

Abstract
Motivation
Mutations (or Single Nucleotide Variants) in folded RiboNucleic Acid structures that cause local or global conformational change are riboSNitches. Predicting riboSNitches is challenging, as it requires making two, albeit related, structure predictions. The data most often used to experimentally validate riboSNitch predictions is Selective 2′ Hydroxyl Acylation by Primer Extension, or SHAPE. Experimentally establishing a riboSNitch requires the quantitative comparison of two SHAPE traces: wild-type (WT) and mutant. Historically, SHAPE data was collected on electropherograms and change in structure was evaluated by ‘gel gazing.’ SHAPE data is now routinely collected with next generation sequencing and/or capillary sequencers. We aim to establish a classifier capable of simulating human ‘gazing’ by identifying features of the SHAPE profile that human experts agree ‘looks’ like a riboSNitch.
Results
We find strong quantitative agreement between experts when RNA scientists ‘gaze’ at SHAPE data and identify riboSNitches. We identify dynamic time warping and seven other features predictive of the human consensus. The classSNitch classifier reported here accurately reproduces human consensus for 167 mutant/WT comparisons with an Area Under the Curve (AUC) above 0.8. When we analyze 2019 mutant traces for 17 different RNAs, we find that features of the WT SHAPE reactivity allow us to improve thermodynamic structure predictions of riboSNitches. This is significant, as accurate RNA structural analysis and prediction is likely to become an important aspect of precision medicine.
Availability and Implementation
The classSNitch R package is freely available at http://classsnitch.r-forge.r-project.org.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
A persistent challenge in the field of structural biology is accurately predicting the conformational and ultimately functional consequences of a mutation on a protein or nucleic acid (Chauhan and Woodson, 2008; Cheng et al., 2005; Churkin et al., 2011; Russell et al., 2002a,b). For both nucleic acids and proteins, accurately predicting the extent of disruption is generally more challenging than predicting the entire structure (Miao et al., 2015; Waldispuhl and Reinharz, 2015; Wan et al., 2014). Indeed it requires making two, albeit related structure predictions. The data most often used in conjunction with RiboNucleic Acid (RNA) structure prediction algorithms are chemical and enzymatic probing experiments (Corley et al., 2015; Ritz et al., 2012; Solem et al., 2015). These experiments, in particular Selective 2′ Hydroxyl Acylation by Primer Extension (SHAPE) provide nucleotide resolution structural information and are exquisitely sensitive to structure change (Cruz et al., 2012; Kutchko et al., 2015; Rice et al., 2014; Siegfried et al., 2014). Recent technological advances enable this data to be collected with unprecedented throughput (Siegfried et al., 2014); traditionally this data was carefully human curated to ensure accuracy, which is simply not possible in the genomic context (Ritz et al., 2012; Rocca-Serra et al., 2011; Sansone et al., 2012).
Chemical and enzymatic probing techniques have long been used in structural, kinetic and thermodynamic characterizations of nucleic acids (Brenowitz et al., 1986a,b; Deras et al., 2000; Sclavi et al., 1997). Until the advent of capillary sequencing and more recently next generation sequencing, the experiments were carried out using traditional gel electrophoresis (Brenowitz et al., 1986a,b; Petri and Brenowitz, 1997). Although informatics tools were developed to rapidly quantify these complex electropherograms, most structural insight was still gleaned by ‘gel gazing;’ for an effect to be robust the scientist had to be able to visualize it (Das et al., 2005; Das et al., 2008; Russell et al., 2002a,b; Takamoto et al., 2004). With high-throughput probing experiments rapidly becoming the norm, it is impossible to systematically visualize all the data.
In this manuscript we are specifically interested in mutation induced structure change in RNA and in particular the detection of riboSNitches using chemical and enzymatic probing data (Corley et al., 2015; Halvorsen et al., 2010; Lokody, 2014; Martin et al., 2012; Ritz et al., 2012; Solem et al., 2015; Wan et al., 2014). Accurately detecting riboSNitches experimentally is essential to establishing robust benchmarks (Corley et al., 2015; Ritz et al., 2012). Moreover, as transcriptome-wide structure probing experiments rapidly become the norm (Martin et al., 2012; Wan et al., 2012, 2014), efficiently detecting riboSNitches is likely to become an important component of personalized medicine (Solem et al., 2015). The main premise for the work presented in this manuscript is in the history of chemical and enzymatic probing techniques and in particular the value of expert human decision making in the determination of whether a structural change is significant. In particular, the distinction between a local structural change affecting several residues and a global structure change affecting a majority of residues.
Human ability to visually detect patterns in data is exceptional; even in the field of RNA structure, humans readily design better RNA folds than purely automated programs (Lee et al., 2014; Rowles, 2013; Treuille and Das, 2014). Interestingly, with enough examples machines can then learn the rules used by humans to make these designs (Lee et al., 2014). In this manuscript, we aim to automate some of the human skills associated with ‘gel gazing’ and apply these to the problem of identifying riboSNitches from high-throughput SHAPE data. We are particularly interested in understanding how humans interpret SHAPE data and what features of the signal they use to classify structure change. We are also interested in determining whether there is a consensus among users of SHAPE data as to what constitutes a small or large change in RNA structure. We therefore created a platform for easily visualizing SHAPE traces and asked experts in the field to classify traces and structures. As will be shown below, there is surprising agreement in human appreciation of the data and from these classifications we are able to identify novel metrics that reproduce the manual classifications. We are therefore able to report a structural classification scheme that quantitatively reproduces the process of ‘gel gazing.’ Our classifier allows us to simulate human eyes on high-throughput datasets and identify important differences in specific RNAs’ sensitivity to mutation.
2 Materials and methods
2.1 Dataset
SHAPE traces for 17 mutate-and-map experiments were obtained from the publicly available RNA Mapping DataBase (RMDB) (Cordero et al., 2012; Kladwang et al., 2011a,b,c). These 17 RNA database entries had a total of 2019 WT and single-point mutant trace pairs (Supplementary Material, Table S1). Of these trace pairs, 200 pairs were chosen for manual evaluation by 14 experts. Due to incomplete survey results we were able to obtain a majority consensus from at least 14 experts on 167 of the pairs.
2.2 Data normalization and noise reduction
Each WT trace was normalized to a mean reactivity of 1.5. A multiplier was used to normalize the respective mutant trace. The multiplier was chosen that minimized the difference between the WT and mutant traces. We reduced noise by setting mutant SHAPE values equal to the WT value, if both reactivities were outliers as defined by (Karabiber et al., 2013). To remove end effects, 8% of the data was trimmed from the 5′ and 3′ ends. Normalization and noise reduction are further explained in Methods Supplementary, Section S2.2.
2.3 Human expert evaluations
An online survey was created for the manual evaluation of 200 WT/mutant trace pairs. A trace pair consisted of a single WT trace and a mutant trace. The same WT trace could be used in multiple pairs with different mutants. The WT structure determined from the mutate-and-map experiments was provided, along with the WT SHAPE trace, the mutant SHAPE trace, the overlay of the WT and mutant traces, and the difference between the WT and mutant trace (Kladwang et al., 2011a,b,c). Survey participants were asked to label each WT/mutant pair as having: (i) no differences or small differences, (ii) local differences or (iii) global differences (Methods Supplementary, Section S2.3). For the purpose of this survey, local differences were considered to be close to the mutation site in sequence space. Under this definition, local changers in secondary structure space may be misclassified as global changers. Similarly, global changers in secondary structure space may be misclassified as local changers. Therefore, it is useful to consider secondary structure in structure change prediction, but the true secondary structure for an RNA is difficult to obtain experimentally. To address this we compared the expert classification to secondary structure prediction guided by SHAPE data. It is important to note that using predicted secondary structures in lieu of experimental structures is imperfect and likely increases the perceived secondary structure classification error by the experts. The experts did occasionally classify local changers in predicted secondary structure as global changers. However, the experts rarely classified global changers in secondary structure as local changers. (Supplementary Material, Table S8). Experts were filtered using a set of questions that gauged their familiarity with the biological sciences, RNA, RNA structure and SHAPE experiments. We identified 14 respondents in our survey results who self-identified as experts.
2.4 Feature and algorithm selection
Twenty-three features were initially used to quantify WT and mutant SHAPE trace differences and are reported in Table 2 and Supplementary Material, Table S2. These features rely solely on the experimental data and are completely independent of any structure prediction. Recursive feature elimination, using the caret package in R (Kuhn, 2008; Saeys et al., 2007) identified 8 features from the set of 23 that optimally classified the human consensus. In addition we used the WEKA suite to execute thirty-five classification algorithms using the default settings with 5-fold cross-validation (Hall et al., 2009). From these algorithms, random forest was selected as the most accurate for classification (Supplementary Material, Table S3) based on the number correctly predicted for non-changers. Assuming a tie at this level, we then selected the most accurate based on local changers and then global changers. We used this ranking because the distinction between change and no change is the most biologically important in our opinion. Further visual analysis of specific traces suggests that the random forest algorithm better distinguishes between local and non-changers than the next best performing algorithms, Multilayer Perceptron and Kstar. This is particularly true for WT/mutant pairs with minimal differences in pattern, but sizeable differences in magnitude such as the G55U mutation in the 16S four-way junction, which we illustrate in Supplementary Material, Figure S1. KStar and Multilayer Perceptron mislabel the pair as a local changer, while Random Forest correctly identified the pair as a non-changer in agreement with the majority vote of experts. Although these minor differences in classification do not indicate that random forest is statistically better than Kstar and Multilayer Perceptron, the correct classification by random forest on these particularly difficult comparisons led us to choose it for implementation in the classSNitch approach. We built a random forest classifier on the set of 167 trace pairs using the randomForest R package with 5001 trees and default settings (Breiman, 2001; Liaw and Wiener, 2002). The random forest classifier was used to predict the classes for the entire set of 2019 normalized and noise reduced WT/mutant trace pairs. Feature selection, algorithm selection, and model building are further explained in Methods Supplementary, Section S2.4. The model’s robustness to noise was tested using both simulated noise and repeated experiments (Supplementary Material, Fig. S2).
Table 2.
Features used to quantify differences between WT and mutant traces
| Feature | Formula | Description |
|---|---|---|
| Pearson CC | PCC(SHAPEref, SHAPEalt) | Pearson correlation coefficient is the covariance between the wild type and mutant trace SHAPE values divided by their standard deviations. Additional descriptions can be found in Supplementary Material, Figure S3 |
| Pattern CC | PCC(Changeref, Changealt) | Pattern correlation coefficient is the Pearson correlation coefficient between wild type and mutant trace patterns. The trace pattern is given by increase (+1), decrease (-1) or no change (0) in SHAPE value moving from one nucleotide to the next across the entire length of the RNAs. The pattern change between wild type and mutant traces are positions where the trace patterns different. Additional descriptions can be found in Supplementary Material, Figure S3 |
| Contiguousness | # of icontiguous | Contiguousness is the number of contiguous stretches of pattern change between wild type and mutant traces. See Pattern CC. Additional descriptions can be found in Supplementary Material, Figure S3 |
| Change Range | max(idiff) – min(idiff) | Change range is the interval containing all pattern changes between wild type and mutant traces. See Pattern CC. Additional descriptions can be found in Supplementary Material, Figure S3 |
| Change Variance | Σi(idiff -mean(idiff))/N | Change variance is the spread of pattern change distances between the wild type and mutant traces. The pattern change distance is the distance away from the mutation site (in nucleotides) that a pattern change occurs. See Pattern CC. Additional descriptions can be found in Supplementary Material, Figure S3 |
| Dynamic time warping | dynamic time warping algorithm | Dynamic time warping is an algorithm to optimally align wild type and mutant traces by ‘warping’ one into the other (Giorgino, 2009). Dynamic time warping aligns two series on the sides of a grid. The distance between each point in the two series is calculated for every position in the grid. Summing over the minimum distance path along the grid gives the overall distance. Additional descriptions can be found in Supplementary Material, Figure S4 |
| eSDC | (1-PCC(SHAPEref, SHAPEalt)*sqrt(N) | Experimental structural disruption coefficient is 1 minus the Pearson correlation coefficient between the wild type and mutant traces, normalized by the square root of the length of the RNA (Ritz, et. al, 2012). See Pearson CC. Additional descriptions can be found in Supplementary Material, Figure S3 |
| Euclidean Norm | Σi(SHAPEref[i]-SHAPEalt[i])2 | Euclidean norm is the L2-norm or distance between the wild type and mutant traces. The distance is calculated as the sum over the squared difference between wild type and mutant traces. Additional descriptions can be found in Supplementary Material, Figure S3 |
Note: Feature formulas and descriptions for the 8 features included in the model. These 8 features were chosen by recursive feature elimination from the total set of 23 features (Methods Supplementary, Section S2.4). The formula symbol descriptions are included in Supplementary Material, Table S5. Additional descriptions for these methods can be found in Supplementary Material, Figures S3 and S4. A list of feature statistics can be found in Supplementary Material, Table S7.
2.5 classSNitch package
An R package was created for the identification of RNA structure change in large amounts of SHAPE data. The package includes methods for normalization, noise reduction, and calculating features. Feature calculations include pattern change, dynamic time warping, change contiguousness, Pearson correlation, Euclidean norm, change variance, eSDC and change range. The package can identify structure change in new SHAPE datasets based on an existing classifier. classSNitch is currently available at R-Forge.
2.6 WT SHAPE improved SNPfold
We modified the SNPfold scoring scheme, which is based on the WT and mutant Pearson correlation coefficient (Halvorsen et al., 2010), to include the WT SHAPE prediction as follows:
| (1) |
where SHAPE{0,1} is 1 if the WT SHAPE reactivity is above the median value of the trace, 0 if it is below; GorC{0,1} is 1 if the WT nucleotide is a G or C, 0 otherwise. SNPfold is further explained in Methods Supplementary, Section S2.6.
3 Results
3.1 The ‘obvious’ riboSNitch
Figure 1A illustrates the published secondary structure of the apo Glycine riboswitch based on multiple probing experiments, phylogenetic analysis and partial crystal structures (Butler et al., 2011; Kladwang et al., 2011a,b,c). The nucleotides are color coded according to SHAPE reactivity (red high, yellow medium and black low). In Figure 1B, the corresponding experimental SHAPE data for the WT RNA is plotted as a black line. A qualitative relationship between the structure and experimental data is evident when the data is presented in this way; in general paired nucleotides have low SHAPE reactivity, while unpaired bases have a ‘peak’ in the profile. In a gel electropherogram, the peaks would be darker, and the paired nucleotides lighter. Figure 1C illustrates the experimental SHAPE data and corresponding SHAPE-directed structure prediction for the A125U mutation in the Glycine riboswitch. The overlay of the two traces reveals no visible difference between the WT (WT, black) and mutant (MUT, blue) trace; the structure prediction is nearly identical to that of the WT. Not surprisingly, mutating A125 in domain 2 (P3) does not affect structure, as this nucleotide is not paired.
Fig. 1.

Structure change patterns in SHAPE trace data for the glycine riboswitch aptamers from Fusobacterium nucleatum. (A) Published WT structure for the apo glycine riboswitch aptamers consistent with the crystal structure and multiple independent structure probing experiments (Butler et al., 2011; Kladwang et al., 2011a,b,c). Red nucleotides indicate high SHAPE reactivity, yellow indicates mid-range reactivity, and black indicates low reactivity. (B) The individual WT trace is shown in black; the colored bars indicate the structural regions for each of the aptamers: P1 (orange), P2 (green) and P3 (blue). (C) The WT trace (black) is overlaid with the mutant SHAPE trace (dark blue), and the absolute difference between the WT and mutant traces is below (dark green). A red bar on the traces shows the mutation site. The A125U mutation is a mutation that leads to no appreciable differences in structure. 100% of experts that classified this mutant labeled it as a non-changer. (D) The A116U mutation leads to a local structure change, where the mutant trace reactivity increases at the mutation site disrupting the P3 region of domain 2. 66% of experts that classified the A116U mutant labeled it as a local changer. (E) The A94U mutation leads to a global structure change, where the mutant trace reactivity increases at both the mutation site and at nucleotides distant in sequence space disrupting both the P1 and P2 regions of domain 2. 66% of experts that classified the A94U mutant labeled it as a global changer (Color version of this figure is available at Bioinformatics online.)
In Figure 1D we report the SHAPE-directed prediction for the A116U mutation, which occurs in the P3 helix of domain 2. In this case we see a local difference in the SHAPE trace, and the predicted structure does not contain this region of P3. This mutation has disrupted a single hairpin. It is important to note that the resulting SHAPE differences are readily visualized with the difference of the two traces (green trace, right panel). Figure 1E shows the effect of disrupting a base in the P2 stem in domain 2 with the A94U mutation. This results in a change in the P1 helix of domain 2 as well and is considered a global change. We chose to illustrate these three mutations from the 158 available for the Glycine riboswitch (Cruz et al., 2012) as they are visually striking. As will be revealed below, not all mutation induced RNA structure change is as clear to visualize.
3.2 Human consensus on local and global structure change
The complexity of interpreting SHAPE traces is illustrated in Figure 2. Here we plot the WT structure for the 16S four-way junction from the E.coli ribosome, as well as the mutant SHAPE data for A26U, A47U (P2b) and U99A (P1c). In each of these cases, it is not visually evident if the structure change is local, global, or if the data is simply inadequate. It is important to note that these SHAPE data are collected in a high throughput fashion, robotically and often not replicated (Cheng et al., 2015; Cordero and Das, 2015; Kladwang et al., 2011a,b,c; Miao et al., 2015). This is one of the main differences in the way in which chemical and enzymatic probing is now collected. Because it can be collected in a very high throughput way, emphasis is placed on multiple experiments (all mutations in an RNA) rather than multiple replicates. Although it would be ideal to replicate these large-scale experiments there is a significant financial cost associated with multiple replicates.
Fig. 2.
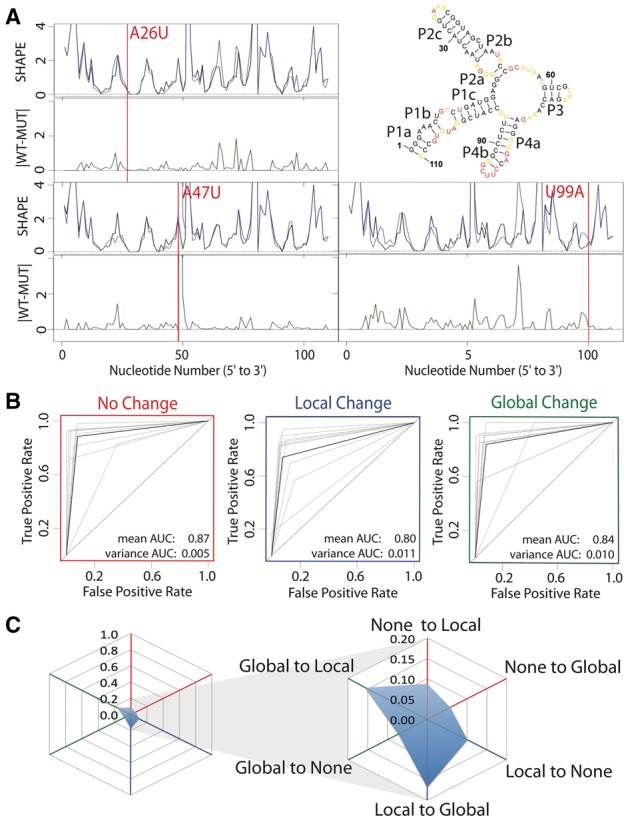
Expert evaluation of RNA structure change in SHAPE data. (A) Accepted WT structure for the 16S four-way junction domain from the E.coli ribosome in agreement with the crystal structure and multiple structure probing experiments (Cordero and Das, 2015; Tian et al., 2014; Zhang et al., 2009). Red nucleotides indicate high SHAPE reactivity, yellow indicates mid-range reactivity, and black indicates low reactivity. For each mutant, the WT trace (black) is overlaid with the mutant SHAPE trace (dark blue). The absolute difference between the WT and mutant traces is depicted below (dark green). 100% of experts that evaluated the A26U WT mutant pair agree that there is no difference or a small difference. 88% of experts agree that the A47U mutation creates a local difference. Experts are split on the U99A mutation. 37.5% of experts indicated that the mutation creates no difference or a small difference, 37.5% of experts indicated that the mutation creates a local difference and 25% of experts indicated the mutation creates a global or distant mutation. (B) ROC curve analysis was used to compare expert classification to the majority vote consensus. The gray curves represent individual expert performances, while the black curves show the average performance among experts. The ROC curves are depicted for performance in identifying non-changers (red), local changers (blue) and global changers (green). (C) Cobweb plots show the percentage of mutants mislabeled by the expert majority vote with non-changers on the red axes, local changers on the blue axes, and global changers on the green axes. Expert classification is least consistent on differences between global and local changers with a higher percentage of global changers mislabeled as local changers, and local changers mislabeled as global changers (Color version of this figure is available at Bioinformatics online.)
In visually inspecting traces like the ones illustrated in Figure 2A, we observed that in general most people in our lab agreed that A26U does not alter structure, A47U causes a local change, and U99A appears to alter the structure globally. We therefore decided to evaluate if RNA scientists, when presented with these types of traces and the accepted secondary structure of the RNA, agree on the classification of these data into none, local and global change. We recruited 14 volunteers from multiple RNA labs to answer an online survey in which each person would classify up to 200 traces (WT/MUT comparisons) into none, local and global changes. In total 1427 comparisons were manually classified, with an average of seven views for each trace (Table 1). From this data we built a consensus human classification of the traces and evaluated each expert’s ROC (receiver operator curve) area under the curve (AUC) to the consensus (Fig. 2B). Since this is a three-way classification we evaluate AUC pairwise for none, local and global change. As can be seen the expert reproducibility is high (AUC average above 0.8) which indicates RNA scientists agree with each other at least with respect to what structure change looks like in a SHAPE trace. We also evaluate human three-way AUC using a cobweb plot (Fig. 2C). This shows that the largest disagreement between self-reported RNA SHAPE experts is in their classification of local versus global change. The average AUC is still 0.8 (blue) suggesting the disagreement is weak. The green AUC curves in Figure 3A, show that for all but distinguishing global vs. none (rightmost graph) eSDC performs quite poorly.
Table 1.
Expert evaluation summary
| Survey statistics | |
|---|---|
| Total Traces | 200 |
| Total Experts | 14 |
| Total Responses | 1427 |
| Mean Trace Coverage | 7.24 |
| SD Trace Coverage | 2.78 |
| Mean Expert Agreement (%) | 79.75 |
| SD Expert Agreement (%) | 0.79 |
| Expert Reproducibility (%) | 79.70 |
| Total Non-Changers (Majority Consensus) | 107 |
| Total Local Changers (Majority Consensus) | 40 |
| Total Global Changers (Majority Consensus) | 20 |
Note: Human survey statistics on WT/mutant SHAPE trace pair classification.
Fig. 3.
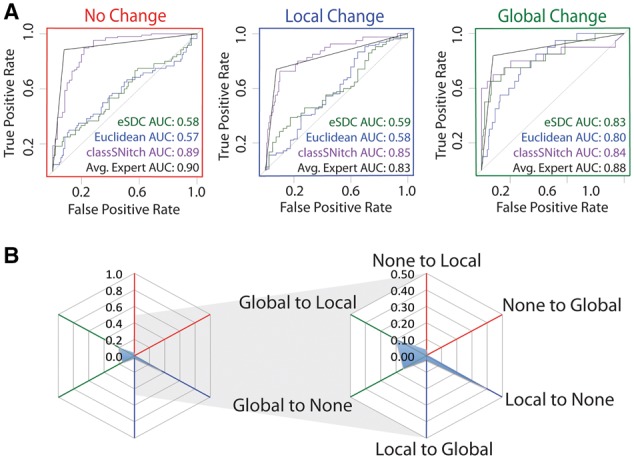
classSNitch performance. (A) ROC curve analysis comparing methods for classifying structure change to the majority consensus by experts. The ROC curves are depicted for performance in identifying non-changers (red), local changers (blue) and global changers (green). The methods used for experimental classification are classSNitch (purple), eSDC (green), Euclidean norm (blue) and the mean expert human performance (black). Consistently, classSNitch performs comparably to the mean expert evaluation. classSNitch outperforms eSDC and the Euclidean norm, which are the current metrics for classifying RNA structure change in SHAPE data. (B) The cobweb plot shows the percentage of traces mislabeled by classSNitch; a higher percentage of local changers are misclassified (Color version of this figure is available at Bioinformatics online.)
We also investigated whether another standard metric, the Euclidean distance (blue AUC) did any better and observed a similar trend. The mean expert performance is shown in black, and is far superior to any single metric. Thus, to achieve consensus, RNA scientists must be looking at other features in the data than simple correlations in the pattern. We set out to discover what these are and to develop an automated classification system of RNA structure change that simulates human consensus calls.
3.3 Automated classification of mutation induced structure change
To develop an automated classifier for identifying mutation induced structure changes in RNA we began by establishing a list of 23 features commonly used to evaluate quantitative differences between two linear datasets (Table 2 and Supplementary Material, Table S2). Using the human survey classification (Table 1) for supervised learning, we trained 38 different algorithms and evaluated their accuracy. The results of this training are provided in Supplementary Materials, Table S3 and suggest the Random Forest classifier performs the best on this data using the eight features found in Table 2. The trained random forest classifier on these eight features is the algorithm used in the classSNitch R package released with this manuscript.
Interestingly no single feature drives the classification, indicating that the human experts are looking at multiple features of the signal to decide what is or is not a change. Nonetheless we performed random feature elimination and did identify that dynamic time warping alone achieves an accuracy of 65% (Supplementary Material, Fig. S5A). Dynamic time warping is less sensitive to distortion caused by local misalignments, a quality that makes the technique useful in speech recognition and likely contributes to the feature’s success in our classifier (Sakoe and Chibe, 1978). We also ranked the eight features by their importance and see that each feature increases accuracy incrementally when added to the model in approximately equal increments. Plotting the WT to mutant Pearson correlation coefficient and contiguousness versus dynamic time warping (Supplementary Material, Fig. S5B) reveals how these features correlate but also illustrates subtle differences in how these different features classify change.
We illustrate the basic dynamic time warping principle in Supplementary Material, Figure S4A and how we score differences based on this trace alignment strategy. The score increases as the two traces differ and is calculated over the entire alignment. Dynamic time warping is visualized on the U99A data in Supplementary Materials, 4B. It identifies the minimum number of insertions and deletions to optimally align the mutant and WT traces. As such, a higher dynamic time warping score indicates greater differences in the traces. It is therefore likely that the expert humans are performing some form of trace alignment combined with pattern matching when evaluating the data. Processing SHAPE data (whether it is obtained by capillary or next generation sequencing) requires an alignment strategy. It is not surprising that humans may choose to ignore small frame shifts in the data (which lead to very high eSDC values) since they know these are most likely errors in trace alignment (Supplementary Material, Fig. S6).
Overall, the classSNitch performance (purple line Fig. 3A) is equivalent to human consensus for none, local and global change. The cobweb plot reveals that the highest error rate in classSNitch classification is false negatives for local change (Fig. 3B). In comparison to eSDC and the Euclidean distance (green and blue AUC, respectively) our classifier performs significantly better. Thus classSNitch is a good approximation of human expert classification of SHAPE trace differences and applying it to high-throughput mutational datasets can simulate human consensus classification of these data.
3.4 classSNitch analysis of experimental structure change
The training data used for the development of the classSNitch classifier (Table 1) represents a small subset of publically available mutational SHAPE data (Cordero et al., 2012). We identified a total of 2019 SHAPE traces for eleven different RNAs (Supplementary Material, Table S1). We classified these using the classSNitch algorithm excluding the training set of 167 RNAs. In this dataset we identified 382 local changers (19%), and 111 global changers (5%). When these data are further broken down by RNA (Fig. 4A) we immediately observe significant differences in the sensitivity of mutation in these RNAs. Some RNAs, like the homeobox (Hox) A9 5′UTR, are more resistant to mutations. The Hox mRNAs are involved in development, and the 5′UTR plays an important role in ribosome-mediated translational control. It is highly structured and folding to a specific conformation is essential to function (Alexander et al., 2009; Xue et al., 2015). Similarly, the phenylalanine-transfer RNA, 16S four-way junction and 5S ribosomal RNA are also relatively resistant to mutation. Other RNAs are more sensitive to mutations, like the synthetic Tebowned aptamer that was designed in the Eterna laboratory as part of their online game (Cordero and Das, 2015; Lee et al., 2014). RNAs folded in different solution conditions, such as aptamers in the absence or presence of their ligand, respond differently to mutation as well (Fig. 4B). For the adenine and glycine riboswitches, ligand binding increases the RNA’s sensitivity to mutations. The synthetic Tebowned aptamer has decreased sensitivity to mutations when in the presence of ligand. The chemical modifier used in chemical mapping experiments also affects the SHAPE data and ultimately sensitivity to structure change (Supplementary Material, Fig. S7). N-methlyiastoic anhydride (NMIA) is less reactive and requires a longer time to react than 1-methyl-7 nitroisatoic anhydride (1M7) (Mortimer and Weeks, 2007). Given the kinetics of the reaction, it is not surprising that 1M7 can detect more subtle differences in structure that could be occurring on a shorter time scale.
Fig. 4.
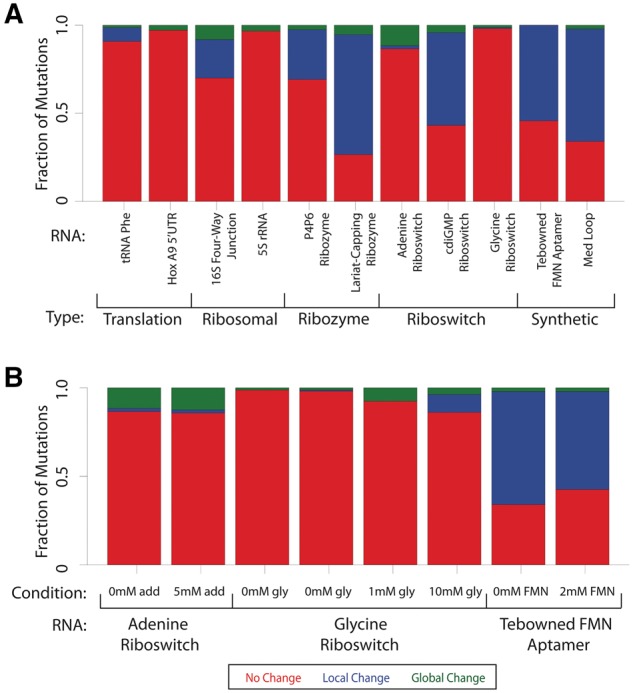
Fraction of disruption for individual RNAs. (A) The fraction of mutations that cause no change (red), local change (blue) or global change (green) for each RNA as classified by classSNitch. The RNAs are grouped by biological function: translation, ribosomal, ribozyme, riboswitch or synthetic. The experimental conditions for each of these RNAs are listed in Supplementary Material, Table S1. (B) The fraction of mutations that cause aptamers to change structure in the absence or presence of differing amounts of ligand for the adenine riboswitch, glycine riboswitch and Tebowned FMN aptamer (Color version of this figure is available at Bioinformatics online.)
Most structure prediction programs have low accuracy when identifying experimental riboSNitches with AUC values ranging from 0.6 to 0.7 (Corley et al., 2015; Ritz et al., 2012). In these benchmark studies, validation of the experimental data is analyzed using simple metrics like eSDC or the Euclidean distance (Corley et al., 2015; Ritz et al., 2012). One possible explanation for the poor predictive performance of the prediction algorithms in these benchmark studies is misclassification of the experimental data with these simple metrics. Indeed, when we observe the performance of SNPfold on data classified with either eSDC or Euclidean difference, the AUC values indicate the algorithm is barely predictive (Fig. 5A). We observe a subtle improvement in performance when we use the classSNitch classification of the experimental data. A similar performance increase is observed for the other published algorithms designed for riboSNitch prediction (Fig. 5B) (Halvorsen et al., 2010; Sabarinathan et al., 2013; Salari et al., 2013). Thus, misclassification of experimental data is likely a confounding factor for the poor performance of riboSNitch prediction algorithms, and the use of classSNitch in future benchmarking studies may improve prediction accuracy. Details on algorithm parameters can be found in Methods Supplementary, Section S3.4.
Fig. 5.
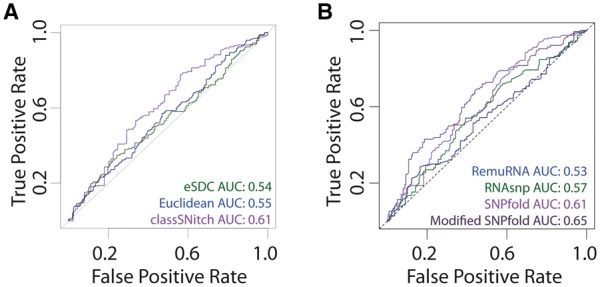
Improving the performance of structure change prediction algorithms (Halvorsen et al., 2010; Sabarinathan et al., 2013; Salari et al., 2013). (A) We performed ROC curve analysis for SNPfold, a structure change prediction algorithm, using classSNitch (purple), eSDC (green) and the Euclidean norm (blue) to classify the experimental data using the 10% tails strategy (Corley et al., 2015). (B) We compare the performance of structure change prediction algorithms on the classSNitch classification for SNPfold (purple), RNAsnp (green) and RemuRNA (blue). Each of these algorithms predicts structure change in RNA using only sequence information. SNPfold, remuRNA and RNAsnp all make ab initio predictions on whether a mutation alters the RNA structure; none of the algorithms benchmarked used SHAPE-directed structure prediction since we are using the WT and mutant SHAPE data for experimental validation. We improved the SNPfold prediction (dark purple) using Equation 1. The ROC curves for local and global change predictions are included in Supplementary Materials, Figure S8 (Color version of this figure is available at Bioinformatics online.)
The mutational strategy data is based primarily on four types of transversion mutations (Kladwang et al., 2011a,b,c) as seen in Supplementary Material, Table S4. The data presented in this table indicates mutating C or G in the WT sequence is more likely to induce structure change than mutating A or U with an odds ratio of 1.9, P < 0.001. We also observed that low SHAPE reactivity in the experimentally predicted WT structure is more likely to lead to structure change when mutated (OR = 1.4, P < 0.05).
3.5 WT SHAPE informed riboSNitch detection
It is well established that incorporating SHAPE into RNA structure folding algorithms improves secondary prediction performance (Diegan et al., 2009). Since we use SHAPE data to detect riboSNitches, it does not make sense to include experimental data for the WT and mutant in structure predictions. Nonetheless our analysis of sequence composition and WT SHAPE data for local and global changers does suggest an alternative. Can the WT SHAPE trace alone inform riboSNitch predictions? This is an attractive strategy since ultra high-throughput techniques exist to collect WT data on a genome-wide scale (Siegfried et al., 2014).
The major bottleneck in collecting systematic mutational information is the molecular biology required to synthesize and validate each mutant. When we modify the SNPfold algorithm scoring to include WT SHAPE data and to take into account the type of mutation (Equation 1), we are able to improve the performance of our algorithm further (Fig. 5B). Thus the WT SHAPE data is useful in increasing the accuracy of riboSNitch prediction.
4 Discussion
Identifying mutations that are likely to lead to changes in RNA structure remains a significant computational and experimental challenge (Chauhan and Woodson, 2008; Cheng et al., 2005; Churkin et al., 2011; Russell et al., 2002a,b). Such predictions are important in the context of personalized medicine since many riboSNitches are now known to be causative of human disease (Solem et al., 2015). Despite the advent of experimental technology enabling us to probe structure on a genome-wide scale, we still rely on structure change prediction algorithms or visual interpretations of the data to detect riboSNitches as there is no ultra-high throughput approach for rapidly mutating an RNA (Ritz et al., 2012; Rocca-Serra et al., 2011; Sansone et al., 2012; Siegfried et al., 2014).
We hypothesized that one reason for the poor performance of RNA structure prediction algorithms (Corley et al., 2015; Ritz et al., 2012) on riboSNitches is the misclassification of the experimental data. We therefore set out to develop novel metrics to evaluate structure change from SHAPE data. This approach did lead to modest improvements in performance suggesting that careful analysis of SHAPE data is essential when using these data as a benchmark. In this age of whole transcriptomic structure probing, manual validation and curation of these datasets is impractical. The classSNitch classifier simulates human consensus on what is and is not a structure change and therefore offers an alternative to simple metrics like eSDC in experimentally describing RNA structure change.
The features that classSNitch uses to classify change reveals some of the subtleties involved in interpreting SHAPE data. Beyond evaluating the magnitude difference between traces, human experts also utilize information on pattern matching and the distribution of change along the length of the RNA (Supplementary Material, Figs S3 and S4). We used those features to develop a classifier that successfully mimics expert classification of structure change (Fig. 3). SHAPE reactivity is correlated with secondary structure, more reactive nucleotides are generally single stranded (Eddy, 2014); however the experiment probes the overall structure of the RNA. The classSNitch classifier does not attempt to model structure, but instead establishes a standard for quantifying change. This is biologically relevant, allowing us to compare different RNAs using a standard vocabulary (Fig. 4). Although only two synthetic RNAs are included in our dataset, there is a striking difference in their sensitivity to mutation (Fig. 4A). Indeed a much larger fraction of the mutations in these RNAs result in conformational rearrangement. Although with only two RNAs it is impossible to draw statistical conclusions, this observation remains biologically interesting and warrants further investigation as more experimental data is obtained on a wide variety of RNAs (both synthetic and naturally occurring). The idea that RNA sequences under natural evolutionary pressure may evolve a general robustness to mutation warrants further investigation.
The data used for training classSNitch was exclusively collected using traditional capillary methods of electrophoresis. The quantification of this type of data from a capillary trace is a challenge, as it requires alignment to a reference ladder (Das et al., 2005; Karabiber et al., 2013; Mitra et al., 2008). Recent algorithmic developments have further automated this process and increased reliability (Yoon et al., 2011). It is interesting that dynamic time warping is the most significant feature used by classSNitch in reproducing expert classification. If alignment errors were to persist in the data, one might expect that experts could be correcting these when gazing at the data. As technology has evolved, in particular with the use of next generation sequencing to collect chemical and enzymatic probing data (Kertesz et al., 2010; Mortimer et al., 2012; Rouskin et al., 2014; Siegfried et al., 2014) alignment artifacts may disappear in the data. As such it may become necessary to retrain classSNitch on these newer types of data. In our lab’s limited experience with these types of data (currently unpublished), classSNitch performance is similar regardless of the type of data analyzed. However, it will be necessary to continue evaluating classSNitch performance as new experimental modalities are used. SHAPE data measures the selective reactivity of a probe for the 2′ OH of the RNA (Diegan et al., 2009). As such, the direct relationship between structure and reactivity is complex and ultimately depends on the 3-D structure of RNA. As a result, differences in SHAPE data due to mutation (or exogenous molecule binding) are notoriously difficult to interpret (Kutchko and Laederach, 2017). This does not however mean that SHAPE data does not contain useful information. Our use of the WT SHAPE data to improve riboSNitch predictions (Equation 1, Fig. 5B) indicates that much as including SHAPE as a free energy term in structure prediction (Diegan et al., 2009), aspects of the reactivity can inform predictions. It is likely that the improvement we observe when using Equation 1, which does not include any free energy terms, is due to the fact that in general, higher SHAPE reactivities are indicative of unpaired nucleotides (Eddy, 2014; Kutchko and Laederach, 2017). The by effectively adjusting the SNPfold score for nucleotides that are likely unpaired in the WT structure, which also are less likely to cause a riboSNitch, we observe a modest improvement in prediction performance. This effect remains modest since the correlation between SHAPE reactivity and base-pair probability is only moderate (Kutchko and Laederach, 2017).
Although classSNitch was trained on riboSNitches and is primarily intended as a tool to evaluate the effect of mutation induced structure change, it is in fact a more general metric for comparing SHAPE data. RNAs will adopt alternative conformations depending on their environment. For example riboswitches adopt different conformations depending on the presence of the ligand. When applied to the WT traces of apo and bound riboswitch data, the algorithm does identify local and global change for a majority of riboswitches, as expected. Protein binding, changes in cellular environment and even counter-ions are known to affect RNA structure (Bai et al., 2005; Frederiksen et al., 2012). The classSNitch classifier provides a common language to describe these differences. For example, it could be used when comparing in vivo and in vitro probing of the RNA to identify regions where the presence of proteins alters structure locally and globally. It also offers an attractive way to quantify these changes in agreement with expert consensus.
Manual classification of traces remains a laborious process, and is the main reason we developed the classSNitch classifier. We limited our training set to 200 traces and were able to recruit 17 experts to classify a majority of these traces. Certainly, a larger number of manual classifications will further improve the performance and precision of our classifier, especially for difficult cases. As such it is important when using the classSNitch classifier to be aware of the limited size of the training set and exercise care in evaluating the predictions on novel data. In particular, the performance of the classifier was with only 5 cross-validation folds in lieu of an independent test set, and as such is likely still somewhat partial. Nonetheless our data do suggest that it will be possible to arrive at a consensus for what a small and large RNA structure change look like and that the approach we present here is viable for developing a community standard.
The agreement between human experts ‘gazing’ at this data is reassuring. Prior to quantitative methods being widely available to life scientists, significant progress was achieved by carefully looking at the data; the structure of group I introns, tRNA and the ribosome were correctly predicted manually years before they were crystallized (Michel and Westhof, 1990). The value of automated systems that reproduce human appreciation of data is underutilized in RNA structural research despite the rich history of success in the field. Developing the classSNitch classifier minimally captures dying expert knowledge, while also making this expertise accessible to the community in an automated package.
Supplementary Material
Acknowledgements
We thank members of the Laederach and Weeks labs for their contribution to our survey questions and for looking at a heroic number of SHAPE traces. Evonne MacArthur, Katrina Kutchko, Lela Lackey, Meredith Corely, Matthew Smola, Sarah Marks, Aaztli Coria, Elizabeth Dethoff, Anthony Mustoe, Benjamin Keith, Amanda Solem and Ruslan Soldatov.
Funding
This work was supported by the U.S. National Institutes of Health [grant numbers HL111527, GM101237 and HG008133 to A.L.].
Conflict of Interest: none declared.
References
- Alexander T. et al. (2009) Hox genes and segmentation of the hindbrain and axial skeleton. Annu. Rev. Cell. Dev. Biol., 25, 431–456. [DOI] [PubMed] [Google Scholar]
- Bai Y. et al. (2005) Probing counterion modulated repulsion and attraction between nucleic acid duplexes in solution. Proc. Natl. Acad. Sci. U. S. A., 102, 1035–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. (2001) Random forests. Mach. Learn., 45, 5–32. [Google Scholar]
- Brenowitz M. et al. (1986a) ‘Footprint’ titrations yield valid thermodynamic isotherms. Proc. Natl. Acad. Sci. U. S. A., 83, 8462–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz M. et al. (1986b) Quantitative DNase footprint titration: a method for studying protein-DNA interactions. Methods Enzymol., 130, 132–181. [DOI] [PubMed] [Google Scholar]
- Butler E. et al. (2011) Structural basis of cooperative ligand binding by the glycine riboswitch. Chem. Biol., 18, 293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S., Woodson S.A. (2008) Tertiary interactions determine the accuracy of RNA folding. J. Am. Chem. Soc., 130, 1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C.Y. et al. (2015) Consistent global structures of complex RNA states through multidimensional chemical mapping. Elife, 4, e07600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z. et al. (2005) Crystal structure and functional analysis of DEAD-box protein Dhh1p. RNA, 11, 1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churkin A. et al. (2011) The RNAmute web server for the mutational analysis of RNA secondary structures. Nucleic Acids Res., 39, W92–W99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero P., Das R. (2015) Rich RNA structure landscapes revealed by mutate-and-map analysis. PLoS Comput. Biol., 11, e1004473.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero P. et al. (2012) An RNA Mapping DataBase for curating RNA structure mapping experiments. Bioinformatics, 28, 3006–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corley M. et al. (2015) Detecting riboSNitches with RNA folding algorithms: a genome-wide benchmark. Nucleic Acids Res., 43, 1859–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz J.A. et al. (2012) RNA-Puzzles: a CASP-like evaluation of RNA three-dimensional structure prediction. RNA, 18, 610–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R. et al. (2005) SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA, 11, 344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R. et al. (2008) Structural inference of native and partially folded RNA by high-throughput contact mapping. Proc. Natl. Acad. Sci. U. S. A., 105, 4144–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deras M.L. et al. (2000) Folding mechanism of the Tetrahymena ribozyme P4-P6 domain. Biochemistry, 39, 10975–10985. [DOI] [PubMed] [Google Scholar]
- Diegan K.E. et al. (2009) Acccurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. U. S. A., 106, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S. (2014) Computational analysis of conserved RNA secondary structure in transcriptomes and genomes. Annu. Rev. Biophys., 43, 433–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen J. et al. (2012) Metal-ion rescue revisited: biochemical detection of site-bound metal ions important for RNA folding. RNA, 18, 1123–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgino T. (2009) Computing and visualizing dynamic time warping alignmnets in R: The dtw Package. J. Stat. Softw., 31, 1–24. [Google Scholar]
- Hall M. et al. (2009) The WEKA data mining software: an update. SIGKDD Explor., 11, [Google Scholar]
- Halvorsen M. et al. (2010) Disease-associated mutations that alter the RNA structural ensemble. PLoS Genet., 6, e1001074.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karabiber F. et al. (2013) QuShape: rapid, accurate, and best-practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. RNA, 19, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M. et al. (2010) Genome-wide measurement of RNA secondary structure in yeast. Nature, 467, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W. et al. (2011a) A mutate-and-map strategy accurately infers the base pairs of a 35-nucleotide model RNA. RNA, 17, 522–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W. et al. (2011b) A two-dimensional mutate-and-map strategy for non-coding RNA structure. Nat. Chem., 3, 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W. et al. (2011c) Understanding the errors of SHAPE-directed RNA structure modeling. Biochemistry, 50, 8049–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M. (2008) Building predictive models in R using the caret package. J. Stat. Softw., 28, [Google Scholar]
- Kutchko K.M., Laederach A. (2017) Transcending the prediction paradigm: novel applications of SHAPE to RNA function and evolution. Wiley Interdiscip Rev RNA., 8, e1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutchko K.M. et al. (2015) Multiple conformations are a conserved and regulatory feature of the RB1 5′ UTR. RNA, 21, 1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. et al. (2014) RNA design rules from a massive open laboratory. Proc. Natl. Acad. Sci. U. S. A., 111, 2122–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A., Wiener M. (2002) Classification and regression by randomForest. R. News, 2, 18–22. [Google Scholar]
- Lokody I. (2014) RNA: riboSNitches reveal heredity in RNA secondary structure. Nat. Rev. Genet., 15, 219.. [DOI] [PubMed] [Google Scholar]
- Martin J.S. et al. (2012) Structural effects of linkage disequilibrium on the transcriptome. RNA, 18, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Z. et al. (2015) RNA-Puzzles Round II: assessment of RNA structure prediction programs applied to three large RNA structures. RNA, 21, 1066–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F., Westhof E. (1990) Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J. Mol. Biol., 216, 585–610. [DOI] [PubMed] [Google Scholar]
- Mitra S. et al. (2008) High-throughput single-nucleotide structural mapping by capillary automated footprinting analysis. Nucleic Acids Res., 36, e63.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer S. et al. (2012) SHAPE-Seq: high-throughput RNA structure analysis. Curr. Protoc. Chem. Biol., 4, 275–297. [DOI] [PubMed] [Google Scholar]
- Mortimer S.A., Weeks K.M. (2007) A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J. Am. Chem. Soc., 129, 4144–4145. [DOI] [PubMed] [Google Scholar]
- Petri V., Brenowitz M. (1997) Quantitative nucleic acids footprinting: thermodynamic and kinetic approaches. Curr. Opin. Biotechnol., 8, 36–44. [DOI] [PubMed] [Google Scholar]
- Rice G.M. et al. (2014) RNA secondary structure modeling at consistent high accuracy using differential SHAPE. RNA, 20, 846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz J. et al. (2012) Evaluating our ability to predict the structural disruption of RNA by SNPs. BMC Genomics, 13, S6.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca-Serra P. et al. (2011) Sharing and archiving nucleic acid structure mapping data. RNA, 17, 1204–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouskin S. et al. (2014) Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature, 505, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowles T.A. (2013) Power to the people: does Eterna signal the arrival of a new wave of crowd-sourced projects?. BMC Biochem., 14, 26.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R. et al. (2002a) Rapid compaction during RNA folding. Proc. Natl. Acad. Sci. U. S. A., 99, 4266–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R. et al. (2002b) Exploring the folding landscape of a structured RNA. Proc. Natl. Acad. Sci. U. S. A., 99, 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabarinathan R. et al. (2013) RNAsnp: efficient detection of local RNA secondary structure changes induced by SNPs. Hum. Mutat. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeys Y. et al. A review of feature selection techniques in bioinformatics. Bioinformatics, 23, 2507–2517. [DOI] [PubMed] [Google Scholar]
- Saeys Y. et al. (2007) A review of feature selection techniques in bioinformatics. Bioinformatics, 2007, 19. [DOI] [PubMed] [Google Scholar]
- Sakoe H., Chibe S. (1978) Dynamic programming algorithm optimization for spoken word recognition. IEEE Trans. Acoust. Speech Signal Process, 26, 43–49. [Google Scholar]
- Salari R. et al. (2013) Sensitive measurement of single-nucleotide polymorphism-induced changes of RNA conformation: application to disease studies. Nucleic Acids Res., 41, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone S.A. et al. (2012) Toward interoperable bioscience data. Nat. Genet., 44, 121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclavi B. et al. (1997) Time-resolved synchrotron X-ray ‘footprinting’, a new approach to the study of nucleic acid structure and function: application to protein-DNA interactions and RNA folding. J. Mol. Biol., 266, 144–159. [DOI] [PubMed] [Google Scholar]
- Siegfried N.A. et al. (2014) RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods, 11, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solem A.C. et al. (2015) The potential of the riboSNitch in personalized medicine. Wiley Interdiscip. Rev. RNA, 6, 517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamoto K. et al. (2004) Principles of RNA compaction: insights from the equilibrium folding pathway of the P4-P6 RNA domain in monovalent cations. J. Mol. Biol., 343, 1195–1206. [DOI] [PubMed] [Google Scholar]
- Tian S. et al. (2014) High-throughput mutate-and-map rescute elevates SHAPE-directed RNA structure and uncovers excited states. RNA, 20, 1815–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuille A., Das R. (2014) Scientific rigor through videogames. Trends Biochem. Sci., 39, 507–509. [DOI] [PubMed] [Google Scholar]
- Waldispuhl J., Reinharz V. (2015) Modeling and predicting RNA three-dimensional structures. Methods Mol. Biol., 1269, 101–121. [DOI] [PubMed] [Google Scholar]
- Wan Y. et al. (2012) Genome-wide measurement of RNA folding energies. Mol. Cell, 48, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y. et al. (2014) Landscape and variation of RNA secondary structure across the human transcriptome. Nature, 505, 706–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S. et al. (2015) RNA regulons in Hox 5′UTRs confer ribosome specificity to gene regulation. Nature, 517, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S. et al. (2011) HiTRACE: high-throughput robust analysis for capillary electrophoresis. Bioinformatics, 27, 1798–1805. [DOI] [PubMed] [Google Scholar]
- Zhang W. et al. (2009) Structures of the ribosome in intermediate states of ratcheting. Science, 325, 1014–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.