

ABSTRACT
Seasonal influenza virus epidemics represent a significant public health burden. Approximately 25% of all influenza virus infections are caused by type B viruses, and these infections can be severe, especially in children. Current influenza virus vaccines are an effective prophylaxis against infection but are impacted by rapid antigenic drift, which can lead to mismatches between vaccine strains and circulating strains. Here, we describe a broadly protective vaccine candidate based on chimeric hemagglutinins, consisting of globular head domains from exotic influenza A viruses and stalk domains from influenza B viruses. Sequential vaccination with these constructs in mice leads to the induction of broadly reactive antibodies that bind to the conserved stalk domain of influenza B virus hemagglutinin. Vaccinated mice are protected from lethal challenge with diverse influenza B viruses. Results from serum transfer experiments and antibody-dependent cell-mediated cytotoxicity (ADCC) assays indicate that this protection is antibody mediated and based on Fc effector functions. The present data suggest that chimeric hemagglutinin-based vaccination is a viable strategy to broadly protect against influenza B virus infection.
IMPORTANCE While current influenza virus vaccines are effective, they are affected by mismatches between vaccine strains and circulating strains. Furthermore, the antiviral drug oseltamivir is less effective for treating influenza B virus infections than for treating influenza A virus infections. A vaccine that induces broad and long-lasting protection against influenza B viruses is therefore urgently needed.
KEYWORDS: hemagglutinin, influenza B virus, vaccine
INTRODUCTION
Influenza B viruses cause significant morbidity and mortality annually on a global scale (1–4). Approximately 20 to 30% of all influenza cases are caused by influenza B viruses, but occasionally, they are the dominant influenza type in a season (5–8). While influenza B viruses are considered to cause less severe infections than H3N2 viruses, they rank above pandemic H1N1 infections in severity (4, 9, 10). Influenza B virus is specifically problematic in children and infants, where it can cause excess mortality. A prime example is the 2010-2011 epidemic, where influenza B viruses caused 25% of all influenza cases but were responsible for 38% of all influenza-related pediatric deaths (11), a trend that was seen worldwide (8, 12, 13). Furthermore, a recent study found that the proportion of deaths in children hospitalized with influenza attributable to influenza B virus is 1.1%, compared to 0.4% for influenza A virus (14). Finally, oseltamivir treatment seems to be less effective against influenza B viruses than against influenza A viruses (15–17), and oseltamivir-resistant mutants emerge (18–20), sometimes without a loss of fitness (21, 22).
Current influenza virus vaccines are an effective prophylactic countermeasure against influenza B virus infections. However, due to the antigenic drift in the globular head domain of viral hemagglutinin (HA), these vaccines have to be reformulated and readministered annually (23). Vaccine strain selection is based on surveillance and predictions, and the selected vaccine strains do not always match the circulating strains. A mismatch between vaccine strains and circulating strains can lead to a sharp drop in vaccine effectiveness (2, 3, 7). This is further complicated by the fact that two antigenically distinct influenza B virus lineages, B/Victoria/2/87-like (Victoria-like) viruses and B/Yamagata/16/88-like (Yamagata-like) viruses, cocirculate in the human population (2, 4, 24). The introduction of quadrivalent influenza virus vaccines (QIVs), which contain vaccine strains of both lineages, alleviates this problem but does not solve the issues of constant antigenic drift, mismatch within a lineage, and waning of vaccine effectiveness within a season (2, 4, 25).
Here, we describe a vaccine approach that refocuses the antibody response toward the conserved stalk domain of HA. In contrast to the immunodominant head domain of HA, the stalk domain is usually not a primary target of the immune system (26, 27). Antibody titers against this domain in humans are low, but previous studies have shown that antibodies that target the stalk can be broadly protective (28). To induce protective antistalk antibody titers, we are using a vaccination strategy based on chimeric HAs (cHAs) (29, 30) that has already been successfully applied as a universal vaccine strategy for group 1 (31–35) and group 2 (36, 37) HA-expressing influenza A viruses. Here, we constructed cHAs with different head domains derived from exotic influenza A virus HA subtypes and stalk domains from influenza B virus HAs. These influenza A/B virus HA chimeras (cHA/B) were used in sequential vaccination regimens in mice to induce protective titers of stalk-reactive antibodies. We show that these vaccine constructs protect mice against lethal challenge with diverse influenza B viruses and that protection is mediated by antibodies, likely via Fc effector functions.
RESULTS
Design and construction of cHA/B HAs.
Chimeric HAs are combinations of globular head domains from an HA of one influenza virus subtype or strain and the stalk domain from an HA of another subtype/strain. This design has been successfully applied to influenza A virus HAs, where a disulfide bond between two conserved cysteines (C52 and C277 [H3 numbering]) was used as a demarcation line between head and stalk regions (Fig. 1A) (29, 30). C52 and C277 are present in all influenza A virus HAs, and their presence makes it relatively easy to graft a head domain from one influenza A virus HA onto the stalk domain of another HA. The sequential presentation of cHAs with conserved stalk domains but very different head domains refocuses the immune response from the immunodominant head domain to the subdominant stalk domain (38). However, the diversity of influenza B virus head domains is low compared to that of influenza A virus head domains (2). We therefore rationalized that the use of diverse influenza A virus HA head domains in combination with influenza B virus stalk domains would be a better choice than making cHAs between different influenza B virus HA head and stalk domains. The obstacle with this approach is that the disulfide bond formed between C52 and C277 at the head/stalk interface, which is conserved in influenza A virus HAs, is not present in influenza B virus HAs. This makes the grafting of influenza A virus head domains onto influenza B virus stalk domains more challenging. By comparing structures and sequences of influenza A and B virus HAs, we identified a pair of alanines (A57 and A306 [B/Yamagata/16/88 numbering, starting with methionine]) in influenza B virus HA in a position homologous to those of the cysteines in influenza A virus HA (Fig. 1B). We therefore designed two construct series. The cHA/Bcys series includes the cysteines from influenza A virus HA with the alanines from influenza B virus HA removed (Fig. 1C). The cHA/Bala series maintains the alanines from influenza B virus HA with the influenza A virus HA cysteines deleted (Fig. 1D). Influenza A virus head domains from H5, H7, and H8 HA were used to construct the cH5/Bcys, cH7/Bcys, and cH8/Bcys and the cH5/Bala, cH7/Bala, and cH8/Bala series of antigens. The constructs where then subcloned into mammalian expression vectors, and their expression after transfection into human embryonic kidney 293T (HEK 293T) cells was confirmed by using immunofluorescence microscopy (Fig. 1E). The cH7/Bcys, cH8/Bcys, cH7/Bala, and cH8/Bala constructs were also expressed in the baculovirus system to create protein antigens for vaccination.
FIG 1.
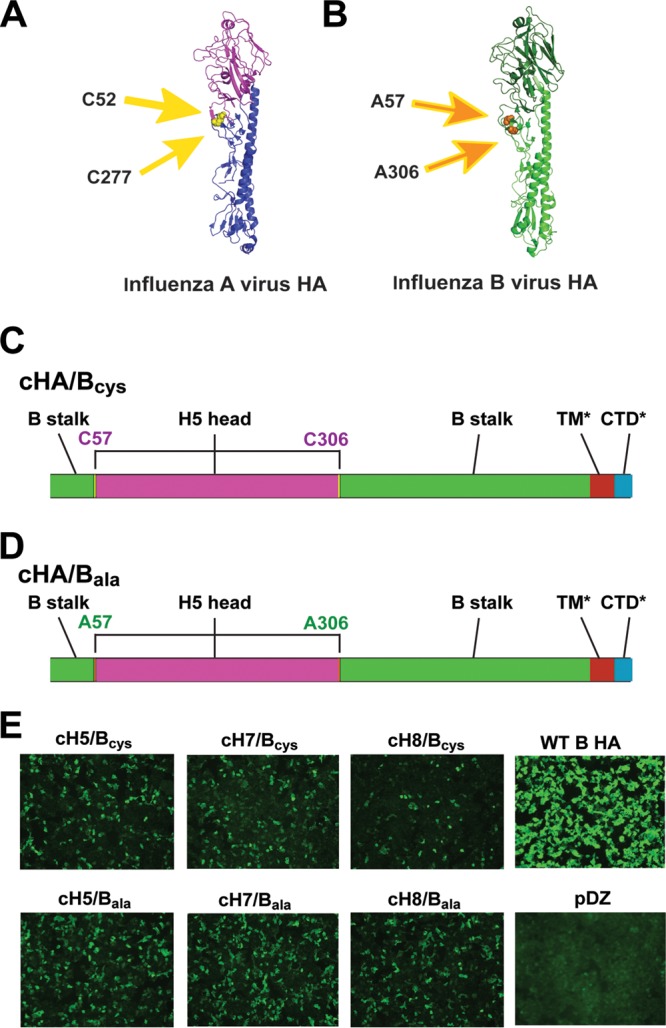
Design of chimeric HA/B constructs. (A) Monomer of the H5 HA structure (based on data reported under Protein Data Bank [PDB] accession number 2FK0 [52]). The head domain is shown in purple, and the stalk domain is shown in blue. The head domain is located between cysteines 52 and 277 (H3 numbering), which form a disulfide bond. The cysteines are indicated by yellow arrows and are highlighted in yellow on the structure. (B) Monomer of an influenza B virus HA (based on data reported under PDB accession number 4M44 [53]). In this case, the stalk domain is shown in light green, and the head domain is shown in dark green. The head domain is located between alanines 57 and 306 (B/Yamagata/16/88 numbering, starting with methionine), which are in positions on the structure similar to those of C52 and C277 on influenza A virus HAs. The alanines are indicated by orange arrows and are highlighted in orange on the structure. (C and D) The two cHA/B constructs that were generated with head domains from influenza A virus HA and stalk domains from influenza B virus. (C) The influenza A virus HA cysteines (now in positions C57/C306, labeled in purple, since they are derived from influenza A virus HA) were maintained as the demarcation line between the head (purple) and stalk (green), and the original influenza B virus alanines were deleted. (D) Similar construct where the alanines (labeled in green since they are derived from influenza B virus HA) were maintained and the cysteines were removed. *, the transmembrane domain (TM) (brown) and cytoplasmic tail domain (CTD) (blue) are derived from A/Puerto Rico/8/34 HA for all constructs for technical reasons. (E) Immunostaining of HEK 293T cells transfected with different cHA/B constructs, wild-type influenza B virus HA (WT B HA) (from B/Yamagata/16/88), or the empty vector (pDZ). cH5/Bcys contains the head domain of H5 HA combined with the stalk domain of B/Yamagata/16/88 HA, with “cys” indicating that the design shown in panel C was used. cH7/Bcys and cH8/Bcys were constructed in the same way, but the head domains are derived from H7 and H8, respectively. cH5/Bala, cH7/Bala, and cH8/Bala are based on the designs shown in panel D. Cells were stained with polyclonal anti-B/Yamagata/16/88 serum.
Sequential vaccination with cHA/B HAs protects from challenge with diverse influenza B virus strains.
Next, we tested the protective effect of sequential vaccination with the cHA/B constructs in the mouse model. Animals were intramuscularly (i.m.) primed with a DNA vaccine expressing cH5/Bcys or cH5/Bala and then vaccinated at 3-week intervals with cH7/Bcys or cH7/Bala proteins, followed by cH8/Bcys or cH8/Bala proteins, respectively (Fig. 2A). Protein antigens were delivered both intranasally (i.n.) and i.m. with poly(I·C) as an adjuvant. Control groups included prime-only animals that received irrelevant protein vaccinations [bovine serum albumin (BSA) with poly(I·C)] and naive animals. In addition, we added “standard-of-care” (SOC) positive-control groups that received two i.m. doses (1 μg of influenza B virus HA per mouse and vaccination) of a human seasonal trivalent vaccine matched to the challenge strain. This regimen is similar to two-dose regimens recommended for the vaccination of children (influenza naive). Animals were then challenged 4 weeks after the last vaccination with a lethal dose of diverse influenza B viruses representing the ancestral (39), Victoria-like, and Yamagata-like (24) lineages.
FIG 2.

Vaccination with cHA/B constructs protects mice from lethal challenge with influenza B viruses. (A) Schematic of the cHA/B-based vaccination regimen (modeled based on data reported under PDB accession number 4M44). Mice were first primed with plasmid DNA expressing cH5/B, followed by protein vaccinations with cH7/B and cH8/B antigens. The animals were then challenged with different influenza B viruses 4 weeks after the last vaccination. (B and C) Weight loss (B) and survival (C) of animals challenged with B/Malaysia/2506/04 (Victoria lineage). cHA/Bcys indicates animals vaccinated with the cHA/B variants that use cysteines as demarcation lines, and cHA/Bala indicates animals vaccinated with constructs that use alanines instead (as shown in Fig. 1). Prime-only (prime-onlycys and prime-onlyala) groups received only the indicated prime but were vaccinated with irrelevant proteins. SOC groups received two vaccinations with a trivalent inactivated human seasonal vaccine containing an influenza B virus strain matched to the challenge virus. A sixth group of mice was naive. The statistical difference between cHA/Bcys-vaccinated and naive mice as well as the difference between cHA/Bala-vaccinated and naive mice were tested and found to be significant (P = 0.0035 [**] and P = 0.0145 [*], respectively). (D and E) Weight loss (D) and survival (E) of animals vaccinated as described above and challenged with B/Florida/04/06 virus (Yamagata lineage). The statistical difference between cHA/Bcys-vaccinated and naive mice as well as the difference between cHA/Bala-vaccinated and naive mice were found to be significant (P = 0.0019 [**] and P = 0.0019 [**], respectively). (F and G) Weight loss (F) and survival (G) after B/Lee/40 challenge of animals vaccinated with cHA/Bcys constructs, prime-only animals, naive animals, or animals vaccinated twice with a trivalent inactivated human seasonal vaccine containing B/Malaysia/2506/04 (SOC-Vic) or B/Florida/04/06 (SOC-Yam) antigens. The statistical difference between the cHA/Bcys-vaccinated and naive mice was found to be significant (P = 0.0019 [**]). *, P ≤ 0.05; **, P ≤ 0.01.
Challenge with the Victoria-like strain B/Malaysia/2506/04 resulted in transient weight loss, with a 7.3% mean maximum weight loss on day 7 postchallenge (Fig. 2B), but full survival in cHA/Bcys-vaccinated animals (Fig. 2C). The cHA/Bala-vaccinated animals showed slightly greater weight loss, with one animal succumbing to infection on day 9 postchallenge (Fig. 2B and C). Positive-control animals vaccinated with the matched standard of care showed weight loss similar to that of cHA/B-vaccinated animals and were completely protected from mortality, as expected. Both prime-only animal groups suffered severe weight loss, with no survival in the prime-onlyala group and 40% survival in the prime-onlycys group. All naive animals succumbed to infection by day 9 postinfection. Differences in survival between cHA/B-vaccinated groups and naive animals were significant, while no statistical difference could be detected between cHA/B-vaccinated and SOC groups.
Lethal challenge with the antigenically distinct Yamagata-like B/Florida/04/06 virus produced similar results. In this case, the percentages of weight loss for cHA/Bcys, cHA/Bala, and matched SOC groups were very similar (approximately 10% maximum weight loss in all groups) and transient, with complete protection from mortality (Fig. 2D and E). Naive animals as well as animals in both prime-only groups succumbed to infection on day 8 and day 9 postchallenge, respectively. Again, differences in survival between cHA/B-vaccinated groups and naive animals were significant, but no statistical difference was detected between cHA/B-vaccinated and SOC groups.
Since the cHA/Bcys constructs worked slightly better in the B/Malaysia/2506/04 challenge study, we moved this construct design forward to test protection against the ancestral B/Lee/40 strain (39), which is antigenically distinct from both the Victoria-like as well as the Yamagata-like lineages. This scenario also offered the opportunity to compare the efficacy of the cHA/B vaccination regimen with that of the current standard of care against an antigenically mismatched virus. In this case, two SOC groups were used, one that received the B/Malaysia/2506/04-containing vaccine (SOC-Vic) and a second one that received the B/Florida/04/06-containing vaccine (SOC-Yam). A prime-only group and a naive control group were added as well. The cHA/Bcys vaccination regimen protected mice completely from morbidity and mortality (Fig. 2F and G). In contrast, animals in both SOC groups lost approximately 20% of their initial body weight. While animals in the SOC-Vic group survived infection, 40% of the SOC-Yam-vaccinated animals died from infection. Furthermore, all naive and prime-only animals succumbed to infection as well. The difference in survival rates between the cHA/Bcys group and the naive group was statistically significant, while there was no statistically significant difference in mortality between the cHA/Bcys group and the two SOC groups. However, the difference in weight loss between animals in the cHA/Bcys group and animals in both SOC groups was significant on several days postchallenge. In summary, cHA/B constructs protect mice from challenge with diverse influenza B viruses.
The cHA/B vaccine regimen induces broadly reactive antibodies.
To determine if sequential vaccination with cHA/B HAs induces broadly reactive antibodies, we tested sera from vaccinated mice in an enzyme-linked immunosorbent assay (ELISA). We chose purified influenza B virions as the ELISA substrate. This approach has the advantage of measuring binding to “real” wild-type virions and avoids problems with reactivity toward the hexahistidine tag or the trimerization domain of purified recombinant HA. These tags/domains are also present in the recombinant cHA/B proteins used as immunogens and might interfere with the assessment of anti-HA titers. However, this strategy also has some caveats, including the variability of antigen content in the virus preparations, which makes it hard to compare results obtained with two different substrates. In addition, the whole-virus preparations contain HA, neuraminidase (NA), and internal viral proteins, and animals that received the standard of care (trivalent inactivated influenza vaccine [TIV]) might react to all of these components, while cHA/B-vaccinated mice react to HA only, which also makes a direct comparison difficult.
ELISAs were performed against the ancestral strain B/Lee/40; the Yamagata lineage strains B/Yamagata/16/88, B/Florida/04/06, and B/Phuket/3073/13; as well as the Victoria lineage strains B/Victoria/2/87, B/Malaysia/2506/04, and B/Brisbane/60/08 (Fig. 3A to G). In all cases, cHA/Bcys and cHA/Bala induced antibody responses that were significantly greater than responses in the prime-only animals, and antibody induction was relatively constant for all tested strains. Sera from animals that received TIV were used as positive controls. Importantly, these sera, as mentioned above, react to HA, NA, and internal proteins of the ELISA substrate, while sera from cHA/B-vaccinated animals react only with the HA stalk. We also analyzed the induction of antibodies over the course of the vaccination regimen and found that the second vaccination induced a higher-level induction than did the first vaccination (Fig. 3H). Overall, the cHA/B-based vaccination strategy led to the induction of broadly reactive antibodies that covered influenza B virus strains from 1940 to 2013.
FIG 3.

Vaccination with cHA/B constructs induces antibody responses against diverse influenza B viruses. (A to G) Quantitative ELISA reactivity reported as endpoint titers for animals vaccinated with the cHA/Bcys and cHA/Bala constructs and their respective prime-only groups (prime-onlycys and prime-onlyala) as well as for naive animals. Sera from animals that received inactivated influenza B virus vaccines were used as positive controls. A panel of purified influenza B viruses representing the ancestral, Victoria (Vic), and Yamagata (Yam) lineages was used as the ELISA substrate. Strains are indicated. Importantly, the reactivity of serum from cHA/B-vaccinated animals is directed against HA only, while positive-control serum is the sum of the reactivity to HA, NA, and conserved internal viral proteins. The geometric mean is indicated for each sample set (n = 10 for all groups except the positive-control groups [n = 5]). (H) Profile of reactivity against one selected substrate (B/Florida/04/06) over the course of the vaccination experiment. Statistical significance is indicated where tested (n.s., not significant [P > 0.05]; *, P ≤ 0.05; **, P ≤ 0.01).
cHA vaccine-based protection from influenza B virus challenge is antibody mediated.
While we were assuming that the protection observed in the mouse model was antibody mediated, we investigated whether antibodies alone could confer protection. Sets of cHA/Bcys, SOC, and naive mice were terminally bled 4 weeks after the last vaccination. Serum was harvested and pooled within groups. The serum was then transferred into naive mice, which were challenged with a lethal dose of B/Malaysia/2506/04 2 h after transfer. Mice that received cHA/Bcys serum suffered only mild transient weight loss and were completely protected from mortality (Fig. 4A and B). Mice that received sera from animals that had been vaccinated with the standard of care matched to the challenge strain were partially protected from mortality, with 60% of mice surviving. All animals that received serum from naive mice succumbed to infection by day 9. We then performed microneutralization assays with B/Malaysia/2506/04 using the same serum pools that were used in the passive-transfer experiment. We found that only the SOC serum had measurable neutralizing activity, while the cHA/Bcys and naive sera did not (Fig. 4C). However, the cHA/Bcys serum had strong activity in an antibody-dependent cell-mediated cytotoxicity (ADCC) reporter assay with B/Malaysia/2506/04, while SOC serum did not show such activity (Fig. 4D). Finally, we determined the effect of cHA/B vaccination on virus replication in the lung. Groups of animals vaccinated with cHA/Bcys, prime-only animals, animals that received the standard of care, as well as naive animals were challenged with a lethal dose of B/Malaysia/2506/04 virus, and their lungs were harvested on day 3 or day 6. Vaccination with cHA/Bcys reduced lung virus titers approximately 10-fold on day 3 and 135-fold on day 6 compared to those in naive animals. Animals that received the standard of care showed only a 2.6-fold reduction on day 3, but virus was not detectable in two out of three animals on day 6 postchallenge (Fig. 4E). In summary, we show that protection by cHA/B-based vaccination is antibody mediated, most likely via Fc effector functions.
FIG 4.

Protection induced by chimeric HA/B HA vaccination is mediated by antibodies, likely via Fc-mediated effector functions. (A and B) Weight loss (A) and survival (B) of mice that received passive transfer of serum from cHA/Bcys, SOC (animals received two vaccinations with a human seasonal inactivated vaccine containing the B/Malaysia/2506/04 influenza B virus component), or naive animals and were subsequently challenged with B/Malaysia/2506/04 (Victoria lineage). Differences in survival between the cHA/Bcys and naive groups were statistically significant (P = 0.0023 [**]). (C) Neutralizing activity of sera from cHA/Bcys, SOC, and naive animals tested in a microneutralization assay with B/Malaysia/2506/04 virus. (D) Activity of the same sera in an ADCC reporter assay. (E) Day 3 and day 6 lung virus titers in animals directly vaccinated with cHA/Bcys and in animals in the prime-only (prime-onlycys), SOC, and naive groups after challenge with B/Malaysia/2506/04. Statistical significance is indicated where tested (**, P ≤ 0.01).
DISCUSSION
Similar to influenza A viruses, both influenza B virus lineages undergo rapid antigenic drift, and vaccines have to be updated and readministered on an annual basis. This is further complicated by the fact that two distinct lineages of influenza B viruses are cocirculating in the human population (24). Vaccine strain selection is based on surveillance/prediction, and selection of a vaccine strain that matches the circulating pathogenic strains is key to achieving high vaccine efficacy and effectiveness. Historically, mismatches between vaccine strains and circulating strains have happened frequently for influenza B viruses, partially due to the unpredictable dynamics of the cocirculation of the two lineages (7). While the inclusion of both influenza B virus lineages in modern quadrivalent influenza virus vaccines alleviated this problem, the issue of antigenic drift remains. Here, a broadly protective vaccination regimen based on chimeric HAs, which has been successfully developed for influenza A viruses (28), was adapted to influenza B virus. Head domains from exotic avian influenza A virus HA subtypes (H5, H7, and H8) were grafted onto influenza B virus HA stalks. These constructs were then used in sequential vaccination experiments in mice to refocus the immune response toward the conserved HA stalk domain. Our experiments show that vaccination with these constructs can induce cross-reactive antibodies against a wider range of diverse influenza B viruses, including isolates from the ancestral, Yamagata-like, and Victoria-like lineages (Fig. 5). The vaccination regimen afforded robust protection against challenge with diverse strains. Through serum transfer challenge experiments, we determined that antibodies alone could protect animals from virus challenge. Interestingly, only negligible neutralizing activity was detected when serum was tested against one of the challenge strains. However, the serum was highly active in an ADCC reporter assay that measures Fc-mediated effector functions, an important mechanism of protection for HA stalk-reactive antibodies (40–46). The nonneutralizing nature of the antibody response combined with its ADCC activity mirror the characteristics of human monoclonal antibody (MAb) CR9114. This monoclonal antibody binds broadly to influenza B virus HAs but is unable to neutralize the virus in vitro (47). However, it shows strong activity in ADCC reporter assays, and it is highly protective against challenge with diverse influenza B virus strains in vivo.
FIG 5.

Phylogenetic tree representing influenza B virus HA diversity. Strains used as challenge strains (B/Malaysia/2506/04 and B/Florida/04/06) or as ELISA substrate in this study are indicated. The bar indicates a 1% change in the amino acid sequence.
Humans have low titers of antibodies that are reactive against the stalk of the influenza B virus HA. A vaccine that induces high titers of these antibodies, either as a stand-alone vaccine or in combination with influenza A virus components, would be a highly valuable prophylactic tool for the control of influenza.
MATERIALS AND METHODS
Cells, viruses, and recombinant proteins.
HEK 293T cells were maintained in Dulbecco's modified Eagle medium (DMEM; Gibco) with 10% fetal bovine serum (FBS; Gibco), 2 mM l-glutamine, and a penicillin (100 U/ml)-streptomycin (100 μg/ml) solution (Pen-Strep; Gibco). Madin-Darby canine kidney (MDCK) cells were grown in DMEM (Gibco) supplemented with 10% FBS and penicillin-streptomycin. Sf9 cells for baculovirus rescue and propagation were grown in Trichoplusia ni medium-formulation Hink (TNM-FH) insect cell medium (Gemini Bioproducts) supplemented with 10% FBS and penicillin-streptomycin. BTI-TN-5B1-4 (High Five) cells for protein expression were grown in serum-free SFX medium (HyClone) supplemented with penicillin-streptomycin. Virus preparations for influenza B virus strains B/Lee/40, B/Yamagata/16/1988, B/Victoria/2/1987, B/Florida/4/2006, B/Malaysia/2506/04, B/Brisbane/60/2008, and B/Phuket/3073/2013 were generated as follows. Viruses were grown in 10-day-old embryonated chicken eggs (Charles River) for 72 h at 33°C. Eggs were then cooled down to 4°C overnight, and allantoic fluid was harvested and cleared by low-speed centrifugation at 2,000 × g for 10 min at 4°C. Viruses were then pelleted through a 30% sucrose cushion (buffered in NTE buffer [100 mM NaCl, 10 mM Tris-HCl, 1 mM EDTA {pH 7.4}]) by centrifugation in a Beckman L7-65 ultracentrifuge at 25,000 rpm for 2 h at 4°C using a Beckman SW28 rotor. Pellets were collected in phosphate-buffered saline (PBS) (pH 7.4), and the protein content was quantified by using the Bradford reagent. Challenge viruses were grown in eggs as described above, and titers of the virus stocks were determined on monolayers of MDCK cells.
Design of chimeric HA vaccine constructs.
Chimeric influenza virus A/B HA (cHA/B) constructs consist of an influenza A virus HA head atop an influenza B virus HA stalk. Constructs were designed to have HA stalks derived from B/Yamagata/16/88 and heads from either H5 (from a low-pathogenic version of A/Vietnam/1203/2004 [48], named cH5/B), H7 (A/mallard/Alberta/24/2001, named cH7/B), or H8 (A/mallard/Sweden/24/2002, named cH8/B) influenza A virus HA. Two designs were assembled for each influenza A virus HA head that was used. The first design includes a pair of cysteines at the head/stalk interface similar to those observed in influenza A virus HAs (at positions 52 and 277 [H3 numbering]). These residues had previously been used as a demarcation line for the head/stalk interface generating influenza A virus chimeric HAs (29, 30). These constructs are referred to as cH5/Bcys, cH7/Bcys, and cH8/Bcys. The second design replaces the pair of cysteines that would naturally encompass the influenza A virus HA head with a pair of alanines that are found in influenza B virus HAs at approximately the same location. These constructs are referred to as cH5/Bala, cH7/Bala, and cH8/Bala. The alanines (positions 57 and 306 [B/Yamagata/16/88 numbering, starting at methionine]) used as a demarcation line were determined by performing sequence alignment between influenza A and influenza B virus HA protein sequences with the BioEdit Sequence Alignment Editor (1997 to 2013; Tom Hall) and using crystal structure visualization programs such as the Molecular Operating Environment (MOE; Chemical Computing Group) and PyMOL (Schrödinger, Inc.). The constructs were then created via overlapping extension PCRs or were synthesized (Thermo Fisher Scientific/Invitrogen) and subcloned into pDZ plasmids for transfection of mammalian cells or into a pHA baculovirus shuttle vector for protein expression and purification (primer sequences are available upon request). As initial attempts to rescue the virus using this cHA/B HA constructs were made by using reverse genetic approaches, all clones were constructed with packaging signals derived from A/Puerto Rico/8/34 (PR8) virus and included the PR8 noncoding regions, signal peptide, transmembrane domain, and cytoplasmic tail domain. For the expression of recombinant proteins, the HA ectodomain open reading frames were cloned into the pHA baculovirus shuttle vector in frame with a C-terminal T4 trimerization domain and a hexahistidine tag, as described previously (49, 50). Recombinant bacmids and, subsequently, baculoviruses were generated, and proteins were expressed in BTI-TN-5B1-4 cells and purified from the culture supernatant according to an established protocol (49). The protein concentration was measured by using the Bradford reagent and a standard curve.
Surface staining.
Twenty-four-well plates were coated with poly-l-lysine (Sigma-Aldrich), and 1.5 × 104 HEK 293T cells were seeded per well. The next day, cells were transfected with 500 ng of each plasmid in optimized minimal essential medium (MEM) (Opti-MEM; Gibco) with Lipofectamine 2000 (Thermo Fisher Scientific) at a 1:2 ratio. Twenty-four hours later, medium was removed, and cells were fixed with 4% paraformaldehyde in PBS for at least 1 h at room temperature (RT). Cells were washed with PBS and blocked with 5% BSA in PBS. Primary anti-B/Yamagata/16/88 murine polyclonal serum was diluted 1:500 in 1% BSA–PBS and incubated on cells for at least 2 h at RT. Cells were washed with PBS and incubated with an anti-mouse Alexa-488 antibody (Life Technologies) diluted 1:1,000 in 1% BSA–PBS for at least 1 h at RT. Fluorescence was visualized and captured by using an Olympus 1X-70 microscope within the Microscopy Core at the Icahn School of Medicine at Mount Sinai.
Animal experiments.
Six- to eight-week-old female BALB/c mice were used for all animal experiments (Jackson Laboratories). Experiments were performed in accordance with protocols approved by the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee (IACUC). For initial experiments, animals were divided into six groups (n = 5) per challenge virus. The six groups included a group that was DNA vaccinated with cH5/Bcys and then vaccinated with cH7/Bcys and cH8/Bcys proteins (cHA/Bcys), a group that was DNA vaccinated with cH5/Bcys and then vaccinated twice with BSA as an irrelevant protein (prime-onlycys), a group that was DNA vaccinated with cH5/Bala and then vaccinated with cH7/Bala and cH8/Bala proteins (cHA/Bala), a group that was DNA vaccinated with cH5/Bala and then vaccinated twice with BSA (prime-onlyala), a group that received a trivalent inactivated vaccine that matched the challenge strain (SOC), and a naive group of mice. DNA vaccines were delivered via intramuscular electroporation in the left calf muscle via a TriGrid electroporation device (Ichor Medical Systems) at a dose of 80 μg DNA per mouse (40 μl at 2 mg/ml in double-distilled water [ddH2O]). DNA priming was included in the experimental design to make the results comparable to data from previous studies with group 1 and group 2 cHA vaccines (31, 36). Protein vaccines (cHA/B HAs) or BSA was administered both i.n. and i.m. at the same time at a dose of 10 μg per mouse (5 μg i.n. and 5 μg i.m.; 50 μl per vaccination site), adjuvanted with 10 μg of poly(I·C) (InvivoGen) per mouse per site. The SOC was given unadjuvanted at a dose of 1 μg of influenza B virus HA per mouse. The Fluzone vaccine (2006 to 2007; Sanofi Pasteur) (includes the B/Malaysia/2506/04 strain) was used for B/Malaysia/2506/04 challenges, and the Flulaval vaccine (2008 to 2009; GlaxoSmithKline) (includes the B/Florida/04/06 strain) was used for B/Florida/04/06 challenge experiments. Vaccinations were given at 3-week intervals. Four weeks after the last vaccination, animals were anesthetized with a ketamine-xylazine cocktail (0.15 mg/kg of body weight of ketamine and 0.03 mg/kg of xylazine per mouse) and then challenged intranasally with 5 murine 50% lethal doses (mLD50) of mouse-adapted B/Malaysia/2506/04 (1 LD50 equals 2.5 × 103 PFU), mouse-adapted B/Florida/04/06 (1 LD50 equals 3.2 × 103 PFU), or B/Lee/40 (1 LD50 equals 2.2 × 105 PFU) virus in a volume of 50 μl. Mouse adaptation was performed prior to the U.S. government “gain-of-function” moratorium in 2014. Animals were monitored for survival and weight loss for 14 days postchallenge and were scored dead and humanely euthanized if they lost more than 25% of their initial body weight. Additional experiments to assess virus replication in the lungs and the protective effect of transferred serum were performed. To determine lung virus titers, animals of selected groups (vaccinated as described above) were challenged as described above with 5 mLD50 of B/Malaysia/2506/04 virus. Lungs from groups of animals were harvested on days 3 and 6 postchallenge (n = 3 per group and time point). Lungs were then homogenized by using a BeadBlaster homogenizer (Benchmark), and lung virus titers were determined by a plaque assay on confluent layers of MDCK cells as described previously (51). For passive-transfer experiments, groups of mice (n = 5) were terminally bled, and serum was harvested, pooled within the groups, and transferred (200 μl per mouse) into groups of naive mice (n = 5) via intraperitoneal (i.p.) injection. Two hours after transfer, the mice were anesthetized and challenged with 5 mLD50 of B/Malaysia/2506/04 virus as described above.
ELISAs.
A quantitative ELISA that uses the reciprocal endpoint titer as a readout was used in this study to evaluate the antibody response to influenza B viruses. ELISA plates (Immulon 4HBX; Thermo Scientific) were coated with 5 μg/ml of the whole-virus preparation per well (50 μl per well) in coating buffer (carbonate-bicarbonate buffer [pH 9.4]) overnight at 4°C. The next day, plates were washed three times with PBS containing 0.1% Tween 20 (PBS-T), blocking solution (3% goat serum, 0.5% nonfat dried milk powder, 96.5% PBS-T) was added to each well, and the plates were incubated for 1 h at RT. Serum samples were serially diluted in blocking solution and added to the plate, followed by a 2-h incubation at RT. ELISA plates were washed three times with PBS-T, and anti-mouse IgG-horseradish peroxidase (HRP)-conjugated antibody (GE Healthcare) was added at a dilution of 1:3,000. Plates were then incubated for 1 h at RT and washed three times with PBS-T. The O-phenylenediamine dihydrochloride (OPD) substrate (SigmaFast OPD; Sigma) was added, and after 10 min of incubation, the reaction was stopped by adding 50 μl of 3 M HCl to the mixture. The optical density (OD) was measured at 490 nm on a Synergy 4 plate reader (BioTek). A cutoff value of the average of the OD values of blank wells plus 3 standard deviations was established for each plate and used for calculating the reciprocal endpoint titer, which was the readout for this assay. The limit of detection of the assay was a titer of 1:100; samples that did not reach this titer were assigned a value of 1:50 for calculation and graphing purposes.
Neutralization assay.
Pooled serum samples were diluted 1:4 and treated with receptor-destroying enzyme (RDE; Denko Seiken) for 18 h at 37°C. RDE treatment was inactivated with 2.5% sodium citrate, resulting in a 1:10 dilution of serum from the starting concentration. RDE-treated serum was then added to wells of a 96-well plate and serially diluted 1:2 in MEM with 1% tosyl phenylalanyl chloromethyl ketone (TPCK) trypsin and penicillin-streptomycin. A total of 1,000 PFU/well of B/Malaysia/2506/04 were added to the wells and left to incubate for 1 h at room temperature. MDCK cells (plated at 500,000 cells per well for 24 h) were then washed one time with 1× PBS and incubated in the virus-serum mixture for 1 h at 37°C. The cells were washed again with 1× PBS and then incubated with serum dilutions for 72 h at 33°C. The cell supernatant was then mixed 1:1 with 0.5% chicken red blood cells and incubated for 40 min at 4°C. Hemagglutination was used as a determination of virus present in the samples. The last serum dilution without hemagglutination was defined as the minimal neutralizing titer.
ADCC reporter assay.
The Promega ADCC Reporter Bioassay kit (catalog number G7010) was used for this experiment. MDCK cells plated onto an opaque 96-well plate (at 50,000 cells per well) were infected 24 h later with B/Malaysia/2506/04 at a multiplicity of infection (MOI) of 5 for a single cycle (no TPCK-treated trypsin was added) overnight. Sera were diluted 1:3 (from a starting dilution of 1:150) in Roswell Park Memorial Institute (RPMI) medium and added to the virus-infected cells, followed by the addition of ADCC mouse effector cells (Promega), and plates were incubated for 6 h at 37°C. Luminescence was read by using a Synergy 4 plate reader and Gen5 2.09 software, and induction over the baseline was calculated and reported.
Statistical analysis and phylogenetic analysis.
ELISA endpoint titers were compared by using Student's t test. Differences in survival were tested for significance by using the Mantel-Cox log rank test. Differences in weight loss were analyzed per day by using one-way analysis of variance (ANOVA) for multiple comparisons. Statistical analysis was performed by using Prism (GraphPad). For phylogenetic analysis, sequences were downloaded from the Global Initiative on Sharing All Influenza Data (GISAID) database with filters to remove duplicates and laboratory-originating strains. The sequences were then aligned by using MUSCLE, and the alignment was edited by using MEGA 6.06 software. Aligned sequences were then translated and randomly subsampled at 30 sequences per year after the year 2001. Subsampling was performed to avoid bias toward recent isolates. Only a few sequences are available per year before 2001, and therefore, subsampling was not applicable for earlier years. A phylogenetic tree was assembled by using the Clustal Omega Web server with a neighbor-joining clustering method using 442 sequences. The tree was visualized by using FigTree software, and annotations were made in Adobe Illustrator.
ACKNOWLEDGMENTS
We thank Zoe Morgan, Ariana Hirsh, and Madhusudan Rajendran for technical assistance. We also thank Irina Margine for help with early pilot projects for this vaccine concept.
This work was partially supported by National Institutes of Health program project grant 1P01AI097092-01A1 (P.P.), the Biomedical Advanced Research and Development Authority (BARDA) (HHSO100201500010C [P.P. and F.K.]), and GlaxoSmithKline (P.P. and F.K.).
REFERENCES
- 1.Paul Glezen W, Schmier JK, Kuehn CM, Ryan KJ, Oxford J. 2013. The burden of influenza B: a structured literature review. Am J Public Health 103:e43–. doi: 10.2105/AJPH.2012.301137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van de Sandt CE, Bodewes R, Rimmelzwaan GF, de Vries RD. 2015. Influenza B viruses: not to be discounted. Future Microbiol 10:1447–1465. doi: 10.2217/fmb.15.65. [DOI] [PubMed] [Google Scholar]
- 3.Tafalla M, Buijssen M, Geets R, Vonk Noordegraaf-Schouten M. 2016. A comprehensive review of the epidemiology and disease burden of influenza B in 9 European countries. Hum Vaccin Immunother 12:993–1002. doi: 10.1080/21645515.2015.1111494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koutsakos M, Nguyen TH, Barclay WS, Kedzierska K. 2016. Knowns and unknowns of influenza B viruses. Future Microbiol 11:119–135. doi: 10.2217/fmb.15.120. [DOI] [PubMed] [Google Scholar]
- 5.Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB. 2007. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 25:5086–5096. doi: 10.1016/j.vaccine.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 6.Dijkstra F, Donker GA, Wilbrink B, Van Gageldonk-Lafeber AB, Van Der Sande MA. 2009. Long time trends in influenza-like illness and associated determinants in The Netherlands. Epidemiol Infect 137:473–479. doi: 10.1017/S095026880800126X. [DOI] [PubMed] [Google Scholar]
- 7.Heikkinen T, Ikonen N, Ziegler T. 2014. Impact of influenza B lineage-level mismatch between trivalent seasonal influenza vaccines and circulating viruses, 1999-2012. Clin Infect Dis 59:1519–1524. doi: 10.1093/cid/ciu664. [DOI] [PubMed] [Google Scholar]
- 8.Brottet E, Vandroux D, Gauzere BA, Antok E, Jaffar-Bandjee MC, Michault A, Filleul L. 2014. Influenza season in Réunion dominated by influenza B virus circulation associated with numerous cases of severe disease, France, 2014. Euro Surveill 19(39):pii=20916 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20916. [DOI] [PubMed] [Google Scholar]
- 9.Pan Y, Zhang Y, Yang P, Qian H, Shi W, Wu S, Cui S, Zhang D, Wang Q. 2015. Epidemiological and phylogenetic characteristics of influenza B infection in severe acute respiratory infection cases in Beijing, 2014 to 2015. Medicine (Baltimore) 94:e2399. doi: 10.1097/MD.0000000000002399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson WW, Moore MR, Weintraub E, Cheng PY, Jin X, Bridges CB, Bresee JS, Shay DK. 2009. Estimating influenza-associated deaths in the United States. Am J Public Health 99(Suppl 2):S225–S230. doi: 10.2105/AJPH.2008.151944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCullers JA, Hayden FG. 2012. Fatal influenza B infections: time to reexamine influenza research priorities. J Infect Dis 205:870–872. doi: 10.1093/infdis/jir865. [DOI] [PubMed] [Google Scholar]
- 12.Chan PK, Chan MC, Cheung JL, Lee N, Leung TF, Yeung AC, Wong MC, Ngai KL, Nelson EA, Hui DS. 2013. Influenza B lineage circulation and hospitalization rates in a subtropical city, Hong Kong, 2000-2010. Clin Infect Dis 56:677–684. doi: 10.1093/cid/cis885. [DOI] [PubMed] [Google Scholar]
- 13.Kosasih H, Roselinda, Nurhayati, Klimov A, Xiyan X, Lindstrom S, Mahoney F, Beckett C, Burgess TH, Blair PJ, Uyeki TM, Sedyaningsih ER. 2013. Surveillance of influenza in Indonesia, 2003-2007. Influenza Other Respir Viruses 7:312–320. doi: 10.1111/j.1750-2659.2012.00403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran D, Vaudry W, Moore D, Bettinger JA, Halperin SA, Scheifele DW, Jadvji T, Lee L, Mersereau T, Canadian Immunization Monitoring Program Active. 2016. Hospitalization for influenza A versus B. Pediatrics 138:e20154643. doi: 10.1542/peds.2015-4643. [DOI] [PubMed] [Google Scholar]
- 15.Sugaya N, Mitamura K, Yamazaki M, Tamura D, Ichikawa M, Kimura K, Kawakami C, Kiso M, Ito M, Hatakeyama S, Kawaoka Y. 2007. Lower clinical effectiveness of oseltamivir against influenza B contrasted with influenza A infection in children. Clin Infect Dis 44:197–202. doi: 10.1086/509925. [DOI] [PubMed] [Google Scholar]
- 16.Kawai N, Ikematsu H, Iwaki N, Tanaka O, Yamanishi Y, Hirotsu N, Kashiwagi S. 2007. Zanamivir treatment is equally effective for both influenza A and influenza B. Clin Infect Dis 44:1666. doi: 10.1086/518385. [DOI] [PubMed] [Google Scholar]
- 17.Kawai N, Ikematsu H, Iwaki N, Maeda T, Satoh I, Hirotsu N, Kashiwagi S. 2006. A comparison of the effectiveness of oseltamivir for the treatment of influenza A and influenza B: a Japanese multicenter study of the 2003-2004 and 2004-2005 influenza seasons. Clin Infect Dis 43:439–444. doi: 10.1086/505868. [DOI] [PubMed] [Google Scholar]
- 18.Elfalki F, Ihazmad H, Bimouhen A, Regragui Z, Benkaroum S, Bakri Y, Barakat A. 2016. Detection of influenza B viruses with reduced sensitivity to neuraminidase inhibitor in Morocco during 2014/15 season. East Mediterr Health J 22:453–459. [PubMed] [Google Scholar]
- 19.Samson M, Pizzorno A, Abed Y, Boivin G. 2013. Influenza virus resistance to neuraminidase inhibitors. Antiviral Res 98:174–185. doi: 10.1016/j.antiviral.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 20.Burnham AJ, Baranovich T, Govorkova EA. 2013. Neuraminidase inhibitors for influenza B virus infection: efficacy and resistance. Antiviral Res 100:520–534. doi: 10.1016/j.antiviral.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pascua PN, Marathe BM, Burnham AJ, Vogel P, Webby RJ, Webster RG, Govorkova EA. 2016. Competitive fitness of influenza B viruses possessing E119A and H274Y neuraminidase inhibitor resistance-associated substitutions in ferrets. PLoS One 11:e0159847. doi: 10.1371/journal.pone.0159847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burnham AJ, Armstrong J, Lowen AC, Webster RG, Govorkova EA. 2015. Competitive fitness of influenza B viruses with neuraminidase inhibitor-resistant substitutions in a coinfection model of the human airway epithelium. J Virol 89:4575–4587. doi: 10.1128/JVI.02473-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerdil C. 2003. The annual production cycle for influenza vaccine. Vaccine 21:1776–1779. doi: 10.1016/S0264-410X(03)00071-9. [DOI] [PubMed] [Google Scholar]
- 24.Rota PA, Wallis TR, Harmon MW, Rota JS, Kendal AP, Nerome K. 1990. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 175:59–68. doi: 10.1016/0042-6822(90)90186-U. [DOI] [PubMed] [Google Scholar]
- 25.Kissling E, Nunes B, Robertson C, Valenciano M, Reuss A, Larrauri A, Cohen JM, Oroszi B, Rizzo C, Machado A, Pitigoi D, Domegan L, Paradowska-Stankiewicz I, Buchholz U, Gherasim A, Daviaud I, Horváth JK, Bella A, Lupulescu E, O'Donnell J, Korczyńska M, Moren A, I-MOVE Case-Control Study Team. 2016. I-MOVE multicentre case-control study 2010/11 to 2014/15: is there within-season waning of influenza type/subtype vaccine effectiveness with increasing time since vaccination? Euro Surveill 21(16):pii=30201. doi: 10.2807/1560-7917.ES.2016.21.16.30201. [DOI] [PubMed] [Google Scholar]
- 26.Andrews SF, Huang Y, Kaur K, Popova LI, Ho IY, Pauli NT, Henry Dunand CJ, Taylor WM, Lim S, Huang M, Qu X, Lee JH, Salgado-Ferrer M, Krammer F, Palese P, Wrammert J, Ahmed R, Wilson PC. 2015. Immune history profoundly affects broadly protective B cell responses to influenza. Sci Transl Med 7:316ra192. doi: 10.1126/scitranslmed.aad0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nachbagauer R, Choi A, Izikson R, Cox MM, Palese P, Krammer F. 2016. Age dependence and isotype specificity of influenza virus hemagglutinin stalk-reactive antibodies in humans. mBio 7:e01996–. doi: 10.1128/mBio.01996-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krammer F, Palese P. 2013. Influenza virus hemagglutinin stalk-based antibodies and vaccines. Curr Opin Virol 3:521–530. doi: 10.1016/j.coviro.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen CJ, Ermler ME, Tan GS, Krammer F, Palese P, Hai R. 2016. Influenza A viruses expressing intra- or intergroup chimeric hemagglutinins. J Virol 90:3789–3793. doi: 10.1128/JVI.03060-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hai R, Krammer F, Tan GS, Pica N, Eggink D, Maamary J, Margine I, Albrecht RA, Palese P. 2012. Influenza viruses expressing chimeric hemagglutinins: globular head and stalk domains derived from different subtypes. J Virol 86:5774–5781. doi: 10.1128/JVI.00137-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krammer F, Pica N, Hai R, Margine I, Palese P. 2013. Chimeric hemagglutinin influenza virus vaccine constructs elicit broadly protective stalk-specific antibodies. J Virol 87:6542–6550. doi: 10.1128/JVI.00641-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nachbagauer R, Miller MS, Hai R, Ryder AB, Rose JK, Palese P, García-Sastre A, Krammer F, Albrecht RA. 2015. Hemagglutinin stalk immunity reduces influenza virus replication and transmission in ferrets. J Virol 90:3268–3273. doi: 10.1128/JVI.02481-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nachbagauer R, Kinzler D, Choi A, Hirsh A, Beaulieu E, Lecrenier N, Innis BL, Palese P, Mallett CP, Krammer F. 2016. A chimeric haemagglutinin-based influenza split virion vaccine adjuvanted with AS03 induces protective stalk-reactive antibodies in mice. NPJ Vaccines 1:16015. doi: 10.1038/npjvaccines.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krammer F, Hai R, Yondola M, Tan GS, Leyva-Grado VH, Ryder AB, Miller MS, Rose JK, Palese P, García-Sastre A, Albrecht RA. 2014. Assessment of influenza virus hemagglutinin stalk-based immunity in ferrets. J Virol 88:3432–3442. doi: 10.1128/JVI.03004-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryder AB, Nachbagauer R, Buonocore L, Palese P, Krammer F, Rose JK. 2015. Vaccination with vesicular stomatitis virus-vectored chimeric hemagglutinins protects mice against divergent influenza virus challenge strains. J Virol 90:2544–2550. doi: 10.1128/JVI.02598-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Margine I, Krammer F, Hai R, Heaton NS, Tan GS, Andrews SA, Runstadler JA, Wilson PC, Albrecht RA, García-Sastre A, Palese P. 2013. Hemagglutinin stalk-based universal vaccine constructs protect against group 2 influenza A viruses. J Virol 87:10435–10446. doi: 10.1128/JVI.01715-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krammer F, Margine I, Hai R, Flood A, Hirsh A, Tsvetnitsky V, Chen D, Palese P. 2014. H3 stalk-based chimeric hemagglutinin influenza virus constructs protect mice from H7N9 challenge. J Virol 88:2340–2343. doi: 10.1128/JVI.03183-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krammer F, Palese P. 2015. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov 14:167–182. doi: 10.1038/nrd4529. [DOI] [PubMed] [Google Scholar]
- 39.Francis T. 1940. A new type of virus from epidemic influenza. Science 92:405–408. doi: 10.1126/science.92.2392.405. [DOI] [PubMed] [Google Scholar]
- 40.Dilillo DJ, Tan GS, Palese P, Ravetch JV. 2014. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat Med 20:143–151. doi: 10.1038/nm.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leon PE, He W, Mullarkey CE, Bailey MJ, Miller MS, Krammer F, Palese P, Tan GS. 2016. Optimal activation of Fc-mediated effector functions by influenza virus hemagglutinin antibodies requires two points of contact. Proc Natl Acad Sci U S A 113:E5944–E5951. doi: 10.1073/pnas.1613225113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cox F, Kwaks T, Brandenburg B, Koldijk MH, Klaren V, Smal B, Korse HJ, Geelen E, Tettero L, Zuijdgeest D, Stoop EJ, Saeland E, Vogels R, Friesen RH, Koudstaal W, Goudsmit J. 2016. HA antibody-mediated FcγRIIIa activity is both dependent on FcR engagement and interactions between HA and sialic acids. Front Immunol 7:399. doi: 10.3389/fimmu.2016.00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He W, Tan GS, Mullarkey CE, Lee AJ, Lam MM, Krammer F, Henry C, Wilson PC, Ashkar AA, Palese P, Miller MS. 2016. Epitope specificity plays a critical role in regulating antibody-dependent cell-mediated cytotoxicity against influenza A virus. Proc Natl Acad Sci U S A 113:11931–11936. doi: 10.1073/pnas.1609316113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mullarkey CE, Bailey MJ, Golubeva DA, Tan GS, Nachbagauer R, He W, Novakowski KE, Bowdish DM, Miller MS, Palese P. 2016. Broadly neutralizing hemagglutinin stalk-specific antibodies induce potent phagocytosis of immune complexes by neutrophils in an Fc-dependent manner. mBio 7:e01624–. doi: 10.1128/mBio.01624-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jegaskanda S, Job ER, Kramski M, Laurie K, Isitman G, de Rose R, Winnall WR, Stratov I, Brooks AG, Reading PC, Kent SJ. 2013. Cross-reactive influenza-specific antibody-dependent cellular cytotoxicity antibodies in the absence of neutralizing antibodies. J Immunol 190:1837–1848. doi: 10.4049/jimmunol.1201574. [DOI] [PubMed] [Google Scholar]
- 46.Jegaskanda S, Weinfurter JT, Friedrich TC, Kent SJ. 2013. Antibody-dependent cellular cytotoxicity is associated with control of pandemic H1N1 influenza virus infection of macaques. J Virol 87:5512–5522. doi: 10.1128/JVI.03030-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC, Lee JH, Metlagel Z, Bujny MV, Jongeneelen M, van der Vlugt R, Lamrani M, Korse HJ, Geelen E, Sahin Ö, Sieuwerts M, Brakenhoff JP, Vogels R, Li OT, Poon LL, Peiris M, Koudstaal W, Ward AB, Wilson IA, Goudsmit J, Friesen RH. 2012. Highly conserved protective epitopes on influenza B viruses. Science 337:1343–1348. doi: 10.1126/science.1222908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steel J, Lowen AC, Pena L, Angel M, Solórzano A, Albrecht R, Perez DR, García-Sastre A, Palese P. 2009. Live attenuated influenza viruses containing NS1 truncations as vaccine candidates against H5N1 highly pathogenic avian influenza. J Virol 83:1742–1753. doi: 10.1128/JVI.01920-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Margine I, Palese P, Krammer F. 2013. Expression of functional recombinant hemagglutinin and neuraminidase proteins from the novel H7N9 influenza virus using the baculovirus expression system. J Vis Exp 81:e51112. doi: 10.3791/51112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krammer F, Margine I, Tan GS, Pica N, Krause JC, Palese P. 2012. A carboxy-terminal trimerization domain stabilizes conformational epitopes on the stalk domain of soluble recombinant hemagglutinin substrates. PLoS One 7:e43603. doi: 10.1371/journal.pone.0043603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nachbagauer R, Wohlbold TJ, Hirsh A, Hai R, Sjursen H, Palese P, Cox RJ, Krammer F. 2014. Induction of broadly reactive anti-hemagglutinin stalk antibodies by an H5N1 vaccine in humans. J Virol 88:13260–13268. doi: 10.1128/JVI.02133-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stevens J, Blixt O, Tumpey TM, Taubenberger JK, Paulson JC, Wilson IA. 2006. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science 312:404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- 53.Ni F, Kondrashkina E, Wang Q. 2013. Structural basis for the divergent evolution of influenza B virus hemagglutinin. Virology 446:112–122. doi: 10.1016/j.virol.2013.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]