

ABSTRACT
Favipiravir (T-705) is a broad-spectrum antiviral agent that has been approved in Japan for the treatment of influenza virus infections. T-705 also inhibits the replication of various RNA viruses, including chikungunya virus (CHIKV). We demonstrated earlier that the K291R mutation in the F1 motif of the RNA-dependent RNA polymerase (RdRp) of CHIKV is responsible for low-level resistance to T-705. Interestingly, this lysine is highly conserved in the RdRp of positive-sense single-stranded RNA (+ssRNA) viruses. To obtain insights into the unique broad-spectrum antiviral activity of T-705, we explored the role of this lysine using another +ssRNA virus, namely, coxsackievirus B3 (CVB3). Introduction of the corresponding K-to-R substitution in the CVB3 RdRp (K159R) resulted in a nonviable virus. Replication competence of the K159R variant was restored by spontaneous acquisition of an A239G substitution in the RdRp. A mutagenesis analysis at position K159 identified the K159M variant as the only other viable variant which had also acquired the A239G substitution. The K159 substitutions markedly decreased the processivity of the purified viral RdRp, which was restored by the introduction of the A239G mutation. The K159R A239G and K159M A239G variants proved, surprisingly, more susceptible than the wild-type virus to T-705 and exhibited lower fidelity in polymerase assays. Furthermore, the K159R A239G variant was found to be highly attenuated in mice. We thus demonstrate that the conserved lysine in the F1 motif of the RdRp of +ssRNA viruses is involved in the broad-spectrum antiviral activity of T-705 and that it is a key amino acid for the proper functioning of the enzyme.
IMPORTANCE In this study, we report the key role of a highly conserved lysine residue of the viral polymerase in the broad-spectrum antiviral activity of favipiravir (T-705) against positive-sense single-stranded RNA viruses. Substitutions of this conserved lysine have a major negative impact on the functionality of the RdRp. Furthermore, we show that this lysine is involved in the fidelity of the RdRp and that the RdRp fidelity influences the sensitivity of the virus for the antiviral efficacy of T-705. Consequently, these results provide insights into the mechanism of the antiviral activity of T-705 and may lay the basis for the design of novel chemical scaffolds that may be endowed with a more potent broad-spectrum antiviral activity than that of T-705.
KEYWORDS: favipiravir, fidelity, mutagenesis, RdRp, CVB3
INTRODUCTION
Favipiravir (T-705) is a broad-spectrum antiviral agent that has been approved in Japan for the treatment of influenza virus infections (1). T-705 has also been reported to inhibit, in vitro and in animal models, the replication of different RNA viruses, including norovirus (2), flaviviruses (3), and hantaviruses (4). The compound has also shown a potential beneficial effect in the treatment of Ebola virus-infected patients in western Africa during the 2014-2015 outbreak (5).
In the cell, T-705 behaves as a purine analogue and is converted into its ribofuranosyl 5′-triphosphate metabolite (T-705-RTP), after which it can be incorporated in the growing RNA strand, as has been shown with the influenza virus RNA-dependent RNA polymerase (RdRp) (1). However, the precise molecular mechanism behind its broad-spectrum antiviral activity has yet to be fully elucidated. Some studies suggest that T-705 inhibits viral RNA synthesis by chain termination of the nascent viral RNA strand (6), while other reports support the induction of lethal mutagenesis (7, 8).
In a previous study from our laboratory, T-705 and its defluorinated analog, T-1105, were shown to inhibit the in vitro replication of chikungunya virus (CHIKV) (9). Treatment of CHIKV-infected AG129 mice with T-705 protected them from severe neurological disease and reduced the mortality rate (9). Low-level T-705-resistant CHIKV variants were selected in vitro and were shown to have acquired the K291R mutation in motif F1 of the RdRp, and a reverse-engineering study corroborated the link between the mutant genotype and the compound-resistant phenotype (9). Motif F of the RdRp of single-stranded, positive-sense RNA (+ssRNA) viruses has been reported to play an important role in nucleoside triphosphate (NTP) binding during viral RNA synthesis (10). It has also been demonstrated that motif F1 of dengue virus is involved in promoter-dependent initiation of RNA synthesis (11). In addition, some residues of motif F of Japanese encephalitis virus have been shown to be involved in GTP recognition (12). Interestingly, the lysine residue of motif F1 that was mutated in the T-705-resistant CHIKV variants is highly conserved among +ssRNA viruses, a group to which many neglected and emerging viruses belong (Fig. 1). The exact role of this lysine in viral RdRp activity remains unclear. Given the need for broad-spectrum antivirals, it is thus of great importance to understand how T-705 precisely interacts with the RdRp of RNA viruses. Such information may potentially provide insight into how more-potent broad-spectrum +ssRNA virus inhibitors can be developed.
FIG 1.

Multiple-sequence alignment of motif F1 of viral RdRp. Part of the multiple-sequence alignment of RdRp of chikungunya virus (ACY09947.1), enteroviruses (coxsackievirus A21 [ABM54541.1], coxsackievirus B3 [AAA74400.1], poliovirus type 1 [AAP37265.1], enterovirus D68 [AAR98503.1], and human rhinovirus B14 [NP_740525.1]), flaviviruses (Zika virus [AMO03410.1] and Japanese encephalitis virus [ABU94628.1]), murine norovirus (AEY83582.1), and hepatitis C virus (CAB46913.1) was generated using the PROMALS server (http://prodata.swmed.edu/promals/promals.php). Motif F1 of the RdRp is indicated by a black box, and the residues corresponding to K291 in CHIKV are highlighted in yellow.
We here explored whether the conserved lysine in the F1 motif of the viral RdRp is involved in the broad-spectrum antiviral activity of T-705. To this end, we used coxsackievirus B3 (CVB3), another +ssRNA virus that is sensitive to T-705 and for which we have the necessary reverse-genetics tools and enzymatic assays available, as the model virus in this study. Using CVB3, we demonstrate for the first time that this highly conserved lysine residue plays a key role in RdRp functionality and fidelity and that a variant of this residue exhibits a highly attenuated phenotype in mice. We next succeeded in elucidating the mechanism of resistance against T-705, thereby demonstrating the role of RdRp fidelity in sensitivity to the antiviral agent.
RESULTS
Viable CVB3 K159 mutants acquired the A239G mutation in the RdRp.
Low-level T-705-resistant CHIKV variants were reported to have the K291R mutation in motif F1 of the RdRp (9). Because this lysine residue is highly conserved in the RdRp of +ssRNA viruses (Fig. 1), we introduced the K-to-R mutation at the corresponding position in motif F1 of the CVB3 RdRp (K159R) to assess the sensitivity of this mutant to the antiviral effect of T-705 (Fig. 2). Initially, no infectious virus could be recovered. However, during the second transfection round, virus-induced cytopathic effect (CPE) was observed and infectious virus was recovered. Sequencing of this virus sample revealed that in addition to the engineered K159R mutation, it had also acquired the alanine-to-glycine substitution at position 239 in motif A of the RdRp (Fig. 2). Interestingly, the single A239G mutant has been reported to be a low-fidelity RdRp CVB3 variant (13).
FIG 2.

Coxsackievirus B3 polymerase structure. (A) Structure of CVB3 polymerase (PDB code 4K4Y) showing the locations of Lys159 (K159) and Ala239 (A239) highlighted in blue and magenta, respectively. (B) Model of CVB3 polymerase structure (fingers, blue; palm, green; thumb, red ribbons) with favipiravir triphosphate (T-705-RTP). The primer RNA strand has a yellow ribbon, the template has a brown ribbon. T-705-RTP has purple carbons. The complementary G is in cyan. The carbons of K159 and A239 residues are highlighted in green. CVB3 hydrogen bonds from the sugar part of T-705-RTP to the N terminus (in cyan) via A239 (green carbons) are shown as black dashed lines.
To explore the importance of the lysine at position 159 in the CVB3 RdRp, a full set of mutations was engineered, each coding for another amino acid residue at this position. From nine of these infectious clones, no virus could be recovered from cell culture after two transfection rounds and no RNA replication could be detected, requiring them to be labeled as “not viable.” Another eight mutants reverted back to wild type (WT) (Table 1). Only transfection with the K159M clone allowed the recovery of infectious virus with a mutation at position 159, but only after it had acquired the A239G substitution (Table 1). The low-fidelity RdRp variant (A239G) (13) and a high-fidelity RdRp variant (A372V) (14) were also generated to be used as controls in the next experiments.
TABLE 1.
Mutation analysis of amino acid 159 of the CVB3 RdRp
| Engineered genotype | Phenotypea | Compensatory mutation | TCID50/mlb |
|
|---|---|---|---|---|
| Vero A cells | HeLa Rh cells | |||
| Lys (wild type) | Viable | 2.2E + 07 | 1.4E + 05 | |
| Arg | Viable, but with mutation | A239G | 6.5E + 06 | 1.9E + 05 |
| Met | Viable, but with mutation | A239G | 7.9E + 07 | 2.5E + 06 |
| Asp | Reverted to WT | |||
| Asn | Reverted to WT | |||
| Gln | Reverted to WT | |||
| Ser | Reverted to WT | |||
| Thr | Reverted to WT | |||
| His | Reverted to WT | |||
| Phe | Reverted to WT | |||
| Val | Reverted to WT | |||
| Ala | Not viable | |||
| Ile | Not viable | |||
| Leu | Not viable | |||
| Trp | Not viable | |||
| Tyr | Not viable | |||
| Glu | Not viable | |||
| Cys | Not viable | |||
| Gly | Not viable | |||
| Pro | Not viable | |||
The phenotype indicates whether the transfected CVB3 variant with the desired mutation was viable, reverted to WT, or did not yield viable virus. Each transfection was performed at least in duplicate.
TCID50, 50% tissue culture infective dose.
K159 mutations in the CVB3 RdRp gene affect polymerase competence.
An enzymatic study was performed to determine how the K159 mutations affect RdRp activity. The in vitro polymerase assays were performed with purified CVB3 RdRp and a homopolymeric primer-template substrate (Fig. 3A), allowing [α-32P]UMP incorporation into RNA after 2, 5, and 10 min. Relative to WT polymerase, the single K159R and K159M variants showed significant overall differences of 100- and 15-fold decreases in polymerase activity, respectively (Fig. 3B and C). Interestingly, introduction of the A239G mutation partially restored the overall polymerase activity of both K159 mutants (Fig. 3B and C). Based on structural analysis of the picornavirus CVB3 (PDB code 3CDW), the Arg159 has a long side chain compared to Lys159, which narrows the access of the incoming nucleotides and impedes correct positioning at the active site of the RdRp (Fig. 4B and E). This may explain the lower efficiency noted in polymerase activity assays. In the case of Met159, it is suggested that the nitrogen group of the original lysine residue has a main role in the binding and positioning of the incoming nucleotide and its substitution by a methyl group in methionine may therefore affect directly the NTP recognition and, in turn, NTP positioning and catalysis. As shown in Fig. 4A and D, the residues Lys159 and Ala239 are spatially close in the channel of the incoming NTP. This channel becomes narrow when the Lys159 is mutated, thereby hindering the access of the incoming NTP to the RdRp active site (Fig. 4B and E). In order to maintain virus viability, the compensatory mutation A239G, with its absence of side chain, appears to restore the open conformation of the NTP channel (Fig. 4C and F). These data are in agreement with the fact that no viable single mutant viruses could be recovered after transfection in cell culture and that the A239G mutation was spontaneously acquired by the K159R and K159M mutants to restore their ability to replicate in vitro.
FIG 3.
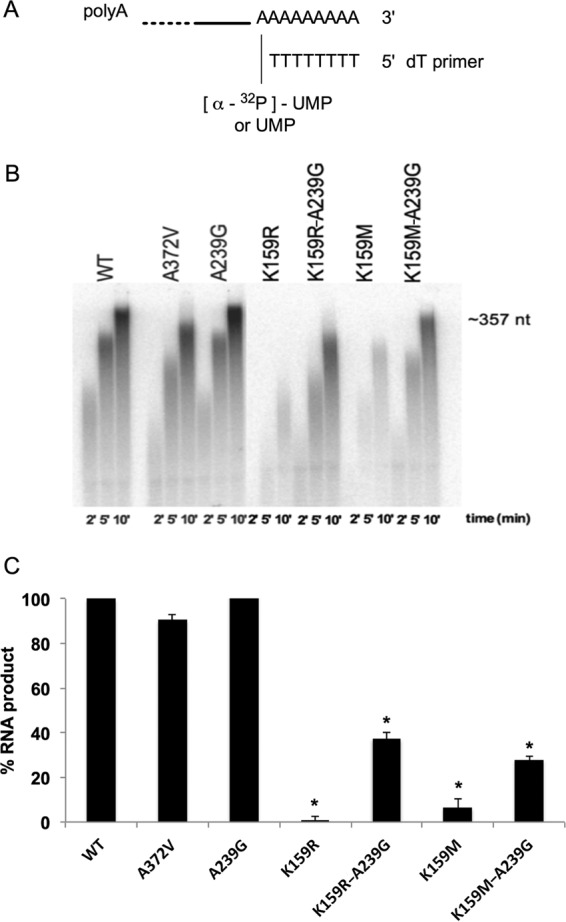
Coxsackievirus B3 RdRp variants exhibit distinct polymerase activities. RNA polymerase assays were performed as described in Materials and Methods. (A) dT15-poly(rA) is a homopolymeric primer-template allowing the incorporation of [α-32P]UMP or UMP at the dT primer 3′ end. (B) RNA products of a representative PAGE gel are shown after 2, 5, and 10 min of quenching. (C) The quantified percentages of RNA products shown are mean values ± SD of the results of two independent experiments. Statistical analysis was performed using two-way analysis of variance (ANOVA) in reference to the WT for the K159R or K159M variant (*, P < 0.05), to the K159R variant for the K159R A239G variant (*, P < 0.05), or to the K159M variant for the K159M A239G variant (*, P < 0.05).
FIG 4.

Top view of the RdRp from CVB3 (PDB code 3CDW). (A) The locations of the residues K159 and A239 are highlighted in purple and red, respectively. The channel from incoming ribonucleoside triphosphates (rNTPs) is shown between these residues. (B) The long chain of the arginine residue R159 mutant is highlighted in purple, narrowing the channel for incoming rNTPs. A239 is shown in red. (C) The G239 mutant, in red, is able to reestablish the access of rNTPs in the channel. The residue R159 is shown in purple. Panels D to F are representative zoom images of panels A to C, respectively.
The K159R and K159M mutations result in an unfavorable phenotype.
The genetic stability of the amino acid substitutions of the K159R A239G and K159M A239G mutants was studied during passaging in HeLa Rh cells. Already after three passages, both K159 mutants had reverted back to the WT lysine. The adaptive mutation at position 239 appeared to be genetically stable, with or without the substitutions at position 159 (Table 2).
TABLE 2.
Genetic stability of the reverse-engineered CVB3 variants in cell culture
| Engineered genotype | Genetic stability after 3 passages |
|
|---|---|---|
| Statusa | Genotypeb | |
| A372V | Stable | V372 |
| A239G | Stable | G239 |
| K159R A239G | Partially stable | K159 G239 |
| K159M A239G | Partially stable | K159 G239 |
The status indicates whether the mutations were stable or partially stable or reverted back to WT over three passages in HeLa Rh cells.
Bold indicates reversion to the WT; underlining indicates mutation stability.
Plaque phenotyping of the reverse-engineered K159 mutants before passaging revealed that the plaque size of the K159M A239G variant was comparable to that of the WT, whereas for the K159R A239G variant, two populations with different plaque sizes were observed (Fig. 5A). Despite numerous attempts, it was not possible to obtain a pure small-plaque variant. In parallel, the in vitro growth kinetics of all variants were determined in Vero A cells by plaque assay. The A239G and K159R A239G variants had slightly slower growth kinetics than the WT virus, whereas the K159M A239G mutant replicated approximately as efficiently as the WT (Fig. 5B).
FIG 5.

Phenotype of reverse-engineered CVB3 variants. (A) Plaque phenotypes of different CVB3 variants were determined by infecting Vero A cells with a 10-fold serial dilution of each variant, followed by the addition of an agarose overlay. After 3 days, viral plaques were visualized by Giemsa staining. A representative image with spread plaques is shown. (B) Growth curves were generated by infecting Vero A cells with the selected CVB3 variant at an MOI of 3, after which the infectious virus titer in the medium was determined at various time points after infection by plaque assay. Infectious virus titers are the means ± SD of the results of two independent experiments. Significant differences with respect to the WT (*, P < 0.05, or ***, P < 0.001) were analyzed by two-way ANOVA. (C) The sensitivity of the reverse-engineered CVB3 variants to the antiviral effect of T-705 was assessed in HeLa Rh cells by a CPE reduction assay. Cell viability was measured using the MTS/PMS method. Data are expressed as percentages of untreated controls and are mean values ± SD of the results of at least three independent experiments.
CVB3 K159 mutants are more susceptible to the antiviral effect of T-705.
The susceptibility to the antiviral effect of T-705 (Fig. 6) of WT virus as well as of RdRp mutants was quantified in a CPE reduction assay (Fig. 5C). The K159R A239G and K159M A239G mutants proved to be, respectively, 3.4- and 2-fold more susceptible to the antiviral effect of T-705 than WT virus (Table 3). Also, the A239G single mutant proved to be 2-fold more susceptible to T-705 than the WT, whereas the high-fidelity polymerase A372V variant was 2-fold less sensitive to T-705 than the WT. The susceptibility of the K159 variants to the antiviral effect of ribavirin (which acts as a mutagenic agent and by depletion of GTP pools) (15) and rupintrivir (an inhibitor of the viral 3C protease) was not significantly altered (Table 3; Fig. 6).
FIG 6.

Chemical structures of favipiravir, ribavirin, and rupintrivir.
TABLE 3.
Antiviral activity of different classes of compounds against CVB3 K159 variants
| CVB3 WT or variant | EC50 (μM) (mean ± SD)a |
||
|---|---|---|---|
| T-705 | Ribavirin | Rupintrivir | |
| WT | 229 ± 24 | 254 ± 37 | 0.6 ± 0.1 |
| K159R A239G variant | 67 ± 6 (0.3)**** | 261 ± 23 | 1 ± 0.4 |
| K159M A239G variant | 116 ± 14 (0.5)*** | 198 ± 9 | 0.7 ± 0.1 |
| A239G variant | 108 ± 2 (0.5)**** | 164 ± 10 (0.6)** | 0.8 ± 0.02 |
| A372V variant | 458 ± 27 (2)**** | 419 ± 16 (1.6)**** | 0.8 ± 0.01 |
The values in parentheses are fold increases in resistance (EC50 for mutant/EC50 for WT), which have significant P values (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001, Student's t test). The 50% cytotoxic/cytostatic concentrations (CC50) for T-705, ribavirin, and rupintrivir were 1,773 ± 9 μM, 914 ± 2 μM, and >200 μM, respectively.
K159 mutations affect polymerase fidelity.
Because low- and high-fidelity variants of the RdRp (A239G and A372V) exhibited different susceptibilities to T-705, in vitro polymerase assays were performed to assess the effect of the K159 mutations on polymerase fidelity (Fig. 7). In these enzymatic assays, insertion of the correct nucleotide (UTP) into RNA was challenged with increasing concentrations of an incorrect (CTP) nucleotide. The low polymerase efficiency of K159R and K159M single variants technically precluded their use in fidelity analysis assays. The A239G variant was used as a low-fidelity control (13, 14). The RNA product formed was quantified and fit as a dose-response curve framing the incorrect nucleotide concentration able to inhibit 50% of RNA product formation (reported as 50% inhibitory concentration [IC50]). For the WT and A239G, K159R A239G, and K159M A239G variants, IC50s of 2.9 ± 0.2 mM (Fig. 7A and B), 2.5 ± 0.3 mM (Fig. 7A and C), 1.7 ± 0.06 mM (Fig. 7A and D), and 0.45 mM ± 0.04 (Fig. 7A and E), respectively, were obtained when 3 μCi [α-32P]UTP or 10 μM unlabeled UTP was challenged with different concentrations of CTP, respectively. Altogether, these results indicate that the K159R A239G and K159M A239G variants are, respectively, 1.7- and 6-fold more sensitive than the WT enzyme to the presence of an incorrect CTP nucleotide (Fig. 7F). These results validate the hypothesis that the variants are indeed affected in their fidelity of nucleotide selection or incorporation. Because loss of fidelity would also favor incorporation of T-705-RTP, these results are in agreement with the observation that the K159 variants are more susceptible to the antiviral effect of this compound.
FIG 7.

K159R A239G and K159M A239G mutants are low-fidelity RdRp variants. Competition assays were performed with 3 μCi [α-32P]UTP or cold 10 μM UTP challenged with CTP concentrations of 1, 2, 3, 4, and 5 mM for the WT (B) and the A239G variant (C) or 0.1, 0.25, 0.5, 1, 2 mM for K159R A239G (D) and K159M A239G (E) variants. The total percentage of RNA product formed represents a mean ± SD of the results of two independent experiments and was measured as a dose-response curve with at least one data point above and below 50% RNA product formed (A) to be able to calculate an interpolated apparent IC50. Data were fit (F) and statistical analyses were performed using two-way ANOVA in reference to the WT for the K159R A239G variant (*, P < 0.05) and for the K159M A239G variant (**, P < 0.01).
T-705 acts as a mutagen during CVB3 replication.
To study whether T-705 acts as a mutagen on CVB3 replication, virus-infected HeLa Rh cells were treated with T-705 or ribavirin at a concentration equal to the 50% effective concentration (EC50) of the corresponding compound (Table 3). This study was performed with the WT virus, the low-fidelity A239G variant, and the high-fidelity A372V variant because, as demonstrated above, the K159 mutants easily revert back to the WT, which would interfere with data analysis. On day 3 postinfection (p.i.), the supernatants were collected and subjected to deep sequencing to determine the mutation frequencies at each nucleotide position after drug treatment. As expected, the high-fidelity A372V variant (Fig. 8B, open bars) has a significantly lower mean basal mutation frequency across its genome (P < 0.0001, paired two-tailed t test, n = 10,749) than the WT virus (Fig. 8A, open bars), whereas the low-fidelity A239G variant showed the highest mean basal mutation frequency across its genome frequencies (P < 0.0001, paired two-tailed t test, n = 10,749) (Fig. 8C, open bars). Treatment with T-705 (Fig. 8, light gray bars) and ribavirin (Fig. 8, dark gray bars) resulted in a significantly higher basal mutation frequency for all CVB3 variants. The data demonstrate that T-705 is a potent RNA mutagen for CVB3, especially for C-to-U and G-to-A transitions, as has been previously described (16, 17). On the other hand, ribavirin increased the mutation frequency for all transition mutations.
FIG 8.
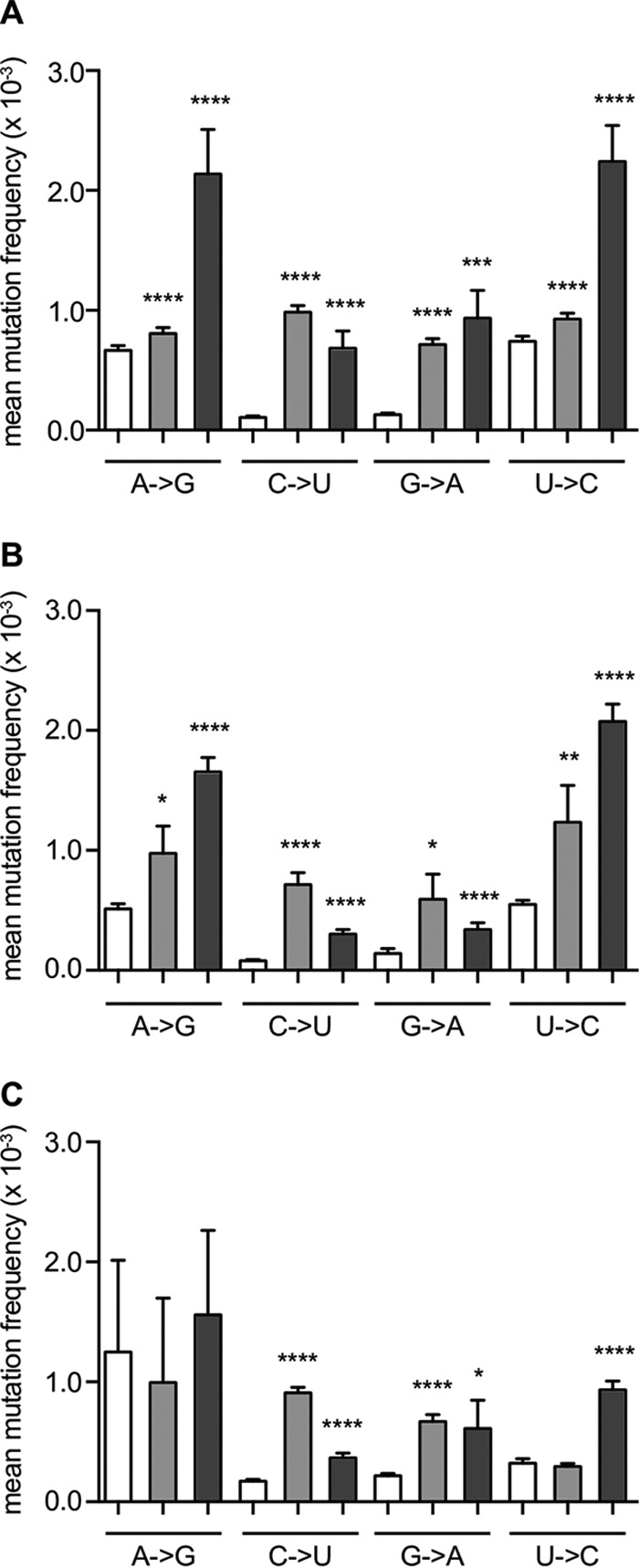
T-705 significantly increases the mutation frequency of CVB3. Wild-type (A), high-fidelity A372V (B), and low-fidelity A239G (C) viruses were either mock treated (open bars) or treated with T-705 (light gray bars) or ribavirin (dark gray bars) at concentrations equal to the corresponding EC50s and subjected to whole-genome deep sequencing. The frequency of transition mutations occurring at each A, C, G, and U was calculated and used to determine the mean frequency and SEM. *, P < 0.05; **, P < 0.01; ***, P = 0.0006; ****, P < 0.0001; two-tailed paired t test; n = 1,416 (A to G), n = 1,169 (C to U); n = 988 (G to A); n = 1,182 (U to C).
The K159R A239G mutant is attenuated in vivo.
The fitness of the CVB3 WT as well as the A239G, K159R A239G, and K159M A239G mutants was studied in immunocompetent SJL mice. The A239G variant was included as a control for in vivo attenuation (13). Three days postinfection, a time at which the highest virus titers are observed in the WT-infected mice, all mice were euthanized and serum samples, hearts, spleens, and pancreases were collected for RNA quantification (Fig. 9). Mice infected with the A239G mutant had lower RNA levels in the serum samples and various organs than those infected with the WT (Fig. 9A to D) as well as fewer infectious virus particles in the heart (Fig. 9E), which is in agreement with previous reports. The K159R A239G mutant was the most attenuated variant in this study, inducing only low or undetectable levels of viral RNA in the heart (Fig. 9B). Furthermore, no infectious virus particles were recovered from this organ (Fig. 9E) in which CVB3 typically replicates to high titers. In contrast, the K159M A239G variant behaved similarly to the WT (Fig. 9A to E). Sequencing of the virus population in serum samples of mice infected with the K159M A239G variant revealed that this variant had reverted back to the WT.
FIG 9.

In vivo fitness of CVB3 K159 variants in SJL mice. SJL mice were infected with 105 PFU of CVB3 WT or A239G, K159R A239G, or K159R A239G variants (n = 5 per variant). On day 3 p.i., all mice were euthanized, after which serum samples and selected organs were collected for RNA extraction. The amount of viral RNA in serum (A), heart (B), pancreas (C), and spleen (D) was determined by qRT-PCR. The number of infectious virus particles in the heart (E) was determined by endpoint titration (*, P < 0.05, and **, P < 0.01, Kruskal-Wallis test).
DISCUSSION
T-705 is a broad-spectrum antiviral compound that has been reported to inhibit the replication of a variety of RNA viruses, in both in vitro and in vivo models. In a previous study, CHIKV variants that are resistant to the antiviral activity of T-705 were selected, and all of them were shown to have acquired the K291R mutation in motif F1 of the RdRp (9). Because this lysine residue is highly conserved in the RdRp of many different +ssRNA viruses, we hypothesized that this residue is essential for the polymerase activity of these viruses and that it is a key interactor with T-705-RTP. To challenge this hypothesis, the K-to-R mutation was inserted at the equivalent position (K159R) in the infectious clone of CVB3. In parallel, two different resistance selection protocols (serial passaging and clonal selection) were used with the aim of selecting T-705-resistant CVB3 variants. However, despite numerous attempts, we did not succeed in selecting such variants, indicating that T-705 has a high barrier to resistance. In contrast to the CHIKV RdRp, the CVB3 RdRp did not tolerate this mutation without the presence of the compensatory A239G amino acid substitution in motif A of the RdRp. Interestingly, a CVB3 variant carrying the single A239G mutation was previously reported to be a low-fidelity polymerase variant (13). Of all CVB3 infectious clones carrying any possible amino acid substitution at position 159, only the K159M variant proved to be viable and this only after the A239G mutation had also been acquired. Interestingly, in vitro polymerase assays revealed that the K159R and K159M mutants exhibit crippled polymerase activity, which can be efficiently restored by the compensatory A239G substitution. As mentioned before, the Lys159 residue is located in motif F of the RdRp, which has been reported to play an important role in NTP binding during viral RNA synthesis (10). NTP binding is known to be a major fidelity checkpoint, and point mutations in this motif could annihilate polymerase activity or slow down catalysis (18). Mutating this lysine residue to an arginine or methionine resulted in reduced polymerization efficiency, which is likely the consequence of less efficient access of the incoming NTPs to the active site of RdRp. According to previous reports (19, 20), the residue Ala239 participates in a tetrahedral hydrogen bond network. This network helps position the Asp238 residue to the active site of the RdRp, providing a direct link between the properly positioned NTP and a set of structural interactions that promote catalysis by stabilizing the closed active site (18). Based on structural analysis of CVB3 polymerase, the residues Lys159 and Ala239 are closely located in the channel of the incoming NTP. This channel becomes narrow when the Lys159 is mutated to a slightly bulkier R or M which is expected to impede access of the incoming NTP to the RdRp active site. With the “removal” of the methyl side chain, the compensatory mutation A239G appears to restore access to the NTP channel. Consequently, the overall polymerase activity and the ability of the virus carrying this compensatory mutation to replicate in vitro are restored. These data indicate first of all that the K159 residue is of key importance for the catalytic activity of the CVB3 RdRp and, second, that the residue at position 239 has an important role in its stability. Surprisingly, the K159R A239G and K159M A239G variants proved, respectively, 3.5- and 2-fold more sensitive (instead of resistant) than the WT to the antiviral activity of T-705. In contrast, no significant differences in sensitivity to ribavirin and rupintrivir were observed, indicating that the K159 residue is specifically involved in the molecular mechanism of action of T-705. Furthermore, the low-fidelity A239G variant was also more sensitive to the antiviral effect of T-705, whereas the high-fidelity A372V variant proved less sensitive. These data thus indicate that the fidelity of the viral RdRp is an important component in the susceptibility of the virus to T-705. Therefore, in vitro polymerase studies were performed to evaluate the fidelity of the K159 mutants. The K159R A239G and K159M A239G variants were, respectively, 1.7- and 6-fold more sensitive than the WT enzyme to the presence of an incorrect nucleotide, and their fidelity was lower than that of the single A239G mutant. This provides an explanation for the increased sensitivity of the double mutants to the antiviral activity of T-705.
To date, the precise mechanism that underlies the broad-spectrum anti-RNA virus activity of T-705 has not been completely unraveled. T-705 is converted intracellularly into its T-705-RTP form, which behaves as a pseudopurine (21, 22). Consequently, incorporation of T-705-RTP into the viral RNA by the RdRp may possibly lead to chain termination or lethal mutagenesis. Some studies support lethal mutagenesis as the mechanism of action of T-705 against, for example, influenza virus (8) and norovirus (7). It was also reported that the incorporation of T-705-RTP by the norovirus RdRp does not result in complete chain termination (23). On the other hand, two studies have shown that incorporation of either a single (6) or two consecutive T-705-RTP molecules (24) into the nascent influenza viral RNA strand prevents further RNA strand extension. Moreover, we demonstrated earlier that the reduction of CHIKV RNA titers by T-705 correlates well with a decrease in infectivity, suggesting that lethal mutagenesis is probably not the mechanism of the antiviral effect of T-705 against CHIKV (9). By deep sequencing, we here determined the mutation frequency in the genome of CVB3 that had been cultured in the presence of T-705. Indeed, the mutation frequency in the presence of ribavirin was increased for all transition mutations; while T-705 had little effect on A-to-G and U-to-C transitions but significantly increased C-to-U and G-to-A mutations between 3- and 10-fold.
In a previous study, low-fidelity RdRP variants of CVB3, such as the A239G variant, were shown to be attenuated in immunocompetent mice (13). Therefore, the fitness of the reverse-engineered K159 mutants was studied in immunocompetent SJL mice. The A239G variant was included in the study as a positive control for in vivo attenuation. Interestingly, the K159R A239G variant had significantly lower viral RNA levels in serum and sampled organs and failed to efficiently infect the heart. In contrast, the infectivity of the K159M A239G variant was comparable to that of the WT. However, sequencing of the collected samples revealed that the K159M A239G variant completely reverted to the WT in infected mice, providing an explanation for the above observation.
To conclude, the conserved lysine residue of the F1 RdRp motif was shown to be key for the antiviral activity of T-705 against both CHIKV and CVB3 and is most likely also a key element in the interaction of T-705 with the RdRp of other +ssRNA viruses. In addition, this lysine is crucial for the proper functioning of the RdRp, which explains most likely the high barrier for antiviral resistance. Moreover, we demonstrate for the first time the role of RdRp fidelity in the mechanism of CVB3 sensitivity for T-705. These results provide more insights into the mechanism of the antiviral activity of T-705 and may help to design novel broad-spectrum +ssRNA virus inhibitors that target this particular region of the RdRp.
MATERIALS AND METHODS
Cells, viruses, and inhibitors.
Vero A cells (ATCC CCL-81) and HeLa Rh cells (kindly provided by K. Andries, Janssen Infectious Diseases, Belgium) were maintained in minimal essential medium (MEM Rega-3; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), 1% l-glutamine (Gibco), and 1% sodium bicarbonate (Gibco). Virus propagation and antiviral assays were performed in the same medium but supplemented with 2% FBS instead of 10% FBS. All cell cultures were maintained at 37°C in an atmosphere of 5% CO2 and 95% to 99% relative humidity.
T-705 was purchased from BOC Sciences (USA). Ribavirin (Virazole) was purchased from Valeant Pharmaceuticals International (Costa Mesa, CA). Rupintrivir was purchased from Axon Medchem (The Netherlands). Compounds were dissolved in dimethyl sulfoxide (DMSO) to yield 10-mg/ml stock solutions and were stored at 4°C until used.
The CVB3 Nancy infectious cDNA clone was kindly provided by F. van Kuppeveld (University of Utrecht, The Netherlands).
Generation of CVB3 mutants.
A full set of CVB3 mutants with mutations at position 159 of the RdRp gene were constructed by introducing the desired mutation in the CVB3 Nancy infectious cDNA clone or in the CVB3 RdRp-pASK3 plasmid in case of prokaryotic protein expression by using a QuikChange mutagenesis kit (Agilent) and the primers described in Table 4. The identity of the mutation in each of the constructs was verified by Sanger sequencing using a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems) and a model 3130 genetic analyzer automatic sequencer (Applied Biosystems). Subsequently, the mutated CVB3 infectious clones were in vitro transcribed into RNA using the RiboMAX large-scale RNA production system-T7 (Promega). An appropriate amount of in vitro-transcribed RNA was transfected into Vero A cells using DMRIE-C reagent (Invitrogen). Following incubation for 3 days at 37°C, the cultures were inspected for CPE. The cultures with clear CPE were used to prepare virus stocks and stored at −80°C. From the cultures for which no CPE could be detected, the cells were harvested using Cells-to-cDNA lysis buffer (Life Technologies) for extraction of intracellular RNA, and quantitative reverse transcription PCR (qRT-PCR) was performed to detect the presence of any replicating virus. In the event that no viral RNA could be detected, the transfection procedure was repeated once more. A virus variant was considered “not viable” when this second round also did not yield a positive PCR signal.
TABLE 4.
CVB3 site-directed mutagenesis primers
| Mutation | Primer | Sequence (5′-3′) | Length (nt) | Position (nt) | Tma |
|---|---|---|---|---|---|
| A239G | MFAT1 | TTACTCTGGGTACGATGGTAGCTTAAGCCCTGTCTG | 39 | 719 | 73.7 |
| MRAT1 | CAGACAGGGCTTAAGCTACCATCGTACCCAGAGTAA | 39 | 719 | 73.7 | |
| A372V | MFAT2 | GTTACCTGGACCAACGTCACTTTCCTAAAGAGG | 33 | 1118 | 69.5 |
| MRAT2 | CCTCTTTAGGAAAGTGACGTTGGTCCAGGTAAC | 33 | 1118 | 69.5 | |
| K159R | MFAT3 | GGTGACTTATGTAAGAGATGAGCTCAGGTC | 30 | 479 | 66.8 |
| MRAT3 | GACCTGAGCTCATCTCTTACATAAGTCACC | 30 | 479 | 66.8 | |
| K159 M | MFAT4 | GGTGACTTATGTAATGGATGAGCTCAGGTC | 30 | 479 | 66.8 |
| MRAT4 | GACCTGAGCTCATCCATTACATAAGTCACC | 30 | 479 | 66.8 |
Tm, melting temperature.
Sanger sequencing.
To confirm the presence of the introduced mutation in the reverse-engineered viruses, viral RNA was extracted using a NucleoSpin RNA isolation kit (Macherey-Nagel). This RNA was used to generate five cDNA fragments that represent the entire coding region by RT-PCR amplification using a OneStep RT-PCR kit (Qiagen). These fragments were purified using a Wizard SV Gel and PCR clean-up system (Promega) and sequenced on both strands (BigDye Terminator v3.1 cycle sequencing kit). The complete nucleotide sequences were assembled in Contig Express (VNTI; Invitrogen) and the genomes of the different variants were compared to the wild-type genome.
qRT-PCR.
One-step qRT-PCR was performed using an ABI 7500 fast real-time PCR system (Applied Biosystems, USA). The sequences of primers and probe and the thermal cycling conditions used in qRT-PCR were as described previously (25). The RNA copy number in each sample was determined by a standard curve using serial dilutions of CVB3 standard cDNA included in the run.
Protein expression and purification.
The CVB3 RdRp-pASK3 plasmid used in this study is a bacterial expression vector that does not use bacteriophage T7 RNA polymerase. It was generated by cloning the region encoding CVB3 RdRp from the CVB3 Nancy infectious cDNA clone into the prokaryote expression vector pASK3 (IBA) by PCR using two primers (forward, 5′-GGAATTCTAAGGAGGTAGAACCATGGGTGAAATAGAATTTATTGAGAGC-3′; reverse, 5′-CCGCCGCTCGAGTTAATGGTGATGGTGATGGTGAAAGGAGTCCAACCACTTCCTG-3′). The CBV3 RdRp contains a 6-histidine tag in the C-terminal end.
E. coli strain Rosetta 2 cells (Novagen), transformed with the CVB3 RdRp-pASK3 WT or variant plasmids, were grown in LB medium at 37°C and induced overnight with 200 μg/liter anhydrotetracycline when the optical density at 600 nm (OD600) reached 0.6. Expression was performed at 17°C for 16 h under light-protected conditions. Soluble fraction and metal ion affinity chromatography (IMAC) purification steps were performed as described previously (26). The pure fractions analyzed on the SDS-PAGE gel were pooled, dialyzed into 20 mM Tris (pH 9), 300 mM NaCl, 5% glycerol, and 0.5 mM Tris (2-carboxyethyl) phosphine hydrochloride (TCEP-HCl; Sigma), and concentrated using Centricon (Millipore) concentrators (30-kDa molecular weight cutoff). All proteins were stored at −20°C under cryoprotection conditions using 50% glycerol.
In vitro polymerase assays.
RNA oligo(dT) primers of 15 nucleotides (nt) and polyriboadenosine [poly(rA)] templates of approximately 357 nt in length were used for elongation assays in an analogous system as previously described (27, 28). Primers and templates were purchased from Thermo Fisher Scientific and Amersham Pharmacia Biotech, respectively. Both were mixed for annealing at a molar ratio of 1:2 (template/primer) in the presence of water. The mixtures were incubated at 70°C for 10 min, cooled down slowly to room temperature, and then left stored at −20°C until further use.
In vitro enzymatic assays were carried out using polymerase assay buffer (20 mM HEPES [pH 7.5], 50 mM KCl, 5 mM dithiothreitol [DTT], 5 mM MgCl2) mixed with 1 μM CVB3 RdRp and 0.5 μM preannealed primer-template substrate in 10 μl of reaction mixture. Thus, the polymerase and its RNA substrate were assembled at 30°C for 15 min to create an active elongation complex. The primer extension was started by the addition of a mixture of 3 μCi [α-32P]UTP and 10 μM unlabeled UTP using the same polymerase assay buffer in an equal volume of reaction mixture. In the case of competition assays, CTP was added at concentrations of 1, 2, 3, 4, or 5 mM for the WT and A239G variants. In the case of the K159R A239G and K159M A239G variants, concentrations were adjusted to 0.1, 0.25, 0.5, 1, or 2 mM CTP. NTPs were purchased from GE Healthcare.
After incubation at 30°C, aliquots of reactions were quenched at various time points by the addition of an equal volume of loading buffer (formamide with 10 mM EDTA). Reaction products were analyzed by PAGE using polyacrylamide sequencing gels of 14% acrylamide-bisacrylamide (19:1), 7 M urea with TTE buffer (89 mM Tris [pH 8.0], 28 mM taurine [2-aminoethanesulfonic acid], 0.5 mM EDTA). RNA bands were visualized using an FLA3000 fluorescent image analyzer (Fuji) and quantified using Image Quant Software (Fuji).
CPE reduction assay.
HeLa Rh cells were seeded in 96-well tissue culture plates (BD Falcon) at a density of 1.8 ×104 cells/well and allowed to adhere overnight. The next day, a 2-fold dilution series of T-705, ribavirin, or rupintrivir (starting concentration of 637, 819, or 8 μM, respectively) was added to the cultures that were then infected with the selected CVB3 variant at a final multiplicity of infection (MOI) of 0.001 PFU/cell. At day 3 p.i., the antiviral effect was quantified using the MTS/PMS method as described by the manufacturer (Promega, The Netherlands). This assay is composed of solutions of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS; inner salt) and phenazine methosulfate (PMS). The 50% effective concentration (EC50), which is defined as the concentration of compound required to inhibit virus-induced cell death by 50%, was determined using logarithmic interpolation.
Genetic stability of the generated mutants.
To assess the genetic stability of the reverse-engineered mutations, HeLa Rh cells were seeded at a density of 4 × 104 cells/well in 48-well plates and infected with the selected CVB3 variant at an MOI of 0.01. Each variant was passaged three times, after which the appropriate region of the RdRp was sequenced.
Virus phenotyping.
Vero A cells were seeded in 6-well plates (BD Falcon) at a density of 1 × 106 cells/well. After 24 h of incubation, the cells were washed with phosphate-buffered saline (PBS) and then infected with 10-fold serial dilutions of the respective virus stock. After 2 h of incubation with gentle shaking, the monolayers were washed with PBS, after which a freshly prepared 1:1 mixture of liquefied 1% low-melting-point agarose (Invitrogen) and 2× medium (i.e., medium in which supplements have double the usual concentration) with 4% FBS was added. On day 3 p.i., the monolayers were carefully washed with PBS, fixed with 4% formaldehyde, and stained with Giemsa solution to visualize the virus-induced plaques, after which they were counted to calculate the number of PFU per ml.
In parallel, the growth kinetics of different CVB3 mutants were determined by infecting Vero A cells (5 × 104 cells/well) in 96-well plates with the respective virus variants at an MOI of 3. After 1 h of infection, the cells were washed with assay medium to remove nonadsorbed virus. At 4, 6, 8, 10, 12, and 24 h p.i., the culture supernatant was collected, after which the infectious virus content was quantified by plaque assay as described before.
The effect of T-705 on the mutation frequency of CVB3 by deep sequencing.
HeLa Rh cells were seeded in a 48-well plate at a cell density of 4 × 104 cells/well and left to adhere overnight. On the next day, the cells were treated with T-705 or ribavirin at a final concentration that equaled the EC50 against the tested variant. The cells were then infected with the CVB3 WT or A239G or A372V variant at a final MOI of 0.001. On day 3 p.i., the culture supernatant in each well was collected and the virus populations in these samples were subjected to a deep-sequencing study. Samples were RNA extracted using TRIzol and RT-PCR amplified by RT (Superscript III) and PCR (Phusion) using primer sets that covered the whole genome in 3- to 4-kb fragments. PCR fragments were purified using the Nucleospin gel and PCR cleanup kit (Macherey-Nagel). PCR products were then fragmented (Fragmentase), linked to Illumina multiplex adapters, and clustered and sequenced using NextSeq500 technology. Sequences were demultiplexed by CASAVA with no mismatches permitted. Clipping was performed using the fastq-mcf tool (https://github.com/ExpressionAnalysis/ea-utils/blob/wiki/FastqMcf.md), removing common adapter contaminants and trimming low-quality bases (Phred score, <30). Clipped reads were aligned to the CVB3 Nancy sequence as the reference with a maximum of 2 mismatches per read and no gaps using BWA v0.5.9. Alignments were processed using SAMTools to obtain a pileup of the called bases at each position. The ViVan pipeline was used to identify statistically significant variants above the background noise due to sequencing error in every sufficiently covered site (>1,000×) (29). Briefly, for each position throughout the viral genome, base identity and their quality scores were gathered. Each variant was determined to be true using a generalized likelihood ratio test, and its allele rate was modified according to its covering read qualities based on a maximum likelihood estimation. Additionally, a confidence interval was calculated for each allele rate. To correct for multiple testing, a Benjamini-Hochberg false-discovery rate of 5% was set. The frequency of transition mutations for compound-treated samples was determined at each position across the genome and compared to that of untreated controls in a two-tailed paired t test.
In vivo experiments.
SJL mice were bred in the Experimental Animal Breeding Facility of the University of Leuven, Leuven, Belgium. Four groups of SJL mice (5 animals/group) were used. Mice were infected with 105 PFU of the selected CVB3 variant (WT or K159R A239G, K159M A239G, or A239G variant) by i.p. injection (200 μl). On day 3 p.i., all animals were euthanized, and serum samples, hearts, spleens, and pancreases were collected. The viral RNA was isolated from serum using a NucleoSpin RNA isolation kit (Macherey-Nagel) and from the homogenized organs using an RNeasy minikit (Qiagen). The amount of viral RNA in serum samples and organs was then quantified by qRT-PCR as described before. In addition, the number of infectious virus particles in the heart was determined by endpoint titration in Vero A cells.
Ethics statement.
The in vivo experiments were performed according to the Belgian guidelines for animal experimentation and with the authorization of the Ethical Committee of University of Leuven (KUL), Belgium (P080-2015).
ACKNOWLEDGMENTS
We thank Barbara Selisko for helpful discussions and for help in the initial phase of this project, François Ferron for help in the analysis of polymerase structures, and Carolien De Keyzer for excellent technical support in the animal experiments.
This work was supported by the BELVIR project from BELSPO (IUAP), the EU-FP7/2011-2014 Project SILVER (GA 260644), and the EU-H2020 Innovative Training Network ANTIVIRALS (GA 642434). A.T.S.D.M. was supported by a postdoctoral fellowship from CNPq (Conselho Nacional de Desenvolvimento Cientifico e Tecnologico—Brasil). L.D. was supported by a postdoctoral fellowship from the FWO (Fund for Scientific Research of Flanders, Belgium).
The authors declare no conflict of interest.
REFERENCES
- 1.Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL. 2013. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res 100:446–454. doi: 10.1016/j.antiviral.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rocha-Pereira J, Jochmans D, Dallmeier K, Leyssen P, Nascimento MSJ, Neyts J. 2012. Favipiravir (T-705) inhibits in vitro norovirus replication. Biochem Biophys Res Commun 424:777–780. doi: 10.1016/j.bbrc.2012.07.034. [DOI] [PubMed] [Google Scholar]
- 3.Zmurko J, Marques RE, Schols D, Verbeken E, Kaptein SJF, Neyts J. 2016. The viral polymerase inhibitor 7-deaza-2′-C-methyladenosine is a potent inhibitor of in vitro zika virus replication and delays disease progression in a robust mouse infection model. PLoS Negl Trop Dis 10:e0004695. doi: 10.1371/journal.pntd.0004695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Safronetz D, Falzarano D, Scott DP, Furuta Y, Feldmann H, Gowen BB. 2013. Antiviral efficacy of favipiravir against two prominent etiological agents of hantavirus pulmonary syndrome. Antimicrob Agents Chemother 57:4673–4680. doi: 10.1128/AAC.00886-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mentré F, Taburet A-M, Guedj J, Anglaret X, Keïta S, de Lamballerie X, Malvy D. 2015. Dose regimen of favipiravir for Ebola virus disease. Lancet Infect Dis 15:150–151. doi: 10.1016/S1473-3099(14)71047-3. [DOI] [PubMed] [Google Scholar]
- 6.Sangawa H, Komeno T, Nishikawa H, Yoshida A, Takahashi K, Nomura N, Furuta Y. 2013. Mechanism of action of T-705 ribosyl triphosphate against influenza virus RNA polymerase. Antimicrob Agents Chemother 57:5202–5208. doi: 10.1128/AAC.00649-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arias A, Thorne L, Goodfellow I. 2014. Favipiravir elicits antiviral mutagenesis during virus replication in vivo. eLife 3:e03679. doi: 10.7554/eLife.03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baranovich T, Wong S-S, Armstrong J, Marjuki H, Webby RJ, Webster RG, Govorkova EA. 2013. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J Virol 87:3741–3751. doi: 10.1128/JVI.02346-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delang L, Segura Guerrero N, Tas A, Quérat G, Pastorino B, Froeyen M, Dallmeier K, Jochmans D, Herdewijn P, Bello F, Snijder EJ, de Lamballerie X, Martina B, Neyts J, van Hemert MJ, Leyssen P. 2014. Mutations in the chikungunya virus non-structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral. J Antimicrob Chemother 69:2770–2784. doi: 10.1093/jac/dku209. [DOI] [PubMed] [Google Scholar]
- 10.Ferrer-Orta C, Agudo R, Domingo E, Verdaguer N. 2009. Structural insights into replication initiation and elongation processes by the FMDV RNA-dependent RNA polymerase. Curr Opin Struct Biol 19:752–758. doi: 10.1016/j.sbi.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Iglesias NG, Filomatori CV, Gamarnik AV. 2011. The F1 motif of dengue virus polymerase NS5 is involved in promoter-dependent RNA synthesis. J Virol 85:5745–5756. doi: 10.1128/JVI.02343-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Surana P, Satchidanandam V, Nair DT. 2014. RNA-dependent RNA polymerase of Japanese encephalitis virus binds the initiator nucleotide GTP to form a mechanistically important pre-initiation state. Nucleic Acids Res 42:2758–2773. doi: 10.1093/nar/gkt1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gnädig NF, Beaucourt S, Campagnola G, Borderia A, Sanz-Ramos VM, Gong P, Blanc H, Peersen OB, Vignuzzi M. 2012. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc Natl Acad Sci U S A 109:E2294–E2303. doi: 10.1073/pnas.1204022109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levi LI, Gnädig NF, Beaucourt S, McPherson MJ, Baron B, Arnold JJ, Vignuzzi M. 2010. Fidelity variants of RNA dependent RNA polymerases uncover an indirect, mutagenic activity of amiloride compounds. PLoS Pathog 6:e1001163. doi: 10.1371/journal.ppat.1001163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paeshuyse J, Dallmeier K, Neyts J. 2011. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 1:590–598. doi: 10.1016/j.coviro.2011.10.030. [DOI] [PubMed] [Google Scholar]
- 16.Vanderlinden E, Vrancken B, Van Houdt J, Rajwanshi VK, Gillemot SAG, Lemey P, Naesens L. 2016. Distinct effects of T-705 (favipiravir) and ribavirin on influenza virus replication and viral RNA synthesis. Antimicrob Agents Chemother 60:6679–6691. doi: 10.1128/AAC.01156-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Ávila AI, Gallego I, Soria ME, Gregori J, Quer J, Esteban JI, Rice CM, Domingo E, Perales C. 2016. Lethal mutagenesis of hepatitis C virus induced by favipiravir. PLoS One 11:e0164691. doi: 10.1371/journal.pone.0164691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peersen OB. 2 February 2017. Picornaviral polymerase structure, function, and fidelity modulation. Virus Res doi: 10.1016/j.virusres.2017.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnold JJ, Vignuzzi M, Stone JK, Andino R, Cameron CE. 2005. Remote site control of an active site fidelity checkpoint in a viral RNA-dependent RNA polymerase. J Biol Chem 280:25706–25716. doi: 10.1074/jbc.M503444200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson AA, Peersen OB. 2004. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J 23:3462–3471. doi: 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furuta Y, Takahashi K, Kuno-Maekawa M, Sangawa H, Uehara S, Kozaki K, Nomura N, Egawa H, Shiraki K. 2005. Mechanism of action of T-705 against influenza virus mechanism of action of T-705 against influenza virus. Antimicrob Agents Chemother 49:981–986. doi: 10.1128/AAC.49.3.981-986.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naesens L, Guddat LW, Keough DT, van Kuilenburg ABP, Meijer J, Vande Voorde J, Balzarini J. 2013. Role of human hypoxanthine guanine phosphoribosyltransferase in activation of the antiviral agent T-705 (favipiravir). Mol Pharmacol 84:615–629. doi: 10.1124/mol.113.087247. [DOI] [PubMed] [Google Scholar]
- 23.Jin Z, Tucker K, Lin X, Kao CC, Shaw K, Tan H, Symons J, Behera I, Rajwanshi VK, Dyatkina N, Wang G, Beigelman L, Deval J. 2015. Biochemical evaluation of the inhibition properties of favipiravir (T-705) and 2′-C-methyl-cytidine triphosphates against human and mouse norovirus RNA polymerases. Antimicrob Agents Chemother 59:7504–7516. doi: 10.1128/AAC.01391-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin Z, Smith LK, Rajwanshi VK, Kim B, Deval J. 2013. The ambiguous base-pairing and high substrate efficiency of T-705 (favipiravir) ribofuranosyl 5′-triphosphate towards influenza A virus polymerase. PLoS One 8:e68347. doi: 10.1371/journal.pone.0068347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Palma AM, Thibaut HJ, Van Der Linden L, Lanke K, Heggermont W, Ireland S, Andrews R, Arimilli M, Al-Tel TH, De Clercq E, Van Kuppeveld F, Neyts J. 2009. Mutations in the nonstructural protein 3A confer resistance to the novel enterovirus replication inhibitor TTP-8307. Antimicrob Agents Chemother 53:1850–1857. doi: 10.1128/AAC.00934-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jabafi I, Selisko B, Coutard B, De Palma AM, Neyts J, Egloff MP, Grisel S, Dalle K, Campanacci V, Spinelli S, Cambillau C, Canard B, Gruez A. 2007. Improved crystallization of the coxsackievirus B3 RNA-dependent RNA polymerase. Acta Crystallogr Sect F Struct Biol Cryst Commun 63:495–498. doi: 10.1107/S1744309107020416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arnold JJ, Cameron CE. 2000. Poliovirus RNA-dependent RNA polymerase (3Dpol). Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub). J Biol Chem 275:5329–5336. [DOI] [PubMed] [Google Scholar]
- 28.Gong P, Campagnola G, Peersen OB. 2009. A quantitative stopped-flow fluorescence assay for measuring polymerase elongation rates. Anal Biochem 391:45–55. doi: 10.1016/j.ab.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Isakov O, Bordería AV, Golan D, Hamenahem A, Celniker G, Yoffe L, Blanc H, Vignuzzi M, Shomron N. 2015. Deep sequencing analysis of viral infection and evolution allows rapid and detailed characterization of viral mutant spectrum. Bioinformatics 31:2141–2150. doi: 10.1093/bioinformatics/btv101. [DOI] [PMC free article] [PubMed] [Google Scholar]