

ABSTRACT
The Cbl E3 ligase has been linked to the down-modulation of surface signaling responses by inducing internalization of surface receptors. The adaptor protein CIN85 is a partner of Cbl that augments many of these interactions. Previously, an interaction was demonstrated between ICP0 and CIN85, which results in the removal of epidermal growth factor receptor (EGFR) from the surface of the infected cells with a concomitant attenuation of EGFR signaling. Here, we examined whether Cbl mediates the removal of the herpes simplex virus 1 (HSV-1) entry receptor Nectin-1 from the surface of infected cells. We found the following: (i) that Cbl, Nectin-1, and the viral glycoprotein D (gD) form a complex in infected cells; (ii) that during infection Nectin-1 is removed from the surface of the infected cells but is retained on the surface of cells that have been depleted of Cbl; and (iii) that in cells infected with a ΔICP0 mutant virus, Nectin-1 remained on the cell surface. Thus, Cbl is necessary but not sufficient for the removal of Nectin-1 from the cell surface. In addition, we observed that in Cbl-depleted cells there was enhanced entry after infection. These cells were susceptible to secondary infections by HSV-1. Viral entry in CIN85-depleted cells was only moderately enhanced compared to that in the Cbl-depleted cells, suggesting that the Cbl–Nectin-1 interaction is likely the key to the downregulation of surface Nectin-1. The removal of the HSV-1 entry receptor Nectin-1 from the surface of the infected cells may be part of the strategy of the virus to efficiently spread to uninfected cells.
IMPORTANCE The Cbl E3 ligase suppresses surface signaling responses by inducing internalization of surface components. The targets of Cbl include such components as immune system receptors, growth factor receptors, adhesion, and cell-to-cell contact molecules. The immediate early protein ICP0 of herpes simplex virus 1 (HSV-1) interacts with CIN85, an adaptor protein that augments Cbl functions. The consequence of this interaction is the removal of the epidermal growth factor receptor (EGFR) from the surface of the infected cells with concomitant suppression of the EGF ligand signaling. The viral entry receptor Nectin-1 is also internalized during HSV-1 infection in a Cbl-dependent mechanism, and that increases the opportunity of the virus to spread to uninfected cells. The diversion of the Cbl/CIN85 endocytic machinery may be a strategy utilized by the virus to alter the cell surface pattern to prevent detrimental host responses.
KEYWORDS: Cbl E3 ligase, Nectin-1, endocytosis, ICP0, HSV-1, cell surface
INTRODUCTION
During herpes simplex virus 1 (HSV-1) infection, the immediate early protein ICP0 (infected cell protein 0) exerts two major sets of functions. First, ICP0 localizes in the nucleus immediately after its production and is involved in the blockage of the gene silencing machinery and provides an impediment to certain branches of innate immunity (1–4). After ICP0 accomplishes its major nuclear functions, it translocates to the cytoplasm, where it interacts with a new set of binding partners and exerts a second set of functions, presumably later in infection (1, 5–8). Although ample literature has been focused on the nuclear functions of ICP0, only a few reports have explored its cytoplasmic roles.
Earlier studies using a yeast two-hybrid screening approach identified two major cytoplasmic binding partners of ICP0. The first is elongation factor 1δ, a subunit of the EF-1 complex, which mediates the elongation of peptide chains during translation of mRNAs. The second is the adaptor protein CIN85, a partner of the E3 ubiquitin ligase Cbl, which ubiquitinates various cell surface receptors and initiates their internalization, endocytic trafficking, and sorting (5, 6, 9–14). Subsequently, it was observed that ICP0 alone, in the absence of other viral proteins, induced a reduction in the surface abundance of epidermal growth factor receptor (EGFR) and decreased the total amount of EGFR in the cells due to internalization and subsequent degradation of the receptor in endocytic compartments (6). Under these conditions EGF-dependent gene activation was blocked.
Our data demonstrate that Cbl interacts with the viral entry receptor Nectin-1. Thus, the question arises as to how Cbl influences the surface abundance of Nectin-1. Several reports have been focused on the fate of the HSV-1 receptor Nectin-1 following infection (15–17). Nectin-1 acts as the main glycoprotein D (gD) receptor in neurons and epithelial cells (18–20). Nectin-1 is a cell adhesion molecule (CAM) that accumulates at adherens junctions of epithelial cells, at contacts between cultured cells, and at synapses of neurons (21). During initial infection, HSV-1 enters either by direct fusion at the plasma membrane or by fusing its envelope with endosomal membranes after endocytosis of the virion (22–24). The pathway used by virions during entry is cell type dependent, but in all cases four essential glycoproteins are required (22). Binding of gD to a receptor is an essential step of entry and cell-to-cell spread, but it is not sufficient to promote fusion of the viral envelope with membranes of the target cell (22, 25, 26). The viral fusion machinery is comprised of three glycoproteins, gB and gH/gL (22, 25, 26). Nectin-1 is one of the few receptors that gD uses for virus entry. All the known receptors are structurally unrelated molecules, but each can mediate entry independently.
In recent years it was demonstrated that the interaction of gD with Nectin-1 is not limited to the early steps of infection. Expression of gD later in infection is correlated with downregulation of Nectin-1 within the infected cells (15–17). Thus, gD accumulates at sites of contact between infected and adjacent noninfected cells but not between infected cells. Three isoforms of human Nectin-1 have been reported and are known as α, β, and γ (27, 28). Nectin-1α and -1β differ in their transmembrane domains and the cytoplasmic tails, but they share identical ectodomains (27–31). The isoform γ lacks a transmembrane domain and is secreted (29–31). Both Nectin-1α and -1β serve as HSV-1 entry receptors, and they are both internalized in infected cells in a gD-dependent manner (15–17, 22).
Here, we present data that Nectin-1 interacts with the Cbl/CIN85 endocytic machinery and is removed from the surface of the infected cells by a Cbl- and ICP0-dependent mechanism. The viral glycoprotein D (gD) is part of the Nectin-1/Cbl complex. The removal of Nectin-1 from the surface of the infected cells increases the opportunity of the virus to spread to noninfected cells. In the situation in which Nectin-1 is not removed from the surface of the infected cells, such as in Cbl-knockdown cells, the spread of progeny virions to neighboring cells is impaired. The removal of viral entry receptors from the surface of infected cells is a common strategy used by other viruses as well to successfully disseminate.
RESULTS
Cbl forms a complex with the major HSV-1 receptor Nectin-1.
In a series of experiments, HEp-2 cells, either mock infected or exposed to 10 PFU of HSV-1(F) per cell, were harvested at 7 h after infection, lysed, and reacted with the Cbl rabbit polyclonal antibody. Control lysates were reacted with antibody directed against an intracellular nonmembrane protein, the RNA polymerase II. The immunoprecipitated complexes were electrophoretically separated on denaturing polyacrylamide gels and reacted with anti-Nectin-1 mouse monoclonal antibody. As shown in Fig. 1A, the Cbl antibody pulled down Nectin-1 but not the control IgG. In the reciprocal immunoprecipitation assay, HEK-293 cells cotransfected with Cbl- and Nectin-1-expressing plasmids were mock infected or infected with HSV-1(F) (10 PFU/cell) at 24 h posttransfection. The cells were harvested at 6 h after infection, lysed, and reacted with the Nectin-1 mouse monoclonal antibody. Control lysates were reacted with a mouse monoclonal antibody directed against RNA polymerase II. The immunoprecipitated complexes were separated on denaturing polyacrylamide gels and reacted with anti-Cbl rabbit polyclonal antibody. As shown in Fig. 1B, the Nectin-1 antibody pulled down Cbl (lanes 3 and 4) but not the control antibody (lane 5). To further confirm this association the glutathione S-transferase (GST)-Cbl fusion protein purified from bacteria or GST alone was incubated with lysates from mock- or HSV-1(F)-infected cells that were harvested at 8 h after exposure to the virus. As shown in Fig. 1C, the Nectin-1 antibody in the input reacts with at least three major bands, ranking in molecular mass from 78 to 120 kDa. These bands most likely correspond to modified forms of Nectin-1. The faster-migrating product of Nectin-1 was specifically pulled down by the GST-Cbl fusion protein and not by the control GST. These results suggest that Cbl associates with Nectin-1.
FIG 1.

Interaction of Cbl with the viral entry receptor Nectin-1. (A) HEp-2 cells seeded on F25 flasks were either mock infected or infected with wild-type virus at 10 PFU/cell. At 7 h after infection the cells were harvested and lysed, and immunoprecipitations (IP) were done either with the Cbl rabbit polyclonal antibody or with the control RNA polymerase II rabbit polyclonal antibody. The Cbl immunocomplexes were electrophoretically analyzed on 9% denaturing polyacrylamide gels, transferred to nitrocellulose sheets, and immunoblotted with the Nectin-1 mouse monoclonal antibody, as described above. (B) HEK-293 cells were cotransfected with a Cbl and a Nectin-1 expression plasmid. At 24 h posttransfection the cells were exposed to HSV-1(F) (10 PFU/cell). The cells were harvested at 6 h postinfection and lysed, and immunoprecipitations were done with a Nectin-1 mouse monoclonal antibody or with the control RNA polymerase II mouse monoclonal antibody. The Nectin-1 immunocomplexes were separated on a 6% denaturing polyacrylamide gel, and immunoblot analysis was done with the Cbl rabbit polyclonal antibody. (C) GST-Cbl or GST-tagged bacterial purified proteins were mixed with lysates from mock- or HSV-1(F)-infected cells (10 PFU/cell, 8 h after infection). The proteins bound on GST or GST-Cbl were separated on denaturing polyacrylamide gels as described above and immunoblotted with the Nectin-1 antibody. The Ponceau S staining depicts the amounts of proteins used in this GST pulldown assay. The arrow b points to the form of Nectin-1 that interacts with Cbl. The line a shows two forms of Nectin-1 that were not pulled down by the GST-Cbl. (D) Infection and GST pulldown reactions were done as described for panel C. The proteins bound on GST or GST-Cbl were separated on denaturing polyacrylamide gels and immunoblotted with the gD mouse monoclonal antibody. WB, Western blotting.
Nectin-1 forms a complex with the HSV-1 glycoprotein D during the entry of the virus in the cells but also postentry. The postentry interaction appears to trigger the internalization and relocalization of Nectin-1. Therefore, we investigated whether GST-Cbl could pull down the viral glycoprotein D along with Nectin-1. For this purpose, a GST pulldown assay was performed similar to the one represented in Fig. 1C, in which the GST-Cbl or the control protein GST was reacted with lysates from HSV-1(F)-infected or uninfected cells. As shown in Fig. 1D, the GST-Cbl protein (lane 6) but not GST alone (lane 4), pulled down glycoprotein D. We conclude that Cbl associates with the complex of Nectin-1 with glycoprotein D.
Nectin-1 is removed from the surface of the HSV-1(F)-infected cells but is retained on the surface of infected cells depleted of Cbl.
To determine whether the interaction of Nectin-1 with Cbl alters the abundance of Nectin-1 on the surface of the infected cells, two series of experiments were performed. First, we derived HEp-2 cell clones from cultures exposed to lentiviruses expressing Cbl-specific short hairpin RNAs (shRNAs) or nontargeting RNAs. For these studies we selected two clones, a clonal line exposed to a nontargeting shRNA and a clonal line exposed to a Cbl-targeted shRNA. The clonal lines were mock infected or exposed to HSV-1(F) (10 PFU/cell). The cells were harvested at 3, 6, 9, or 18 h and analyzed as described above for the presence of Cbl. The results were as follows. The levels of Cbl contained in cells exposed to the nontargeting shRNA remained stable throughout the 18-h interval after infection (Fig. 2A, lanes 1 to 5). The clones inoculated with lentiviruses expressing a Cbl-specific shRNA contained significantly less Cbl than the nontargeting shRNA-treated cells (Fig. 2A, lanes 6 to 10). β-Actin contained in cell lysates served as a loading control.
FIG 2.
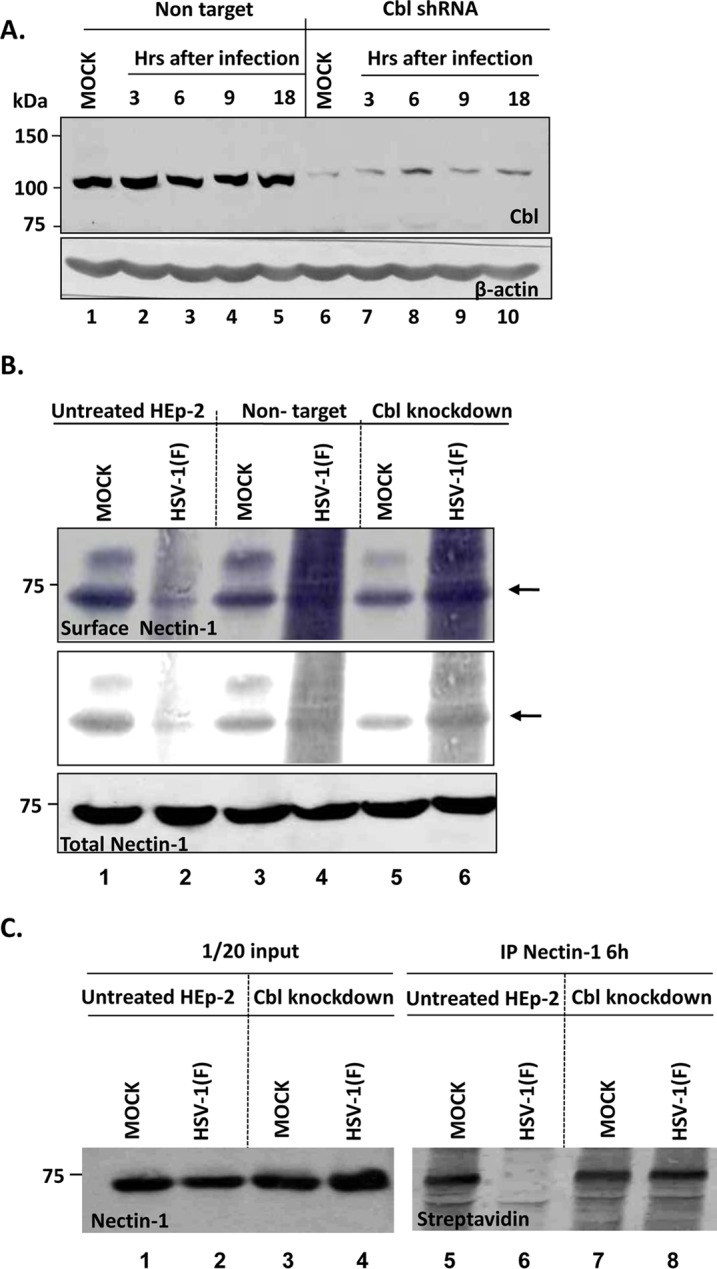
Nectin-1 is removed from the surface of the Cbl-expressing but not from the Cbl-depleted cells during HSV-1 infection. (A) Characterization of a Cbl-depleted HEp-2 cell line. HEp-2 cells were infected with lentiviral vectors carrying a Cbl shRNA or control nontargeting shRNA. Puromycin-resistant clones were obtained as described in Materials and Methods. The Cbl-depleted cells and the control derivatives were infected with HSV-1(F) at 10 PFU/cell. The cells were harvested at 3 h, 6 h, 9 h, and 18 h after infection, equal amounts of total proteins were analyzed as described above, and immunoblotting was done with the Cbl rabbit polyclonal antibody. β-Actin was used as a loading control. (B) HEp-2 cells, Cbl-knockdown cells, and the nontarget controls seeded in F150 flasks were infected with wild-type virus at 10 PFU/cell. At 9 h after infection, the cell surface proteins were labeled with biotin as indicated in the Materials and Methods section. The cells were harvested at 30 min postlabeling and lysed, Nectin-1 was immunoprecipitated with the Nectin-1 mouse monoclonal antibody, and immunoblotting was done with the streptavidin-HRP-conjugated anti-mouse IgG, as described above. (C) HEp-2 cells or their Cbl-knockdown derivatives seeded in F150 flasks were infected with wild-type virus at 5 PFU/cell or mock infected. At 6 h after exposure of the cells to the virus, surface proteins were labeled with biotin as described above, and surface Nectin-1 was detected as described for panel B.
In the second series of experiments the Cbl-depleted and control cells were mock infected or exposed to 10 PFU of HSV-1(F) per cell. At 9 h after infection the surface proteins were labeled with a nonpermeable form of biotin. The reaction was then quenched, the unbound biotin was rinsed off, and the cells were lysed. Lysates of replicate cultures were reacted with antibody to Nectin-1. The immunoprecipitated biotin-bound Nectin-1 was detected with antibody to streptavidin. The results are shown in Fig. 2B. The Nectin-1 bound by biotin is shown under normal exposure (top panel). It is noteworthy that in this and virtually all experiments performed at 9 h postinfection, we observed a “smear” in lanes containing electrophoretically separated proteins from infected cells. One hypothesis that could explain the smear is that proteins made during infection nonspecifically bind to Nectin-1 and are pulled down. This phenomenon is accentuated at 9 h, or later, after infection. To enable a better evaluation of the results, the same data but at a much higher contrast are shown in the middle panel in Fig. 2B. As indicated by the arrow, the amount of Nectin-1 pulled down from the cell surface was significantly higher in Cbl-depleted cells. It is noteworthy that, as shown in the bottom panel, the total amount of Nectin-1 did not change, suggesting that the internalized Nectin-1 was not necessarily degraded during HSV-1 infection.
To explore whether the reduction in the surface amount of Nectin-1 is apparent at earlier times after exposure to the virus, we repeated the experiment described above except that the cells were biotinylated at 6 h postinfection, that is, before significant amounts of progeny viruses were released from infected cells, and we exposed the cells to 5 PFU/cell instead of 10 PFU/cell. As shown in Fig. 2C the Nectin-1 was removed from the surface of the infected cells by 6 h after exposure to the wild-type virus (Fig. 2C, compare lanes 1 and 6). The elimination of Nectin-1 from the cell surface was blocked in Cbl-knockdown cells (compare lanes 6 and 8).
Removal of Nectin-1 from the cell surface requires the presence of ICP0.
HEp-2 cells were mock infected or exposed to 5 PFU per cell of HSV-1(F) or the ΔICP0 mutant virus. The assessment of cell surface Nectin-1 was done at 6 h postinfection, according to the procedures described above. The results shown in Fig. 3 suggest that the amount of Nectin-1 is not reduced in cells infected with wild-type or mutant virus (compare lanes 1 to 3). Moreover, these results indicate that the amount of Nectin-1 on the surface of wild-type virus-infected cells was reduced (Fig. 3, compare lanes 4 and 5) but not on the surface of the ΔICP0 mutant virus-infected cells (compare lanes 4 to 6). We conclude that ICP0 is involved in the internalization of Nectin-1 during HSV-1 infection.
FIG 3.
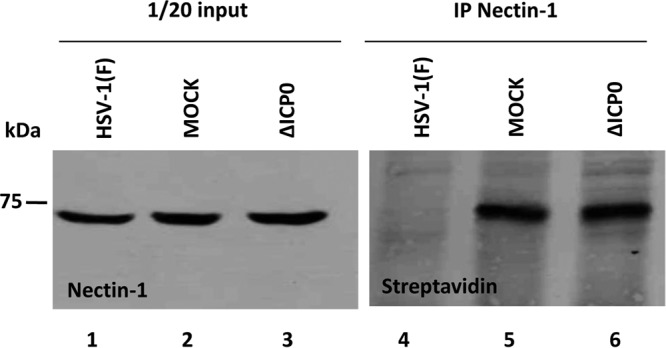
ICP0 is required for the removal of Nectin-1 from the surface of the infected cells. The ICP0-null mutant or the wild-type virus was used to infect HEp-2 cells (5 PFU/cell) seeded in F150 flasks, as described above. The cells were harvested at 6 h postinfection. Detection of the surface Nectin-1 was done as described in the legend of Fig. 2.
Cbl and Nectin-1 accumulate in the perinuclear space in infected cells.
The distribution of Cbl was monitored in mock-infected and infected HEp-2 cells. The HEp-2 cells were exposed to 10 PFU of HSV-1 per cell. The mock-infected and infected cells were fixed 8 h after infection and reacted with anti-Cbl polyclonal antibody and anti-ICP0 monoclonal antibody. The results shown in Fig. 4 indicate that in mock-infected cells Cbl formed small granules that were dispersed throughout the cytoplasm (panel a). In infected cells Cbl retained its granular appearance but appeared to be concentrated in perinuclear space (Fig. 4, compare panel a to panels b to d and compare panel e to panels f to h).
FIG 4.
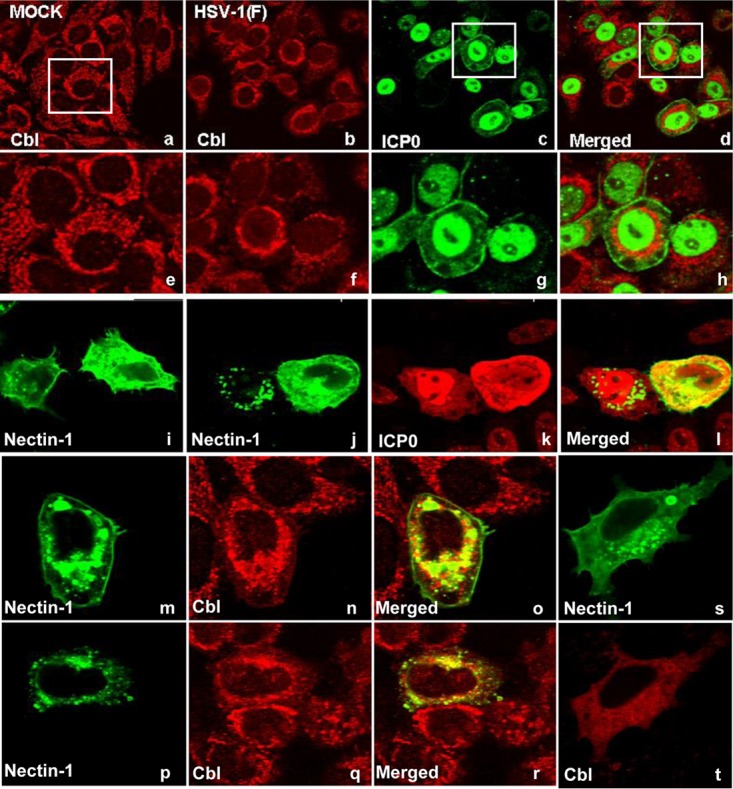
Perinuclear accumulation of Cbl and colocalization with Nectin-1 during herpes infection. (a to h) HEp-2 cells seeded in four-well slides were either mock infected or exposed to the wild-type virus (10 PFU/cell). The cells were fixed at 8 h after infection and costained with the Cbl rabbit polyclonal antibody and the ICP0 mouse monoclonal antibody, as described in Materials and Methods. (i) Nectin-1 transfected HEp-2 cells stained with the Nectin-1 mouse monoclonal antibody. (j t o l) HEp-2 cells seeded in four-well slides were infected with HSV-1(F) (10 PFU/cell) at 24 h following transfection with a Nectin-1-expressing plasmid. The cells were fixed 8 h after exposure to the virus and costained with the Nectin-1 mouse monoclonal antibody and the ICP0 rabbit polyclonal antibody, as described above. (m to r) HEp-2 cells seeded in four-well slides were transfected with a Nectin-1-expressing plasmid for 24 h prior to infection with HSV-1(F) (10 PFU/cell). The cells were fixed at 8 h after infection and costained with the Nectin-1 mouse monoclonal antibody and the Cbl rabbit polyclonal antibody. (s and t) Nectin-1 transfected HEp-2 cells stained with the Nectin-1 mouse monoclonal antibody and the Cbl rabbit polyclonal antibody.
Endogenous Nectin-1 could not be detected by immunofluorescence. To determine the effects of infection, we first transfected the cells with a Nectin-1-expressing plasmid and then infected them with HSV-1(F) (10 PFU/cell), as detailed in the legend of Fig. 4. As shown in Fig. 4i, Nectin-1 was dispersed throughout the cytoplasm in uninfected cells. In infected cells it tended to aggregate in granular structures in the perinuclear space (panels j to l). In addition, we investigated potential colocalization of Nectin-1 with Cbl in cells transfected with a Nectin-1-expressing plasmid and infected with HSV-1(F). As shown in Fig. 4, Nectin-1 (panels m and p) and Cbl (panels n and q) following HSV-1(F) infection accumulate in the perinuclear area, where they partially colocalize (panels o and r), while in uninfected cells no particular perinuclear accumulation or colocalization was noticed (panels s and t). We conclude that HSV-1 infection alters the localization of Nectin-1 and Cbl and that the proteins colocalize in the perinuclear area.
To further confirm that the localization of Nectin-1 is altered in a Cbl-dependent mechanism, we performed a lipid floatation assay. HEp-2 cells or their Cbl-knockdown derivatives were either mock infected or exposed to HSV-1(F) (10 PFU/cell). The cells were harvested at 8 h postinfection, and detergent-resistant membranes were dissolved in a heavy sucrose solution of 1.5 M and overlaid with two lighter layers of sucrose of 1.3 M and 0.15 M. Following ultracentrifugation, samples were collected from the top to the bottom of the gradient, and the distribution of Nectin-1 in the gradient was examined by immunoblot analysis. The results shown in Fig. 5 may be summarized as follows: in uninfected cells and HSV-1(F)-infected cells the distribution of Nectin-1 in the sucrose gradient appears similar although a reproducible reduction in the amounts of Nectin-1 could be detected in the light-density sucrose fractions in HSV-1(F)-infected cells. In uninfected Cbl-depleted cells, we observed a scattering of Nectin-1 to heavier sucrose fractions. Finally, a significant loss of Nectin-1 from the light-density sucrose fractions was observed in Cbl-depleted cells that were exposed to HSV-1(F). In the absence of Cbl, Nectin-1 appears to translocate from detergent-insoluble membranes to membranes that accumulate in high-density sucrose fractions. We conclude that Cbl influences the localization of Nectin-1.
FIG 5.
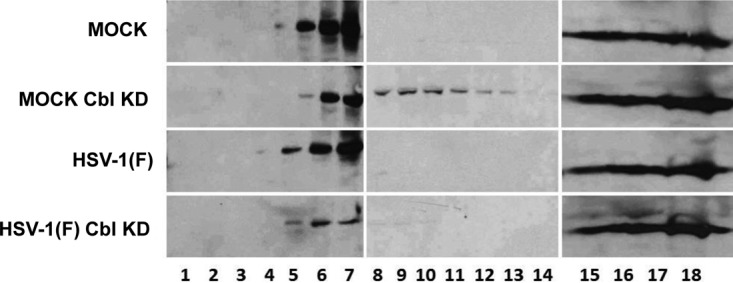
Changes in the membrane distribution of Nectin-1 in Cbl-depleted cells. HEp-2 cells and their Cbl-depleted derivatives were either mock infected or exposed to HSV-1(F) (10 PFU/cell). The cells were harvested at 8 h postinfection, and detergent-insoluble membranes were adjusted to 1.5 M sucrose and overlaid with 5 ml of 1.2 M sucrose, followed by a 2-ml layer of 0.15 M sucrose. Following ultracentrifugation as detailed in the Materials and Methods section, fractions (500 μl) were collected from the top to the bottom of the gradient. Equal volumes from each fraction were used for protein analysis in 9% denaturing polyacrylamide gels. Immunoblotting was done with the Nectin-1 antibody.
In Cbl-depleted cells viral entry is enhanced, and the infected cells remain susceptible to virus superinfection.
The objective of another series of experiments was to test the hypothesis that in infected cells ICP0 mediates the removal of Nectin-1 from the cell surface by the Cbl-CIN85 complex in order to reduce the entry into the infected cells of virus progeny made in that cell.
In this series of experiments we exposed HEp-2 cells, the Cbl-knockdown clonal derivatives, and nontarget controls to 30 PFU of HSV-1(F) per cell. At 0, 2, 5, or 8 h after infection with HSV-1(F), the cells were exposed to 1 PFU/cell of a second recombinant herpesvirus. To differentiate between the two viruses, the second virus was tagged by enhanced green fluorescent protein (EGFP) fused to ICP0. All infected cells were harvested at 2 h after exposure to the second, tagged virus, and the amounts of EGFP DNA were measured by quantitative PCR (qPCR) and normalized to the amounts of EGFP DNA present in HEp-2 cells at 2 h after exposure of the cells to the second virus at 0 h (time zero) after infection with HSV-1(F). The results are described below.
As shown in Fig. 6, the amount of second virus that entered Cbl-knockdown cells at time zero was approximately 10-fold higher than that in HEp-2 cells or in the nontarget controls. From these results we conclude that the amount of Nectin-1 normally present in untreated infected cells was limited and may not have been sufficient for entry of all of the virus particles to which the cells were exposed. We estimate a ratio of 40 to 100 physical particles per PFU (32). By this calculation, the cells were exposed to approximately 1,200 to 3,000 particles per cell. The amount of Nectin-1 present on the cell surface appears to be tightly regulated in as much as significantly more virus entered cells in Cbl-knockdown cells. We also noticed that the ability of the second virus to infect cells was diminished with time in both untreated and Cbl-knockdown cells. It may be that additional changes occur to Nectin-1 during infection that prevent it from functioning as a receptor despite its presence on the cell surface. The data suggest that Cbl is necessary but not sufficient to down-modulate the amount of Nectin-1 available for entry of HSV-1 into cells.
FIG 6.
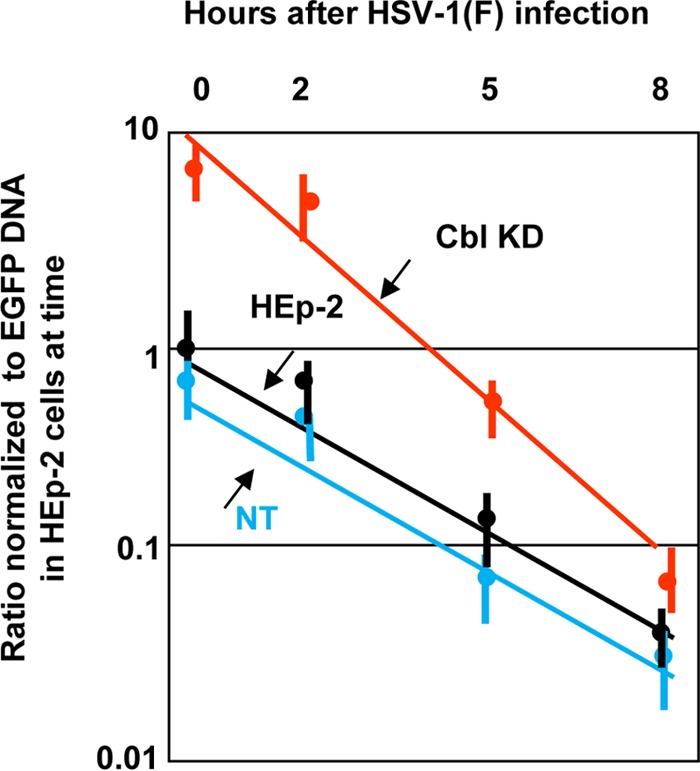
Susceptibility of cells depleted of Cbl to virus superinfection. HEp-2 cells, the Cbl-knockdown derivatives, and control shRNA-treated cells were exposed to the wild-type virus at 30 PFU/cell. At 0 h, 2 h, 5 h, or 8 h postinfection, the cells were exposed to the EGFP-ICP0 virus (1 PFU/cell) for 2 h. Then the cells were harvested, total DNA was extracted as described in Materials and Methods, and quantification was done by qPCR analysis using primers for EGFP. Amplification of β-actin DNA served as an internal control for the normalization process.
The role of CIN85 in the HSV-1(F) entry process.
The objective of another series of experiments concerned the adapter protein CIN85. Since CIN85 is the adaptor that is recruited by Cbl during the receptor internalization process, we sought to determine the contribution of CIN85 to the viral entry process. For this purpose HEp-2 cells were depleted of CIN85 with the aid of lentiviral vectors, as described in Materials and Methods. Of the four cell lines that were developed, we selected clone 22 for further studies (Fig. 7A), but similar analysis was performed using clone 20, which yielded similar results (data not shown). Specifically, the CIN85-depleted cells, the Cbl knockdown (KD) cells, HEp-2 cells, and the nontargeting shRNA-treated cells were exposed to 30 PFU of HSV-1(F) per cell. At 0, 2, 5, or 8 h after wild-type virus infection, the cells were superinfected with the EGFP-ICP0 virus as described above. As in the experiment shown in Fig. 6, EGFP-ICP0 virus entry was quantified by qPCR. The results shown in Fig. 7B demonstrate that although Cbl depletion enhanced viral entry by approximately 10-fold, the CIN85-depleted cells show only a small increase in viral entry. In both instances the susceptibility of cells depleted of CIN85 to superinfection gradually declined.
FIG 7.
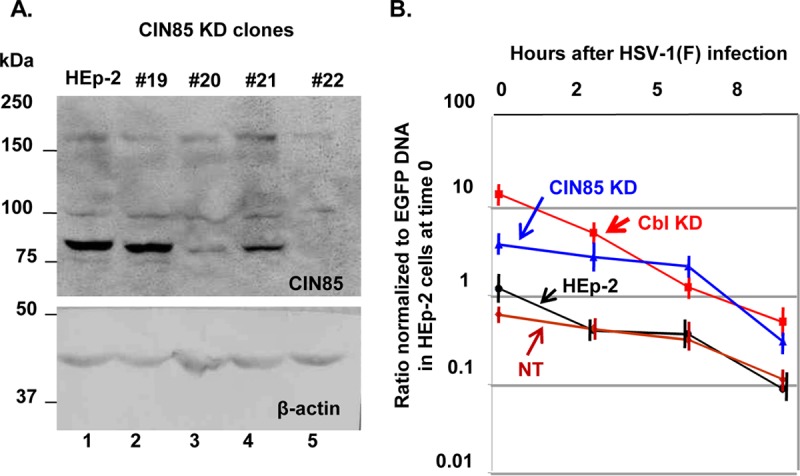
(A) Development of CIN85-depleted HEp-2 cells. HEp-2 cells were exposed to lentiviruses carrying different shRNAs against CIN85, which were propagated as described in Materials and Methods. After cells were selected in puromycin, the efficiency of depletion of CIN85 was evaluated by Western blot analysis using equal amounts of lysates from the shRNA-treated cell lines and the parental HEp-2 cells. Clone 22 demonstrated more efficient depletion of CIN85. β-Actin was used as a loading control. (B) The Cbl KD cells are more susceptible to virus superinfection than the CIN85 KD cells. HEp-2, the Cbl KD, CIN85 KD derivatives, and nontargeted shRNA-treated cells were exposed to the wild-type virus and the EGFP-ICP0 virus, as described in the legend of Fig. 6. The entry of the EGFP-ICP0 virus was quantified by qPCR as described in the legend of Fig. 6.
DISCUSSION
Here, we have discussed a strategy that HSV has evolved to successfully disseminate to uninfected cells that involves the removal of the HSV entry receptor Nectin-1 from the surface of the cells which the virus enters and in which it replicates. Previously, the Cbl/CIN85 complex was shown to mediate the elimination of the EGFR from the surface of the HSV-1-infected cells. We show here that the Cbl/CIN85 complex is also involved in the elimination of the viral entry receptor Nectin-1 from the surface of the infected cells (21). In cells depleted of Cbl, the Nectin-1 receptor remained on the cell surface, and the progeny virions entered cells in which they were produced. A combination of pulldown and immunoprecipitation assays demonstrated that Nectin-1 and Cbl form a complex. Moreover, the viral glycoprotein D, which binds to Nectin-1 during the entry of the virus into the cells, was also found in the Nectin-1/Cbl complex. Cbl usually recognizes posttranslational modifications, such as tyrosine phosphorylation in the cytoplasmic tail of surface molecules before internalization (13, 33–36). Whether such a modification is introduced to Nectin-1 during HSV-1 infection and how the Cbl/CIN85 complex is recruited to eliminate Nectin-1 following the entry of the virus into the cells are currently unknown. Nectin-1 was detected in three forms by immunoblot analysis using two different antibodies. Interestingly, only the faster-migrating form was pulled down by Cbl. The nature of the other two forms is unknown. Nectin-1 exists in three isoforms containing 517 amino acids (aa), 458 aa, and 352 aa (28). Each isoform undergoes posttranslational modifications, including glycosylation, phosphorylation, and perhaps ubiquitination (28). Moreover, nectins form cis and trans dimers with each other to form cell-cell adhesions (28). They also form heterophilic complexes with other immunoglobulin-like CAMs, especially with nectin-like molecules (28). Also interactions of the different Nectin forms with different matrix metalloproteinases have been reported that may result in the formation of different multiprotein complexes and may generate very specific signals at cell-to-cell junctions (28). Cbl may recognize certain posttranslational modifications introduced to Nectin-1 following entry of the virus into the cells. Also, the formation of the Nectin-1/gD/Cbl complex may occur in particular submembrane compartments. Finally, Cbl may associate with Nectin-1 when it is in the monomeric stage and not when it forms dimers.
Indeed, following infection, a fraction of Nectin-1 colocalizes with Cbl in the perinuclear area. Additionally, an analysis of detergent-insoluble membranes in a sucrose gradient demonstrated that the depletion of Cbl followed by wild-type virus infection significantly reduced the amounts of Nectin-1 present in the detergent-insoluble membranes, which float in light-density fractions. These results confirmed that Cbl influences the localization of Nectin-1.
Our data also indicated that ICP0 is required for the internalization of Nectin-1. Possible modifications of surface components mediated by ICP0 cannot be excluded. The elimination of Nectin-1 from infected cells occurs approximately 6 h following inoculation of cells with virus, which coincides with the time that ICP0 accumulates in the cytoplasm (37). ICP0 is known to interact with CIN85 in the cytoplasm (6). Depletion of CIN85 did not increase the HSV-1 entry levels compared to levels in the Cbl-depleted cells. These data imply that Cbl is the major player in the receptor elimination process. This is one more example which shows that the virus hijacks the partners of key regulators, the other examples being the interaction of ICP0 with BMAL-1 to recruit CLOCK to remodel the viral chromatin and the interaction of ICP0 with cyclin D3 to recruit the CDK4/CDK6 kinases to optimize viral gene expression (38, 39).
Upon internalization, Nectin-1 remains stable in intracellular compartments, which is in contrast to the EGFR, which is rapidly degraded following internalization (6). After internalization, many surface components remain stable and can signal from within the intracellular compartments (40). This has been linked to signal amplification. Many internalized components are targeted to recycling endosomes and from there again to the cell surface or the tight junctions (40–44). The functions and the fate of internalized Nectin-1 remain to be identified.
In the cells in which Nectin-1 was not internalized during HSV infection, we observed a 10-fold increase in the amount of virus entering the cells. Reentry was monitored for several hours after the cells were exposed to the first virus. However, we observed that entry gradually declined even though Nectin-1 was retained on the cell surface. One possible explanation is that Nectin-1 is modified by a virus-induced mechanism later during infection, and therefore it does not function any more as an entry receptor. This modified form of Nectin-1 might actually be the one internalized with the aid of Cbl.
This study underscores the significance of the ICP0-CIN85 interaction in the cytoplasm and suggests that ICP0 has important cytoplasmic functions in addition to its role in the nucleus immediately after infection. The repertoire of receptors targeted for internalization during HSV-1 infection could identify novel antiviral factors.
MATERIALS AND METHODS
Cells and recombinant viruses.
The sources and propagation of HEp-2 and HEK-293 cells were reported elsewhere (38). HSV-1(F), a limited-passage isolate, is the prototype strain used in this laboratory (45). The properties of R7910, a ΔICP0 null mutant virus, and of the EGFP-ICP0 virus, which expresses an EGFP fusion form of ICP0, were described before (46, 47).
Development of stable cell lines depleted of Cbl or CIN85 using shRNA lentiviral vectors.
The PLKO.1 shRNAs for Cbl, CIN85, and the nontargeting control shRNA were purchased from ThermoFisher. For the development of the respective lentiviral vectors, HEK-293 cells seeded in an F25 (25-cm2) flask at 60% confluence were cotransfected with 8 μg of the plasmid carrying the shRNA, 8 μg of the Gag-Pol-expressing plasmid, and 1 μg of a vesicular stomatitis virus G protein (VSV-G)-encoding plasmid with TurboFect, according to the manufacturer's instructions. At 48 h after transfection the supernatant from the cultures was collected, filtered through 0.45-μm-pore-size filters, and used to infect HEp-2 cells. Puromycin selection initiated 24 h after cell exposure to lentiviruses and continued until only resistant clones survived. The clones of HEp-2 cells with the greatest reduction in Cbl or CIN85 expression were used for further experimentation.
Immunoprecipitation.
Cells seeded on an F25 flask were lysed in 1 ml of lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1% NP-40, 1 mM Na3VO4, 1 mM NaF, 0.5% sodium deoxycholate, and protease inhibitor cocktail) and incubated for 30 min on ice, and the cell debris was removed by centrifugation at 7,000 rpm for 10 min. The supernatant fluids were reacted with protein A-agarose 50% slurry (Sigma) at 4°C for 20 min to clear the lysates. Upon removal of the beads, lysates were reacted either with 2 μg of rabbit polyclonal Cbl antibody (Santa Cruz) or with the control RNA polymerase II rabbit polyclonal antibody (Santa Cruz) overnight at 4°C. The lysate-antibody mixtures were incubated with 50 μl of protein A-agarose 50% slurry at 4°C for 2 h. The beads were then rinsed four times with the same buffer and eluted by 1× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer at 95°C for 5 min. The precipitates were loaded on a 9% SDS-PAGE gel and examined by Western blotting.
Purification of the GST-Cbl fusion protein and GST-Cbl pulldown assays.
The full-length Cbl was PCR amplified from pRK5-Cbl, digested with EcoR V, and inserted into the SmaI site of pGEX-4T2 in frame with glutathione S-transferase (GST) (6). The purification of Cbl was done as previously described (39). Briefly, Escherichia coli XL1-Blue cells transformed with the pGEX-4T2- or with the pGEX-4T2-Cbl-expressing plasmid were induced with 0.3 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h after the optical density at 600 nm reached a value of 0.5. The harvested cells were lysed by sonication in phosphate-buffered saline (PBS) supplemented with Triton X-100 and sodium lauroyl sarcosinate at a final concentration of 1%. The cell debris was removed by centrifugation, and the GST or GST-Cbl proteins were adsorbed to the glutathione-agarose beads (Sigma). Purified proteins were eluted after extensive rinsing of the beads with PBS–1% Triton X-100. For the GST pulldown assays, 2 × 107 HEp-2 cells, mock infected or infected with the wild-type virus at 10 PFU/cell, were lysed in HEPES–1% Triton X-100 buffer consisting of 50 mM HEPES (pH 7.4), 250 mM NaCl, 10 mM MgCl2, 1 mM PMSF, and protease cocktail inhibitor (Sigma) supplemented with 1% Triton X-100. The lysates were reacted overnight with equal amounts of GST or GST-Cbl proteins immobilized on glutathione beads and rinsed three times with the HEPES–1% Triton X-100 buffer, and the bound protein complexes were dissolved in loading buffer composed of 4% SDS, 100 mM Tris-Cl (pH 6.8), 20% glycerol, and 0.2% bromophenol blue, supplemented with β-mercaptoethanol, and boiled for 5 min. After centrifugation to remove the beads, the eluted proteins were subjected to electrophoretic analysis on 9% polyacrylamide gels.
Immunoblot analysis.
The procedures for immunoblot analysis were described elsewhere (39). Briefly, the cells were harvested at the times after infection discussed in the figure legends, rinsed with PBS, solubilized in triple detergent buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1% Nonidet P-40, 0.5% sodium deoxycholate, 100 μg ml−1 of phenylmethylsulfonyl fluoride) supplemented with phosphatase inhibitors (10 mM NaF, 10 mM β-glycerophosphate, 0.1 mM sodium vanadate) and protease inhibitor cocktail (Sigma), as specified by the manufacturer, and briefly sonicated. The protein concentration in total cell lysates was determined with the aid of a Bio-Rad protein assay (Bio-Rad Laboratories). Approximately 60 μg of proteins per sample was subjected to further analysis. Proteins were electrophoretically separated in denaturing polyacrylamide gels, transferred to a nitrocellulose sheet electrically, blocked with PBS supplemented with 0.02% (vol/vol) Tween 20 (PBST) and 5% nonfat milk, and reacted overnight at 4°C with the appropriate primary antibodies diluted in PBST–1% nonfat milk. The rabbit polyclonal antibody to Cbl (Santa Cruz) and the mouse monoclonal antibodies to CIN85 (Santa Cruz), Nectin-1 (CK6; Santa Cruz), and Nectin-1 (R&D systems) were used at a dilution of 1:500. The mouse monoclonal antibody to β-actin (Sigma) and the horseradish peroxidase (HRP)-streptavidin anti-mouse IgG (Pierce) were used at a 1:1,000 dilution. The mouse monoclonal antibody to gD (kindly provided by B. Roizman, University of Chicago) was used at a 1:1,000 dilution. After several rinses with PBST–1% nonfat milk, the membranes were reacted with the appropriate secondary antibody conjugated to either alkaline phosphatase or horseradish peroxidase. Finally, protein bands were visualized with 5-bromo-4-chloro-3-indolylphosphate (BCIP)-nitroblue tetrazolium (Denville Scientific, Inc.) or with ECL Western blotting detection reagents (Amersham Biosciences) according to the manufacturer's instructions.
Biotinylation and purification of cell surface proteins.
HEp-2 cells (2 × 107) were washed three times with ice-cold PBS and incubated with 10 ml of ice-cold sulfo-NHS-LC-biotin (sulfosuccinimidyl-6-biotinamido-hexanoate), freshly dissolved in PBS (pH 8) to 0.5 mg/ml, for 30 min at 4°C. Next, the cells were washed three times with PBS supplemented with 100 mM glycine to quench the reaction and remove excess biotin reagent and by-products. Cells were then collected, lysed in radioimmunoprecipitation assay (RIPA) buffer (100 mM Tris [pH 8.0], 140 mM NaCl, 1% Triton X-100/0.1% SDS, 1% deoxycholic acid, 0.5 mM EDTA, protease and phosphatase inhibitors), and briefly sonicated. Immunoprecipitation of total Nectin-1 was done with 3 μg of mouse monoclonal Nectin-1 antibody, as detailed above, and immunoblotting was done with the HRP-streptavidin anti-mouse IgG diluted 1:1,000 (Pierce). Protein bands were visualized with 5-bromo-4-chloro-3-indolylphosphate (BCIP)-nitroblue tetrazolium (Denville Scientific, Inc.), as described above.
Superinfection assay.
HEp-2 cells, their Cbl- or CIN85-knockdown derivatives, and the nontargeting control shRNA-treated cells, seeded in T25 flasks, were exposed to 30 PFU of HSV-1(F). At 0 h, 2 h, 5 h, or 8 h after infection with the wild-type virus, the cells were exposed to 1 PFU of the EGFP-ICP0 virus in 199V medium, composed of medium 199 (Sigma catalog no. M0393) supplemented with 1% fetal bovine serum, for 2 h at 37°C. For the extraction of total DNA, the cells were harvested, lysed in buffer containing 50 mM Tris-HCl [pΗ 8.0], 100 mM EDTA [pH 8.0], and 0.5% (wt/vol) SDS and incubated with 100 μg/ml proteinase K overnight at 55°C. Then the samples were incubated with RNase A (50 μg/ml) for 2 h at 37°C prior to phenol-chloroform extraction and ethanol precipitation. The entry of the EGFP-ICP0 virus was measured by quantitative real-time PCR analysis using the following EGFP primers: forward, 5′-CCA AGC TTG AAT TCA CCA TGG TGA GCA-3; reverse, 5′-GGA AGC TTG ATA TCC TTG TAC AGC TCG TC-3′. Primers for the β-actin DNA used for the normalization process were the following: forward, 5′-CTA TCC CTG TAC GCC TCT GG-3′; reverse, 5′-TGG TGG TGA AGC TGT AGC C-3′.
Isolation of detergent-insoluble membranes.
For isolation of detergent-insoluble membrane domains, approximately 1 × 108 cells per sample were resuspended in 1 ml of ice-cold detergent lysis buffer (25 mM Tris, pH 7.6, 150 mM NaCl, 5 mM EDTA, 40 mM Na3VO4, 200 mM NaF, 0.05% Triton X-100, and protease inhibitor cocktail), followed by 30 min of incubation on ice before ultracentrifugation. The samples were then adjusted to 1.5 M sucrose in 20 mM Tris, pH 7.5, in a total volume of 2 ml, placed in the bottom of an ultracentrifuge tube, and overlaid with 5 ml of 1.2 M sucrose, followed by a 2-ml layer of 0.15 M sucrose. Samples were centrifuged at 38,000 rpm in a Beckman SW41 rotor for 18 h at 4°C. Fractions (500 μl) were withdrawn from top to bottom and analyzed by immunoblot analysis.
Transfection/infection assays.
HEK-293 cells seeded in six-well plates at 70 to 80% confluence were transfected with 1 μg/well of Nectin-1-expressing plasmid, kindly provided by R. Longnecker (Northwestern University), and 1 μg/well of Cbl-expressing plasmid, kindly provided by I. Dikic (Goethe University, Germany), with the aid of Lipofectamine 3000 (Life Technologies), according to the instructions of the suppliers. At 24 h posttransfection the cells were exposed to HSV-1(F) (10 PFU/cell). The cells were harvested at 6 h postinfection, and immunoprecipitations with a Nectin-1 mouse monoclonal antibody (R&D Systems) or a control RNA polymerase II mouse monoclonal antibody (Santa Cruz) were done as described above.
Immunofluorescence.
The procedures for immunofluorescence have been described elsewhere (39). Briefly, HEK-293 cells seeded in four-well slides (Erie Scientific) were transfected with 200 ng/well of Nectin-1-expressing plasmid, kindly provided by R. Longnecker (Northwestern University), in mixtures with 1 μl of Lipofectamine and 1.5 μl of Plus reagents per well, as specified by the supplier (Invitrogen). At 24 h posttransfection, the cells were exposed to 10 PFU of virus per cell and fixed in 4% paraformaldehyde at the times indicated in Results. The cells were permeabilized, blocked with PBS-TBH solution consisting of 0.1% Triton X-100 in PBS, 10% horse serum, and 1% bovine serum albumin (BSA), and reacted with primary antibodies diluted in PBS-TBH. The ICP0 mouse monoclonal antibody (Goodwin Institute for Cancer Research, Plantation, FL) was used at a dilution of 1:800. The mouse monoclonal antibody to Nectin-1 was used at a dilution of 1:1,000. The Cbl rabbit polyclonal antibody was used at a dilution of 1:500. The four-well cultures were then rinsed several times with PBS-TBH and reacted with Alexa-Fluor 594-conjugated goat anti-rabbit or Alexa-Fluor 488-conjugated goat anti-mouse, diluted 1:1,000 in PBS-TBH. After several rinses, first with PBS-TBH and then with PBS, the samples were mounted and examined with a Zeiss confocal microscope equipped with software provided by Zeiss.
ACKNOWLEDGMENTS
This research was initiated at the University of Chicago in Bernard Roizman's laboratory. We thank Bernard Roizman for his long-term support, mentorship, and generosity in providing materials and other resources. We thank Richard Longnecker (Northwestern University Feinberg School of Medicine) for kindly providing the Nectin-1-expressing plasmid. We also thank Edward Stephens (University of Kansas Medical Center) for editing the manuscript.
M.K. is funded through start-up funds from the University of Kansas Medical Center and COBRE grant P20GM113117.
REFERENCES
- 1.Hagglund R, Roizman B. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J Virol 78:2169–2178. doi: 10.1128/JVI.78.5.2169-2178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roizman B, Gu H, Mandel G. 2005. The first 30 minutes in the life of a virus: unREST in the nucleus. Cell Cycle 4:1019–1021. doi: 10.4161/cc.4.8.1902. [DOI] [PubMed] [Google Scholar]
- 3.Roizman B, Zhou G, Du T. 2011. Checkpoints in productive and latent infections with herpes simplex virus 1: conceptualization of the issues. J Neurovirol 17:512–517. doi: 10.1007/s13365-011-0058-x. [DOI] [PubMed] [Google Scholar]
- 4.Roizman B. 2011. The checkpoints of viral gene expression in productive and latent infection: the role of the HDAC/CoREST/LSD1/REST repressor complex. J Virol 85:7474–7482. doi: 10.1128/JVI.00180-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawaguchi Y, Bruni R, Roizman B. 1997. Interaction of herpes simplex virus 1 α regulatory protein ICP0 with elongation factor 1δ: ICP0 affects translational machinery. J Virol 71:1019–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang Y, Kurakin A, Roizman B. 2005. Herpes simplex virus 1 infected cell protein 0 forms a complex with CIN85 and Cbl and mediates the degradation of EGF receptor from cell surfaces. Proc Natl Acad Sci U S A 102:5838–5843. doi: 10.1073/pnas.0501253102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paladino P, Collins SE, Mossman KL. 2010. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS One 5:e10428. doi: 10.1371/journal.pone.0010428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor KE, Chew MV, Ashkar AA, Mossman KL. 2014. Novel roles of cytoplasmic ICP0: proteasome-independent functions of the RING finger are required to block interferon-stimulated gene production but not to promote viral replication. J Virol 88:8091–8101. doi: 10.1128/JVI.00944-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jozic D, Cardenes N, Deribe YL, Moncalian G, Hoeller D, Groemping Y, Dikic I, Rittinger K, Bravo J. 2005. Cbl promotes clustering of endocytic adaptor proteins. Nat Struct Mol Biol 12:972–979. doi: 10.1038/nsmb1000. [DOI] [PubMed] [Google Scholar]
- 10.Jozic D, Cardenes N, Deribe YL, Moncalian G, Hoeller D, Groemping Y, Dikic I, Rittinger K, Bravo J. 2008. Reply to “The binding stoichiometry of CIN85 SH3 domain A and Cbl-b.” Nat Struct Mol Biol 15:891–892. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt MH, Dikic I. 2005. The Cbl interactome and its functions. Nat Rev Mol Cell Biol 6:907–918. doi: 10.1038/nrm1762. [DOI] [PubMed] [Google Scholar]
- 12.Sorkin A, Goh LK. 2009. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res 315:683–696. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 13.Swaminathan G, Tsygankov AY. 2006. The Cbl family proteins: ring leaders in regulation of cell signaling. J Cell Physiol 209:21–43. doi: 10.1002/jcp.20694. [DOI] [PubMed] [Google Scholar]
- 14.Xiu X, Lu ZM. 2005. Ubiquitination-mediated degradation of epidermal growth factor receptor. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 27:120–127. (In Chinese.) [PubMed] [Google Scholar]
- 15.Stiles KM, Milne RS, Cohen GH, Eisenberg RJ, Krummenacher C. 2008. The herpes simplex virus receptor nectin-1 is down-regulated after trans-interaction with glycoprotein D. Virology 373:98–111. doi: 10.1016/j.virol.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stiles KM, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C. 2010. Herpes simplex virus glycoprotein D interferes with binding of herpesvirus entry mediator to its ligands through downregulation and direct competition. J Virol 84:11646–11660. doi: 10.1128/JVI.01550-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stiles KM, Krummenacher C. 2010. Glycoprotein D actively induces rapid internalization of two nectin-1 isoforms during herpes simplex virus entry. Virology 399:109–119. doi: 10.1016/j.virol.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hung SL, Cheng YY, Wang YH, Chang KW, Chen YT. 2002. Expression and roles of herpesvirus entry mediators A and C in cells of oral origin. Oral Microbiol Immunol 17:215–223. doi: 10.1034/j.1399-302X.2002.170403.x. [DOI] [PubMed] [Google Scholar]
- 19.Mata M, Zhang M, Hu X, Fink DJ. 2001. HveC (nectin-1) is expressed at high levels in sensory neurons, but not in motor neurons, of the rat peripheral nervous system. J Neurovirol 7:476–480. doi: 10.1080/135502801753170336. [DOI] [PubMed] [Google Scholar]
- 20.Simpson SA, Manchak MD, Hager EJ, Krummenacher C, Whitbeck JC, Levin MJ, Freed CR, Wilcox CL, Cohen GH, Eisenberg RJ, Pizer LI. 2005. Nectin-1/HveC Mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J Neurovirol 11:208–218. doi: 10.1080/13550280590924214. [DOI] [PubMed] [Google Scholar]
- 21.De Regge N, Nauwynck HJ, Geenen K, Krummenacher C, Cohen GH, Eisenberg RJ, Mettenleiter TC, Favoreel HW. 2006. Alpha-herpesvirus glycoprotein D interaction with sensory neurons triggers formation of varicosities that serve as virus exit sites. J Cell Biol 174:267–275. doi: 10.1083/jcb.200510156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campadelli-Fiume G, Menotti L. 2007. Entry of alphaherpesviruses into the cell, p 93–111. In Arvin A, Campadelli-Fiume G, Mocarski E, Morre PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 23.Wittels M, Spear PG. 1991. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res 18:271–290. doi: 10.1016/0168-1702(91)90024-P. [DOI] [PubMed] [Google Scholar]
- 24.Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol 77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gianni T, Forghieri C, Campadelli-Fiume G. 2006. The herpesvirus glycoproteins B and H · L are sequentially recruited to the receptor-bound gD to effect membrane fusion at virus entry. Proc Natl Acad Sci U S A 103:14572–14577. doi: 10.1073/pnas.0606127103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J Biol Chem 284:17370–17382. doi: 10.1074/jbc.M109.005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez M, Cocchi F, Avitabile E, Leclerc A, Adelaide J, Campadelli-Fiume G, Dubreuil P. 2001. Novel, soluble isoform of the herpes simplex virus (HSV) receptor nectin1 (or PRR1-HIgR-HveC) modulates positively and negatively susceptibility to HSV infection. J Virol 75:5684–5691. doi: 10.1128/JVI.75.12.5684-5691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takai Y, Ikeda W, Ogita H, Rikitake Y. 2008. The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu Rev Cell Dev Biol 24:309–342. doi: 10.1146/annurev.cellbio.24.110707.175339. [DOI] [PubMed] [Google Scholar]
- 29.Cocchi F, Menotti L, Mirandola P, Lopez M, Campadelli-Fiume G. 1998. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J Virol 72:9992–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 31.Krummenacher C, Nicola AV, Whitbeck JC, Lou H, Hou W, Lambris JD, Geraghty RJ, Spear PG, Cohen GH, Eisenberg RJ. 1998. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J Virol 72:7064–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith KO. 1963. Physical and biological observations on herpesvirus. J Bacteriol 86:999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmidt MH, Furnari FB, Cavenee WK, Bogler O. 2003. Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc Natl Acad Sci U S A 100:6505–6510. doi: 10.1073/pnas.1031790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng N, Wang P, Jeffrey PD, Pavletich NP. 2000. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell 102:533–539. doi: 10.1016/S0092-8674(00)00057-X. [DOI] [PubMed] [Google Scholar]
- 35.Cooper JA, Kaneko T, Li SS. 2015. Cell regulation by phosphotyrosine-targeted ubiquitin ligases. Mol Cell Biol 35:1886–1897. doi: 10.1128/MCB.00098-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohapatra B, Ahmad G, Nadeau S, Zutshi N, An W, Scheffe S, Dong L, Feng D, Goetz B, Arya P, Bailey TA, Palermo N, Borgstahl GE, Natarajan A, Raja SM, Naramura M, Band V, Band H. 2013. Protein tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim Biophys Acta 1833:122–139. doi: 10.1016/j.bbamcr.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez P, Van SC, Roizman B. 2001. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J Virol 75:3832–3840. doi: 10.1128/JVI.75.8.3832-3840.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalamvoki M, Roizman B. 2010. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc Natl Acad Sci U S A 107:17721–17726. doi: 10.1073/pnas.1012991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalamvoki M, Roizman B. 2010. Interwoven roles of cyclin D3 and cdk4 recruited by ICP0 and ICP4 in the expression of herpes simplex virus genes. J Virol 84:9709–9717. doi: 10.1128/JVI.01050-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miaczynska M. 2013. Effects of membrane trafficking on signaling by receptor tyrosine kinases. Cold Spring Harb Perspect Biol 5:a009035. doi: 10.1101/cshperspect.a009035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eaton S, Martin-Belmonte F. 2014. Cargo sorting in the endocytic pathway: a key regulator of cell polarity and tissue dynamics. Cold Spring Harb Perspect Biol 6:a016899. doi: 10.1101/cshperspect.a016899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldenring JR. 2015. Recycling endosomes. Curr Opin Cell Biol 35:117–122. doi: 10.1016/j.ceb.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott CC, Vacca F, Gruenberg J. 2014. Endosome maturation, transport and functions. Semin Cell Dev Biol 31:2–10. doi: 10.1016/j.semcdb.2014.03.034. [DOI] [PubMed] [Google Scholar]
- 44.Taguchi T. 2013. Emerging roles of recycling endosomes. J Biochem 153:505–510. doi: 10.1093/jb/mvt034. [DOI] [PubMed] [Google Scholar]
- 45.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 46.Kawaguchi Y, Van SC, Roizman B. 1997. Herpes simplex virus 1 α regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol 71:7328–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gu H, Poon AP, Roizman B. 2009. During its nuclear phase the multifunctional regulatory protein ICP0 undergoes proteolytic cleavage characteristic of polyproteins. Proc Natl Acad Sci U S A 106:19132–19137. doi: 10.1073/pnas.0910920106. [DOI] [PMC free article] [PubMed] [Google Scholar]