakabayashi K, Nakatani K, Shimizu K, Tachibana M, Mizuguchi H. 2016. NF-kappaB promotes leaky expression of adenovirus genes in a replication-incompetent adenovirus vector. Sci Rep 6:19922. doi: 10.1038/srep19922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sakurai F, Narii N, Tomita K, Togo S, Takahashi K, Machitani M, Tachibana M, Ouchi M, Katagiri N, Urata Y, Fujiwara T, Mizuguchi H. 2016. Efficient detection of human circulating tumor cells without significant production of false-positive cells by a novel conditionally replicating adenovirus. Mol Ther Methods Clin Dev 3:16001. doi: 10.1038/mtm.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shayakhmetov DM, Gaggar A, Ni S, Li ZY, Lieber A. 2005. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J Virol 79:7478–7491. doi: 10.1128/JVI.79.12.7478-7491.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]