

Abstract
Objective:
We aimed to generate a review and description of the phenotypic and genotypic spectra of ARHGEF9 mutations.
Methods:
Patients with mutations or chromosomal disruptions affecting ARHGEF9 were identified through our clinics and review of the literature. Detailed medical history and examination findings were obtained via a standardized questionnaire, or if this was not possible by reviewing the published phenotypic features.
Results:
A total of 18 patients (including 5 females) were identified. Six had de novo, 5 had maternally inherited mutations, and 7 had chromosomal disruptions. All females had strongly skewed X-inactivation in favor of the abnormal X-chromosome. Symptoms presented in early childhood with delayed motor development alone or in combination with seizures. Intellectual disability was severe in most and moderate in patients with milder mutations. Males with severe intellectual disability had severe, often intractable, epilepsy and exhibited a particular facial dysmorphism. Patients with mutations in exon 9 affecting the protein's PH domain did not develop epilepsy.
Conclusions:
ARHGEF9 encodes a crucial neuronal synaptic protein; loss of function of which results in severe intellectual disability, epilepsy, and a particular facial dysmorphism. Loss of only the protein's PH domain function is associated with the absence of epilepsy.
X-chromosomal mutations are a common cause of intellectual disability (X-linked intellectual disability [XLID]) in males accounting for 10%–12% of ID.1,2 XLID is highly heterogeneous and is usually divided into syndromic and nonsyndromic forms, depending on the association with particular clinical findings.1,2 Epileptic seizures accompany XLID in almost half the disorders, in some beginning in infancy prior to the developmental delay being evident. Recent next-generation sequencing (NGS) approaches have expedited the identification of XLID genes, including those associated with epilepsy,1–3 many of which encode proteins involved in synaptic function.4 ARHGEF9 is one such gene,5–9 encoding collybistin (Cb), a brain-specific guanine nucleotide exchange factor with an essential role in inhibitory synaptic transmission.5 Cb interacts with the inhibitory receptor–anchoring protein gephyrin and is required for the formation of gephyrin and gephyrin-dependent GABAA clusters in the postsynaptic membrane (figure 1).5,8,10 Cb knockout mice exhibit increased anxiety, impaired spatial learning, and convulsions.10,11
Figure 1. Interaction of collybistin in the postsynaptic membrane.

Schematic representation of the interactions of collybistin in the formation of gephyrin and gephyrin-dependent GABAA clusters in the postsynaptic membrane.
Over 10 patients with ARHGEF9 mutations have been reported,5–9,12–15 indicating that this will likely be one of the more common XLIDs. Descriptions of the published cases vary from scant (in large NGS projects) to case reports with substantial detail. In the present study, we combine detailed descriptions of the phenotypes of 18 patients with ARFGH9 mutations. In 4 cases, adequate detail was present at first published description. In 6 cases, we went back to the patients mentioned in tables of NGS results and worked with their physicians to obtain clinical details. The remaining are new unpublished patients.
METHODS
Two brothers (patients P10 and P11) with ID and epilepsy and unremarkable MRIs were tested by clinical whole-exome sequencing and found to have mutations in the ARHGEF9 gene. Further 16 patients (including 5 females) with mutations or chromosomal disruptions affecting ARHGEF9 were identified through a literature review and by contacting genetic centers in North America, Asia, and Europe.
Standard protocol approvals, registrations, and patient consents.
A detailed developmental epilepsy and general medical history together with examination findings were obtained for each patient via a standardized questionnaire filled by their treating physician, or if this was not possible by a review of the described phenotypic features in publications. No samples were obtained from patients for research purposes. Written consent was obtained from patients' parents for the publication of recognizable photographs.
X-chromosome inactivation analysis.
In the unpublished cases (patients P1, P3, and P4) X-chromosome inactivation analysis was performed with the human androgen receptor assay: amplification of the CAG repeat in exon 1 of the androgen receptor gene was performed by PCR with fluorescence-tagged primers. Subsequently, digestion with the methylation-sensitive enzyme HpaII was performed, and fragments were analyzed with an automated capillary sequencer (ABI 3100; Applied Biosystems, Foster City, CA). Genescan and Genotyper Software (Applied Biosystems) were used to determine fragment sizes and intensities, and the degree of X-inactivation was calculated.
RESULTS
Genetic findings.
Tables 1 and 2 summarize the genetic findings of the 18 patients. Eleven had hemizygous ARHGEF9 mutations, including premature stop codon, splicing, and point mutations. Six were de novo mutations; the 2 above brothers (P10 and 11) and patients P8, P15, and P18 showed a maternal inheritance, the latter with 3 affected brothers with a similar phenotype. Seven had chromosomal disruptions. The 11 mutations were in exons 8 (3), 9 (2), 1 (1), 3 (1), 4 (1), 5 (1), and 7 (1), and in intron 9 (1) (exon numbering based on the longest ARHGEF9 transcript NM_015185.2). The patients with chromosomal disruptions had deletions (4), translocations (2), and a paracentric inversion (1). The 5 female patients all showed skewed X-inactivation of the normal chromosome.
Table 1.
Genetic findings and phenotypic features of patients with ARHGEF9 mutations or disruptions (females)
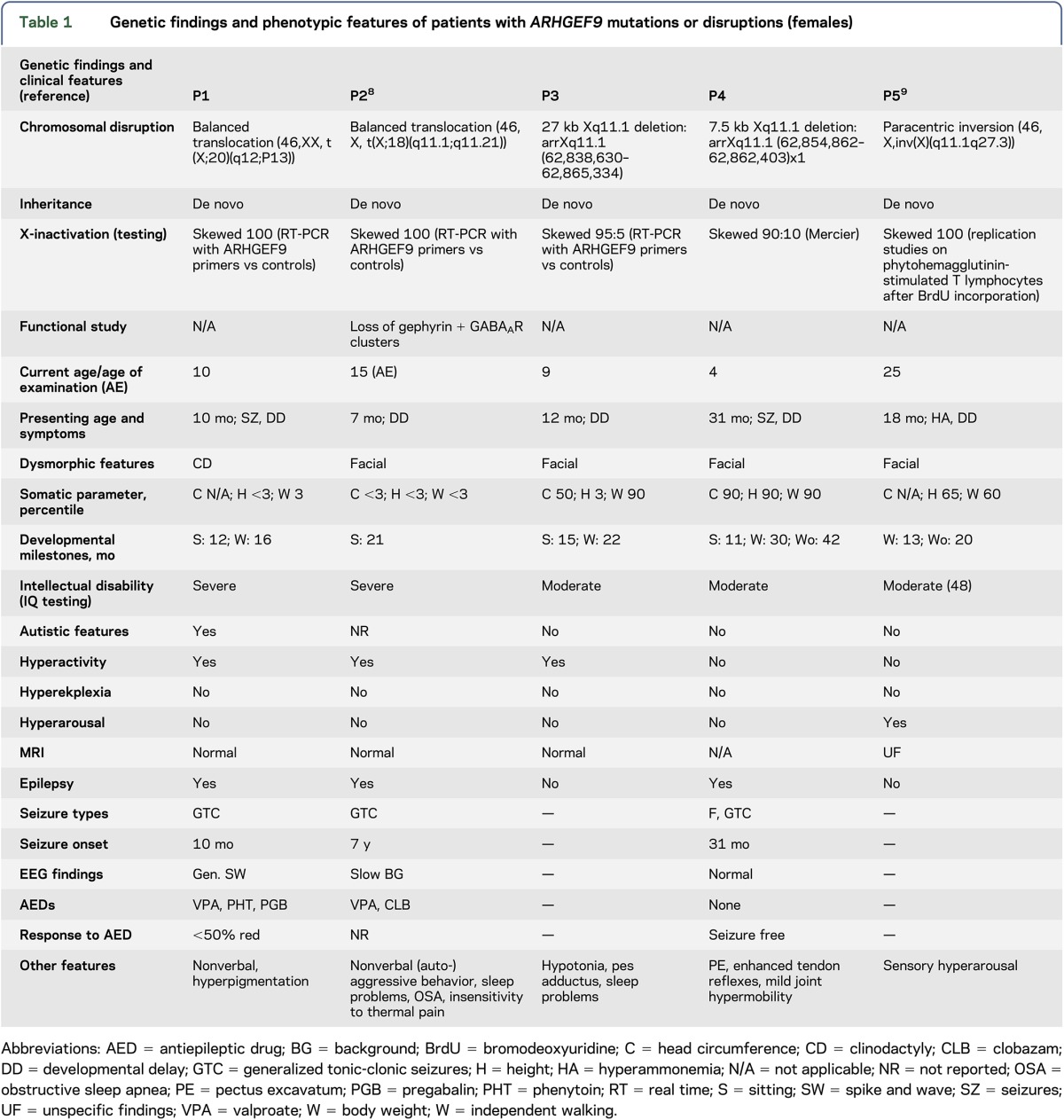
Table 2.
Genetic findings and phenotypic features of patients with ARHGEF9 mutations or disruptions (males)



Clinical features.
Tables 1 and 2 summarize the clinical features of the 18 patients.
Demographics.
Current age or age at last examination ranged from 4 to 57 years. Presenting symptoms: onset of symptoms occurred at a mean age of 15 months (median 9 months, range 1 day–7 years). Presenting symptoms varied from seizures (5), developmental delay in combination with seizures (4), and developmental delay/intellectual disability alone (6). One patient presented with hyperekplexia and seizures shortly after birth and 1 patient with hyperarousal and developmental delay (with no seizures).
Intellectual disability.
The majority of patients (13) showed delayed early developmental milestones. All patients had ID ranging from mild (1) to moderate (7) to severe (10). Of the 10 patients with severe ID, 7 were nonverbal.
Neuropsychiatric features.
Autistic features were reported in 4 patients. Five patients had hyperactivity.
Dysmorphic features.
Photographs for review of dysmorphic features were available for 9 of the 18 patients. Eight patients showed mild facial dysmorphic features. Out of this group, it appears that the male patients who are most severely affected with the overall neurologic syndrome (i.e., most severe ID and seizures) show more distinctive and consistent facial features, including enlarged, fleshy earlobes, a sunken appearance of the middle face (midface hypoplasia) in combination with a protruding of the jaw (prognathism) (figure 2). Two patients showed 5-digit clinodactyly.
Figure 2. Facial dysmorphism of 2 severely affected male patients.

Patients P6 (A) and P11 (B) from tables 1 and 2. Note enlarged fleshy earlobes, midface hypoplasia, and prognathism.
Neuroimaging findings.
MRI data were available in 15 patients. Findings were none (7), hippocampal sclerosis (1), hypoplastic frontal lobe (1), brain atrophy (1), polymicrogyria (1), delayed myelination (1), or nonspecific (T2 hyperintensities, accentuated perivascular spaces) (3).
Seizure/epilepsy details.
Thirteen patients had epilepsy with onset at a mean age of 20 months (median 12 months, range 1 week–7 years). Seizure types were variable, including generalized tonic-clonic (9), focal dyscognitive (5), focal (4), myoclonic (1), and tonic (2) seizures. EEG findings were available for 11 of the 13 patients and showed generalized (3), bilateral (1), multifocal (1), and focal (1) epileptiform discharges and were normal in 2 patients. One patient with polymicrogyria showed continuous spike-and-wave discharges in slow-wave sleep. Two patients had only moderate background slowing.
Epilepsy treatment.
Four patients had medically refractory epilepsy, 4 showed 50% seizure reduction with antiepileptic drugs (AEDs), 1 showed seizure reduction of less than 50%, and 2 became completely seizure free without further AED treatment. AEDs consisted mainly of combination therapy (10); only 1 patient was on monotherapy (1).
Additional features included sleep disorder (4), ataxic gait (1), aggressive behavior (2), fetal finger and toe pads (2), pigmentation abnormalities (2), pectus excavatum (2), hyperreflexia (1), tachypnea (1), macro-orchidism (1), and insensitivity to thermal pain (1).
DISCUSSION
We describe and review the phenotypes of 18 patients with ARHGEF9 mutations and chromosomal disruptions. Symptoms usually present in early childhood with delayed motor development alone or in combination with seizures. ID later on is usually moderate to severe with the majority of severe cases being nonverbal.
Epilepsy is common (13/18), starting usually at an early age (median 12 months). Seizure semiology is variable with a majority of patients showing generalized tonic-clonic seizures and many showing multiple seizure types. Most of the epilepsies were difficult to treat with 10 patients on combination AED therapy.
Facial dysmorphism, in particular, enlarged earlobes, midface hypoplasia, and prognathism appear to be a constant feature of the neurologically severely affected male patients.
The overall picture, therefore, is one of a moderate-to-severe XLID (with severe cases nonverbal), moderate-to-severe epilepsy (with severe cases poorly responsive or intractable to AEDs), and a facial dysmorphism in the severe cases.
Mothers of the various male patients in our study do not have XLID, indicating that random skewing of X-inactivation protects female carriers of ARHGEF9 mutations. We did, however, find 5 female patients. X-inactivation studies in all 5 showed skewing in favor of the abnormal X-chromosome (table 1), rendering them equivalent to male counterparts with hemizygous mutations. The female phenotype would invariably be affected by remaining expression from the normal allele in cells in which it is active, and by differences in the extent of X-inactivation skewing in the brain compared to leucocytes.
Close analysis of males in the genotype and phenotype tables (1 and 2, respectively) suggests a tentative genotype—epilepsy phenotype correlation related to the region of the protein affected by the mutation. To clarify this, we briefly review some aspects of Cb structure and function. Inhibitory neurotransmitter receptor channels (GABAA and glycine) imbedded in the postsynaptic membrane are anchored and clustered therein by a submembrane lattice composed of the gephyrin protein. The gephyrin lattice itself binds the plasma membrane via intermediary proteins critical among which is Cb. Cb binds gephyrin through its DH domain and attaches to the membrane through both its termini as follows: the N-terminal SH3 domain binds the membrane-integral neuroligin-2 protein, and the C-terminal PH domain binds the phosphoinositol 3-phosphate (PI3P) constituent of the plasma membrane (figure 1).5,8,10 In male patients P6 and P7, the entire ARHGEF9 gene is deleted, and in patient P8, there is a premature stop in the second codon (table 1). These 3 patients therefore lack Cb altogether, and their phenotypes would be representative of the complete loss of Cb function. Table 2 shows that all 3 have severe ID and epilepsy. Patients 9 through 15 have various missense mutations affecting different regions of the protein, but not the C-terminal PI3P-interacting PH domain. All have epilepsy. Where functional experiments were performed (patients 9, 14, and 15), it was shown that the mutations result in loss of gephyrin clusters.5,12,16 The 3 remaining patients (16 through 18) all have mutations that affect the PH domain. None of these patients have epilepsy (and all have mild-to-moderate ID). Therefore, tentatively, mutations affecting only the PI3P-interacting C-terminus of the protein are not associated with epilepsy. Future functional experiments for these and subsequent PH domain mutations may uncover differences from the effects of the epileptogenic mutations and a possible understanding of epileptogenesis in this disease.
An additional important observation gleaned from our summary tables is comparing male patients P8 and P9 (table 2). Patient P8 has a stop mutation, p.Q2*, in the very first exon, and his symptom constellation is very severe and similar to patients P6 and P7 in whom the entire gene is deleted. This first exon is coding, and therefore mutation p.Q2* truncating, in a shorter transcript (NM_001173479) that skips over the next 2 exons utilized by the longest transcript (NM_015185.2) of the gene. The shorter transcript, NM_001173479, was confirmed to be highly expressed in both the developing and adult human brain and may be the dominant transcript.6 Patient P9 is the very first ARHGEF9 patient described, and a striking feature of his presentation was hyperekplexia. His mutation is in exon 2, of the longer transcript, which is not utilized by the shorter transcript. Hyperekplexia is not found in any of the patients described after that first patient (tables 1 and 2), which suggests that this symptom may result from a particular function of the slightly longer protein encoded by the longer transcript.
As in so many other neurogenetic disorders, our review reveals that the ARHGEF9 disease is not uniform and varies with the gene mutation and likely other genetic and extragenetic factors. Therefore, the diagnostic approach in candidate patients still requires chromosomal microarray testing as the first-line genetic testing because of the substantial diagnostic yield and low relative cost, followed by a gene panel or whole-exome sequencing approach as second tier.
There is, however, a core phenotype of moderate-to-severe ID and epilepsy with a facial dysmorphism (large earlobes, midface hypoplasia, and prognathism) in patients with complete loss of the gene's function.
This phenotype is moderated with lesser mutations—less severe intellectual disability and seizures, and absence of facial dysmorphism. As mentioned, mutations affecting the last exon alone appear to be nonepiletogenic.
ACKNOWLEDGMENT
The authors thank the patients and their families. They thank Ms. Wendy Gu (University Toronto) for generating the graphic of figure 1.
GLOSSARY
- AED
antiepileptic drug
- Cb
collybistin
- NGS
next-generation sequencing
- XLID
X-linked intellectual disability
Footnotes
AUTHOR CONTRIBUTIONS
Michael Alber: acquisition of data, analysis and interpretation of data, and study supervision. Vera M. Kalscheuer, Elysa Marco, Elliott Sherr, Gaetan Lesca, Marianne Till, Gyri Gradek, Antje Wiesener, Christoph Korenke, Sandra Mercier, Felicitas Becker, Toshiyuki Yamamoto, Stephen W. Scherer, and Christian R. Marshall: acquisition of data and critical revision of manuscript for intellectual content. Susan Walker: acquisition of data, analysis of data, and critical revision of manuscript for intellectual content. Usha R. Dutta, Ashwin B. Dalal, Vanessa Suckow, Payman Jamali, Kimia Kahrizi, and Hossein Najmabadi: acquisition of data and critical revision of manuscript for intellectual content. Berge A. Minassian: acquisition of data, analysis and interpretation of data, and critical revision of manuscript for intellectual content.
STUDY FUNDING
Study funded by Genome Canada and the Ontario Brain Institute. This work was supported by the Ontario Brain Institute, Genome Canada, The McLaughlin Foundation, The Centre for Applied Genomics and the EU FP7 project GENCODYS, grant number 241995 (to V.M.K., H.N., and K.K.). B.A.M. holds the University of Toronto Michael Bahen chair in Epilepsy Research.
DISCLOSURE
Dr. Alber has received travel funding/speaker honoraria from AES Fellows Program 2015. Dr. Kalscheuer reports no disclosures. Dr. Marco has been a consultant for Grand Rounds Second Opinions (Clinical opinions through UCSF) and has received research support from the Wallace Research Foundation. Dr. Sherr has served on the scientific advisory board of InVitae; holds a patent for Blood-based diagnostic for autism; has been a consultant for Personalis; has received research support from National Institute of Neurological Disorders and Stroke, Simons Foundation, CURE Foundation, and the John and Marsha Goldman Foundation; holds stock/stock options in InVitae and ChemoCentryx; has worked with the following law firms: Sheuerman, Martini & Tabari; Galloway and Lucchese; Zuger Kirmis & Smith; Donahoe & Kearney; LaFollette, Johnson; Huff Powell & Bailey; and Bell, Roper and Kohlmyer; and has served on the Board of Directors of the National Organization of Disorders of the Corpus Callosum. Dr. Lesca, Dr. Till, Dr. Gradek, Dr. Wiesener, Dr. Korenke, Dr. Mercier, and Dr. Becker report no disclosures. Dr. Yamamoto has received research support from the Japan Ministry of Education, Science, Sports and Culture. Dr. Scherer has served on the scientific advisory board of Population Diagnostics; has served on the editorial boards of Genomic Medicine; Genes, Genomes, Genetics; Journal of Personalized Medicine; The Open Genomics Journal; The Hugo Journal; Genome Medicine; the Journal of Neurodevelopmental Disorders; Autism Research; PathoGenetics; Comparative and Functional Genomics; BMC Medical Genomics; and Cytogenetics and Genome Research; and has received research support from Genome Canada/Ontario Genomics Institute, Canadian Institutes of Health Research, Canadian Institute for Advanced Research, McLaughlin Centre, Canada Foundation for Innovation, the government of Ontario, Autism Speaks, and SickKids Foundation. Dr. Marshall has received travel funding from Affymetrix and Life Technologies and serves on the editorial board of Genes, Genomes, Genetics. Dr. Walker reports no disclosures. Dr. Dutta has received research support from the Department of Biotechnology—India (University of Mysore) and SERB. Dr. Dalal has served on the editorial board of Official Publication of Society for Indian Academy of Medical Genetics and has received research support from the Department of Science and Technology (Govt. of India) and the Indian Council of Medical Research (Govt. of India). Dr. Suckow, Dr. Jamali, Dr. Kahrizi, and Dr. Najmabadi report no disclosures. Dr. Minassian holds patents for diagnostic testing of the following genes: EPM2A, EPM2B, MECP2, VMA21; has received research support from the National Institute of Neurological Disorders and Stroke of the NIH under award number P01 NS097197; and receives license fee payments/royalty payments from patents for diagnostic testing of the following genes: EPM2A, EPM2B, MECP2, VMA21. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet 2010;11:167–187. [DOI] [PubMed] [Google Scholar]
- 2.Stevenson RE, Charles E, Schwartz R, Rogers RC. Atlas of X-linked Intellectual Disability Syndromes, 2nd ed New York: Oxford University Press; 2012. [Google Scholar]
- 3.Hu H, Haas SA, Chelly J, et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol Psychiatry 2016;21:133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humeau Y, Gambino F, Chelly J, Vitale N. X-linked mental retardation: focus on synaptic function and plasticity. J Neurochem 2009;109:1–14. [DOI] [PubMed] [Google Scholar]
- 5.Harvey K, Duguid IC, Alldred MJ, et al. The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J Neurosci 2004;24:5816–5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimojima K, Sugawara M, Shichiji M, et al. Loss-of-function mutation of collybistin is responsible for X-linked mental retardation associated with epilepsy. J Hum Genet 2011;56:561–565. [DOI] [PubMed] [Google Scholar]
- 7.Lesca G, Till M, Labalme A, et al. De novo Xq11.11 microdeletion including ARHGEF9 in a boy with mental retardation, epilepsy, macrosomia, and dysmorphic features. Am J Med Genet A 2011;155A:1706–1711. [DOI] [PubMed] [Google Scholar]
- 8.Kalscheuer VM, Musante L, Fang C, et al. A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum Mutat 2009;30:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marco EJ, Abidi FE, Bristow J, et al. ARHGEF9 disruption in a female patient is associated with X linked mental retardation and sensory hyperarousal. J Med Genet 2008;45:100–105. [DOI] [PubMed] [Google Scholar]
- 10.Papadopoulos T, Soykan T. The role of collybistin in gephyrin clustering at inhibitory synapses: facts and open questions. Front Cell Neurosci 2011;24:5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papadopoulos T, Korte M, Eulenburg V, et al. Impaired GABAergic transmission and altered hippocampal synaptic plasticity in collybistin-deficient mice. EMBO J 2007;26:3888–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long P, May MM, James VM, et al. Missense mutation R338W in ARHGEF9 in a family with X-linked intellectual disability with variable macrocephaly and macro-orchidism. Front Mol Neurosci 2016;8:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson JP, Nelson R, Schwartz CE. A family with mental retardation, variable macrocephaly and macro-orchidism, and linkage to Xq12-q21. J Med Genet 1998;35:1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012;367:1921–1929. [DOI] [PubMed] [Google Scholar]
- 15.Lemke JR, Riesch E, Scheurenbrand T, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012;53:1387–1398. [DOI] [PubMed] [Google Scholar]
- 16.Papadopoulos T, Schemm R, Grubmüller H, Brose N. Lipid binding defects and perturbed synaptogenic activity of a collybistin R290H mutant that causes epilepsy and intellectual disability. J Biol Chem 2015;290:8256–8270. [DOI] [PMC free article] [PubMed] [Google Scholar]