

Abstract
Discriminative stimulus and other drug effects are determined by the concentration of drug at its target receptor and by the pharmacodynamic consequences of drug-receptor interaction. For in vivo procedures such as drug discrimination, drug concentration at receptors in a given anatomical location (e.g., the brain) is determined both by the dose of drug administered and by pharmacokinetic processes of absorption, distribution, metabolism, and excretion that deliver drug to and from that anatomical location. Drug discrimination data are often analyzed by strategies of dose-effect analysis to determine parameters such as potency and efficacy. Pharmacokinetic-Pharmacodynamic (PKPD) analysis is an alternative to conventional dose-effect analysis, and it relates drug effects to a measure of drug concentration in a body compartment (e.g., venous blood) rather than to drug dose. PKPD analysis can yield insights on pharmacokinetic and pharmacodynamic determinants of drug action. PKPD analysis can also facilitate translational research by identifying species differences in pharmacokinetics and providing a basis for integrating these differences into interpretation of drug effects. Examples are discussed here to illustrate the application of PKPD analysis to the evaluation of drug effects in rhesus monkeys trained to discriminate cocaine from saline.
Keywords: Acute tolerance, Cocaine, Drug discrimination, Hysteresis, Pharmacodynamics, Pharmacokinetics, Prodrug
1 Introduction
Drugs produce their effects by interacting with receptor targets, and drug discrimination is one behavioral procedure that is useful for investigating determinants of this interaction. In conceptualizing drug-receptor interactions in whole organisms, it is convenient to think of the receptors as relatively fixed in anatomical space, whereas each dug molecule embarks on a journey from its site of administration, through the body to the receptor upon which it acts, and then back out of the body. Pharmacokinetics and pharmacodynamics are subdisciplines within the field of pharmacology that address two facets of this journey. Pharmacokinetics (PK) is concerned with the processes that govern a drug’s path through the body and its resulting concentration in different body compartments. Pharmacodynamics (PD), in contrast, is concerned with the physiological and behavioral consequences produced by that subset of drug molecules that find and occupy receptors during their journey through the body.
The relationship between PK and PD is described by PKPD analysis that relates drug concentration to drug effect. This type of analysis provides an alternative to conventional “dose-effect” analysis of drug effects, and they have value for at least three reasons [1]. First, drug effects are ultimately determined by drug concentration at the receptors upon which the drug acts, and that concentration is determined not only by the drug dose administered, but also by the PK processes that deliver that dose to and from the receptors. “Dose” is a measure of the amount of drug determined prior to its delivery, often in units of drug mass relative to the mass of the organism (e.g., mg/kg). Dose is precisely controlled by the experimenter, and it often serves as the principal independent variable in analysis of data from in vivo studies. For example, the “dose-effect curve” is a common mode of data presentation used to estimate critical drug features such as potency and efficacy. However, after a dose is administered, the drug must be absorbed into the body from the site of its administration (e.g., absorbed from gastrointestinal tract into the blood stream after oral delivery) and distributed from that site to the sites where receptors are located (e.g., distributed by the circulatory system from the gastrointestinal tract to brain). Moreover, drug molecules are subject to degradation via metabolism by enzymes and to removal from the body via excretion by routes such as urine, feces, or exhaled air. Together, these PK processes of absorption, distribution, metabolism, and excretion convert a drug dose administered at a single anatomical site and a single point in time into a dynamic tide of drug concentrations that rises and then falls throughout the body over time. These changing drug concentrations through time can then be related to changing drug effects through time to yield a richer data set than can be achieved by a reference to only a single drug dose administered at the beginning of an experiment. The most precise assessment of this relationship between drug concentration and drug effect would ideally measure drug concentrations at the site of receptors that mediate the measured effect. In practice, measurement of drug concentration at the receptor is often difficult, and the site of receptors might be unknown or broadly distributed. Accordingly, a common compromise is to measure drug concentrations in more accessible compartments (e.g., venous blood or cerebrospinal fluid) that usefully approximate drug concentrations across broad areas within the organism.
A second advantage of PKPD analysis is that it permits evaluation of the relationship between drug effect and concentrations not only of the administered drug, but also of drug metabolites. All drugs are subject to at least some degree of metabolism in the body, and in many cases, these metabolites are active and may contribute to the overall effect produced by an administered drug dose. An extreme example of this phenomenon is prodrugs, which are compounds designed to be metabolized in the body to active metabolites that then produce the drug’s intended effect [2]. When samples of blood or cerebrospinal fluid are collected and analyzed for concentrations of the administered drug, they can also be analyzed for concentrations of known or suspected metabolites, and changing drug effects over time can be related to changing concentrations of the metabolites as well as of the parent drug.
A third advantage of PKPD analysis is that it provides a basis not only for evaluating changing drug effects over time within an organism, but also for evaluating variable drug effects between organisms [3]. Thus, the administration of a given drug dose in mg/kg units often produces different effects across subjects within a species or across subjects of different species in translational studies. One factor that may contribute to such between-subject or between-species variability in drug effect is variability in PK processes. For example, metabolism may proceed at different rates or yield different metabolites in different subjects, and these differences in metabolism will result in different temporal profiles of drug and metabolite concentrations and associated behavioral and physiological effects despite use of the same administered dose. Use of drug and metabolite concentration, rather than drug dose, as the primary independent variable can reveal PK differences across subjects or species and provide a basis for integrating these differences into interpretation of drug effects.
The remainder of this chapter will illustrate strategies for using PKPD analysis in drug discrimination research using results from studies in rhesus monkeys trained to discriminate cocaine from saline.
2 PKPD Analysis of the Discriminative Stimulus Effects of Cocaine
2.1 PKPD Analysis in Rhesus Monkeys
Cocaine produces reliable discriminative stimulus effects in rhesus monkeys and other species, and these effects are both dose- and time-dependent. As one example, Fig. 1a shows the time course of the cocaine training dose in rhesus monkeys trained to discriminate 0.4 mg/kg intramuscular cocaine from saline in a two-key, food-reinforced drug discrimination procedure [4]. During training sessions, either cocaine or saline was administered 10 min before a 5-min response period, and only responding on the injection-appropriate lever produced food. During time-course test sessions (separate test sessions for each pretreatment time), the cocaine training dose was administered 1, 3, 5, 10, 20, 30, 60, or 100 min before 5-min response periods, during which responding on either key produced food. Under these conditions, the discriminative stimulus effects of cocaine displayed a rapid onset of action, peaking within 3 min, and had a relatively short duration of action, with effects declining after 20 min and no longer apparent after 100 min. Figure 1b shows venous plasma levels of cocaine from these same monkeys. Samples were collected separately from behavioral studies, and for plasma collection, subjects were anesthetized with ketamine, equipped with a temporary catheter in the saphenous vein, and placed into a primate restraint chair. The training dose of 0.4 mg/kg cocaine was administered intramuscularly as in behavioral sessions, and samples were collected at the same times as the onset of response periods in behavioral sessions. Venous cocaine levels peaked after 10 min and then declined. Figure 1c directly compares the time course of discriminative stimulus effects and venous cocaine levels after administration of 0.4 mg/kg cocaine, and for this figure, “Time” on the X-axis is represented on a log scale to facilitate comparison of effects that occurred early as well as later after cocaine administration. This comparison shows that both the onset and offset of cocaine-induced discriminative stimulus effects occurred earlier than the rise and fall in venous cocaine levels. Lastly, Fig. 1d shows a plot of discriminative stimulus effect as a function of venous cocaine levels over time, and arrows show the sequence in which data points were collected from first to last. This plot shows a variable relationship over time between venous cocaine levels and levels of cocaine-appropriate responding. For example, similar venous cocaine levels of 35–40 ng/ml were associated with nearly 100% cocaine-appropriate responding after 3 min but with less than 25% cocaine-appropriate responding after 60 min. This type of data display is known as a “hysteresis loop,” with the term “hysteresis” denoting a changing relationship over time between drug concentration and drug effect, and the term “loop” denoting the circular shape of the graph. Moreover, the direction of the loop can also be specified, and in this case, the loop is clockwise (i.e., the trajectory of data points over time flows in a clockwise direction).
Fig. 1.
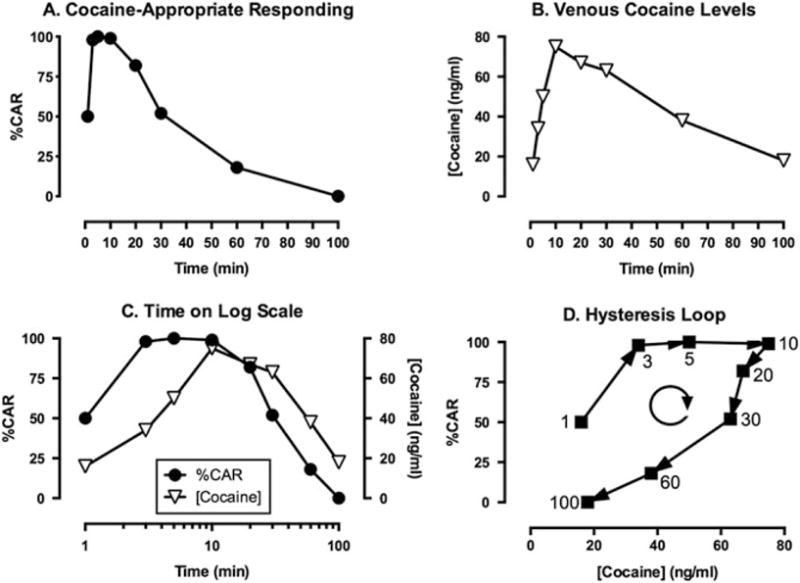
PKPD analysis of discriminative stimulus effects produced by 0.4 mg/kg intramuscular cocaine in rhesus monkeys. (a) Time course of discriminative stimulus effects expressed as % Cocaine-Appropriate Responding (%CAR). (b) Time course of venous cocaine concentrations expressed in units of ng/ml of plasma. (c) Comparison of the time course of discriminative stimulus effects and venous cocaine levels with time expressed on a log scale. (d) Concentration-effect relationship between venous cocaine levels and %CAR and the resulting clockwise hysteresis loop. Numbers indicate the time in minutes after cocaine injection, and the clockwise circular arrow indicates the clockwise flow of data in the hysteresis loop. Adapted from Lamas et al. [4]
2.2 Relationship to PKPD Analysis in Humans
Hysteresis loops are common in PKPD analyses, and the presence and direction of the loop (clockwise or counterclockwise) can be used to draw inferences about PK and PD processes that contribute to drug effects in whole organisms [5]. At the most superficial level of analysis, the clockwise hysteresis loop observed in Fig. 1d for the relationship between venous cocaine levels and cocaine-induced discriminative stimulus effects in monkeys indicates that discriminative stimulus effects declined faster than venous drug concentrations. Before addressing the implications of this finding in more depth, it is first useful to note that these results in monkeys agree with the observation of clockwise hysteresis loops for cocaine-induced subjective effects in humans [6, 7]. For example, Evans et al. evaluated the time course of venous cocaine levels and a range of subjective effects after either smoked cocaine (25 or 50 mg) or intravenous cocaine (16 or 32 mg) in human subjects experienced with both routes of cocaine administration [7]. Of relevance to this review, both smoked and intravenous cocaine yielded clockwise hysteresis loops relating venous cocaine levels to subjective effects such as “Stimulated,” “High,” and “Drug Liking.” In addition to this qualitative similarity, results of these studies in monkeys and humans could also be compared quantitatively. Specifically, intramuscular administration of 0.4 mg/kg cocaine in monkeys (equivalent to 28 mg in a 70 kg human) produced venous cocaine concentrations that were similar in magnitude, though with a slightly delayed time course, to those produced by 25–50 mg of smoked cocaine in humans, and both produced about half of the peak venous cocaine concentrations produced in humans by intravenous 16–32 mg cocaine. The delayed time course is consistent with the slower rate of drug absorption by the intramuscular route of cocaine administration used in monkeys than by the inhalation and intravenous routes used in humans. However, the finding that similar venous cocaine levels produced discriminative stimulus effects in monkeys and subjective effects in humans provides additional evidence for similarities between discriminative effects of drugs in animals and subjective drug effects in humans.
2.3 PK Factors in the Clockwise Hysteresis Loop for Cocaine
In addition to providing a nuanced basis for evaluating translation of drug effects across species, concentration-effect curves and hysteresis loops can also provide additional insights into the pharmacological determinants of drug effects in general and discriminative stimulus effects in particular. Table 1 lists some of the processes that may contribute to clockwise or counterclockwise hysteresis loops. In the case of cocaine discrimination shown in Fig. 1, at least two factors appear to contribute to the clockwise hysteresis loop that relates venous cocaine levels to discriminative stimulus effects.
Table 1.
Factors that may contribute to clockwise and counterclockwise hysteresis loop
| I. Clockwise hysteresis |
| A. Pharmacokinetic factors |
| 1. Slower distribution to site of drug concentration measurement than to site of drug action |
| 2. Generation of an active antagonistic metabolite |
| B. Pharmacodynamic factors (acute tolerance/tachyphylaxis)a |
| 1. Rates of receptor binding or signal transduction much faster than rates of drug distribution |
| 2. Desensitization/downregulation of receptors or downstream signaling pathways over time |
| 3. Recruitment of negative feedback processes |
|
|
| II. Counterclockwise hysteresis |
| A. Pharmacokinetic factors |
| 1. Faster distribution to site of drug concentration measurement than to site of drug action |
| 2. Generation of an active agonist metabolite (e.g., by a prodrug) |
| B. Pharmacodynamic factorsa |
| 1. Rates of receptor binding or signal transduction much slower than rates of drug distribution |
| 2. Sensitization/upregulation of receptors or downstream signaling pathways over time |
| 3. Recruitment of positive feedback processes |
Listed PD factors apply for drugs that are agonists at their target receptor. For drugs that function as antagonists or inhibitors, different mechanisms would apply. See text for a discussion of mechanisms that might apply for cocaine
First, regarding PK, recall that a drug is absorbed from its site of administration and distributed through a circuit of compartments before it is ultimately metabolized and/or excreted. For example, Fig. 2 shows that intramuscular cocaine is absorbed into the blood stream in muscle and transferred by veins to the heart and cardiopulmonary circulatory system where blood is oxygenated. After oxygenated blood containing cocaine returns to the heart, it is pumped via the aorta and systemic arteries to sites throughout the body, including sites of drug action such as brain, before being collected in veins and returned to the heart and cardiopulmonary system. Metabolism and excretion can occur at multiple points along this circuit. For cocaine, metabolism occurs largely via esterases in blood and liver, and excretion of cocaine and metabolites occurs largely via the kidneys into urine [9]. For the study shown in Fig. 1, cocaine-induced discriminative stimulus effects are thought to be mediated largely by binding of cocaine to dopamine transporters at one location (i.e., brain; [10,11]), and cocaine concentrations were determined in plasma isolated from a different location (i.e., venous blood samples collected from the saphenous vein). As depicted in Fig. 2, intramuscular cocaine would be distributed to its site of action in brain before it would reach the saphenous vein and other systemic veins, and this lag in drug distribution from the site of cocaine action to the site of blood collection could contribute to the lag between expression of discriminative stimulus effects and the later increases in venous cocaine levels. Results from the study by Evans et al. in humans support this possibility [7]. In addition to measuring subjective effects and venous cocaine levels after cocaine administration in humans, these authors also measured arterial cocaine levels, and thereby sampled cocaine concentrations from compartments that would be reached before as well as after access of cocaine to its sites of action in brain. Arterial cocaine levels peaked at approximately tenfold higher concentrations than venous levels, these peaks were reached more quickly in arteries than in veins (15 s vs. 4 min for both smoked and intravenous cocaine), and the arterial concentration-effect curve resulted in a counterclockwise rather than a clockwise hysteresis loop. A similar finding for counterclockwise vs. clockwise hysteresis loops has also been found for arterial vs. venous concentration-effect curves for the short-acting opioid remifentanil, which is also rapidly metabolized by esterases in blood [12]. These results are consistent with drug distribution first to arteries, then to sites of action in brain, and lastly to veins. Additionally, rapid metabolism to inactive metabolites contributes to markedly lower concentrations reaching venous blood, thereby accentuating the expression of clockwise hysteresis loops that relate centrally mediated drug effects to venous drug levels.
Fig. 2.
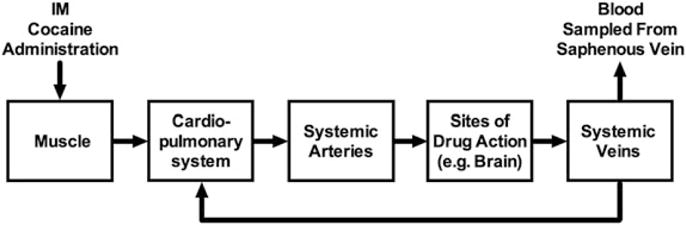
Schematic of drug distribution after intramuscular drug injection. For the studies shown in Figs. 1 and 3, drug and metabolite concentrations were determined from plasma of blood samples collected from the saphenous vein
2.4 PD Factors in the Clockwise Hysteresis Loop for Cocaine
Although PK factors likely bear primary responsibility for the clockwise hysteresis loop relating venous cocaine levels to discriminative stimulus subjective effects for cocaine, PD factors may also contribute. In particular, clockwise hysteresis loops are suggestive of acute tolerance to drug effects. “Tolerance” is a descriptive rather than an explanatory term, and in the context of PKPD analysis, it indicates a decrease in effect produced by a given drug concentration over time without implicating a particular mechanism. Possible mechanisms that may contribute to the phenomenon of acute tolerance (also known as “tachyphylaxis”) are listed in Table 1. Insofar as cocaine produces its discriminative stimulus effects primarily by blocking dopamine transporters and increasing extracellular dopamine levels in brain regions such as nucleus accumbens [10,11,13], possible mechanisms of acute tolerance to the discriminative stimulus effects of cocaine could result from upregulation of dopamine transporters to facilitate dopamine clearance, decreases in dopamine release due to feedback inhibition of dopamine neuronal activity, and/or desensitization or downregulation of postsynaptic dopamine receptors responding to elevated dopamine levels. The precise mechanisms that might confer acute tolerance during the time course of effects pursuant to a single cocaine administration remain to be fully elucidated. However, acute tolerance has been observed for many cocaine effects in experimental designs that involve two sequential cocaine treatments. For example, pretreatment with an active cocaine dose in humans decreased the cardiovascular and subjective effects of a second cocaine dose administered 60 min later [14], and in rhesus monkeys, two similarly spaced cocaine injections resulted in a smaller increase in extracellular dopamine levels in nucleus accumbens after the second injection [15].
3 PKPD Analysis of the Cocaine-Like Discriminative Stimulus Effects of Lisdexamfetamine and Phendimetrazine
3.1 Lisdexamfetamine
Lisdexamfetamine is a prodrug for D-amphetamine in which the amino acid L-lysine is coupled to the nitrogen of amphetamine [16, 17]. It is approved for treatment of attention-deficit hyperactivity disorder and binge-eating disorder, and it is also under consideration as a maintenance medication for treatment of cocaine abuse [8, 18]. Lisdexamfetamine is thought to be inactive as a parent drug, but it is metabolized in blood to lysine and the active metabolite amphetamine by peptidase enzymes associated with red blood cells [19]. Administration of amphetamine itself substitutes for the discriminative stimulus effects of cocaine across a wide range of conditions (e.g., [20]), and Fig. 3 shows results from a study that examined effects of lisdexamfetamine in rhesus monkeys trained to discriminate 0.32 mg/kg intramuscular cocaine from saline in a procedure otherwise identical to the one described above for studies with cocaine [8]. Lisdexamfetamine produced a dose- and time-dependent substitution for cocaine, and Fig. 3a shows the time course of cocaine-like discriminative stimulus effects produced by a dose of 3.2 mg/kg lisdexamfetamine, together with venous plasma levels of lisdexamfetamine and D-amphetamine. The discriminative stimulus effects of lisdexamfetamine had a slow onset and long duration of action. Venous levels of lisdexamfetamine were highest at the initial measurement at 10 min and declined rapidly to low levels, whereas venous amphetamine levels peaked more slowly and declined more gradually over a period of 2 days. The delayed appearance of amphetamine is consistent with the conclusion that amphetamine is a metabolite of lisdexamfetamine.
Fig. 3.
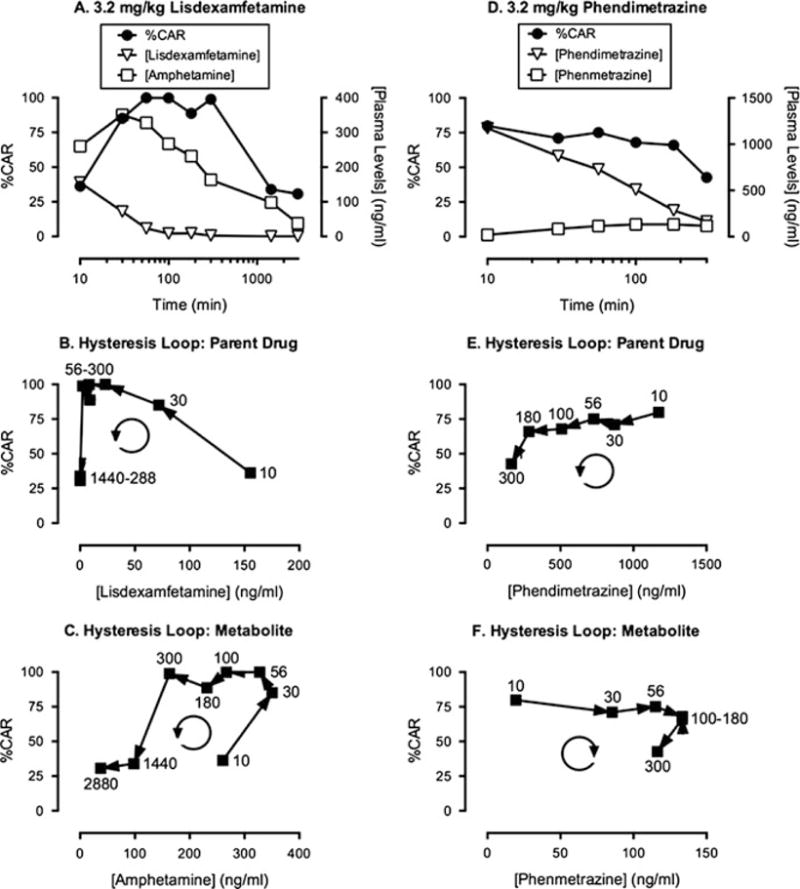
PKPD analysis of cocaine-like discriminative stimulus effects produced by intramuscular lisdexamfetamine and phendimetrazine in rhesus monkeys. (a, d) Time course of discriminative stimulus effects (expressed as % Cocaine-Appropriate Responding; %CAR) and venous plasma levels of the parent drug and metabolite (ng/ml) for lisdexamfetamine (a) or phendimetrazine (d). Note that time in minutes is shown on a log scale. (b, e) Hysteresis loops for venous levels of the parent drug and %CAR for lisdexamfetamine (b) and phendimetrazine (e). (c, f) Hysteresis loops for venous levels of the metabolite and % CAR for lisdexamfetamine (c, metabolite = amphetamine) and phendimetrazine (f, metabolite = phenmetrazine). Numbers in (b, c, e, and f) indicate the time in minutes after parent drug injection, and the circular arrows indicate the clockwise or counterclockwise flow of data in the hysteresis loop. Adapted from Banks et al. [8]
Figure 3b shows the hysteresis loop relating discriminative stimulus effects to venous levels of the parent compound lisdexamfetamine, and this hysteresis loop differs from that for cocaine in Fig. 1d in two ways. First, the initial rise in %CAR was not associated with a parallel rise in venous lisdexamfetamine levels. Rather, the highest lisdexamfetamine levels measured at 10 min were associated with low levels of cocaine-appropriate responding, and the onset of discriminative stimulus effects was associated with a drop in venous lisdexamfetamine levels. Second, the hysteresis loop flowed in a counterclockwise rather than in a clockwise direction. These two phenomena together are consistent with the conclusion that lisdexamfetamine is an inactive prodrug being converted to an active metabolite [5].
Figure 3c shows the hysteresis loop relating discriminative stimulus effects to venous plasma levels of amphetamine. In contrast to the plot for the parent drug, the plot for amphetamine did show rising plasma levels during the onset of discriminative stimulus effects during the first 30 min after drug administration, suggesting that amphetamine is indeed functioning as an active metabolite of lisdexamfetamine. However, as with the parent drug, the overall hysteresis loop for amphetamine also flowed in a counterclockwise direction. A counterclockwise hysteresis loop was also reported for the relationship for venous amphetamine levels to locomotor activity and mesolimbic dopamine release in rats after lisdexamfetamine administration [21]. This observation has been interpreted to suggest that amphetamine levels accumulate in systemic vasculature in general, and systemic veins in particular, more quickly than in brain to produce centrally mediated effects [8, 21]. More specifically, in reference to Fig. 2, these findings suggest that most of the conversion of lisdexamfetamine to amphetamine occurs in systemic veins. This conclusion would be consistent with (1) the requirement for peptidases in red blood cells to accomplish this metabolism, (2) the higher percentage of total blood volume in veins vs. arteries, and (3) the consequent longer residence time for any one circulating blood constituent (e.g., a red blood cell or drug molecule) in veins vs. arteries. Any amphetamine generated from lisdexamfetamine in veins would then require recirculation for delivery to brain. Moreover, rates of amphetamine delivery from vasculature across the blood-brain barrier and into neural tissue may also be limited [21], and this would produce a further delay between the time course of venous amphetamine levels and the time course of discriminative stimulus effects.
Two other points warrant mention. First, the venous amphetamine levels associated with cocaine-like discriminative stimulus effects in monkeys are much higher after lisdexamfetamine administration than after administration of amphetamine itself. For example, Fig. 3a shows that the dose of 3.2 mg/kg lisdexamfetamine sufficient to produce full substitution yielded a peak venous amphetamine levels of more than 300 ng/ml, whereas a dose of 0.32 mg/kg amphetamine sufficient to produce full substitution produced peak venous amphetamine levels of less than 100 ng/ml (M.L. Banks and S.S. Negus; unpublished results), and an oral dose of 20 mg amphetamine sufficient to produce significant subjective effects in humans yielded peak venous plasma levels of approximately 40 ng/ml [22]. One likely explanation for this difference is that venous levels after amphetamine administration likely underestimate the arterial drug levels initially delivered to the site of action (e.g., see above for cocaine), whereas venous levels after lisdexamfetamine are likely very similar to arterial levels delivered to the site of action (because amphetamine is generated largely in the systemic venous compartment). Direct evaluation of this hypothesis would be useful by comparing venous and arterial levels of amphetamine after lisdexamfetamine administration. In a second and related point, counterclockwise hysteresis loops relating venous amphetamine levels and centrally mediated behavioral effects after lisdexamfetamine administration differ from the finding of clockwise hysteresis loops after administration of amphetamine itself. For example, oral amphetamine in humans results in clockwise hysteresis loops that relate venous amphetamine levels to subjective effects [22], and we have similarly found that intramuscular amphetamine in rhesus monkeys produces clockwise hysteresis loops that relate venous amphetamine levels to cocaine-like discriminative stimulus effects (M.L. Banks and S.S. Negus, unpublished results). This distinction in rotational direction for hysteresis loops for amphetamine administered either directly or generated via metabolism of lisdexamfetamine illustrates one manifestation of PK differences that can be produced by different formulations of the same drug. In this case, the implication is that administration of amphetamine itself results in distribution of drug to sites of drug action before delivery to systemic veins, whereas administration of lisdexamfetamine results in generation of amphetamine in systemic veins prior to its delivery to sites of drug action.
3.2 Phendimetrazine
Phendimetrazine is approved for clinical use as an appetite suppressant for the treatment of obesity [23], and like lisdexamfetamine, it is also under consideration as a maintenance medication for the treatment of cocaine use disorder [24]. Phendimetrazine is metabolized to the compound phenmetrazine, and although both drugs interact with dopamine and norepinephrine transporters, the metabolite has high potency and functions as an amphetamine-like transporter substrate that promotes release of dopamine and norepinephrine, whereas the parent compound is more than 100-fold less potent and functions as a cocaine-like transporter inhibitor that prevents dopamine and norepinephrine reuptake [25]. The low potency of phendimetrazine at monoamine transporters suggested that it might function as a relatively inactive prodrug for the active metabolite phenmetrazine, similar to the function of lisdexamfetamine as a prodrug for amphetamine. This hypothesis was tested in PKPD studies in cocaine-discriminating rhesus monkeys [26]. For the purposes of the discussion below, phendimetrazine will be referred to as PDM, and phenmetrazine will be referred to as PM, because the spellings of the full drug names are similar and easily confused.
Initial studies indicated that administration of PM directly produced dose- and time-dependent substitution for cocaine and increases in venous PM levels, and the hysteresis plot relating venous PM concentration to cocaine-appropriate responding rotated in a clockwise direction similar to that described above for cocaine and amphetamine. PDM also produced dose- and time-dependent substitution for cocaine, and Fig. 3d shows results with a dose of 3.2 mg/kg PDM. Figure 3d also shows that this PDM dose produced time-dependent increases in venous levels of both PDM and PM. PDM levels peaked at the earliest time point at levels greater than 1,000 ng/ml, whereas PM levels rose more slowly and peaked at tenfold lower levels of approximately 100 ng/ml. The delayed emergence of PM after PDM administration is consistent with the status of PM as a metabolite of PDM. Moreover, venous PM levels were similar after administration of behaviorally active doses either of PM itself or of PDM, consistent with the conclusion that PM was functioning as an active metabolite sufficient to mediate behavioral effects of PDM. However, the PKPD profile of PDM and its metabolite PM differed from the profile for lisdexamfetamine and its metabolite amphetamine in two ways as illustrated by the hysteresis plots.
First, Fig. 3e shows the hysteresis loop that relates venous PDM levels to cocaine-appropriate responding. As with lisdexamfetamine, the direction of rotation for this hysteresis loop was counterclockwise; however, in contrast to results with lisdexamfetamine, the highest venous levels of PDM were associated with the highest levels of cocaine-appropriate responding. Although earlier time points were not assessed, these results indicate that the onset of cocaine-appropriate responding was associated with the period of rising PDM levels.
Second, Fig. 3f shows the hysteresis loop that relates PM levels to cocaine-appropriate responding. In contrast to the findings for amphetamine after lisdexamfetamine administration, the hysteresis loop for PM after PDM administration rotated in a clockwise direction. Of particular importance, high levels of cocaine-appropriate responding were observed at the earliest time point when PM levels were low, and the period of rising PM levels was associated not with onset of cocaine-appropriate responding, but rather with a period of sustained cocaine-appropriate responding. At later time points, there was a decrease in both venous PM levels and in cocaine-appropriate responding.
Taken together, these results were not consistent with the conclusion that PDM was an inactive parent drug for the active metabolite PM. Rather, these findings suggest that both PDM and PM were active, and the time course of cocaine-like discriminative stimulus effects after PDM administration reflected an initial phase of cocaine-like effects mediated by the parent drug PDM followed by a later phase of cocaine-like effects mediated by the metabolite PM.
4 Conclusions
PKPD analysis is an alternative to conventional dose-effect analysis of in vivo drug effects, and it focuses on the relationship of drug-induced behavioral or physiological effects to drug and metabolite concentrations in the body rather than to drug dose. Hysteresis loops are one manifestation of PKPD analysis, and these loops describe the time course of the potentially variable relationship between drug/metabolite concentration and drug effect over time. PKPD analysis, including analysis of hysteresis loops, can play a valuable role in interpretation of drug effects and PKPD relationships for the purposes of drug assessment and translational research in pharmacology. This chapter provided examples of the application of PKPD analysis to studies of the discriminative stimulus effects of drugs.
Acknowledgments
Supported by NIH Grant R01DA026946.
References
- 1.van der Graaf PH, Benson N. Systems pharmacology: bridging systems biology and pharmacokinetics-pharmacodynamics (PKPD) in drug discovery and development. Pharm Res. 2011;28:1460–1464. doi: 10.1007/s11095-011-0467-9. [DOI] [PubMed] [Google Scholar]
- 2.Huttunen KM, Raunio H, Rautio J. Prodrugs–from serendipity to rational design. Pharmacol Rev. 2011;63:750–771. doi: 10.1124/pr.110.003459. [DOI] [PubMed] [Google Scholar]
- 3.Bueters T, Ploeger BA, Visser SA. The virtue of translational PKPD modeling in drug discovery: selecting the right clinical candidate while sparing animal lives. Drug Discov Today. 2013;18:853–862. doi: 10.1016/j.drudis.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Lamas X, Negus SS, Hall E, Mello NK. Relationship between the discriminative stimulus effects and plasma concentrations of intramuscular cocaine in rhesus monkeys. Psychopharmacology (Berl) 1995;121:331–338. doi: 10.1007/BF02246072. [DOI] [PubMed] [Google Scholar]
- 5.Louizos C, Yanez JA, Forrest ML, Davies NM. Understanding the hysteresis loop conundrum in pharmacokinetic/pharmacodynamic relationships. J Pharm Pharm Sci. 2014;17:34–91. [PMC free article] [PubMed] [Google Scholar]
- 6.Ellefsen KN, Concheiro M, Pirard S, Gorelick DA, Huestis MA. Pharmacodynamic effects and relationships to plasma and oral fluid pharmacokinetics after intravenous cocaine administration. Drug Alcohol Depend. 2016 doi: 10.1016/j.drugalcdep.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Evans SM, Cone EJ, Henningfield JE. Arterial and venous cocaine plasma concentrations in humans: relationship to route of administration, cardiovascular effects and subjective effects. J Pharmacol Exp Ther. 1996;279:1345–1356. [PubMed] [Google Scholar]
- 8.Banks ML, Hutsell BA, Blough BE, Poklis JL, Negus SS. Preclinical assessment of lisdexamfetamine as an agonist medication candidate for cocaine addiction: effects in rhesus monkeys trained to discriminate cocaine or to self-administer cocaine in a cocaine versus food choice procedure. Int J Neuropsychopharmacol. 2015;18 doi: 10.1093/ijnp/pyv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones RT. Pharmacokinetics of cocaine: considerations when assessing cocaine use by urinalysis. NIDA Res Monogr. 1997;175:221–234. [PubMed] [Google Scholar]
- 10.Kleven MS, Anthony EW, Woolverton WL. Pharmacological characterization of the discriminative stimulus effects of cocaine in rhesus monkeys. J Pharmacol Exp Ther. 1990;254:312–317. [PubMed] [Google Scholar]
- 11.Witkin JM, Nichols DE, Terry P, Katz JL. Behavioral effects of selective dopaminergic compounds in rats discriminating cocaine injections. J Pharmacol Exp Ther. 1991;257:706–713. [PubMed] [Google Scholar]
- 12.Hermann DJ, Egan TD, Muir KT. Influence of arteriovenous sampling on remifentanil pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 1999;65:511–518. doi: 10.1016/S0009-9236(99)70070-6. [DOI] [PubMed] [Google Scholar]
- 13.Desai RI, Paronis CA, Martin J, Desai R, Bergman J. Monoaminergic psychomotor stimulants: discriminative stimulus effects and dopamine efflux. J Pharmacol Exp Ther. 2010;333:834–843. doi: 10.1124/jpet.110.165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischman MW, Schuster CR, Javaid J, Hatano Y, Davis J. Acute tolerance development to the cardiovascular and subjective effects of cocaine. J Pharmacol Exp Ther. 1985;235:677–682. [PubMed] [Google Scholar]
- 15.Bradberry CW. Acute and chronic dopamine dynamics in a nonhuman primate model of recreational cocaine use. J Neurosci. 2000;20:7109–7115. doi: 10.1523/JNEUROSCI.20-18-07109.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blick SK, Keating GM. Lisdexamfetamine. Paediatr Drugs. 2007;9:129–135. doi: 10.2165/00148581-200709020-00007. discussion 136–128. [DOI] [PubMed] [Google Scholar]
- 17.Heal DJ, Buckley NW, Gosden J, Slater N, France CP, Hackett D. A preclinical evaluation of the discriminative and reinforcing properties of lisdexamfetamine in comparison to d-amfetamine, methylphenidate and modafinil. Neuropharmacology. 2013;73C:348–358. doi: 10.1016/j.neuropharm.2013.05.021. [DOI] [PubMed] [Google Scholar]
- 18.Mooney ME, Herin DV, Specker S, Babb D, Levin FR, Grabowski J. Pilot study of the effects of lisdexamfetamine on cocaine use: A randomized, double-blind, placebo-controlled trial. Drug Alcohol Depend. 2015;153:94–103. doi: 10.1016/j.drugalcdep.2015.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutson PH, Pennick M, Secker R. Preclinical pharmacokinetics, pharmacology and toxicology of lisdexamfetamine: a novel D-amphetamine pro-drug. Neuropharmacology. 2014;87:41–50. doi: 10.1016/j.neuropharm.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 20.de la Garza RD, Johanson CE. The discriminative stimulus properties of cocaine in the rhesus monkey. Pharmacol Biochem Behav. 1983;19:145–148. doi: 10.1016/0091-3057(83)90323-4. [DOI] [PubMed] [Google Scholar]
- 21.Rowley HL, Kulkarni R, Gosden J, Brammer R, Hackett D, Heal DJ. Lisdexamfetamine and immediate release d-amfetamine - differences in pharmacokinetic/pharmacodynamic relationships revealed by striatal microdialysis in freely-moving rats with simultaneous determination of plasma drug concentrations and locomotor activity. Neuropharmacology. 2012;63:1064–1074. doi: 10.1016/j.neuropharm.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Brauer LH, Ambre J, De Wit H. Acute tolerance to subjective but not cardiovascular effects of D-amphetamine in normal, healthy men. J Clin Psychopharmacol. 1996;16:72–76. doi: 10.1097/00004714-199602000-00012. [DOI] [PubMed] [Google Scholar]
- 23.Bray GA, Ryan DH. Drug treatment of obesity. Psychiatr Clin North Am. 2011;34:871–880. doi: 10.1016/j.psc.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 24.Howell LL, Negus SS. Monoamine transporter inhibitors and substrates as treatments for stimulant abuse. Adv Pharmacol. 2014;69:129–176. doi: 10.1016/B978-0-12-420118-7.00004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothman RB, Katsnelson M, Vu N, Partilla JS, Dersch CM, Blough BE, Baumann MH. Interaction of the anorectic medication, phendimetrazine, and its metabolites with monoamine transporters in rat brain. Eur J Pharmacol. 2002;447:51–57. doi: 10.1016/s0014-2999(02)01830-7. [DOI] [PubMed] [Google Scholar]
- 26.Banks ML, Blough BE, Fennell TR, Snyder RW, Negus SS. Role of phenmetrazine as an active metabolite of phendimetrazine: evidence from studies of drug discrimination and pharmacokinetics in rhesus monkeys. Drug Alcohol Depend. 2013;130:158–166. doi: 10.1016/j.drugalcdep.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]