

Abstract
Heparanase, the sole heparan sulfate degrading endoglycosidase, regulates multiple biological activities that enhance tumor growth, angiogenesis and metastasis. Heparanase expression is enhanced in almost all cancers examined including various carcinomas, sarcomas and hematological malignancies. Numerous clinical association studies have consistently demonstrated that upregulation of heparanase expression correlates with increased tumor size, tumor angiogenesis, enhanced metastasis and poor prognosis. In contrast, knockdown of heparanase or treatments of tumor-bearing mice with heparanase-inhibiting compounds, markedly attenuate tumor progression further underscoring the potential of anti-heparanase therapy for multiple types of cancer. Heparanase neutralizing monoclonal antibodies block myeloma and lymphoma tumor growth and dissemination; this is attributable to a combined effect on the tumor cells and/or cells of the tumor microenvironment. In fact, much of the impact of heparanase on tumor progression is related to its function in mediating tumor-host crosstalk, priming the tumor microenvironment to better support tumor growth, metastasis and chemoresistance. The repertoire of the physiopathological activities of heparanase is expanding. Specifically, heparanase regulates gene expression, activates cells of the innate immune system, promotes the formation of exosomes and autophagosomes, and stimulates signal transduction pathways via enzymatic and non-enzymatic activities. These effects dynamically impact multiple regulatory pathways that together drive inflammatory responses, tumor survival, growth, dissemination and drug resistance; but in the same time, may fulfill some normal functions associated, for example, with vesicular traffic, lysosomal-based secretion, stress response, and heparan sulfate turnover. Heparanase is upregulated in response to chemotherapy in cancer patients and the surviving cells acquire chemoresistance, attributed, at least in part, to autophagy. Consequently, heparanase inhibitors used in tandem with chemotherapeutic drugs overcome initial chemoresistance, providing a strong rationale for applying anti-heparanase therapy in combination with conventional anti-cancer drugs. Heparin-like compounds that inhibit heparanase activity are being evaluated in clinical trials for various types of cancer. Heparanase neutralizing monoclonal antibodies are being evaluated in pre-clinical studies, and heparanase-inhibiting small molecules are being developed based on the recently resolved crystal structure of the heparanase protein. Collectively, the emerging premise is that heparanase expressed by tumor cells, innate immune cells, activated endothelial cells as well as other cells of the tumor microenvironment is a master regulator of the aggressive phenotype of cancer, an important contributor to the poor outcome of cancer patients and a prime target for therapy.
Keywords: Heparanase, tumorigenesis, inflammation, exosomes, autophagy, heparanase-inhibiting compounds, chemoresistance, clinical trials
1. Introduction/Preface
Heparanase is an endoglucuronidase that cleaves heparan sulfate (HS) thereby altering the structure and function of heparan sulfate proteoglycans (HSPG) and contributing to tumor-mediated remodeling of both cell surfaces and the extracellular matrix (Bernfield et al., 1999; Hammond et al., 2014; Ilan et al., 2006; Iozzo and Sanderson, 2011; Rivara et al., 2016). These actions dynamically impact multiple regulatory pathways, most notably by accelerating cell invasion and augmenting the bioavailability of growth factors and cytokines bound to HS (Bernfield et al., 1999; Iozzo and Sanderson, 2011). Immunohistochemistry, in situ hybridization, PCR and Western blot analyses demonstrate that heparanase expression is enhanced in almost all cancers examined including, for example, ovarian, pancreatic, gastric, renal, head & neck, colon, bladder, brain, prostate, breast and liver carcinomas (Hammond et al., 2014; Ilan et al., 2006; Rivara et al., 2016; Vlodavsky et al., 2012; Vreys and David, 2007; Zhang et al., 2011) as well as Ewing’s sarcoma (Cassinelli et al., 2016; Cassinelli et al., 2013; Shafat et al., 2011), multiple myeloma (Kelly et al., 2003; Ramani et al., 2013; Ritchie et al., 2011) and B-lymphomas (Weissmann et al., 2016). Numerous clinical association studies have consistently demonstrated that upregulation of heparanase expression correlates with increased tumor size, tumor angiogenesis, enhanced metastasis and poor prognosis (Hammond et al., 2014; Ilan et al., 2006; Rivara et al., 2016; Vlodavsky et al., 2012; Vreys and David, 2007).
A causal role of heparanase in tumor metastasis was demonstrated by the increased lung, liver and bone colonization of cancer cells following overexpression of the heparanase gene, and by a marked decrease in the metastatic potential of cells subjected to heparanase gene silencing. (Edovitsky et al., 2004; Lerner et al., 2008). The pro-tumorigenic effect of heparanase is attributed primarily to its HS degrading activity, facilitating cell invasion and ‘priming’ the tumor microenvironment. This notion is reinforced by in vivo studies indicating a marked inhibition of tumor growth in mice treated with heparanase-inhibiting heparin-like compounds (i.e., PI-88 = Mupafostat, Roneparstat = SST0001, Necuparanib = M402, PG545) which are currently being tested in clinical trials in cancer patients (Pala et al., 2016; Pisano et al., 2014; Rivara et al., 2016). A marked inhibition of tumor growth and dissemination was also exerted by heparanase neutralizing monoclonal antibodies in xenograft models of lymphoma and myeloma, emphasizing the significance of the enzymatic activity of heparanase in promoting tumorigenesis (Weissmann et al., 2016). In addition, both enzymatically active and inactive heparanase promotes signal transduction, including Akt, STAT, Src, Erk, HGF-, IGF- and EGF-receptor signaling (Barash et al., 2010; Ilan et al., 2006; Sanderson and Iozzo, 2012), highlighting the notion that non-enzymatic activities of heparanase play a significant role in heparanase-driven tumor progression (Fux et al., 2009a; Fux et al., 2009b). Moreover, heparanase induces the transcription of pro-angiogenic (i.e., VEGF-A, VEGF-C, COX-2, MMP-9), pro-thrombotic (i.e., tissue factor), pro-inflammatory (i.e., TNFα, IL-1, IL-6), pro-fibrotic (i.e., TGFβ), mitogenic (i.e., HGF), osteolyic (RANKL) and various other genes (Cohen-Kaplan et al., 2008b; Goodall et al., 2014; Ilan et al., 2006; Nadir et al., 2006; Parish et al., 2013; Purushothaman et al., 2008), thus significantly expanding its functional repertoire and mode of action in promoting aggressive tumor behavior (Barash et al., 2010; Ilan et al., 2006; Sanderson and Iozzo, 2012). Recent studies (detailed later in this review) emphasize the involvement of heparanase in autophagy (Shteingauz et al., 2015), exosome formation (Thompson et al., 2013), activation of the immune system (Goodall et al., 2014; Hermano et al., 2014) and chemoresistance (Ramani et al., 2015; Shteingauz et al., 2015), further highlighting its significance in mediating the crosstalk between tumor cells and the tumor microenvironment and in dictating the tumor response to stress, anti-cancer drugs and host factors. Thus, the published data supporting a role for heparanase in tumor progression is strong and well supported by in vitro and pre-clinical studies as well as by extensive research focusing on human cancer patients.
Several up-to-date reviews successfully describe basic and translational aspects related to the involvement of heparanase in cancer and inflammation (Hermano et al., 2012; Li and Vlodavsky, 2009; McKenzie, 2007; Vreys and David, 2007). The present review focuses primarily on recent new developments in the ‘heparanase field’, including its function in the tumor microenvironment, hematological malignancies, exosome formation, autophagy, chemoresistance, polarization of macrophages and toll-like receptor activation. Recently resolved structural features and translational aspects are also addressed.
1.1. Heparan sulfate proteoglycans
HSPG exert their multiple functional impacts via several distinct mechanisms that combine structural, biochemical and regulatory aspects. By interacting with other macromolecules such as laminin, fibronectin, and collagens I and IV, HSPG contribute to the structural integrity, self-assembly and insolubility of the extracellular matrix (ECM) and basement membrane, thus intimately modulating cell-ECM interactions (Bernfield et al., 1999; Timpl and Brown, 1996; Udo Hacker, 2005). HSPG also directly transfer information from the extracellular space to intracellular kinases and cytoskeletal elements and thus affect cell signaling, adhesion and motility (Aalkjaer and Boedtkjer, 2009). The biosynthesis of HS takes place in the Golgi apparatus and has been studied in great detail. Briefly, the polysaccharide chains are modified at various positions by sulfation, epimerization and N-acetylation, yielding clusters of sulfated disaccharides separated by low or non-sulfated regions (Iozzo and Sanderson, 2011; Lindahl and Li, 2009). The compositions of HS chains are adapted to their function and can differ between cells and tissues even when the core HSPG protein is the same (Wu et al., 2015). The modular nature and heterogeneity of HS structure provide interaction sites for a large number of different binding partners and is central to the proper biological function of HS (Bernfield et al., 1999; Lindahl and Li, 2009) in the control of normal and pathological processes, among which are morphogenesis, tissue repair, cancer metastasis, inflammation, vascularization, atherosclerosis, thrombosis, diabetic nephropathy and diabetes (Iozzo and Sanderson, 2011; Lindahl and Li, 2009). HSPGs not only provide a storage depot for heparin-binding molecules, but also decisively regulate their accessibility, function and mode of action. Cleavage of HSPGs would ultimately release these proteins and convert them into bioactive mediators, ensuring rapid tissue response to local or systemic cues. This function of HS provides the cell with a rapidly accessible reservoir, precluding the need for de novo synthesis when the requirement for a particular protein is increased (Vlodavsky et al., 1991; Vlodavsky et al., 2011).
1.2. Heparanase
Unlike the well resolved biosynthetic pathway of HS, the mode of HS breakdown is less characterized. While synthesis and modification of HS chains require the activity of an array of enzymes, degradation of mammalian HS is primarily carried out by one enzyme, heparanase (HPSE), which cleaves the HS side chains of HSPGs into fragments of 10–20 sugar units (Vlodavsky et al., 1999). Enzymatic activity capable of cleaving glucuronidic linkages and converting macromolecular heparin to physiologically active fragments was first identified by Ogren and Lindahl (Ogren and Lindahl, 1975). Subsequent studies revealed that the same endo-β-D-glucuronidase (heparanase) is critically involved in various pathologies such as cancer (Arvatz et al., 2011b; Ilan et al., 2006; Parish et al., 2001; Vlodavsky et al., 2011; Vlodavsky and Friedmann, 2001), inflammation (Lerner et al., 2011; Li and Vlodavsky, 2009), thrombosis (Baker et al., 2012; Nadir et al., 2010), atherosclerosis (Blich et al., 2012; Osterholm et al., 2012; Planer et al., 2011) and kidney dysfunction (Gil, 2011; van den Hoven et al., 2006). As a direct result of these studies, heparanase was advanced from being an obscure enzyme with a poorly understood function to a highly promising drug target, offering new treatment strategies for various cancers and other diseases.
Heparanase is initially translated as a preproenzyme containing a signal sequence spanning Met1–Ala35. Cleavage of this signal sequence by signal peptidase yields an inactive 65-kDa pro-heparanase, which must undergo further processing for activity. Proteolytic removal by cathepsin L (Abboud-Jarrous et al., 2008) of a linker spanning Ser110–Gln157 liberates an N-terminal 8-kDa subunit and a C-terminal 50-kDa subunit, which remain associated as a noncovalent heterodimer in mature active heparanase (Levy-Adam et al., 2003). Heparanase present in late endosomes and lysosomes plays an essential housekeeping role in catabolic processing of internalized HSPGs (Escobar Galvis et al., 2007a), and, as discussed below, in autophagy (Shteingauz et al., 2015) and exosome formation (Thompson et al., 2013). Importantly, heparanase can be trafficked to the cell surface or released into the ECM, where it affects breakdown of extracellular pools of HS (Nadav et al., 2002). Heparanase-mediated breakdown of HS in the ECM has several effects on the behavior of nearby cells. Weakening of structural HS networks in the ECM directly facilitates cell motility and invasion into surrounding tissues (Sasaki et al., 2004). Latent pools of growth factors stored by HS are released upon breakdown by heparanase and subsequently promote increased cell proliferation, motility and angiogenesis (Elkin et al., 2001; Vlodavsky et al., 1991). HS fragments generated by heparanase activity can also activate downstream signaling cascades (Goodall et al., 2014). Whereas controlled heparanase activity plays an important part in physiological processing of the ECM, tissue repair (Zcharia et al., 2005b), hair follicle growth (Zcharia et al., 2005a) and immune surveillance, aberrant heparanase expression is associated with inflammation and cancer, strongly correlating with metastasis and worsened clinical prognoses (Hammond et al., 2014; Ilan et al., 2006; Rivara et al., 2016; Vlodavsky et al., 2012). Only one gene with heparanase catalytic activity has been identified in mammals, suggesting that loss of heparanase activity may not easily be compensated for by the cell. Accordingly, as discussed below, heparanase inhibition has attracted intense interest as an anticancer strategy and oligosaccharide-like HS mimetics are being tested in clinical trials for various types of cancer (Hammond et al., 2014; Pisano et al., 2014; Rivara et al., 2016; Vlodavsky et al., 2012).
Heparanase is a member of the glycoside hydrolase 79 (GH79) family of carbohydrate-processing enzymes (Hulett et al., 2000; Wu et al., 2015). It catalyzes hydrolysis of internal GlcUA (β1→4) GlcNS linkages in HS, with net retention of anomeric configuration (Wilson et al., 2014). Heparanase-mediated breakdown of HS is not indiscriminate but instead is restricted to a small subset of GlcUAs, thus reflecting a requirement for specific N- and O-sulfation patterns on neighboring sugars (Okada et al., 2002; Peterson and Liu, 2012; Pikas et al., 1998). The GH79 family of carbohydrate-processing enzymes belongs to the larger GH-A clan, which is characterized by a (β/α)8 domain containing the catalytic site (Cantarel et al., 2009). The crystal structure of human heparanase has been recently resolved, revealing how an endo-acting binding cleft is exposed by proteolytic activation of latent pro-heparanase (Wu et al., 2015). A cleft spanning ~10 Å in the (β/α)8 domain of the apo-enzyme was identified, suggesting that the HS-binding site is contained within this part of the enzyme (Wu et al., 2015). The cleft contained residues Glu343 and Glu225, which have been identified as the catalytic nucleophile and acid-base of heparanase (Hulett et al., 2000) required for retaining the catalytic mechanism (Davies and Henrissat, 1995). In accordance with the negatively charged nature of its HS substrate, the heparanase binding cleft is lined by basic side chains contributed by Arg35, Lys158, Lys159, Lys161, Lys231, Arg272, Arg273 and Arg303 (Wu et al., 2015). The positioning of the 8-kDa-subunit C terminus and 50-kDa-subunit N terminus clearly indicates that the excised Ser110–Gln157 linker of pro-heparanase should lie very near or even within the active site cleft of the (β/α)8 domain (Abboud-Jarrous et al., 2008; Abboud-Jarrous et al., 2005; Wu et al., 2015). This positioning would hinder incoming HS substrates and is consistent with a previously proposed steric-block mechanism for pro-heparanase inactivation by its own linker (Abboud-Jarrous et al., 2008; Abboud-Jarrous et al., 2005; Nardella et al., 2004). A single-chain constitutively active heparanase was obtained by connecting the 8 and 50 kDa subunits with a spacer of three glycine-serine pairs. This preparation (HepGS3) is comparable to the processed, heterodimeric enzyme with regard to specific activity, profile of hydrolysis products, and inhibition by heparin (Nardella et al., 2004). Our own crystal structure of the HepGS3 protein (Livnah, Golan, et al., unpublished results) revealed structural features (i.e., TIM-barrel domain that contains the two catalytic glutamic acids and two heparin-binding domains, attached to a β-sandwich C-terminal domain) (Fig. 1) identical to the recently resolved crystal structure of the native enzyme (Wu et al., 2015). The newly resolved 3D structure of heparanase and its interaction with HS substrates provides a long-anticipated structural rationale that ties together the results of numerous biochemical studies on the HS sulfation patterns required for heparanase cleavage (Peterson and Liu, 2010; Pikas et al., 1998). The crystal structure confirmed that sulfation is key for heparanase interaction with HS and that the recognized cleavage site is a trisaccharide accommodated in the heparanase binding cleft. Structurally, −2 N-sulfate and +1 6O-sulfate appear to be the main determinants for recognition, because these directly contact the enzyme through hydrogen-bonding networks (Wu et al., 2015).
Figure 1.
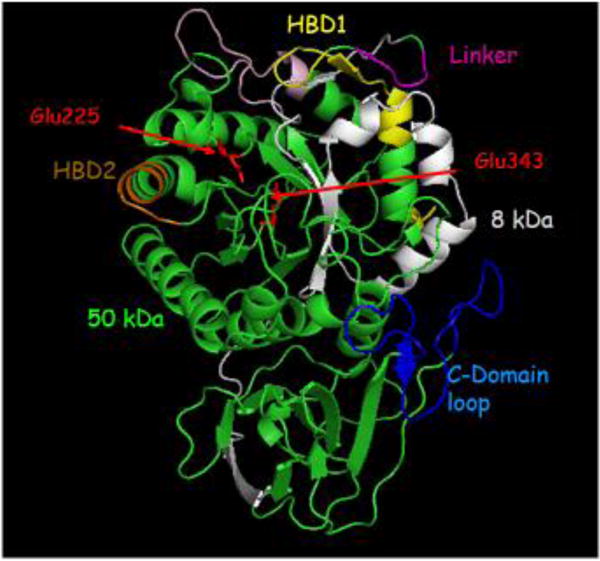
Crystal structure of heparanase. Heparanase is composed of a TIM-barrel fold which contains the active site of the enzyme and a C-terminus domain required for secretion and signaling function of the protein. Heparanase has a catalytic mechanism which includes a putative proton donor at Glu225 and a nucleophile at Glu343 (red) as well as the heparin/heparan sulfate binding domains (HBD1 = Lys158-Asp171 & HBD2 = Gln270-Lys280; yellow and brown, respectively) that are located in close proximity to the active site micro-pocket fold. The C-domain is comprised of 8 β-strands arranged in two sheets, as well as a flexible, unstructured loop that lies in-between (blue). Notably, the 8 kDa subunit (gray) enfolds the 50 kDa subunit (green), contributing β/α/β unit to the TIM-barrel fold and, moreover, one of the 8 β strands that comprise the C-domain.
2. Heparanase and the tumor microenvironment
2.1. Impact of host heparanase on tissue susceptibility to carcinogenesis and tumor initiation
Compelling evidence tie heparanase with human cancer, but the timing of its induction and the significance of heparanase in the early phases of tumor initiation and development were not elucidated. To address this issue, we have utilized non-transformed human mammary epithelial cell line (MCF10A). Ectopic overexpression of heparanase resulted in expanded and disorganized acinar structures coincident with increased cell proliferation (Boyango et al., 2014). These features were far more dramatic when heparanase was overexpressed in MCF10AT1 cells that acquired the ability for xenograft growth after transfection with aberrant T24 Ha-Ras. Importantly, unlike the parental cells, xenograft lesions formed by heparanase overexpressing MCF10AT1 cells, were diagnosed as invasive carcinoma, indicating that heparanase cooperates with Ras to promote mammary tumor progression (Boyango et al., 2014). In subsequent studies, we exposed heparanase overexpressing transgenic (Hpa-Tg) mice and wt mice to two-steps DMBA/TPA skin carcinogenesis because more than 90% of skin cancer initiated by DMBA carries Ha-Ras activating mutations (Schwarz et al., 2013). In this 2-stage carcinogenesis model, DMBA induces mutations in dermal epithelium at an early stage, while TPA elicits an inflammatory response that drives further transformation, resulting in papilloma development. We found the Hpa-Tg model significantly more sensitive to carcinogen induced tumor formation. Indeed, the Hpa-Tg mice experienced 10-fold increase in tumor burden and rampant macrophage infiltration consistent with aggressive and invasive carcinomas. Strikingly, genetic ablation of heparanase resulted in a dramatic attenuation of tumorigenesis upon carcinogen exposure. Pharmacological inhibition of heparanase with the heparanase-inhibiting compound PG545 (Dredge et al., 2011) potently inhibited tumor establishment and the invasion of macrophages into the tumor per se, clearly depicting a critical role of heparanase in skin tumorigenesis (Boyango et al., 2014). Mechanistically, heparanase and Ras cooperatively promoted Met phosphorylation and the activation of various downstream oncogenic relays, such as Erk, Src, and Akt. Concurrent with increased Akt activation, FOXO1 was sequestered within the cytosol upon heparanase expression. However, inhibition with PG545 attenuated signaling and permitted nuclear occupancy of FOXO1 concomitant with a substantial decrease in phosphorylated Akt. These results uncover a critical role of heparanase and Ras in promoting early phases of tumorigenesis, indicating that pharmacological intervention targeting the enzymatic and signaling activities of heparanase may provide a valid avenue for combatting initial tumor development (Boyango et al., 2014).
2.2. Heparanase in hematological malignancies
The vast majority of studies on the mode of heparanase action and its impact on cancer progression focused on solid tumors (Arvatz et al., 2011b; Hammond et al., 2014; Ilan et al., 2006; Rivara et al., 2016). With the exception of multiple myeloma that exemplify a tumor that closely depends on the tumor microenvironment (Sanderson and Yang, 2008; Ramani et al., 2013), little is known about the involvement of heparanase in hematological malignancies. Recent studies on myeloma (Ramani et al., 2016; Ramani et al., 2015) and lymphomas (Weissmann et al., 2016) contribute to our basic understanding of the multifaceted nature of heparanase functions in cancer and in particular its pronounced impact on the cross talk between the tumor and the tumor microenvironment.
Multiple myeloma (MM) is the second most prevalent hematologic malignancy in the United States (Vincent Rajkumar, 2014). The emergence of novel, targeted therapeutics (e.g., bortezomib, thalidomide) has greatly improved survival rates in patients with myeloma (Laubach et al., 2014); however, there is still an unmet need in identifying and developing therapies designed to further prevent the progression of this disease without sacrificing patient quality of life. Recognition that the myeloma tumor microenvironment helps drive the aggressive nature of myeloma has led to new strategies for attacking the tumor microenvironment (Meads et al., 2008). Heparanase has been shown to promote an aggressive malignant phenotype, in part by synergizing with syndecan-1 (CD138) to create a niche within the bone marrow microenvironment further driving myeloma growth and dissemination (Iozzo and Sanderson, 2011; Ramani et al., 2013; Sanderson and Iozzo, 2012). Moreover, active heparanase can be detected in the bone marrow of most myeloma patients and the presence of high levels of this enzyme correlates with enhanced angiogenic activity, an important promoter of myeloma growth and progression (Kelly et al., 2003). Many of the pro-tumorigenic effects of heparanase in myeloma have been shown to rely upon increased expression and shedding of syndecan-1 (Ramani et al., 2013). This proteoglycan is expressed on most myeloma tumor cells and is a critical determinant of myeloma cell survival and growth (Beauvais et al., 2016). Heparanase enhances syndecan-1 shedding by up-regulating the expression of matrix metalloproteinase 9 (MMP9) and urokinase plasminogen activator (uPA) that are recognized as syndecan sheddases (Purushothaman et al., 2008; Ramani et al., 2016). Heparanase-mediated shortening of HS chains on syndecan-1 also contributes to shedding, presumably by providing increased access of sheddases to the syndecan-1 core protein (Ramani et al., 2012). Importantly, heparanase-induced shedding of syndecan-1 was recently shown to couple VEGF receptor-2 with α4β1 integrin on both myeloma and endothelial cells. This receptor coupling led to activation of VEGF receptor-2 signaling that promoted myeloma cell invasion and endothelial tube formation (Jung et al., 2016). In addition, heparanase enhances the expression of HGF that can associate with shed syndecan-1 and facilitate paracrine signaling via its high affinity receptor, c-Met (Ramani et al., 2011). This discovery reveals for the first time how via a single mechanism, heparanase can stimulate both tumor metastasis and angiogenesis. It was further demonstrated that heparanase stimulates expression of VEGF, MMP-9 and RANKL, it promotes ERK signaling and represses expression of CXCL10 (Barash et al., 2014; Yang et al., 2010). Through these actions, heparanase dynamically impacts multiple regulatory pathways within the tumor microenvironment that together drive myeloma survival, growth, dissemination and osteolysis. Moreover, heparanase plays a role in modulating the sensitivity of myeloma cells to therapy. Increased expression and secretion of heparanase was detected in myeloma cells following treatment with bortezomib or melphalan and high expression of heparanase by these cells rendered them less susceptible to the cytotoxic effects of bortezomib or melphalan (Ramani et al., 2015). These data are clinically relevant as they are consistent with our finding that heparanase gene expression increases dramatically in myeloma patients following chemotherapy (Ramani et al., 2015). Moreover, heparanase expression within the bone marrow microenvironment is associated with shorter event-free survival of patients with newly diagnosed multiple myeloma treated with high dose chemotherapy and stem cell transplantation (Mahtouk et al., 2007). Together, these findings imply that heparanase is a desirable and druggable therapeutic target whose inhibition will disrupt the myeloma microenvironment and diminish tumor growth and chemoresistance, while causing minimal side effects in patients. These results prompted a First in Man, multicenter phase I clinical study, currently ongoing in advanced heavily pre-treated refractory myeloma patients who have exhausted currently available lines of therapy (Rivara et al., 2016). The drug of choice is Roneparstat (SST0001), a potent inhibitor of heparanase enzyme activity (IC50~3 nM). It consists of a chemically modified heparin (ROH = reduced oxidized N-acetylated heparin) that is non-anticoagulant and is not degraded by the enzyme (Naggi et al., 2005). We have extensively studied Roneparstat in several models where human myeloma tumor cells were injected either subcutaneously, into fragments of human fetal bone implanted in mice (SCID-hu), or intravenously into the mouse tail vein. Roneparstat significantly inhibited growth, angiogenesis and bone metastasis of myeloma tumors in these models (Ritchie et al., 2011). Moreover in a model of dexamethasone resistant MM, the combination of Roneparstat with dexamethasone inhibited tumor growth. Consistent with the compound’s ability to inhibit heparanase activity, analysis of subcutaneous tumors from animals treated with Roneparstat revealed that the level of VEGF, HGF and MMP-9 were dramatically reduced and angiogenesis was repressed compared to animals treated with vehicle (Ritchie et al., 2011). When human myeloma cells expressing a high level of heparanase were injected intravenously, they home to and grew almost exclusively, in bone. In this model, Roneparstat monotherapy did not significantly inhibit tumor growth; however, it was highly effective when used in combination with bortezomib or melphalan (Ramani et al., 2015). The ability of Roneparstat to dramatically reduce tumor growth in bone when used in combination with either bortezomib or melphalan indicates that blocking heparanase diminishes drug resistance in myeloma (Ramani et al., 2015).
2.3. Heparanase enhances myeloma progression via CXCL10 down regulation
Applying an inducible model system we have demonstrated that heparanase induction markedly promotes tumor development by CAG myeloma, in agreement with the decisive role of heparanase in myeloma progression (Ramani et al., 2013; Ritchie et al., 2011). Utilizing gene array methodology we found that heparanase induction associates with CXCL10 down-regulation (Barash et al., 2014). Moreover, heparanase gene silencing resulted in elevation of CXCL10 transcription, collectively implying that heparanase levels inversely associate with CXCL10 expression. CXCL10 is therefore a member of a growing list of genes (i.e., VEGF, VEGF-C, TF, RANKL, HGF, and MMP9) regulated by heparanase and associated with tumorigenesis. A novel set of genes under heparanase regulation has been characterized in T cells (He et al., 2012). In this context, nuclear heparanase was shown to regulate the transcription of a cohort of inducible immune response genes by controlling histone H3 methylation (He et al., 2012), further expanding the transcriptional potential of heparanase (Parish et al., 2013).
Importantly, CXCL10 gene silencing resulted in tumor xenografts that were bigger than control myeloma tumors, while overexpression of CXCL10 or its injection into tumor bearing mice resulted in a marked decrease in tumor development, clearly depicting anti-cancer properties of CXCL10 in myeloma. Taken together, we identified a novel molecular mechanism that underlines the pro-tumorigenic function of heparanase in myeloma and involves down regulation of CXCL10. This chemokine appears to operate in three independent manners to suppress myeloma progression, directly attenuating myeloma cell proliferation; attenuating endothelial cell proliferation and tumor angiogenesis; and chemoattracting anti-tumor immune cells. Hence, CXCL10 can be regarded as a tumor suppressor in this malignancy. Our results indicate that CXCL10 and especially the CXCL10-Ig fusion protein, may be applied as a novel therapeutic modality in myeloma (Barash et al., 2014).
2.4. Heparanase-neutralizing antibodies attenuate lymphoma and myeloma tumor growth and metastasis by targeting heparanase in the tumor microenvironment
Lymphomas are a heterogeneous group of cancers that arise from developing lymphocytes and produce tumors predominantly in lymphoid structures (i.e., bone marrow) but also in extra-nodal tissues. Collectively, lymphomas constitute the fifth most common cancer in North America, with more than 90% of the patients being affected by lymphomas of B cell origin (Scott and Gascoyne, 2014). Despite overall improvements in outcomes of lymphoma, approximately 30–40% of patients have disease that is either refractory or relapses after standard therapy (Ansell, 2015). The microenvironment of B cell lymphomas contains variable numbers of immune cells (i.e., T-cells, macrophages, neutrophils), stromal cells (i.e., fibroblasts), and endothelial cells (Blonska et al., 2015; Scott and Gascoyne, 2014; Shain et al., 2015), many of which have been reported to express heparanase (Matzner et al., 1985; Vlodavsky et al., 1992). A complex cross-talk between lymphoma cells and their microenvironment is bidirectional and multiple secreted factors and cell surface molecules contribute to the activation of signaling pathways in both the lymphoma and stromal cells (Blonska et al., 2015), many of which are targets for the development of lymphoma therapeutics (Mehta-Shah and Younes, 2015). In a recent study, we provided evidence that heparanase is expressed by B-lymphomas, and that heparanase inhibitors restrain tumor growth. Furthermore, we describe the development of novel heparanase-neutralizing monoclonal antibodies (mAbs) that attenuate lymphoma growth by targeting heparanase in the tumor microenvironment (Weissmann et al., 2016). Our results with the newly developed mAbs are described below, further highlighting the potential of heparanase as a valid target for drug development.
Four carbohydrate-based heparanase inhibitors have reached clinical trials (Pala et al., 2016; Pisano et al., 2014; Rivara et al., 2016). These compounds apparently work by binding to the heparin/HS-substrate binding domain of the enzyme, thus blocking its accessibility to natural HS substrates. Owing to their heparin-based nature, these compounds can bind, in addition, many heparin-binding proteins in vivo leaving open the question as to how much of their anti-tumor effect is due specifically to blocking heparanase activity. Monoclonal antibodies against cancer-related targets have met with considerable success due to their specificity and long half-life in humans, yet none have been tested against heparanase in preclinical cancer models towards further clinical development. We have identified three potential heparin-binding domains of heparanase (Levy-Adam et al., 2005). Particular attention was given to the Lys158-Asp171 heparin binding domain (designated HBD1) since a peptide (termed KKDC) corresponding to this sequence physically interacts with heparin and HS, with high affinity, and inhibits heparanase enzymatic activity (Levy-Adam et al., 2005). We have followed this rationale and generated a panel of monoclonal antibodies (mAbs) attempting to target the interaction of heparanase with its HS substrate. We have selected two mAbs (9E8, H1023) that neutralize heparanase enzymatic activity (Weissmann et al., 2016) and demonstrated that both antibodies substantially decreased the cellular uptake of latent heparanase, a HS-dependent mechanism that limits extracellular retention of the enzyme and enables intracellular processing of the latent enzyme into its active form (Gingis-Velitski et al., 2004b; Ilan et al., 2006). Thus, the newly generated antibodies not only neutralize the enzyme extracellularly, but also diminish uptake and accumulation of heparanase inside the cell (Fig. 2) (Weissmann et al., 2016).
Figure 2.
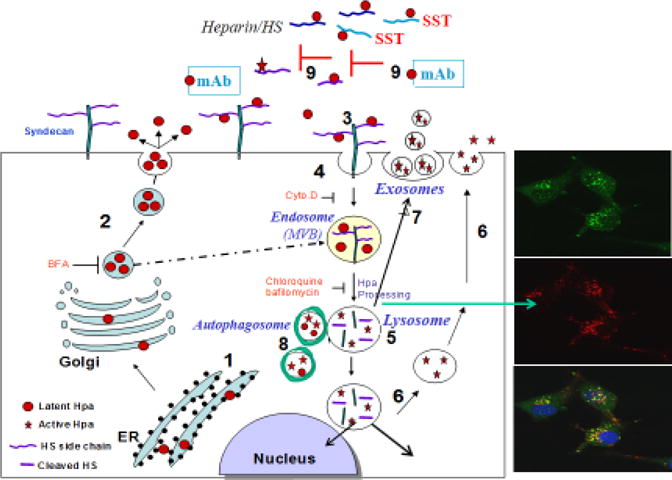
Schematic model of heparanase trafficking. Pre-pro-heparanase is first targeted to the ER lumen via its signal peptide (1). The 65 kDa pro-heparanase is then shuttled to the Golgi apparatus, and is subsequently secreted via vesicles that bud from the Golgi (2), a step that is inhibited by Brefeldin A (BFA). Once secreted, heparanase rapidly interacts with cell membrane HSPGs (i.e., syndecan family members) (3), followed by a rapid endocytosis of the heparanase-syndecan complex that accumulates in late endosomes (4). These steps are inhibited by heparin, glycol-split heparin (SST) and heparanase neutralizing mAb (9), all of which bind to the heparin/HS binding domains of the enzyme, resulting in extracellular accumulation of the latent enzyme and depletion of the intracellular content of the active 50 + 8 kDa enzyme. Fusion of endosomes to lysosomes results in heparanase processing and activation (primarily by cathepsin L) (5) that, in turn, participates in the turnover of HS side chains in the lysosome. Heparanase processing is inhibited by chloroquine and Bafilomycin A, inhibitors of lysosomal proteinases via alkalinization of the lysosomal pH. Typically, heparanase appears in perinuclear lysosomal vesicles (right panels: heparanase-green; lysotracker-red). Lysosomal heparanase may translocate to the nucleus, where it affects gene transcription, and/or can get secreted via secretory lysosomes (6) and exosomes (7). Heparanase promotes the formation of exosomes and autophagosomes (8), resulting in accelerated tumor growth and chemoresistance.
Both the 9E8 (IgM) and H1023 (IgG) mAbs markedly inhibited cell invasion and tumor metastasis, the hallmarks of heparanase function. Moreover, both mAbs inhibited the spontaneous metastasis of ESb mouse lymphoma cells from the subcutaneous primary lesion to the liver (Weissmann et al., 2016). Decreased blood vessel density in Raji Burkitt’s lymphoma treated with mAb 9E8 and H1023 (Weissmann et al., 2016) critically stamps the pro-angiogenic capacity of heparanase (Ilan et al., 2006). This is likely due to reduced release by heparanase of angiogenic-promoting factors sequestered in the bone marrow ECM (Elkin et al., 2001). Importantly, treatment with mAb 9E8 or mAb H1023, as a single agent, attenuated the growth of CAG myeloma and Raji lymphoma tumors, and even greater inhibition was observed when combining the two mAbs together (Fig. 3) (Weissmann et al., 2016), in agreement with the notion that combining two different mAbs increases the inhibitory outcome. Not surprising, mAb 9E8 and H1023 are not cytotoxic to lymphoma or myeloma cells, implying that the mAbs do not exert a direct effect on tumor cells but rather affect the tumor microenvironment. This is best demonstrated with Raji Burkitt’s lymphoma cells that lack intrinsic heparanase activity due to gene methylation (Shteper et al., 2003), yet tumor xenografts produced by the same cells exhibit typical heparanase activity (Fig. 3) (Weissmann et al., 2016). Thus, the ability of mAbs 9E8 and H1023 to attenuate the growth of these tumors is due primarily to neutralization of heparanase contributed by the tumor microenvironment (Weissmann et al., 2016). This implies that the host contributes a significant amount of heparanase, and that inhibition of this fraction is sufficient to attenuate tumor growth. This notion was further supported by showing that mouse El-4 lymphoma cells developed larger (3.5-fold) tumor xenografts when transplanted in heparanase overexpressing transgenic (Hpa-tg) mice vs. wild type C57BL mice. In contrast, smaller (8.5-fold) tumors developed when the same cells were inoculated into heparanase knockout (Hpa-KO) mice vs. wild type mice, further implying that host heparanase plays a decisive role in tumor progression. The finding that EL4 lymphoma cells grew aggressively in Hpa-tg but not wt animals is particularly instructive because EL4 lymphoma cells lack heparanase expression, leaving only host cells to contribute heparanase. (Gutter-Kapon et al., unpublished data).
Figure 3.
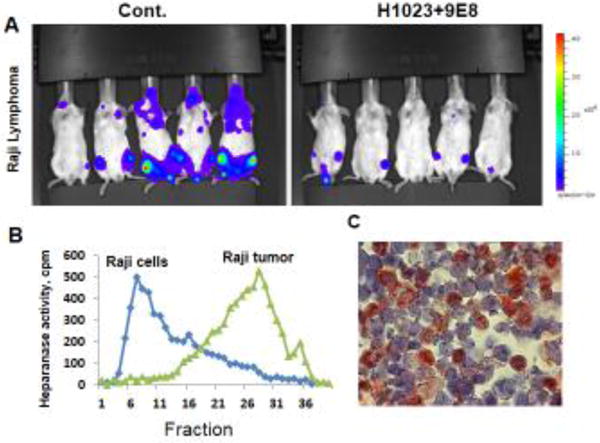
Heparanase neutralizing mAbs attenuate tumor growth. NOD/SCID mice were inoculated (i.v.) with luciferase-labelled Raji-lymphoma cells (A). Mice were treated with mAb 9E8 and mAb H1023 (500 μg/mouse every other day) and tumor growth was evaluated by IVIS imaging. Notably, Raji cells lack heparanase activity (B, Raji cells) whereas tumors developed by Raji cells exhibit typical heparanase activity (B, Raji tumors, fraction 20–35). Bone marrow cells are positively stained for heparanase (C), suggesting that the neutralizing mAbs attenuate lymphoma growth by targeting heparanase activity contributed by the tumor microenvironment.
3. Heparanase in acute and chronic inflammation
HS is known to control inflammatory responses at multiple levels, including sequestration of cytokines/chemokines in the extracellular space, modulation of leukocyte interactions with endothelium and ECM, and initiation of innate immune responses through interactions with toll-like receptors 4 (TLR4) (Akbarshahi et al., 2011; Axelsson et al., 2012; Bode et al., 2012; Brunn et al., 2005; Gotte, 2003; Johnson et al., 2002; Parish, 2006; Wang et al., 2005). Thus, HS remodeling by heparanase may affect several aspects of inflammatory reactions, such as leukocyte recruitment, extravasation and migration towards inflammation sites; release of cytokines and chemokines anchored within the ECM or cell surfaces, as well as activation of innate immune cells (Goldberg et al., 2013; Li and Vlodavsky, 2009; Zhang et al., 2014). The link between inflammation and heparanase was first demonstrated more than two decades ago, prior to cloning of the heparanase gene, when HS-degrading activity was discovered in immunocytes (neutrophils, activated T-lymphocytes) and found to contribute to their ability to extravasate and accumulate in target organs (Lider et al., 1989; Lider et al., 1990; Matzner et al., 1985; Naparstek et al., 1984; Vlodavsky et al., 1992). In addition, non-enzymatic functions of the enzyme were found to promote pro-inflammatory cell adhesion and signal transduction (Riaz et al., 2013). Anti-inflammatory effects demonstrated for heparanase-inhibiting substances (i.e., heparin, synthetic heparin-mimicking compounds) in animal and clinical studies (Edovitsky et al., 2004; Floer et al., 2010; Goldberg et al., 2013; Hershkoviz et al., 1995; Parish et al., 1998; Young, 2008) further support the involvement of the enzyme in inflammatory reactions.
Heparanase expression is up-regulated in the colon of patients suffering from Crohn’s disease and ulcerative colitis (Doviner et al., 2006; Lerner et al., 2011; Waterman et al., 2007), and in the synovial fluid and tissue of patients suffering from rheumatoid arthritis, but not in unaffected individuals (Li et al., 2008). Heparanase has also been implicated in the severity of atherosclerosis, as its expression is increased in vulnerable coronary plaques as opposed to stable plaques and controls (Blich et al., 2013).
Notably, heparanase gene silencing resulted in decreased delayed-type hypersensitivity (DTH) reaction (Edovitsky et al., 2006), and heparanase knockout mice showed reduced airway and acute lung injury responses in models of allergy and sepsis (Poon et al., 2014; Schmidt et al., 2012). Furthermore, transgenic mice overexpressing heparanase are endowed with increased DTH (Edovitsky et al., 2006), colon (colitis) (Lerner et al., 2011), pancreas (unpublished data) and skin (psoriasis-like) (Lerner et al., 2014) inflammation, collectively implying that heparanase is an important player in the inflammatory reaction (Goldberg et al., 2013; Li and Vlodavsky, 2009; Zhang et al., 2014). With the accumulation of experimental data, a complex picture of the versatile role of heparanase in inflammation is evolving, whereby heparanase may act either in facilitating or limiting inflammatory responses, depending on the cellular/extracellular context. Below we discuss several emerging mechanistic concepts reflecting the involvement of heparanase in inflammatory reactions, primarily via modulating the activity of innate immune cells.
3.1. Neutrophils and sepsis
Cumulative evidence suggests that heparanase affects the activities of several types of innate immunocytes, including neutrophils, macrophages, dendritic and mast cells (Benhamron et al., 2006; Benhamron et al., 2012; Blich et al., 2013; Lerner et al., 2011; Massena et al., 2011; Schmidt et al., 2012; Wang et al., 2011). Of those, neutrophils represent the important effectors in the acute inflammatory responses. The consequence of heparanase action on neutrophil behavior was highlighted in a study by Schmidt et al., focusing on enzymatic degradation of endothelial glycocalyx in sepsis-associated lung injury. Glycocalyx, the thin gel-like endothelial layer that coats the luminal surface of blood vessels, is composed of HS and other glycosaminoglycans, proteoglycans and glycoproteins (Constantinescu et al., 2003; Weinbaum et al., 2007). It serves as a barrier to circulating cells and controls availability of endothelial surface adhesion molecules to circulating leukocytes (Constantinescu et al., 2003; Weinbaum et al., 2007). In the mouse model of sepsis-associated inflammatory lung disease, rapid induction of heparanase activity (through TNFα-dependent mechanism) was demonstrated in pulmonary microvascular endothelial cells (Schmidt et al., 2012). Heparanase induction was also found in biopsies of human inflammatory lung disease (Schmidt et al., 2012). Moreover, sepsis-associated loss of pulmonary glycocalyx and endothelial hyperpermeability were attenuated in heparanase-null mice and in mice treated with inhibitors of heparanase enzymatic activity (Schmidt et al., 2012). It was suggested that the lung microvasculature is masked by HS-rich glycocalyx that has to be removed by lung expressed heparanase in order for neutrophils to entrap in the pulmonary vasculature in response to LPS septic signals (Schmidt et al., 2012). Unlike this work, applying the same heparanase-null mice as well as ICAM-1 and ICAM-2 double knockout mice, we have demonstrated that lung entrapment of neutrophils in response to systemic LPS does not require either heparanase mediated degradation of the pulmonary HS rich glycocalyx or neutrophil recognition of vascular ICAM-1 and 2 (Petrovich et al., 2016). Thus, although lung endothelial heparanase may be triggered by LPS and other systemic inflammatory signals to unmask the pulmonary vasculature (Schmidt et al., 2012), it appears that this heparanase-dependent step is not a rate-limiting step in LPS-triggered neutrophil entrapment in the pulmonary vasculature. We therefore favor the possibility that septic conditions modeled by systemic LPS trigger a deformation of neutrophils by their cytoskeletal stiffening resulting in ICAM-, β2 integrin- and heparanase- independent mechanical entrapment inside the thin capillary vessels (Mizgerd et al., 1997; Petrovich et al., 2016; Worthen et al., 1989). Our results are consistent with a study by Morris et al., which has ruled out a role for lung heparanase in neutrophil infiltration to lungs exposed to intranasal LPS (Morris et al., 2015). Likewise, neutrophil emigration from blood to the inflamed skin or peritoneal cavity was shown to be heparanase independent (Stoler-Barak et al., 2015). We also found no role for T cell-based heparanase in effector T cell entrapment inside this vasculature although these T cells express high levels of the enzyme (Stoler-Barak et al., 2015). Overall, our results indicate that effector T cells use their constitutively adhesive LFA-1 to readily recognize pulmonary ICAM-1 and 2 independently of heparanase mediated glycocalyx degradation, probably because their large and stiff nucleus allows these cells to readily penetrate the thick glycocalyx barrier without HS degradation.
3.2. Heparanase is critical for neutrophil entry to lungs exposed to tobacco smoke
Given that heparanase activity is implicated in multiple chronic inflammatory processes (Hermano et al., 2012; Lerner et al., 2011; Naggi et al., 2005), we searched for a potential role of the enzyme in neutrophil emigration to lungs subjected to a prolonged smoke exposure. A daily exposure of wt mice to high levels of tobacco smoke for 2 weeks resulted in a significant infiltration of neutrophils to the lung parenchyma and a small fraction of these neutrophils also penetrated the bronchoalveolar space (Petrovich et al., 2016). Strikingly, heparanase deficiency resulted in dramatically reduced numbers of neutrophils entrapped either in the lung or recovered in the BAL (Petrovich et al., 2016). The contributions of heparanase to neutrophil migration and retention in smoke exposed lungs are probably more complex than those of LPS induced neutrophil migration to lungs. For example, in a model of ear inflammation, endothelial heparanase was suggested to remodel the HS-rich vascular basement membrane (Benhamron et al., 2006). A similar function may be mediated by pulmonary heparanase during prolonged exposure to smoke and potentially other air irritants. Moreover, lung heparanase may be also involved in transcriptional regulation of neutrophil attracting chemokines, as evident from the reduced levels of smoke triggered CXCL5 transcription detected in heparanase null lungs. These results collectively suggest a role of lung heparanase in neutrophil recruitment to smoke exposed lungs, which could be partially due to aberrant transcription of neutrophil attracting chemokines by smoke inflamed lungs (Petrovich et al., 2016).
3.3. Mode of action
In some experimental settings heparanase was noted to inhibit, rather than promote, inflammation. Thus, constitutive overexpression of heparanase in Hpa-tg mice attenuated intraluminal crawling of neutrophils in the cremaster muscle microvessels toward an extravascular chemokine source, reportedly due to reduction in endothelial surface HS chain length and altered ability of truncated HS to serve as a ligand for chemokines (Massena et al., 2011). Likewise, exploring acute inflammatory phenotypes of Hpa-tg mice in models of inflammatory (Li et al., 2012b) and neuroinflammation (Zhang et al., 2014; Zhang et al., 2012b) revealed that neutrophil recruitment and activation were attenuated in the presence of constitutively increased levels of heparanase in Hpa-tg mice. This was reportedly due to cleavage of HS on the endothelial cell surface and the resulting disruption of chemokine gradient(s) which is critical for directional migration of immune cells and infection resolution (Massena et al., 2011). Consequently, crawling of neutrophils toward chemokine-releasing gel was impaired in Hpa-tg mice; instead, Hpa-tg neutrophils exhibited random crawling, ultimately leading to severely reduced ability to clear bacterial infection (Massena et al., 2011). A similar situation may occur in pleural empyema, an inflammatory condition that progresses from acute to chronic, life-threatening phase. Heparanase expression and activity were markedly increased in patients with chronic vs. acute pleural empyema and in a mouse model of empyema (Lapidot et al., 2015). Unexpectedly, however, Hpa-tg mice exposed to empyema exhibited prolonged survival, supporting the occurrence of a protective anti-inflammatory mechanism in this model (Lapidot et al., 2015), likely due to impaired chemokine gradient and the attenuated recruitment of neutrophils.
In light of the reported anti-inflammatory effects of heparin (Young, 2008), increased levels of highly sulfated, “heparin-resembling” HS fragments, which are constantly present in Hpa-tg, as compared to wt mice (Escobar Galvis et al., 2007b; Li et al., 2005), may offer an explanation for the inhibitory effects of continuous heparanase overexpression on neutrophils in these settings (Li et al., 2012a; Zhang et al., 2012a). In addition, it was noted that the thickness of the glycocalyx varies greatly between microvessels from different tissues: it is almost 3 times thinner in cremaster (systemic) microvessels endothelium than in pulmonary endothelium (Schmidt et al., 2012). Thus, the overall effect of heparanase on neutrophil behavior may depend on the proportional contribution of glycocalyx removal (which is expected to facilitate neutrophil access to the blood vessel wall (Schmidt et al., 2012) versus the disturbance of chemokine gradients at the endothelial cell surface (which attenuates neutrophil recruitment (Li et al., 2012a; Massena et al., 2011; Zhang et al., 2012b). Additionally, effects of heparanase on acute inflammatory reactions in different organ/tissue settings may reflect different roles of the enzyme through distinctive phases of the inflammatory cascade or may be a consequence of tissue-specific patterns reported for HS (Allen et al., 2001; Guo et al., 2007; Ledin et al., 2004). Altogether, depending on the context and experimental system, overexpression of heparanase may exert opposing effect on acute inflammatory responses. Taking into account the complex nature of inflammation and the multifaceted action of heparanase, the end result depends on the intricate balance between its pro-inflammatory (i.e., tissue damage, infiltration of leukocytes) and anti-inflammatory (i.e., disruption of chemokine gradients) effects. The above examples illustrate the complexity and duality of heparanase function in inflammatory conditions. Regardless, increased heparanase levels in the course of Crohn’s disease, ulcerative colitis (Waterman et al., 2007), empyema (Lapidot et al., 2015), psoriasis (Lerner et al., 2014), pancreatitis (Koliopanos et al., 2001) and arthritis (Li et al., 2008), and the reduced severity of sepsis (Schmidt et al., 2012) and diabetic nephropathy (Gil et al., 2012) in heparanase knockout mice implies that heparanase inhibitors may prove beneficial in some of these pathological conditions. The ability of heparanase inhibitors (i.e., PG545, SST0001, PI-88) to restrain experimental acute pancreatitis (Khamaysi et al., unpublished data), diabetic nephropathy (Gil et al., 2012), and autoimmune type 1 diabetes (Parish et al., 2013; Ziolkowski et al., 2012) provides hope that these and other heparanase inhibitors will restrain the expanding variety of inflammation-based disorders.
3.4. Heparanase effects on macrophages in chronic inflammation
When acute inflammation is not properly resolved, the composition of the infiltrating leukocytes changes from neutrophils to macrophages, dominant cellular players in chronic inflammation. Heparanase ability to modulate macrophage responses was highlighted in studies focusing on inflammatory bowel disease (IBD). Ulcerative colitis and Crohn’s disease, the two chronic disorders collectively known as IBD, result from inappropriate persistent activation of the mucosal immunity by normal gut flora in genetically susceptible individuals (Xavier and Podolsky, 2007). In patients with IBD, an equilibrium between the immune response to pathogens and tolerance to the normal flora becomes unbalanced, leading to the uncontrolled uptake of proinflammatory substances (i.e., bacteria, bacterial products) from the gut lumen and triggering immune activation, cytokine release, and dysfunction of the epithelial barrier (Clayburgh et al., 2004; Xavier and Podolsky, 2007). Given the important role of HS in maintaining the integrity of the gut wall (Belmiro et al., 2005; Bode et al., 2008), enzymatic degradation of HS is thought to significantly affect colon permeability and inflammatory reactions. Analysis of glycosaminoglycan content in normal colonic tissue and colons of IBD patients revealed loss of HS from the subepithelial BM and from the vascular endothelium in the submucosa (Belmiro et al., 2005; Day and Forbes, 1999; Murch et al., 1993). In agreement, preferential expression of heparanase was reported in colonic epithelium of IBD patients during both acute and chronic phases of the disease (Lerner et al., 2011; Waterman et al., 2007). Of note, immunocytes in the involved areas exhibit little heparanase expression during all phases (Waterman et al., 2007). A similar temporo-spatial pattern of heparanase expression was observed in experimental dextran sodium sulfate (DSS)-induced chronic colitis in mice (Lerner et al., 2011). It appears that in the setting of colitis, heparanase of epithelial origin modulates inflammatory phenotype of macrophages, preventing inflammation resolution and switching macrophage responses to chronic inflammation pattern (Lerner et al., 2011). In support of this notion, exacerbated chronic inflammatory phenotype and augmented recruitment and activation of macrophages were detected in colonic mucosa of Hpa-tg mice following induction of DSS colitis (Lerner et al., 2011). Moreover, recombinant heparanase strongly augmented in vitro activation of macrophages, resulting in increased production of pro-inflammatory cytokines (Blich et al., 2013; Goodall et al., 2014; Hermano et al., 2014; Lerner et al., 2011). Activated macrophages can in turn induce epithelial heparanase expression (via a TNFα-dependent mechanism) and post-translational processing of the pro-enzyme via increased secretion of cathepsin L (Lerner et al., 2011) (Fig. 4), fueling a self-sustaining inflammatory circuit which could partially explain the so called “multiplier” effect in IBD, in which small elevation in initiating inflammatory stimuli leads to a large increase in downstream cytokines. Notably, augmented macrophage activation in response to heparanase was also reported in a model of neointimal lesions following vascular injury (Baker et al., 2009) and in atherosclerotic plaque progression toward vulnerability (Blich et al., 2013), further supporting a role of the enzyme in modulating macrophage responses. Moreover, our studies on the involvement of heparanase in diabetic nephropathy indicate that activation of kidney-infiltrating macrophages may represent an important mechanism through which heparanase contributes to chronic low-grade inflammation and renal damage in diabetic kidney disease (Goldberg et al., 2014).
Figure 4.
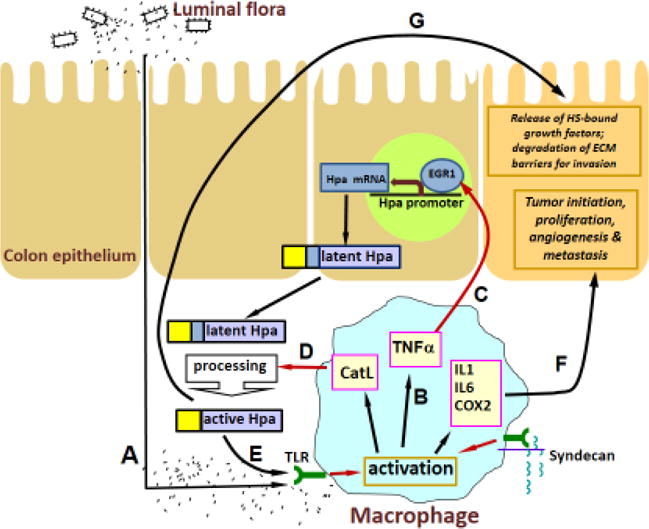
A model of heparanase-driven vicious cycle that powers colitis and the associated tumorigenesis. Macrophages activated by excessive exposure to the luminal flora due to epithelial barrier dysfunction and activation of TLR4 (A) produce increased levels of TNF-α (B) and induce heparanase expression in the colon epithelium via EGR1 dependent mechanism (C). D. The secreted 65-kDa latent heparanase is processed into its enzymatically-active (8+50 kDa) form by cathepsin L (CatL), supplied by the activated macrophages, and in turn sensitizes macrophages to further activation by luminal flora (E), thus preventing inflammation resolution, switching macrophage responses to the chronic inflammation pattern and creating a tumor initiating inflammatory environment (F). In addition, heparanase promotes tumor take and progression via stimulation of angiogenesis, release of ECM-bound growth factors and bioactive HS fragments, and removal of extracellular barriers for invasion (G).
3.5. Heparanase in inflammation-associated cancer
Chronic inflammatory conditions present in the microenvironment of most tumors (Mantovani et al., 2008) contribute to cancer progression (Grivennikov et al., 2010; Hanahan and Weinberg, 2011), among other mechanisms, through mobilization of tumor-supporting immunocytes (e.g., tumor associated macrophages, neutrophils) which supply bioactive molecules that foster survival, angiogenesis, invasion and metastasis (Hanahan and Weinberg, 2011; Mantovani et al., 2008; Qian and Pollard, 2010). Moreover, in several anatomic sites chronic inflammation is crucially implicated in tumor initiation, producing a mutagenic environment through release of reactive oxygen/nitrogen species from infiltrating immune cells, generating cytokines, chemokines, growth factors, and anti-apoptotic proteins and activating tumor stimulating signaling pathways (e.g., NF-kB, STAT3) (Grivennikov and Karin, 2010; Hanahan and Weinberg, 2011; Mantovani et al., 2008; Qian and Pollard, 2010). Progression of Barrett’s esophagus to adenocarcinoma (Picardo et al., 2012); chronic gastritis to intestinal-type gastric carcinoma, chronic hepatitis C to hepatocellular carcinoma (Chiba et al., 2012); pancreatitis to pancreatic adenocarcinoma (Lowenfels et al., 1993) and colitis to colorectal cancer (Gupta et al., 2007) are well-known examples of inflammation-driven tumorigenesis. Remarkably, induction of heparanase prior to the appearance of malignancy was reported in essentially all of the above-mentioned inflammatory conditions, i.e., Barrett’s esophagus (Brun et al., 2009; Sonoda et al., 2010), hepatitis C infection (El-Assal et al., 2001), chronic pancreatitis (Koliopanos et al., 2001), Crohn disease and ulcerative colitis (Waterman et al., 2007 and Lerner et al., 2011). Given the causal role of heparanase in tumor progression in tissues in which cancer-related inflammation typically occurs [i.e., gastrointestinal tract, pancreas, liver (Brun et al., 2009; El-Assal et al., 2001; Hoffmann et al., 2008; Koliopanos et al., 2001; Naomoto et al., 2005; Nobuhisa et al., 2005; Sonoda et al., 2010; Xiong et al., 2012), it is conceivable that inflammation-induced heparanase may be involved in coupling inflammation and cancer. This notion is supported by a study utilizing a mouse model of colitis-associated colon carcinoma (Lerner et al., 2011), showing that heparanase promotes polarization of innate immunocytes toward pro-inflammatory and/or pro-tumorigenic phenotype (Fig. 4). By sustaining continuous activation of macrophages that supply cancer-promoting cytokines (i.e., TNFα, IL-1, IL-6), heparanase (produced primarily by inflamed epithelium and activated by cathepsin L contributed by tumor associated macrophages) (Fig. 4) participates in creating a tumorigenic microenvironment characterized by enhanced NFκB and STAT3 signaling, augmented levels of cyclooxygenase 2 and increased vascularization (Lerner et al., 2011).
Blich et al., have reported that heparanase stimulates the release of cytokines from primary mouse peritoneal macrophages and the mouse macrophage cell line J774 (Blich et al., 2013). Similarly, Goodhall et al., showed that heparanase promotes the release of pro-inflammatory cytokines from both human peripheral blood mononuclear cells (IL-1β, IL-6, IL-8, IL-10 and TNFα) and mouse splenocytes (MCP-1, IL-6, TNFα), likely by generating soluble HS fragments that activate TLR-dependent pathway(s) (Goodall et al., 2014) (Fig. 5). This mechanism was supported by the significant inhibition of cytokine release upon inhibition of heparanase enzymatic activity and by showing that heparanase-induced cytokine release was dependent on MyD88, a key TLR adaptor molecule, and mediated in part through TLR-4 via the NF-κB pathway (Blich et al., 2013; Goodall et al., 2014). Notably, cytokine expression in response to heparanase is transcriptionally regulated, consistent with HS fragments acting through TLRs via NF-κB (Fig. 5). Indeed, heparanase-mediated cytokine release is reduced when cells are treated with the NF-κB inhibitor BAY117082 (Blich et al., 2013). Furthermore, it is interesting to note that resistin has been identified as a new interacting partner for heparanase (Novick et al., 2014). Resistin has been shown to bind and signal through TLR4, leading to an increase in the pro-inflammatory protein MCP-1 (Kopp et al., 2009). Together, these results identify a novel role for heparanase in the regulation of an inflammatory response that is distinct from the previously described physical breakdown of the ECM to promote efficient migration of leukocytes to sites of inflammation.
Figure 5.
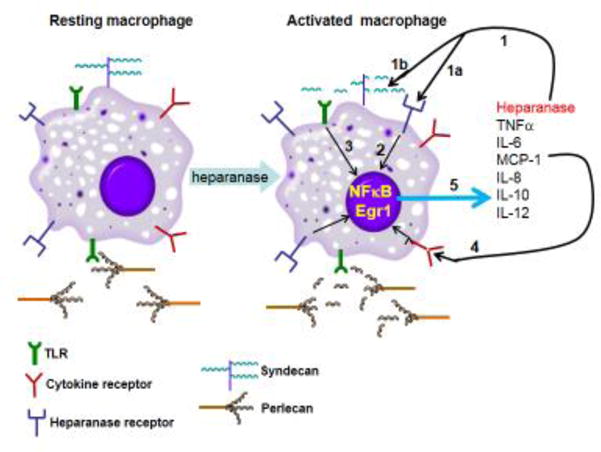
Schematic model of the function of heparanase in activation of macrophages. Heparanase binds to a putative receptor that mediates its non-enzymatic signaling activity (1a). Heparanase also cleaves HS at the cell surface (syndecan) and ECM (perlecan) and releases HS fragments that then signal through MyD88-dependent tall-like receptors (1b) (e.g.,TLR4), leading to NF-κB cleavage and activation (3). An intriguing possibility is that intact extracellular HS inhibits TLR4 responses and macrophage activation, while its removal/degradation by heparanase relieves this inhibition, most likely by increasing accessibility of TLRs to their ligands. NF-κB upregulates the production and release of pro-inflammatory cytokine, including: IL-8, IL-10, IL12, IL-6, TNF, IL-1β and MCP-1 (5). These cytokines promote (via cell surface receptors) inflammatory events and may also increase heparanase production via the Egr1 transcription factor. Altogether, these events contribute to a vicious cycle that induces inflammatory responses.
3.6. Involvement of heparanase in coupling aseptic inflammation and tumorigenesis
Pancreatic tumor microenvironment and in particular infiltrating inflammatory cells (largely macrophages), represents an important contributing factor to pancreatic ductal adenocarcinoma (PDAC) aggressiveness and resistance to treatment (Baumgart et al., 2013; Feig et al., 2012). We have highlighted a novel action of heparanase in coupling inflammation and cancer during PDAC progression (as a prototype of aseptic inflammation-associated tumor). We found that heparanase polarizes macrophages, stimulated by necrotic cells (which are abundantly present in both clinical and experimental PDAC (Hiraoka et al., 2010; Ijichi et al., 2011) toward tumor-promoting phenotype, characterized by augmented expression of several key cytokines driving pancreatic tumorigenesis. Utilizing a mouse model of heparanase-overexpressing pancreatic carcinoma, we demonstrated that the enzyme directs protumorigenic behavior of TAM. Overexpression of heparanase resulted in increased TAM infiltration. Moreover, well-recognized markers of protumorigenic macrophage population (i.e., MSR-2, CCL-2, IL-10, VEGF, IL-6) that are tightly implicated in the pathogenesis of PDAC (Jones et al., 2011; Lesina et al., 2011) were significantly upregulated in TAM isolated from heparanase-overexpressing mouse tumors (Hermano et al., 2014).
We hypothesized that heparanase facilitates tumor-promoting action of TAM activated by endogenous ligands originating in the PDAC microenvironment. In fact, several elements in the tumor microenvironment (e.g., ECM components, necrotic cell-derived substances) may affect macrophage behavior through TLR activation (Yu et al., 2013)). In particular, it is increasingly apparent that necrotic cancer cells may lead to more aggressive tumors because of the stimulatory role of necrosis-induced inflammation (Proskuryakov and Gabai, 2010). Importantly, treatment of necrotic cell-stimulated macrophages with active heparanase resulted in induction of key cytokines implicated in PDAC (including IL-6) and facilitated their ability to activate STAT3, a cornerstone of pancreatic carcinogenesis acting downstream IL-6 (Baumgart et al., 2013). Notably, activation of STAT3 was observed in experimental and clinical specimens of PDAC with increased heparanase levels (Hermano et al., 2014).
The exact mechanism of heparanase-mediated change in macrophage phenotype is not fully elucidated. One intriguing possibility is that intact extracellular HS inhibits TLR4 responses and macrophage activation, while its removal relieves this inhibition (Brunn et al., 2005), most likely by increasing accessibility of TLRs to their ligands. On the other hand, soluble HS fragments themselves were found to stimulate TLR (in particular TLR4) signaling in vitro (Brunn et al., 2005; Johnson et al., 2002; Yu et al., 2010) and in pancreatic tissue in vivo (Yu et al., 2010). Importantly, heparanase releases bioactive HS degradation fragments from the ECM/cell surface (Elkin et al., 2001; Kato et al., 1998). While further investigation is warranted, our results reveal a novel role of heparanase in the molecular decision-making process that guides changes in the TAM phenotype during tumor development. Moreover, it implies that heparanase expression status may become highly relevant in defining target patient subgroup that is likely to benefit the most from treatment modalities that target TAM/IL-6 signaling, which are currently under intensive preclinical/clinical validation in PDAC and other tumor types (Fang and Declerck, 2013). Taken together, our findings describe a role of heparanase in powering cancer-related aseptic inflammation, highly relevant to the pathogenesis of PDAC and additional tumor types (Hermano et al., 2014). Heparanase inhibiting compounds and neutralizing monoclonal antibodies are expected to exert therapeutic benefits in both inflammation and malignancy by disrupting heparanase-driven heterotypic interactions between epithelial, endothelial, and immune cells.
4. Cellular uptake and trafficking of heparanase
4.1. Processing of heparanase is mediated by syndecan 1 cytoplasmic domain and involves syntenin and α-actinin
A number of studies have shown that secreted or exogenously added latent heparanase rapidly interacts with normal and tumor-derived cells, followed by internalization and processing into a highly active enzyme (Ben-Zaken et al., 2008; Gingis-Velitski et al., 2004a; Gingis-Velitski et al., 2004b; Nadav et al., 2002; Vreys et al., 2005; Zetser et al., 2004), collectively defined as heparanase uptake. Several approaches, including HS-deficient cells, addition of heparin or xylosides, and deletion of HS-binding domains of heparanase, provided compelling evidence for the involvement of HS in heparanase uptake (Gingis-Velitski et al., 2004b; Levy-Adam et al., 2005). Heparanase uptake is regarded a pre-requisite for the delivery of latent 65 kDa heparanase to lysosomes and its subsequent proteolytic processing and activation into 8 and 50 kDa protein subunits. Briefly, following uptake, heparanase was noted to reside primarily intracellularly within endocytic vesicles, assuming a polar, peri-nuclear localization and co-localizing with lysosomal markers (Goldshmidt et al., 2002; Nadav et al., 2002). Notably, heparanase processing was blocked by chloroquine and bafilomycin A1 which inhibit lysosomal proteases by raising the lysosome pH (Zetser et al., 2004; Zhitomirsky and Assaraf, 2015; 2016). Subsequent studies employing site-directed mutagenesis, gene silencing, and pharmacological inhibitors have identified cathepsin L as the primary lysosomal protease responsible for heparanase processing and activation (Abboud-Jarrous et al., 2008; Abboud-Jarrous et al., 2005). We have noted that syndecan-1 and 4 are internalized by cells following addition of exogenous heparanase, co-localizing with heparanase in endocytic vesicles (Gingis-Velitski et al., 2004b; Levy-Adam et al., 2010). Structurally, all syndecans are composed of an extracellular domain, membrane domain, and a conserved short C-terminal cytoplasmic domain divided into the first conserved region (C1), the variable domain (V), and the second conserved region (C2). Each of these cytoplasmic domains has been shown to interact with specific adaptor molecules and to mediate cellular function (Beauvais and Rapraeger, 2004; Tkachenko et al., 2005).
Heparanase interacts with syndecans by virtue of the typical high affinity that exists between an enzyme and its substrate. This high affinity interaction directs rapid and efficient cellular uptake of the heparanase-syndecan complex (Gingis-Velitski et al., 2004b; Shteingauz et al., 2014). Peptide corresponding to the heparin/HS binding domain of heparanase (Lys158-Asp171, termed KKDC) similarly associates with the plasma membrane and induces clustering of syndecans, resulting in improved cell spreading (Levy-Adam et al., 2005; Levy-Adam et al., 2008). Notably, syndecan clusters formed by the KKDC peptide were exceptionally large and failed to be internalized (Levy-Adam et al., 2005; Levy-Adam et al., 2008). Likewise, heparanase-2 (Hpa2), a close homolog of heparanase, binds HS with high affinity but fails to undergo internalization (Levy-Adam et al., 2010), implying that interaction and clustering per se are not sufficient for the internalization of syndecan and its cargo. We examined the role of syndecan-1 cytoplasmic domain in heparanase processing (Shteingauz et al., 2014). To this end, we transfected cells with a full length mouse syndecan-1 or deletion constructs lacking the entire cytoplasmic domain (delta), the conserved (C1 or C2) or variable (V) regions. Heparanase uptake was markedly increased following syndecan-1 overexpression, thus challenging the notion that cell surface HS is at saturation and does not limit ligand binding. In contrast, heparanase was retained at the cell membrane and its processing was impaired in cells overexpressing syndecan-1 deleted for the entire cytoplasmic tail (Shteingauz et al., 2014). Subsequent studies revealed that the C2 and V regions of syndecan-1 cytoplasmic tail mediate heparanase processing. Furthermore, syntenin, known to interact with syndecan C2 domain, and α actinin are essential for heparanase processing (Shteingauz et al., 2014). These results illustrate the tight regulation of heparanase activation, and shed light on syndecan-mediated endocytosis. Interestingly, syndecans and syntenin, via interaction with ALIX, have been implicated in regulating the biogenesis of exosomes (Baietti et al., 2012). Importantly, as discussed below, heparanase facilitates the production of exosomes and regulates their secretion and composition (Roucourt et al., 2015b), implying that heparanase-syndecan-syntenin establish a linear axis that regulates exosome formation (Shteingauz et al., 2014) and the related effects on tumor progression (Shteingauz et al., 2014).
4.2. Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes
Exosomes are membrane-bound vesicles which are released into the extracellular space when a multivesicular endosome housing the exosomes fuses with the plasma membrane (Simons and Raposo, 2009; Tkach and Théry, 2016). Exosomes are powerful mediators of intercellular communication that drive tumor progression by regulating the behavior of tumor and host cells both locally within the tumor microenvironment and distally throughout the body (Peinado et al., 2011; Zhang and Grizzle, 2011). Exosomes accomplish this regulatory function by docking to recipient cells and delivering their cargo of protein, DNA, mRNA and miRNA (Simons and Raposo, 2009). When exosomes come in contact with recipient cells, the cells can undergo immediate stimulation through cell signaling mediated by exosomal proteins, or cell behavior can be modified by the proteins, microRNA or mRNA delivered by the exosome. Most cells secrete exosomes, but their secretion is upregulated in disease states. In cancer, secretion of exosomes often increases as tumors transit toward a more aggressive phenotype. The tumor-host crosstalk mediated by exosomes has multiple effects that can influence processes such as formation of the pre-metastatic niche, angiogenesis, host immune function as well as conferring anticancer drug resistance (Di Marzo et al., 2016; Peinado et al., 2012; Peinado et al., 2011; Wojtuszkiewicz et al., 2016; Zhang and Grizzle, 2011). Several members of the Rab protein family have been shown to control exosome formation and more recently it was reported that syndecan proteoglycans, through their interaction with the syntenin-ALIX complex, influence the biogenesis of exosomes (Baietti et al., 2012; Ostrowski et al., 2010; Peinado et al., 2011). Briefly, syntenin interacts with the syndecan core protein via two PDZ domains as well as with ALIX via three LYPXnL motifs (Baietti et al., 2012). ALIX then binds to ESCRT-III (endosomal-sorting complex required for transport), the machinery responsible for intraluminal vesicle formation at multivesicular endosomes (MVEs). Importantly, relevant to the current review, it was further demonstrated that heparanase activates the syndecan-syntenin-ALIX exosome pathway (Baietti et al., 2012; Roucourt et al., 2015b). Briefly, heparanase activity in endosomes trims long HS chains into shorter ones, allowing clustering of syndecans through lateral interactions between their HS polysaccharide chains (David and Zimmermann, 2016; Roucourt et al., 2015a). Heparanase-induced clustering is thought to stimulate the binding of syndecan cytoplasmic domains to the tandem PDZ domains of syntenin, driving ALIX-ESCRT-mediated sorting into exosomes (David and Zimmermann, 2016; Friand et al., 2015; Roucourt et al., 2015a). Notably, heparanase activity also facilitated the recruitment of CD63 into exosomes, in a syntenin-dependent manner (David and Zimmermann, 2016; Roucourt et al., 2015a). All in all, a complex picture is emerging, in which syndecans, CD63 and possibly other membrane proteins that associate with endosomal syndecan and/or tetraspanin-enriched microdomains, are sorted into exosomes by a shared heparanase-syndecan-syntenin-ALIX pathway machinery (David and Zimmermann, 2016; Friand et al., 2015). Heparanase effects on exosome biogenesis seem best explained by heparanase activity on syndecan, promoting the assembly of syndecan in complexes that, by recruiting syntenin and ALIX–ESCRT, promote endosomal membranes to bud (Friand et al., 2015). From that perspective, heparanase inhibitors (Vreys and David, 2007; Vreys et al., 2005) as well as specific syntenin-PDZ inhibitors might be of particular interest for cancer, where both exosome release and heparanase are often elevated in the more aggressive forms of disease (Ramani et al., 2013; Thompson et al., 2013).
In support of the above considerations, the first evidence that heparanase was involved in exosome biogenesis came when we discovered that enhanced heparanase expression in myeloma cells, or exposure of cells to exogenous heparanase, dramatically increased exosome secretion (Thompson et al., 2013) (Fig. 2). Thus, heparanase released by a tumor cell or by host cells (e.g., macrophages) could diffuse within the microenvironment and impact neighboring tumor cells and enhance, among other effects, their secretion of exosomes. Heparanase enzyme activity is required for enhanced exosome secretion because enzymatically inactive forms of heparanase, even when present in high amounts, do not substantially increase exosome secretion (Thompson et al., 2013). Heparanase also impacts exosome protein cargo as reflected by higher levels of syndecan-1, VEGF and HGF in exosomes secreted by heparanase-high expressing cells as compared to heparanase-low expressing cells (Thompson et al., 2013). In functional assays, exosomes from cells with high expression of heparanase stimulated spreading of tumor cells on fibronectin and invasion of endothelial cells through extracellular matrix, better than did exosomes secreted by cells expressing low levels of heparanase, suggesting a role in promoting tumor cell spreading and angiogenesis (Thompson et al., 2013).
Our finding that heparanase is present in exosomes (Thompson et al., 2013) raises the possibility that the enzyme is delivered to distal locations via exosomes (Fig. 2). Because of the known role of heparanase in promoting angiogenesis and metastasis, delivery of the enzyme via exosomes may play a role in establishing niches to which tumor cells eventually home and grow. Our results indicate that heparanase can lead to secretion of exosomes that interact with both tumor and host cells and drive them toward behaviors associated with an aggressive tumor phenotype which includes drug resistance. Notably, because heparanase enhances exosome secretion, the tumor microenvironment will be bathed in much higher levels of exosomes when tumors are expressing high levels of heparanase. Emerging data indicate that exosomes can act as barriers to anti-cancer therapy by interacting with tumor cells and enhancing their chemoresistance (Au Yeung et al., 2016). Indeed, our ongoing studies reveal that heparanase enhances both exosome docking (Purushothaman et al., 2016) and exosome-mediated transfer of chemoresistance to tumor cells. Collectively, our studies reveal that heparanase helps drive exosome secretion, alters exosome composition and facilitates production and docking of exosomes that impact both tumor and host cell behavior thereby promoting tumor progression and chemoresistance (Thompson et al., 2013).
4.3. Heparanase enhances tumor growth and chemoresistance by augmenting autophagy
As noted above, heparanase resides primarily within endocytic vesicles, assuming a polar, peri-nuclear localization and co-localizing with lysosomal markers (Gingis-Velitski et al., 2004b; Goldshmidt et al., 2002; Levy-Adam et al., 2010; Nadav et al., 2002; Shteingauz et al., 2014; Zetser et al., 2004) (Fig. 2). It was further demonstrated that heparanase processing is blocked by chloroquine and bafilomycin A1 which inhibit lysosomal proteases by raising the lysosome pH (Zetser et al., 2004; Zhitomirsky and Assaraf, 2015; 2016). In spite of its localization in a highly active protein degradation environment such as the lysosome, heparanase appears stable (Goldshmidt et al., 2002; Zetser et al., 2004) and exhibits a half-life of about 30 hours (Gingis-Velitski et al., 2004b), relatively long compared with a t1/2 of 2-6 hours, and 25 minutes for transmembrane and GPI-anchored HSPGs, respectively (Egeberg et al., 2001). The residence and accumulation of heparanase in lysosomes indicate that the enzyme may function under normal physiological conditions in this organelle. In a search for such a physiological function we revealed a role of lysosomal heparanase in modulating autophagy (Shteingauz et al., 2015). Autophagy is an evolutionarily conserved catabolic pathway through which cytoplasmic components, including macromolecules such as proteins and lipids as well as whole organelles, are sequestered into double-membrane vesicles called autophagosomes (Fig. 2). Autophagosomes are subsequently fused with lysosomes, wherein these macromolecules and organelles are degraded and recycled. This process occurs in every cell at a basal level and is required to remove unfolded proteins and damaged organelles, thus maintaining cellular homeostasis. Autophagy is further induced by starvation and stress, promoting cancer cell survival by providing their metabolic needs (Rosenfeldt and Ryan, 2011; White, 2012). Our results indicate that heparanase promotes autophagy and that enhanced tumor growth and chemoresistance conferred by heparanase is mediated, at least in part, by enhancing autophagy (Shteingauz et al., 2015). This was concluded because reduced LC3-II (an autophagy protein biomarker that specifically associates with autophagosomes) levels are found in cells and tissues obtained from heparanase knockout mice as opposed to elevated LC3-II levels found in transgenic mice that overexpress heparanase. Even enhanced induction of autophagy was evident in head and neck carcinoma and glioma cells overexpressing heparanase (Shteingauz et al., 2015), in accordance with a strong pre-clinical and clinical significance of heparanase in the progression of these malignancies (Arvatz et al., 2011a; Cohen-Kaplan et al., 2008a; Cohen-Kaplan et al., 2012; Cohen-Kaplan et al., 2008b; Doweck et al., 2006; Fux et al., 2009a; Zetser et al., 2006). Notably, electron microscopy analyses of cells overexpressing heparanase revealed not only a higher number of autophagic vacuoles, but also abundant release of vesicles, likely exosomes, from the cell surface, further supporting the notion that heparanase enhances exosome secretion that contributes to tumor growth (Roucourt et al., 2015a; Thompson et al., 2013).
The mechanism underlying autophagy induction by heparanase is not entirely clear, but likely involves mTOR1 that plays a pivotal role in nutrient-sensing, autophagy regulation as well as biogenesis of lysosomes (Dunlop and Tee, 2014; Napolitano and Ballabio, 2016; Settembre et al, 2012; Zhitomirsky and Assaraf, 2015; 2016). mTOR1 activity inhibits autophagy but under starvation, its activity is repressed, leading to autophagy induction. We found that heparanase overexpression is associated with reduced mTOR1 activity, evident by decreased levels of p70 S6-kinase phosphorylation, an mTOR1 substrate. In contrast, heparanase-KO cells exhibited increased mTOR1 activity and p70 S6-kinase phosphorylation (Shteingauz et al., 2015). Notably, mTOR1 appeared more diffusely scattered in control cells, whereas in cells with high content of heparanase, mTOR1 is found mostly in peri-nuclear regions, co-localizing with heparanase and LysoTracker that labels acidic lysosomal vesicles (Dunlop and Tee, 2014; Napolitano and Ballabio, 2016; Settembre et al., 2012; Zhitomirsky and Assaraf, 2015; 2016). This is consistent with the notion that activation of mTOR1 by nutrients is associated with peripheral lysosomes, whereas starvation leads to peri-nuclear clustering of lysosomes and decreased mTOR1 activity (Korolchuk et al., 2011). Our results imply that induction of autophagy contributes to the pro-tumorigenic function of heparanase. This emerges from in vitro and in vivo experiments utilizing inhibitors of autophagy (chloroquine) and heparanase (PG545) alone or in combination (Shteingauz et al., 2015). Thus, combining PG545 and chloroquine in a tumor xenograft model resulted in significantly smaller and more differentiated tumors, suggesting that heparanase activity drives cancer cell de-differentiation as part of its pro-tumorigenic properties. Equally important is the ability of heparanase overexpression to confer resistance to stress, chemotherapy and targeted drugs (Ramani et al., 2016), mediated, at least in part, by enhancing autophagy (Shteingauz et al., 2015). Indeed, diverse classes of anticancer drugs induce autophagy (Larrue et al., 2016) as well as lysosomal biogenesis (Zhitomirsky and Assaraf, 2015; 2016) thus attenuating tumor cell elimination, while autophagy inhibitors overcome chemo-resistance (Levy and Thorburn, 2011; Wu et al., 2012). Based on this concept, chloroquine is currently evaluated in several clinical trials in combination with different classes of chemotherapeutic agents (Levy and Thorburn, 2011).
5. Chemotherapy induces expression and release of heparanase leading to alterations associated with an aggressive tumor phenotype
In spite of significant advances in the clinical use of chemotherapy, it often doesn’t eradicate cancers. Especially in aggressive cancers, even after several rounds and lines of chemotherapy involving concurrent administration of distinct drugs, tumor cells persist and regrow leading to relapse. In addition, chemotherapy frequently has undesirable side effects and in some cases chemotherapy has been shown to actually lead to enhanced tumor growth and provoke chemoresistance (Ramani et al., 2016). Chemotherapy often dramatically reduces tumor burden in newly diagnosed cancer patients, yet many will eventually succumb due to development of drug resistance (Ramani et al., 2016). Although little is known regarding a role for heparanase in chemoresistance, high heparanase expression promotes cell survival and is an indicator of poor patient prognosis (Vlodavsky et al., 2012). In addition, heparanase enhances cell spreading and activates macrophages, two characteristics associated with environment-mediated drug resistance (EMDR) (Shain et al., 2015; Zheng et al., 2013). High heparanase expression in cancer patients was found to be closely associated with high expression of NEK2, a chromosome instability gene that correlates highly with drug resistance, rapid relapse, and poor outcome in multiple cancers (Zhou et al., 2013). We have previously shown that cells with high levels of heparanase have enhanced Akt, STAT, P38, Erk and EGF receptor signaling activity, each of which can provide survival signals to thwart chemotherapeutic attack (Cohen-Kaplan et al., 2008a; Cohen-Kaplan et al., 2012; Gingis-Velitski et al., 2004a). These findings led us to ask whether or not heparanase plays a role in chemoresistance. Briefly, we found that heparanase-high cells (i.e., myeloma, glioma, head & neck) were more resistant to killing by cytotoxic drugs than were heparanase-low cells (Ramani et al., 2016; Shteingauz et al., 2015). In addition, exposure to chemotherapeutic drugs dramatically increased heparanase synthesis and secretion in tumor cells that survive treatment (Ostrovsky et al., 2014; Ramani et al., 2016; Ramani et al., 2015). The secreted heparanase is likely performing an important pro-survival function by priming the tumor microenvironment to better support tumor growth. This is consistent with the well-established finding that exogenous heparanase can be taken up by cells and modulate their behavior (Gingis-Velitski et al., 2004a; Gingis-Velitski et al., 2004b). These data indicate that heparanase enhances resistance to chemotherapy and that the surviving cells, because they have high heparanase expression, will be more aggressive following treatment.
Focusing on hematological malignancies, we have noted that tumor cells derived from myeloma patients following chemotherapy, had an elevated level of heparanase expression compared to cells harvested prior to therapy. This led us to speculate that treatment of myeloma cells with chemotherapeutic drugs may have the undesirable effect of stimulating the expression of heparanase. In trying to elucidate the underlying mechanism we found that anti-myeloma drugs activate the NF-κB pathway thereby elevating heparanase expression in tumor cells (Ramani et al., 2016). NF-κB has been shown to regulate heparanase promoter activity and thereby heparanase expression in different pathologies (Cao et al., 2005; Hadigal et al., 2015; Wu et al., 2010). Thus, it appears that in patients undergoing chemotherapy, NF-κB undergoes activation, leading to upregulation of heparanase. This is consistent with our finding that in the majority of multiple myeloma patients studied, the tumor cells which survived chemotherapy had elevated heparanase expression compared to baseline levels measured prior to therapy (Ostrovsky et al., 2014; Ramani et al., 2015) (Fig. 6C). In addition, chemotherapy led to release of a soluble form of heparanase from tumor cells, followed by its endocytosis by both macrophages and tumor cells in a HS-dependent manner. The endocytosed, therapy-induced, soluble heparanase stimulated the expression of pro-tumorigenic genes (HGF, MMP-9, and VEGF) and activated ERK and Akt signaling, resulting in a substantial impact on tumor regrowth (Ramani et al., 2016). Our finding that heparanase drives ERK signaling to promote drug resistance is consistent with the role of ERK in regulating myeloma cell proliferation, survival, angiogenesis and drug resistance (Purushothaman et al., 2008; Ramani et al., 2016; (Ramani et al., 2016).
Fig. 6.
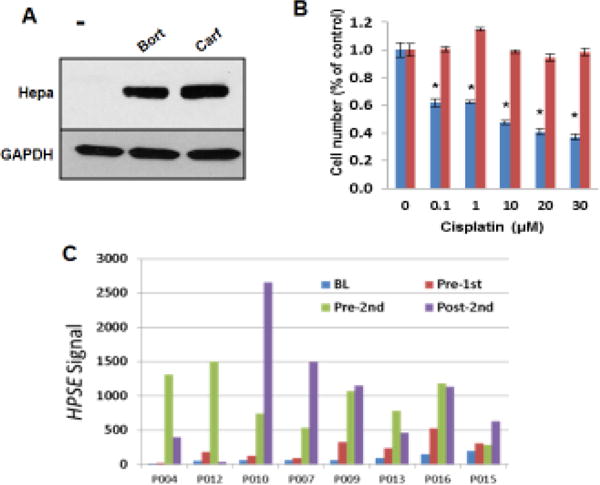
Effect of chemotherapy on heparanase expression and drug resistance. A. Anti-myeloma therapy elevates heparanase expression in CAG cells. Western blot for heparanase in cell extracts following treatment (14 h, 25 nM) with bortezomib (Bort) or carfilzomib (Carf). B. Chemoresistance. Control mock (blue) and heparanase over expressing (red) FaDu pharyngeal carcinoma cells were treated (24 h) without (0) or with the indicated concentration of cisplatin and cell survival was evaluated by MTT. Note resistance to cisplatin of cells overexpressing heparanase. C. Chemotherapy elevates heparanase expression in myeloma patients. Myeloma cells were collected from each patient at baseline (BL), just before first round of chemotherapy (Pre-1st), just before 2nd chemotherapy (Pre-2nd) and after the second round of chemotherapy (Post-2nd) and heparanase gene expression was determined. Data generated in collaboration with F. Zhan, University of Iowa (Ramani et al., 2016).
It has been well-described that cells can take up and activate exogenously supplied heparanase (Gingis-Velitski et al., 2004b; Ilan et al., 2006). To determine if soluble heparanase can be taken up by cells, myeloma cells were exposed to medium obtained from heparanase-high cells after bortezomib (BTZ) treatment (BTZ conditioned medium, BTZ CM). It was found that the myeloma cells take up the pro-form of heparanase present in the BTZ CM and process it into the active 50 kDa heparanase molecule (Ramani et al., 2016). Digesting cell surface HS using bacterial heparinase III enzyme (that extensively degrades HS) or addition of cytochalasin D, an inhibitor of actin-mediated endocytosis, both diminished sHpa uptake and processing, demonstrating a role for HS-mediated endocytosis (Ramani et al., 2016). To further confirm the involvement of HS in heparanase uptake, BTZ CM was added to human lymphoblastoid cells expressing either wild-type human syndecan-1 or a mutated form of human syndecan-1 lacking all glycosylation attachment sites including glycosaminoglycan attachment sites. Western blot analysis revealed that uptake of heparanase was diminished in the mutant cells (Ramani et al., 2016), further confirming that uptake of heparanase was HS-dependent. Importantly, heparanase uptake was inhibited in the presence of SST0001 (Roneparstat), a modified heparin that is 100% N-acetylated and 25% glycol split (Naggi et al., 2005), shown to compete with HS and thereby abolish heparanase cellular uptake (Fig. 2). Of particular significance is the up-regulation of TNFα, a pro-inflammatory cytokine the transcription of which is regulated by NF-κB and is known to promote myeloma progression (Hideshima et al., 2009). We found that incubation of macrophages with the BTZ CM significantly upregulated TNFα secretion and this effect was lost if sHpa was immunodepleted from the BTZ CM prior to its addition to the macrophages (Ramani et al., 2016). As discussed earlier, recombinant heparanase promotes TNFα production by macrophages (Blich et al., 2013; Goodall et al., 2014; Lerner et al., 2011) and possibly other cell types, via direct and/or indirect activation of tall-like receptors (Blich et al., 2013; Goodall et al., 2014). These results further establish the critical significance of heparanase in the crosstalk between tumor cells, immune cells and other cells of the tumor microenvironment (Ramani et al., 2013; Vlodavsky et al., 2012).
We have previously demonstrated that chemotherapy stimulates shedding of syndecan-1 from tumor cells (Ramani et al., 2016) and that large amounts of syndecan-1 accumulate within the marrow of myeloma patients post-chemotherapy (Bayer-Garner et al., 2001). This marrow-bound syndecan-1 could act to create reservoirs of heparanase that would be available to facilitate growth of the tumor cells that survived the therapy. This growth would be further supported by the impact of heparanase on gene expression, cell signaling and angiogenesis (Purushothaman et al., 2010; Yang et al., 2010). Taken together our findings demonstrate how chemotherapy, as opposed to its intended purpose, could potentially fuel aggressive tumor behavior by stimulating heparanase expression (Fig. 6). Moreover, these results are consistent with reports that other types of cancer therapies can induce heparanase. For example, radiotherapy enhances expression of the transcription factor Egr1 leading to elevation of heparanase in pancreatic cancer (Meirovitz et al., 2011). Also, treatment of prostate cancer cells with the methylation inhibitor 5′-aza-2-deoxycytidine was found to upregulate heparanase expression (Ogishima et al., 2005). Importantly, heparanase is up-regulated (both at the mRNA and protein levels) in patients with various hematological malignancies following high dose chemotherapy (conditioning) prior to bone marrow transplantation (Ostrovsky et al., 2014; Ramani et al., 2015) (Fig. 6C). Hence, our results indicate that: i) in tumor cells expressing little or no heparanase, anti-cancer drugs can induce the synthesis and accumulation of heparanase (Fig. 6A,C), ii) mechanistically, this chemotherapy-induced increase in heparanase expression is traced to activation of the NF-kB pathway, iii) heparanase induced by chemotherapy is released by the tumor cells and can be taken up by tumor cells and host (macrophage) cells, and iv) following uptake of heparanase, it is activated and can promote the expression of pro-tumorigenic factors and stimulate cell signaling. These effects are blocked by the anti-heparanase drug Roneparstat (Ramani et al., 2016). It appears that heparanase facilitates chemoresistance (Fig. 6B) and that heparanase inhibitors used in tandem with chemotherapeutic drugs will overcome initial chemoresistance and enhance tumor killing, providing a strong rationale for testing anti-heparanase therapy in combination with anti-cancer drugs. In fact, we have demonstrated a novel use for the heparanase inhibitor Roneparstat in blocking the uptake of therapy-induced heparanase (Ramani et al., 2016). Similarly, as discussed above, the heparanase neutralizing monoclonal antibodies not only neutralize the enzyme extracellularly, but also diminish uptake and accumulation of heparanase inside the cell (Weissmann et al., 2016). The finding that Roneparstat blocks heparanase-induced stimulation of gene expression and cell signaling indicates that this inhibitor could be used in combination with anti-myeloma drugs to improve their efficacy. In recent preclinical studies we tested this concept by treating animals harboring established multiple myeloma tumors with Roneparstat in combination with either bortezomib or melphalan. We found that the combination inhibited tumor growth significantly better than did the use of these drugs as single agents (Ramani et al., 2015). Likewise, the combination of Roneparstat with irinotecan was highly effective and well tolerated in human A204 rhabdoid sarcoma xenografts (Cassinelli et al., 2016).
Similar results were obtained when employing a model of brain metastatic breast cancer where Roneparstat was found to overcome lapatinib resistance (Zhang et al., 2015). Moreover, ERK inhibition was also observed when lapatinib-resistant breast cancer cells were treated with Roneparstat (Zhang et al., 2015), consistent with our finding that Roneparstat blocks heparanase-induced ERK signaling (Ramani et al., 2016; Ramani et al., 2015). Similarly, PG545 was recently shown to enhance the anticancer activity of chemotherapies in animal models of ovarian and pancreatic cancer (Jung et al., 2014; Winterhoff et al., 2015). Together, these studies support the notion that heparanase contributes to the promotion of drug resistance in various types of cancer. These results and the finding that chemotherapy upregulates heparanase expression and release, provide a strong rationale for a clinical trial designed to test Roneparstat, PG545 and/or the anti-heparanase antibodies in combination with anti-cancer drugs.
6. Heparanase inhibitors in cancer therapy
There is currently strong evidence that heparanase is a viable target for cancer therapy, a view widely held by researchers in both academia and pharma and the topic of multiple reviews (Barash et al., 2010; Hammond et al., 2014; Pisano et al., 2014; Ramani et al., 2013; Rivara et al., 2016; Zhang et al., 2011). There is only a single, enzymatically active form of heparanase in humans, which is expressed at very low levels in normal tissues and heparanase knock-out animals exhibit no obvious phenotype or deficits. These characteristics imply that inhibition of heparanase will cause minimal side effects in cancer patients, together stirring heparanase as a highly desirable druggable target for anti-cancer therapy. This indication is further supported by the fact that there are four ongoing clinical trials in cancer patients testing novel anti-heparanase drugs. The four drugs being tested are all heparin/HS mimetics that presumably block the active site of the enzyme by interacting with heparin-binding domains that flank the active site (Pala et al., 2016).
Heparin is one of the closest mimics of HS and is a natural choice as heparanase inhibitor. Heparin, however, is not clinically useful as an anti-cancer/anti-inflammatory drug because of its potent anticoagulant activity. Considerable efforts have thus been expended in the development of modified heparins and related polysulfated compounds with reduced anticoagulant activity. Modified heparins and heparin mimetics are the best studied heparanase inhibitors and some compounds of this class are currently in various stages of clinical development (Hammond et al., 2014; Pisano et al., 2014; Rivara et al., 2016). Chemically modified heparins have been developed in an attempt to abolish the anticoagulant activity and to retain or enhance their affinity for heparanase (Hammond et al., 2014; Pisano et al., 2014; Rivara et al., 2016). These features are critical requirements for their clinical use, because high doses and protracted treatment schedules should be used to fully exploit the therapeutic potential of heparin derivatives. Currently there are four carbohydrate-based heparanase inhibitors (PI-88, Roneparstat, M402, and PG545) that have reached clinical trials (Hammond et al., 2014; Pisano et al., 2014; Rivara et al., 2016). Because these inhibitors are heparin-based, they can bind to many heparin-binding proteins in vivo (which could be good or bad) leaving open the question as to how much of their anti-tumor activity is due specifically to blocking heparanase activity.
Muparfostat (formally PI-88), a phosphomannopentaose that displays anti-metastatic and anti-angiogenic activity (Ferro et al., 2007), is the most extensively studied HS mimetic in clinical trials. It has progressed to phase III clinical trial (NCT01402908) as adjuvant treatment of subjects with hepatitis virus related hepatocellular carcinoma after surgical resection (Kudchadkar et al., 2008; Liu et al., 2014). The study has been completed and a final report is expected in the near future. In some patients PI-88 can induce a dangerous antibody-induced thrombocytopenia, a side effect that could impact its clinical utility (Kudchadkar et al., 2008). Whilst developed primarily for cancer indications, PI-88 and maltohexaose sulfate display promising anti-inflammatory activity. Building on the clinical progress of PI-88, a new series of HS mimetics was developed, having improved properties (Dredge et al., 2010). Of particular note is PG545, a fully sulfated synthetic tetrasaccharide functionalized with a cholestanyl aglycon (Dredge et al., 2011). PG545 has demonstrated promising in vivo antiangiogenic and antimetastatic activity and potent anti-tumor activity in multiple preclinical models that are resistant to PI-88 treatment (Hammond et al., 2012; Jung et al., 2015; Ostapoff et al., 2013). Likely unrelated to its anti-heparanase activity, PG545 exerts its major anti-lymphoma effects through activation of the innate immune system via natural killer (NK) cells (Brennan et al., 2016). Unlike PI-88 and other sulfated oligosaccharides which are mixtures, PG545 and related compounds are single chemical entities that contain lipophilic modifications which endow them with significantly improved pharmacokinetic properties and in vivo activity in preclinical cancer models. PG545 is a potent heparanase inhibitor (Ki = 6 nM) (Hammond et al., 2011) and is being subjected to phase I clinical trials against multiple solid tumors (NCT02042781). In initial clinical studies, patients had reactions at the site of subcutaneous injection thus necessitating reformulation of the compound for intravenous injection.
Roneparstat (formally SST0001) is ending a phase I clinical trial in myeloma patients (NCT01764880). It is a modified heparin with a mean molecular mass of 20 kDa and is 100% N-acetylated and 25% glycol split (Vlodavsky, 2007; Naggi et al., 2005; Pala et al., 2016; Ritchie et al., 2011). The N-acetylation is responsible for the loss of the anticoagulant activity and glycol splitting likely contributes to the enhanced affinity for heparanase (Vlodavsky, 2007; Pala et al., 2016). Roneparstat was found to be effective as inhibitor of heparanase in vivo and as modulator of levels of growth factors, including VEGF and HGF (Ritchie et al., 2011). The antiangiogenic activity of Roneparstat reflects the downregulation of angiogenic heparin-binding factors and the impact of heparanase inhibition on tumor-microenvironment interactions. Roneparstat also inhibits shedding of syndecan-1, a process regulated by heparanase and known to be implicated in signaling pathways in both tumor and endothelial cells that promote tumor cell invasion and angiogenesis (Purushothaman et al., 2010; Jung et al., 2016). Preclinical studies indicate that Roneparstat is effective in the treatment of selected human tumor models including myeloma, pediatric sarcoma and pancreatic carcinoma (Cassinelli et al., 2016; Cassinelli et al., 2013; Meirovitz et al., 2011; Ritchie et al., 2011). The good tolerability of Roneparstat in protracted treatment schedules is consistent with the selective heparanase inhibition and lack of anticoagulant activity (Pala et al., 2016; Rivara et al., 2016).
Necuparanib (formally M402) is very similar to Roneparstat in that it is a glycol-split heparin having smaller molecular mass (6 kDa) (Zhou et al., 2011). This compound is N-sulfated instead of N-acetylated, which may equip it with a broader growth factor binding activity. M402 showed efficacy in metastasis models (Zhou et al., 2011) and progressed in 2012 to phase I/II clinical trial in combination with gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (NCT01621243) (Rivara et al., 2016). Both Roneparstat and Necuparanib have showed early signs of efficacy in some patients.
Another drug undergoing clinical evaluation is defibrotide, a polydisperse oligonucleotide isolated from porcine mucosa that has multiple biological effects including inhibition of heparanase gene expression and enzymatic activity (Mitsiades et al., 2009). Defibrotide was evaluated in patients with relapsed/refractory multiple myeloma in combination with multi-agent chemotherapy (Palumbo et al., 2010). In addition to the above described well advanced heparin/HS mimetics, there are other viable drug development possibilities, with monoclonal antibodies and small molecule inhibitors being the foremost options. Monoclonal antibodies against cancer related targets have displayed considerable success due to their specificity and long half-life in humans. Heparanase neutralizing monoclonal antibodies have been generated and found effective in preclinical cancer models (Weissmann et al., 2016), yet none have been tested clinically against heparanase. The drug discovery of small molecule inhibitors of heparanase has been delayed due to lack of structural information. These inhibitors include iminosugars and other putative transition-state analogues, substrate analogues, inhibitors discovered via screening of compound libraries, and natural products and their derivatives (Li et al., 2013; Rivara et al., 2016; Simizu et al., 2004; Tayel et al., 2014). Now that the crystal structure of human heparanase has been resolved (Wu et al., 2015), studies are ongoing to develop structure-based heparanase-inhibiting small molecules. These directions are in early stages of development, but are beyond the scope of the current review.
7. Conclusions and perspectives
Compelling evidence tie heparanase with all steps of tumor formation including tumor initiation, growth, metastasis, and chemoresistance (Arvatz et al., 2011a; Barash et al., 2014; Boyango et al., 2014; Hammond et al., 2014; Ramani et al., 2016; Ramani et al., 2015; Shteingauz et al., 2015). Mechanistic studies on heparanase action have focused primarily on its expression by tumor cells and revealed that heparanase promotes an aggressive tumor behavior via multiple mechanisms. However, non-tumor (host) cells including T lymphocytes, B lymphocytes, neutrophils, monocyte/macrophages, endothelial cells, osteoclasts and fibroblasts can also upregulate heparanase expression upon activation and thereby contribute not only to cancer progression (Arvatz et al., 2011b; Barash et al., 2014; Edovitsky et al., 2004; Lerner et al., 2011; Ramani et al., 2016; Vlodavsky et al., 2012), but also to acute and chronic inflammation (Goldberg et al., 2013; Li et al., 2008; Vlodavsky et al., 2012), autoimmunity (de Mestre et al., 2007; Li et al., 2008), atherosclerosis (Vlodavsky et al., 2013), tissue fibrosis (Secchi et al., 2015), kidney dysfunction (Garsen et al., 2016a; Garsen et al., 2016b; van den Hoven et al., 2007), ocular surface dysfunction (McKown et al., 2009; Zhang et al., 2010) diabetes (Parish et al., 2013) and diabetic complications (Gil et al., 2012; Wang et al., 2013). Noteworthy, heparanase appears to activate cells of the innate immune system and soluble HS fragments generated by heparanase trigger the expression and secretion of pro-inflammatory cytokines through toll-like receptors (TLR) (Blich et al., 2013; Brunn et al., 2005; Goodall et al., 2014; Hermano et al., 2012). Moreover, we have recently revealed a linear cascade by which heparanase activates Erk, p38 and JNK signaling in macrophages, leading to increased c-Fos levels and induction of cytokine expression (Gutter-Kapon et al., unpublished results). These results identify heparanase as a key mediator of macrophage activation and function, paving the way for the development of heparanase-based treatment modalities that either stimulate or inhibit macrophage activation and function in a broad range of human diseases, including cancer, inflammation, kidney failure, diabetes and atherosclerosis. Heparanase also tailors syndecan for exosome formation (David and Zimmermann, 2016) and is found as cargo within exosomes, providing a potential mechanism for the crosstalk between the tumor, the host and the tumor microenvironment and for distributing heparanase among cells within a tumor (Roucourt et al., 2015a; Thompson et al., 2013). While heparanase up-regulation by tumor cells is well documented, the pro-tumorigenic contribution of heparanase provided by the host and by cells composing the tumor microenvironment has not been sufficiently explored. The significance of heparanase residing in the tumor microenvironment has been demonstrated by showing that heparanase-neutralizing antibodies attenuate the growth of lymphoma cells that do not express heparanase, implying that neutralization of heparanase contributed by the tumor microenvironment may be sufficient to restrain tumor growth (Weissmann et al., 2016). Heparanase resides primarily within endocytic vesicles, assuming a polar, peri-nuclear localization and co-localizing with lysosomal markers. Residence and accumulation of heparanase in lysosomes indicate that this enzyme may function in the normal physiology of this organelle. In a search for such function we identified a role of lysosomal heparanase in augmenting autophagy, a physiological process required to remove unfolded proteins and damaged organelles, thus maintaining cellular homeostasis. Autophagy is further induced by starvation and stress, including chemotherapy, thereby promoting cancer cells survival and chemoresistance by providing their metabolic needs (Shteingauz et al., 2015). While the traditional thinking envisions heparanase as an enzyme that functions extracellularly to cleave HS and facilitate remodeling and ‘priming’ of the ECM, our results indicate that heparanase may also function from inside the cells (Ilan et al., 2015). From a translational point of view, targeting heparanase in the lysosome may be as important as its inhibition extracellularly, but the ability of currently available heparanase inhibitors to cross the plasma membrane and enter the cell is unclear. Alternatively, the pro-autophagy function of heparanase can be attenuated by inhibiting its cellular uptake and hence decreasing its lysosomal content (Ilan et al., 2015). This opens the way for the development of a new class of highly specific inhibitors (i.e., monoclonal antibodies) that prevent heparanase uptake by targeting its heparin-binding domain. Involvement of heparanase in exosome formation, autophagy and activation of innate immune cells indicate that it fulfills normal functions associated, for example, with vesicular traffic, lysosomal secretion, stress response, HS turnover and immune surveillances. Unraveling these aspects of heparanase biology is ongoing and critical to our understanding of its multiple roles in health and disease. A related aspect is the identification of a putative natural inhibitor of heparanase, a common feature of ECM degrading enzymes. Such a role appears to be fulfilled by heparanase 2 (Hpa2), a close homolog of heparanase that lacks intrinsic HS-degrading activity, the hallmark of heparanase, but retains the capacity to bind HS with high affinity (Levy-Adam et al., 2010; McKenzie et al., 2000). In head and neck cancer patients, Hpa2 expression correlates with prolonged time to disease recurrence and inversely correlates with tumor cell dissemination to regional lymph nodes (Gross-Cohen et al., 2016a), suggesting that Hpa2 functions as a tumor suppressor. Similarly, Hpa2 expression inversely correlates with bladder carcinoma grade and stage (Gross-Cohen et al., 2016b). The molecular mechanism associated with favorable prognosis following Hpa2 induction is unclear, but likely involves reduced Id1 expression, a transcription factor highly implicated in VEGF-A and VEGF-C gene regulation (Gross-Cohen et al., 2016a).
The long term research focusing on the causal involvement of heparanase in cancer progression together with strong preclinical data validated heparanase as a promising target for cancer therapy. Optimal efficacy is achieved when compounds are administered during the early phase of tumor growth following tumor cell inoculation. Indeed, the inhibition of release/activation of growth factors that provide favorable “soil” conditions for tumor growth might be the critical event implicated in the control of primary tumor growth. This consideration would suggest a therapeutic potential of heparanase inhibitors in the control of minimal residual disease, which requires support of the microenvironment, rather than in the treatment the bulky progressive disease. Heparin/HS mimicking compounds are being tested in clinical trials for various cancers and the development of heparanase-inhibiting small molecules and neutralizing antibodies is ongoing. Noteworthy, heparanase is up-regulated in patients with various hematological malignancies following high dose chemotherapy. It therefore appears that heparanase facilitates chemoresistance and that heparanase inhibitors used in tandem with chemotherapeutic drugs will overcome initial chemoresistance and enhance tumor killing, providing a strong rationale for testing anti-heparanase therapy in combination with anti-cancer drugs. Thus, the efficacy of heparanase inhibitors can best be exploited in rational combination therapy with other antitumor agents. Yet, only few studies address this aspect of the therapeutic applications of heparanase inhibitors.
Acknowledgments
This study was supported by research grants awarded to I.V. by the Israel Science Foundation (ISF grant 601/14), the ISF-UGB joint research program (grant No. 2277/15), and the Israel Cancer Research Fund (ICRF); to RS by the National Institutes of Health (CA138340) and jointly to I.V. and R.S. by the United States-Israel Binational Science Foundation (BSF); and to M.E. by grants from the Israel Science Foundation (ISF grants 806/14 and 663/16) and the Mizutani Foundation for Glycoscience. I. Vlodavsky is a Research Professor of the ICRF. We gratefully acknowledge the contribution, motivation, and assistance of the research teams in the Cancer and Vascular Biology Research Center of the Rappaport Faculty of Medicine (Technion, Haifa). We apologize for not citing several relevant articles, due to space limitation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
I.V. has a research contract with sigma-tau Research Switzerland and R.S. is a member of the Scientific Advisory Board of sigma-tau Research Switzerland.
References
- Aalkjaer C, Boedtkjer DB. Getting neointimal: the emergence of heparanase into the vascular matrix. Circulation Res. 2009;104:277–279. doi: 10.1161/CIRCRESAHA.108.192625. [DOI] [PubMed] [Google Scholar]
- Abboud-Jarrous G, Atzmon R, Peretz T, Palermo C, Gadea BB, Joyce JA, Vlodavsky I. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J Biol Chem. 2008;283:18167–18176. doi: 10.1074/jbc.M801327200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abboud-Jarrous G, Rangini-Guetta Z, Aingorn H, Atzmon R, Elgavish S, Peretz T, Vlodavsky I. Site-directed mutagenesis, proteolytic cleavage, and activation of human proheparanase. J Biol Chem. 2005;280:13568–13575. doi: 10.1074/jbc.M413370200. [DOI] [PubMed] [Google Scholar]
- Akbarshahi H, Axelsson JB, Said K, Malmstrom A, Fischer H, Andersson R. TLR4 dependent heparan sulphate-induced pancreatic inflammatory response is IRF3-mediated. J Transl Med. 2011;9:219. doi: 10.1186/1479-5876-9-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL, Filla MS, Rapraeger AC. Role of heparan sulfate as a tissue-specific regulator of FGF-4 and FGF receptor recognition. J Cell Biol. 2001;155:845–858. doi: 10.1083/jcb.200106075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansell SM. Non-Hodgkin Lymphoma: Diagnosis and Treatment. Mayo Clin Proc. 2015;90:1152–1163. doi: 10.1016/j.mayocp.2015.04.025. [DOI] [PubMed] [Google Scholar]
- Arvatz G, Barash U, Nativ O, Ilan N, Vlodavsky I. Post-transcriptional regulation of heparanase gene expression by a 3′ AU-rich element. Faseb J. 2011a;24:4969–4976. doi: 10.1096/fj.10-156372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvatz G, Shafat I, Levy-Adam F, Ilan N, Vlodavsky I. The heparanase system and tumor metastasis: is heparanase the seed and soil? Cancer Metastasis Rev. 2011b;30:253–268. doi: 10.1007/s10555-011-9288-x. [DOI] [PubMed] [Google Scholar]
- Au Yeung CL, Co NN, Tsuruga T, Yeung TL, Kwan SY, Leung CS, Li Y, Lu ES, Kwan K, Wong KK, Schmandt R, Lu KH, Mok SC. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun. 2016;7:11150. doi: 10.1038/ncomms11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson J, Xu D, Kang BN, Nussbacher JK, Handel TM, Ley K, Sriramarao P, Esko JD. Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood. 2012;120:1742–1751. doi: 10.1182/blood-2012-03-417139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baietti MF, Zhang Z, Mortier E, Melchior A, Degeest G, Geeraerts A, Ivarsson Y, Depoortere F, Coomans C, Vermeiren E, Zimmermann P, David G. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nature cell Biol. 2012;14:677–685. doi: 10.1038/ncb2502. [DOI] [PubMed] [Google Scholar]
- Baker AB, Gibson WJ, Kolachalama VB, Golomb M, Indolfi L, Spruell C, Zcharia E, Vlodavsky I, Edelman ER. Heparanase regulates thrombosis in vascular injury and stent-induced flow disturbance. J Am Coll Cardiol. 2012;59:1551–1560. doi: 10.1016/j.jacc.2011.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AB, Groothuis A, Jonas M, Ettenson DS, Shazly T, Zcharia E, Vlodavsky I, Seifert P, Edelman ER. Heparanase alters arterial structure, mechanics, and repair following endovascular stenting in mice. Circulation Res. 2009;104:380–387. doi: 10.1161/CIRCRESAHA.108.180695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash U, Cohen-Kaplan V, Dowek I, Sanderson RD, Ilan N, Vlodavsky I. Proteoglycans in health and disease: new concepts for heparanase function in tumor progression and metastasis. FEBS J. 2010;277:3890–3903. doi: 10.1111/j.1742-4658.2010.07799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash U, Zohar Y, Wildbaum G, Beider K, Nagler A, Karin N, Ilan N, Vlodavsky I. Heparanase enhances myeloma progression via CXCL10 downregulation. Leukemia. 2014;28:2178–2187. doi: 10.1038/leu.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart S, Ellenrieder V, Fernandez-Zapico ME. Oncogenic transcription factors: cornerstones of inflammation-linked pancreatic carcinogenesis. Gut. 2013;62:310–316. doi: 10.1136/gutjnl-2011-301008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer-Garner IB, Sanderson RD, Dhodapkar MV, Owens RB, Wilson CS. Syndecan-1 (CD138) immunoreactivity in bone marrow biopsies of multiple myeloma: shed syndecan-1 accumulates in fibrotic regions. Mod Pathol. 2001;14:1052–1058. doi: 10.1038/modpathol.3880435. [DOI] [PubMed] [Google Scholar]
- Beauvais DM, Jung O, Yang Y, Sanderson RD, Rapraeger AC. Syndecan-1 (CD138) suppresses apoptosis in multiple myeloma by activating IGF1 receptor: prevention by synstatinIGF1R inhibits tumor growth. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-16-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvais DM, Rapraeger AC. Syndecans in tumor cell adhesion and signaling. Reprod Biol Endocrinol. 2004;2:3. doi: 10.1186/1477-7827-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmiro CL, Souza HS, Elia CC, Castelo-Branco MT, Silva FR, Machado RL, Pavao MS. Biochemical and immunohistochemical analysis of glycosaminoglycans in inflamed and non-inflamed intestinal mucosa of patients with Crohn’s disease. Int J Colorectal Dis. 2005;20:295–304. doi: 10.1007/s00384-004-0677-2. [DOI] [PubMed] [Google Scholar]
- Ben-Zaken O, Shafat I, Gingis-Velitski S, Bangio H, Kelson IK, Alergand T, Amor Y, Maya RB, Vlodavsky I, Ilan N. Low and high affinity receptors mediate cellular uptake of heparanase. Int J Biochem & Cell Biol. 2008;40:530–542. doi: 10.1016/j.biocel.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamron S, Nechushtan H, Verbovetski I, Krispin A, Abboud-Jarrous G, Zcharia E, Edovitsky E, Nahari E, Peretz T, Vlodavsky I, Mevorach D. Translocation of active heparanase to cell surface regulates degradation of extracellular matrix heparan sulfate upon transmigration of mature monocyte-derived dendritic cells. J Immunol. 2006;176:6417–6424. doi: 10.4049/jimmunol.176.11.6417. [DOI] [PubMed] [Google Scholar]
- Benhamron S, Reiner I, Zcharia E, Atallah M, Grau A, Vlodavsky I, Mevorach D. Dissociation between mature phenotype and impaired transmigration in dendritic cells from heparanase-deficient mice. PloS one. 2012;7:e35602. doi: 10.1371/journal.pone.0035602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Ann Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- Blich M, Golan A, Arvatz G, Sebbag A, Shafat I, Sabo E, Cohen-Kaplan V, Petcherski S, Avniel-Polak S, Eitan A, Hammerman H, Aronson D, Axelman E, Ilan N, Nussbaum G, Vlodavsky I. Macrophage Activation by Heparanase Is Mediated by TLR-2 and TLR-4 and Associates With Plaque Progression. Arterioscler Thromb & Vasc Biol (ATVB) 2012;33:e56–65. doi: 10.1161/ATVBAHA.112.254961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blonska M, Agarwal NK, Vega F. Shaping of the tumor microenvironment: Stromal cells and vessels. Semin Cancer Biol. 2015;34:3–13. doi: 10.1016/j.semcancer.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode JG, Ehlting C, Haussinger D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal. 2012;24:1185–1194. doi: 10.1016/j.cellsig.2012.01.018. [DOI] [PubMed] [Google Scholar]
- Bode L, Salvestrini C, Park PW, Li JP, Esko JD, Yamaguchi Y, Murch S, Freeze HH. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J Clin Invest. 2008;118:229–238. doi: 10.1172/JCI32335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyango I, Barash U, Naroditsky I, Li JP, Hammond E, Ilan N, Vlodavsky I. Heparanase cooperates with Ras to drive breast and skin tumorigenesis. Cancer Res. 2014;74:4504–4514. doi: 10.1158/0008-5472.CAN-13-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan TV, Lin L, Brandstadter JD, Rendell VR, Dredge K, Huang X, Yang Y. Heparan sulfate mimetic PG545-mediated antilymphoma effects require TLR9-dependent NK cell activation. J Clin Invest. 2016;126:207–219. doi: 10.1172/JCI76566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun R, Naroditsky I, Waterman M, Ben-Izhak O, Groisman G, Ilan N, Vlodavsky I. Heparanase expression by Barrett’s epithelium and during esophageal carcinoma progression. Mod Pathol. 2009;22:1548–1554. doi: 10.1038/modpathol.2009.115. [DOI] [PubMed] [Google Scholar]
- Brunn GJ, Bungum MK, Johnson GB, Platt JL. Conditional signaling by Toll-like receptor 4. Faseb J. 2005;19:872–874. doi: 10.1096/fj.04-3211fje. [DOI] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao HJ, Fang Y, Zhang X, Chen WJ, Zhou WP, Wang H, Wang LB, Wu JM. Tumor metastasis and the reciprocal regulation of heparanase gene expression by nuclear factor kappa B in human gastric carcinoma tissue. World J Gastroenterology. 2005;11:903–907. doi: 10.3748/wjg.v11.i6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassinelli G, Favini E, Dal Bo L, Tortoreto M, De Maglie M, Dagrada G, Pilotti S, Zunino F, Zaffaroni N, Lanzi C. Antitumor efficacy of the heparan sulfate mimic roneparstat (SST0001) against sarcoma models involves multi-target inhibition of receptor tyrosine kinases. Oncotarget. 2016 doi: 10.18632/oncotarget.10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassinelli G, Lanzi C, Tortoreto M, Cominetti D, Petrangolini G, Favini E, Zaffaroni N, Pisano C, Penco S, Vlodavsky I, Zunino F. Antitumor efficacy of the heparanase inhibitor SST0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochem Pharmacol. 2013;85:1424–1432. doi: 10.1016/j.bcp.2013.02.023. [DOI] [PubMed] [Google Scholar]
- Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology. 2012;143:550–563. doi: 10.1053/j.gastro.2012.07.009. [DOI] [PubMed] [Google Scholar]
- Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest. 2004;84:282–291. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- Cohen-Kaplan V, Doweck I, Naroditsky I, Vlodavsky I, Ilan N. Heparanase augments epidermal growth factor receptor phosphorylation: correlation with head and neck tumor progression. Cancer Res. 2008a;68:10077–10085. doi: 10.1158/0008-5472.CAN-08-2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Kaplan V, Jrbashyan J, Yanir Y, Naroditsky I, Ben-Izhak O, Ilan N, Doweck I, Vlodavsky I. Heparanase Induces Signal Transducer and Activator of Transcription (STAT) Protein Phosphorylation: Preclinical and clinical significance in head and neck cancer. J Biol Chem. 2012;287:6668–6678. doi: 10.1074/jbc.M111.271346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Kaplan V, Naroditsky I, Zetser A, Ilan N, Vlodavsky I, Doweck I. Heparanase induces VEGF C and facilitates tumor lymphangiogenesis. Int J Cancer. 2008b;123:2566–2573. doi: 10.1002/ijc.23898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu AA, Vink H, Spaan JA. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arteriosclerosis Thromb & Vascular Biol (ATVB) 2003;23:1541–1547. doi: 10.1161/01.ATV.0000085630.24353.3D. [DOI] [PubMed] [Google Scholar]
- David G, Zimmermann P. Heparanase tailors syndecan for exosome production. Mol Cell Oncol. 2016;3:e1047556. doi: 10.1080/23723556.2015.1047556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G, Henrissat B. Structures and mechanisms of glycosyl hydrolases. Structure. 1995;3:853–859. doi: 10.1016/S0969-2126(01)00220-9. [DOI] [PubMed] [Google Scholar]
- Day R, Forbes A. Heparin, cell adhesion, and pathogenesis of inflammatory bowel disease. Lancet. 1999;354:62–65. doi: 10.1016/S0140-6736(98)09267-8. [DOI] [PubMed] [Google Scholar]
- Di Marzo L, Desantis V, Solimando AG, Ruggieri S, Annese T, Nico B, Fumarulo R, Vacca A A, Frassanito MA. Microenvironment drug resistance in multiple myeloma: emerging new players. Oncotarget. 2016 doi: 10.18632/oncotarget.10849. 2016 Jul 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mestre AM, Staykova MA, Hornby JR, Willenborg DO, Hulett MD. Expression of the heparan sulfate-degrading enzyme heparanase is induced in infiltrating CD4+ T cells in experimental autoimmune encephalomyelitis and regulated at the level of transcription by early growth response gene 1. J Leukocyte Biol. 2007;82:1289–1300. doi: 10.1189/jlb.0507315. [DOI] [PubMed] [Google Scholar]
- Doviner V, Maly B, Kaplan V, Gingis-Velitski S, Ilan N, Vlodavsky I, Sherman Y. Spatial and temporal heparanase expression in colon mucosa throughout the adenoma-carcinoma sequence. Mod Pathol. 2006;19:878–888. doi: 10.1038/modpathol.3800603. [DOI] [PubMed] [Google Scholar]
- Doweck I, Kaplan-Cohen V, Naroditsky I, Sabo E, Ilan N, Vlodavsky I. Heparanase localization and expression by head and neck cancer: correlation with tumor progression and patient survival. Neoplasia. 2006;8:1055–1061. doi: 10.1593/neo.06577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dredge K, Hammond E, Davis K, Li CP, Liu L, Johnstone K, Handley P, Wimmer N, Gonda TJ, Gautam A, Ferro V, Bytheway I. The PG500 series: novel heparan sulfate mimetics as potent angiogenesis and heparanase inhibitors for cancer therapy. Invest New drugs. 2010;28:276–283. doi: 10.1007/s10637-009-9245-5. [DOI] [PubMed] [Google Scholar]
- Dredge K, Hammond E, Handley P, Gonda TJ, Smith MT, Vincent C, Brandt R, Ferro V, Bytheway I. PG545, a dual heparanase and angiogenesis inhibitor, induces potent anti-tumour and anti-metastatic efficacy in preclinical models. Br J Cancer. 2011;104:635–642. doi: 10.1038/bjc.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop EA, Tee AR. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014;36:121–129. doi: 10.1016/j.semcdb.2014.08.006. [DOI] [PubMed] [Google Scholar]
- Edovitsky E, Elkin M, Zcharia E, Peretz T, Vlodavsky I. Heparanase Gene Silencing, Tumor Invasiveness, Angiogenesis, and Metastasis. J Natl Cancer Inst. 2004;96:1219–1230. doi: 10.1093/jnci/djh230. [DOI] [PubMed] [Google Scholar]
- Edovitsky E, Lerner I, Zcharia E, Peretz T, Vlodavsky I, Elkin M. Role of endothelial heparanase in delayed-type hypersensitivity. Blood. 2006;107:3609–3616. doi: 10.1182/blood-2005-08-3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeberg M, Kjeken R, Kolset SO, Berg T, Prydz K. Internalization and stepwise degradation of heparan sulfate proteoglycans in rat hepatocytes. Biochim Biophys Acta. 2001;1541:135–149. doi: 10.1016/s0167-4889(01)00132-x. [DOI] [PubMed] [Google Scholar]
- El-Assal ON, Yamanoi A, Ono T, Kohno H, Nagasue N. The Clinicopathological Significance of Heparanase and Basic Fibroblast Growth Factor Expressions in Hepatocellular Carcinoma. Clin Cancer Res. 2001;7:1299–1305. [PubMed] [Google Scholar]
- Elkin M, Ilan N, Ishai-Michaeli R, Friedmann Y, Papo O, Pecker I, Vlodavsky I. Heparanase as mediator of angiogenesis: mode of action. Faseb J. 2001;15:1661–1663. doi: 10.1096/fj.00-0895fje. [DOI] [PubMed] [Google Scholar]
- Escobar Galvis ML, Jia J, Zhang X, Jastrebova N, Spillmann D, Gottfridsson E, van Kuppevelt TH, Zcharia E, Vlodavsky I, Lindahl U, Li JP. Transgenic or tumor-induced expression of heparanase upregulates sulfation of heparan sulfate. Nat Chem Biol. 2007a;3:773–778. doi: 10.1038/nchembio.2007.41. [DOI] [PubMed] [Google Scholar]
- Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. 2013;73:4965–4977. doi: 10.1158/0008-5472.CAN-13-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro V, Dredge K, Liu L, Hammond E, Bytheway I, Li C, Johnstone K, Karoli T, Davis K, Copeman E, Gautam A. PI-88 and novel heparan sulfate mimetics inhibit angiogenesis. Semin Thromb Hemost. 2007;33:557–568. doi: 10.1055/s-2007-982088. [DOI] [PubMed] [Google Scholar]
- Floer M, Gotte M, Wild MK, Heidemann J, Gassar ES, Domschke W, Kiesel L, Luegering A, Kucharzik T. Enoxaparin improves the course of dextran sodium sulfate-induced colitis in syndecan-1-deficient mice. Am J Pathol. 2010;176:146–157. doi: 10.2353/ajpath.2010.080639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friand V, David G, Zimmermann P. Syntenin and syndecan in the biogenesis of exosomes. Biol Cell. 2015;107:331–341. doi: 10.1111/boc.201500010. [DOI] [PubMed] [Google Scholar]
- Fux L, Feibish N, Cohen-Kaplan V, Gingis-Velitski S, Feld S, Geffen C, Vlodavsky I, Ilan N. Structure-function approach identifies a COOH-terminal domain that mediates heparanase signaling. Cancer Res. 2009a;69:1758–1767. doi: 10.1158/0008-5472.CAN-08-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fux L, Ilan N, Sanderson RD, Vlodavsky I. Heparanase: busy at the cell surface. Trends Biochem Sci. 2009b;34:511–519. doi: 10.1016/j.tibs.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsen M, Benner M, Dijkman HB, van Kuppevelt TH, Li JP, Rabelink TJ, Vlodavsky I, Berden JH, Rops AL, Elkin M, van der Vlag J. Heparanase Is Essential for the Development of Acute Experimental Glomerulonephritis. Am J Pathol. 2016a;186:805–815. doi: 10.1016/j.ajpath.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Garsen M, Lenoir O, Rops AL, Dijkman HB, Willemsen B, van Kuppevelt TH, Rabelink TJ, Berden JH, Tharaux PL, van der Vlag J. Endothelin-1 Induces Proteinuria by Heparanase-Mediated Disruption of the Glomerular Glycocalyx. J Am Society Nephrol (JASN) 2016b doi: 10.1681/ASN.2015091070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil N, Goldberg R, Neuman T, Garsen M, Zcharia E, Rubinstein AM, van Kuppevelt T, Meirovitz A, Pisano C, Li JP, van der Vlag J, Vlodavsky I, Elkin M. Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes. 2012;61:208–216. doi: 10.2337/db11-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil N, Goldberg R, Neuman T, Garsen M, Zcharia E, Rubinstein A, van Kuppevelt T, Meirovitz A, Pisano C, Li JP, van der Vlag J, Vlodavsky I, Elkin M. Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes. 2012;61:208–2016. doi: 10.2337/db11-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingis-Velitski S, Zetser A, Flugelman MY, Vlodavsky I, Ilan N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J Biol Chem. 2004a;279:23536–23541. doi: 10.1074/jbc.M400554200. [DOI] [PubMed] [Google Scholar]
- Gingis-Velitski S, Zetser A, Kaplan V, Ben-Zaken O, Cohen E, Levy-Adam F, Bashenko Y, Flugelman MY, Vlodavsky I, Ilan N. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J Biol Chem. 2004b;279:44084–44092. doi: 10.1074/jbc.M402131200. [DOI] [PubMed] [Google Scholar]
- Goldberg R, Meirovitz A, Hirshoren N, Bulvik R, Binder A, Rubinstein AM, Elkin M. Versatile role of heparanase in inflammation. Matrix Biol. 2013;32:234–240. doi: 10.1016/j.matbio.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg R, Rubinstein AM, Gil N, Hermano E, Li JP, van der Vlag J, Atzmon R, Meirovitz A, Elkin M. Role of heparanase-driven inflammatory cascade in pathogenesis of diabetic nephropathy. Diabetes. 2014;63:4302–4313. doi: 10.2337/db14-0001. [DOI] [PubMed] [Google Scholar]
- Goldshmidt O, Nadav L, Aingorn H, Irit C, Feinstein N, Ilan N, Zamir E, Geiger B, Vlodavsky I, Katz BZ. Human heparanase is localized within lysosomes in a stable form. Exp Cell Res. 2002;281:50–62. doi: 10.1006/excr.2002.5651. [DOI] [PubMed] [Google Scholar]
- Goodall KJ, Poon IK, Phipps S, Hulett MD. Soluble Heparan Sulfate Fragments Generated by Heparanase Trigger the Release of Pro-Inflammatory Cytokines through TLR-4. PloS one. 2014;9:e109596. doi: 10.1371/journal.pone.0109596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotte M. Syndecans in inflammation. Faseb J. 2003;17:575–591. doi: 10.1096/fj.02-0739rev. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Karin M. Inflammation and oncogenesis: a vicious connection. Curr Opin Genet & Develop. 2010;20:65–71. doi: 10.1016/j.gde.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross-Cohen M, Feld S, Doweck I, Neufeld G, Hasson P, Arvatz G, Barash U, Naroditsky I, Ilan N, Vlodavsky I. Heparanase 2 attenuates head and neck tumor vascularity and growth. Cancer Res. 2016a;76:2791–2801. doi: 10.1158/0008-5472.CAN-15-1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross-Cohen M, Feld S, Naroditsky I, Nativ O, Ilan N, Vlodavsky I. Heparanase 2 expression inversely correlates with bladder carcinoma grade and stage. Oncotarget. 2016b;7:22556–22565. doi: 10.18632/oncotarget.8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo CH, Koo CY, Bay BH, Tan PH, Yip GW. Comparison of the effects of differentially sulphated bovine kidney- and porcine intestine-derived heparan sulphate on breast carcinoma cellular behaviour. Int J Oncol. 2007;31:1415–1423. [PubMed] [Google Scholar]
- Gupta RB, Harpaz N, Itzkowitz S, Hossain S, Matula S, Kornbluth A, Bodian C, Ullman T. Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: a cohort study. Gastroenterology. 2007;133:1099–1105. doi: 10.1053/j.gastro.2007.08.001. quiz 1340–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadigal SR, Agelidis AM, Karasneh GA, Antoine TE, Yakoub AM, Ramani VC, Djalilian AR, Sanderson RD, Shukla D. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat Commun. 2015;6:6985. doi: 10.1038/ncomms7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond E, Brandt R, Dredge K. PG545, a heparan sulfate mimetic, reduces heparanase expression in vivo, blocks spontaneous metastases and enhances overall survival in the 4T1 breast carcinoma model. PloS one. 2012;7:e52175. doi: 10.1371/journal.pone.0052175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond E, Khurana A, Shridhar V, Dredge K. The Role of Heparanase and Sulfatases in the Modification of Heparan Sulfate Proteoglycans within the Tumor Microenvironment and Opportunities for Novel Cancer Therapeutics. Front Oncol. 2014;4:195. doi: 10.3389/fonc.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond E, Li CP, Ferro V. Development of a colorimetric assay for heparanase activity suitable for kinetic analysis and inhibitor screening. Analytical biochemistry. 2011;396:112–116. doi: 10.1016/j.ab.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- He YQ, Sutcliffe EL, Bunting KL, Li J, Goodall KJ, Poon IK, Hulett MD, Freeman C, Zafar A, McInnes RL, Taya T, Parish CR, Rao S. The endoglycosidase heparanase enters the nucleus of T lymphocytes and modulates H3 methylation at actively transcribed genes via the interplay with key chromatin modifying enzymes. Transcription. 2012;3:130–145. doi: 10.4161/trns.19998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermano E, Lerner I, Elkin M. Heparanase enzyme in chronic inflammatory bowel disease and colon cancer. Cell Mol Life Sci. 2012;69:2501–2513. doi: 10.1007/s00018-012-0930-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermano E, Meirovitz A, Meir K, Nussbaum G, Appelbaum L, Peretz T, Elkin M. Macrophage polarization in pancreatic carcinoma: role of heparanase enzyme. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershkoviz R, Mor F, Miao HQ, Vlodavsky I, Lider O. Differential effects of polysulfated polysaccharide on experimental encephalomyelitis, proliferation of autoimmune T cells, and inhibition of heparanase activity. J Autoimmun. 1995;8:741–750. doi: 10.1006/jaut.1995.0055. [DOI] [PubMed] [Google Scholar]
- Hideshima T, Ikeda H, Chauhan D, Okawa Y, Raje N, Podar K, Mitsiades C, Munshi NC, Richardson PG, Carrasco RD, Anderson KC. Bortezomib induces canonical nuclear factor-kappa B activation in multiple myeloma cells. Blood. 2009;114:1046–1052. doi: 10.1182/blood-2009-01-199604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka N, Ino Y, Sekine S, Tsuda H, Shimada K, Kosuge T, Zavada J, Yoshida M, Yamada K, Koyama T, Kanai Y. Tumour necrosis is a postoperative prognostic marker for pancreatic cancer patients with a high interobserver reproducibility in histological evaluation. Br J Cancer. 2010;103:1057–1065. doi: 10.1038/sj.bjc.6605854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AC, Mori R, Vallbohmer D, Brabender J, Drebber U, Baldus SE, Klein E, Azuma M, Metzger R, Hoffmann C, Hoelscher AH, Danenberg KD, Prenzel KL, Danenberg PV. High expression of heparanase is significantly associated with dedifferentiation and lymph node metastasis in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA and via HIF1a to HB-EGF and bFGF. J Gastrointest Surg. 2008;12:1674–1681. doi: 10.1007/s11605-008-0628-2. [DOI] [PubMed] [Google Scholar]
- Hulett MD, Hornby JR, Ohms SJ, Zuegg J, Freeman C, Gready JE, Parish CR. Identification of active-site residues of the pro-metastatic endoglycosidase heparanase. Biochemistry. 2000;39:15659–15667. doi: 10.1021/bi002080p. [DOI] [PubMed] [Google Scholar]
- Ijichi H, Chytil A, Gorska AE, Aakre ME, Bierie B, Tada M, Mohri D, Miyabayashi K, Asaoka Y, Maeda S, Ikenoue T, Tateishi K, Wright CV, Koike K, Omata M, Moses HL. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. The Journal of clinical investigation. 2011;121:4106–4117. doi: 10.1172/JCI42754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilan N, Elkin M, Vlodavsky I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. The international journal of biochemistry & cell biology. 2006;38:2018–2039. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Ilan N, Shteingauz A, Vlodavsky I. Function from within: Autophagy induction by HPSE/heparanase-new possibilities for intervention. Autophagy. 2015;11:2387–2389. doi: 10.1080/15548627.2015.1115174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo RV, Sanderson RD. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J Cell Mol Med. 2011;15:1013–1031. doi: 10.1111/j.1582-4934.2010.01236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J immunol. 2002;168:5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375–3383. doi: 10.1172/JCI57158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung DB, Yun M, Kim EO, Kim J, Kim B, Jung JH, Wang E, Mukhopadhyay D, Hammond E, Dredge K, Shridhar V, Kim SH. The heparan sulfate mimetic PG545 interferes with Wnt/beta-catenin signaling and significantly suppresses pancreatic tumorigenesis alone and in combination with gemcitabine. Oncotarget. 2015;6:4992–5004. doi: 10.18632/oncotarget.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung O, Trapp-Stamborski V, Purushothaman A, Jin H, Wang H, Sanderson RD, Rapraeger AC. Heparanase-induced shedding of syndecan-1/CD138 in myeloma and endothelial cells activates VEGFR2 and an invasive phenotype: prevention by novel synstatins. Oncogenesis. 2016;5:e202. doi: 10.1038/oncsis.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Wang H, Kainulainen V, Fitzgerald ML, Ledbetter S, Ornitz DM, Bernfield M. Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2. Nature Med. 1998;4:691–697. doi: 10.1038/nm0698-691. [DOI] [PubMed] [Google Scholar]
- Kelly T, Miao HQ, Yang Y, Navarro E, Kussie P, Huang Y, MacLeod V, Casciano J, Joseph L, Zhan F, Zangari M, Barlogie B, Shaughnessy J, Sanderson RD. High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res. 2003;63:8749–8756. [PubMed] [Google Scholar]
- Koliopanos A, Friess H, Kleeff J, Shi X, Liao Q, Pecker I, Vlodavsky I, Zimmermann A, Buchler MW. Heparanase expression in primary and metastatic pancreatic cancer. Cancer Res. 2001;61:4655–4659. [PubMed] [Google Scholar]
- Kopp A, Buechler C, Neumeier M, Weigert J, Aslanidis C, Scholmerich J, Schaffler A. Innate immunity and adipocyte function: ligand-specific activation of multiple Toll-like receptors modulates cytokine, adipokine, and chemokine secretion in adipocytes. Obesity (Silver Spring) 2009;17:648–656. doi: 10.1038/oby.2008.607. [DOI] [PubMed] [Google Scholar]
- Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, O’Kane CJ, Deretic V, Rubinsztein DC. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011;13:453–460. doi: 10.1038/ncb2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudchadkar R, Gonzalez R, Lewis KD. PI-88: a novel inhibitor of angiogenesis. Expert Opin Investig Drugs. 2008;17:1769–1776. doi: 10.1517/13543784.17.11.1769. [DOI] [PubMed] [Google Scholar]
- Lapidot M, Barash U, Zohar Y, Geffen Y, Naroditsky I, Ilan N, Best LA, Vlodavsky I. Involvement of Heparanase in Empyema: Implication for Novel Therapeutic Approaches. J Clin Cell Immunol. 2015;6(1) doi: 10.4172/2155-9899.1000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrue C, Saland E, Boutzen H, Vergez F, David M, Joffre C, Hospital MA, Tamburini J, Delabesse E, Manenti S, Sarry JE, Recher C. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood. 2016;127:882–892. doi: 10.1182/blood-2015-05-646497. [DOI] [PubMed] [Google Scholar]
- Laubach JP, Voorhees PM, Hassoun H, Jakubowiak A, Lonial S, Richardson PG. Current strategies for treatment of relapsed/refractory multiple myeloma. Expert Rev Hematol. 2014;7:97–111. doi: 10.1586/17474086.2014.882764. [DOI] [PubMed] [Google Scholar]
- Ledin J, Staatz W, Li JP, Gotte M, Selleck S, Kjellen L, Spillmann D. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J Biol Chem. 2004;279:42732–42741. doi: 10.1074/jbc.M405382200. [DOI] [PubMed] [Google Scholar]
- Lerner I, Baraz L, Pikarsky E, Meirovitz A, Edovitsky E, Peretz T, Vlodavsky I, Elkin M. Function of heparanase in prostate tumorigenesis: potential for therapy. Clin Cancer Res. 2008;14:668–676. doi: 10.1158/1078-0432.CCR-07-1866. [DOI] [PubMed] [Google Scholar]
- Lerner I, Hermano E, Zcharia E, Rodkin D, Bulvik R, Doviner V, Rubinstein AM, Ishai-Michaeli R, Atzmon R, Sherman Y, Meirovitz A, Peretz T, Vlodavsky I, Elkin M. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J Clin Invest. 2011;121:1709–1721. doi: 10.1172/JCI43792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner I, Zcharia E, Neuman T, Hermano E, Rubinstein AM, Vlodavsky I, Elkin M. Heparanase is preferentially expressed in human psoriatic lesions and induces development of psoriasiform skin inflammation in mice. Cell Mol Life Scien (CMLS) 2014;71:2347–2357. doi: 10.1007/s00018-013-1496-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, Yoshimura A, Reindl W, Sipos B, Akira S, Schmid RM, Algul H. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Levy-Adam F, Abboud-Jarrous G, Guerrini M, Beccati D, Vlodavsky I, Ilan N. Identification and characterization of heparin/heparan sulfate binding domains of the endoglycosidase heparanase. J Biol Chem. 2005;280:20457–20466. doi: 10.1074/jbc.M414546200. [DOI] [PubMed] [Google Scholar]
- Levy-Adam F, Feld S, Cohen-Kaplan V, Shteingauz A, Gross M, Arvatz G, Naroditsky I, Ilan N, Doweck I, Vlodavsky I. Heparanase 2 interacts with heparan sulfate with high affinity and inhibits heparanase activity. J Biol Chem. 2010;285:28010–28019. doi: 10.1074/jbc.M110.116384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Adam F, Feld S, Suss-Toby E, Vlodavsky I, Ilan N. Heparanase facilitates cell adhesion and spreading by clustering of cell surface heparan sulfate proteoglycans. PloS one. 2008;3:e2319. doi: 10.1371/journal.pone.0002319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Adam F, Miao HQ, Heinrikson RL, Vlodavsky I, Ilan N. Heterodimer formation is essential for heparanase enzymatic activity. Biochem Biophys Res Commun. 2003;308:885–891. doi: 10.1016/s0006-291x(03)01478-5. [DOI] [PubMed] [Google Scholar]
- Levy JM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Ther. 2011;131:130–141. doi: 10.1016/j.pharmthera.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JP, Galvis ML, Gong F, Zhang X, Zcharia E, Metzger S, Vlodavsky I, Kisilevsky R, Lindahl U. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc Nat Academy Sci USA. 2005;102:6473–6477. doi: 10.1073/pnas.0502287102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JP, Vlodavsky I. Heparin, heparan sulfate and heparanase in inflammatory reactions. Thromb Haemost. 2009;102:823–828. doi: 10.1160/TH09-02-0091. [DOI] [PubMed] [Google Scholar]
- Li L, Wang B, Gao T, Zhang X, Hao JX, Vlodavsky I, Wiesenfeld-Hallin Z, Xu XJ, Li JP. Heparanase overexpression reduces carrageenan-induced mechanical and cold hypersensitivity in mice. Neurosci Lett. 2012a;511:4–7. doi: 10.1016/j.neulet.2011.12.038. [DOI] [PubMed] [Google Scholar]
- Li RW, Freeman C, Yu D, Hindmarsh EJ, Tymms KE, Parish CR, Smith PN. Dramatic regulation of heparanase activity and angiogenesis gene expression in synovium from patients with rheumatoid arthritis. Arthritis Rheum. 2008;58:1590–1600. doi: 10.1002/art.23489. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu H, Huang YY, Pu LJ, Zhang XD, Jiang CC, Jiang ZW. Suppression of endoplasmic reticulum stress-induced invasion and migration of breast cancer cells through the downregulation of heparanase. Int J Mol Med. 2013;31:1234–1242. doi: 10.3892/ijmm.2013.1292. [DOI] [PubMed] [Google Scholar]
- Lider O, Baharav E, Mekori YA, Miller T, Naparstek Y, Vlodavsky I, Cohen IR. Suppression of experimental autoimmune diseases and prolongation of allograft survival by treatment of animals with low doses of heparins. J Clin Invest. 1989;83:752–756. doi: 10.1172/JCI113953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lider O, Mekori YA, Miller T, Bar-Tana R, Vlodavsky I, Baharav E, Cohen IR, Naparstek Y. Inhibition of T lymphocyte heparanase by heparin prevents T cell migration and T cell-mediated immunity. Eur J Immunol. 1990;20:493–499. doi: 10.1002/eji.1830200306. [DOI] [PubMed] [Google Scholar]
- Lindahl U, Li JP. Interactions between heparan sulfate and proteins-design and functional implications. Int Rev Cell Mol Biol. 2009;276:105–159. doi: 10.1016/S1937-6448(09)76003-4. [DOI] [PubMed] [Google Scholar]
- Liu CJ, Chang J, Lee PH, Lin DY, Wu CC, Jeng LB, Lin YJ, Mok KT, Lee WC, Yeh HZ, Ho MC, Yang SS, Yang MD, Yu MC, Hu RH, Peng CY, Lai KL, Chang SS, Chen PJ. Adjuvant heparanase inhibitor PI-88 therapy for hepatocellular carcinoma recurrence. World J Gastroenterology. 2014;20:11384–11393. doi: 10.3748/wjg.v20.i32.11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andren-Sandberg A, Domellof L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. New Eng J Med. 1993;328:1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Massena S, Christoffersson G, Hjertstrom E, Zcharia E, Vlodavsky I, Ausmees N, Rolny C, Li JP, Phillipson M. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood. 2011;116:1924–1931. doi: 10.1182/blood-2010-01-266072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahtouk K, Hose D, Raynaud P, Hundemer M, Jourdan M, Jourdan E, Pantesco V, Baudard M, De Vos J, Larroque M, Moehler T, Rossi JF, Reme T, Goldschmidt H, Klein B. Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood. 2007;109:4914–4923. doi: 10.1182/blood-2006-08-043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzner Y, Bar-Ner M, Yahalom J, Ishai-Michaeli R, Fuks Z, Vlodavsky I. Degradation of heparan sulfate in the subendothelial extracellular matrix by a readily released heparanase from human neutrophils. Possible role in invasion through basement membranes. The J Clin Invest. 1985;76:1306–1313. doi: 10.1172/JCI112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie E, Tyson K, Stamps A, Smith P, Turner P, Barry R, Hircock M, Patel S, Barry E, Stubberfield C, Terrett J, Page M. Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochem Biophys Res Commun. 2000;276:1170–1177. doi: 10.1006/bbrc.2000.3586. [DOI] [PubMed] [Google Scholar]
- McKenzie EA. Heparanase: a target for drug discovery in cancer and inflammation. Br J Pharmacol. 2007;151:1–14. doi: 10.1038/sj.bjp.0707182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKown RL, Wang N, Raab RW, Karnati R, Zhang Y, Williams PB, Laurie GW. Lacritin and other new proteins of the lacrimal functional unit. Exp Eye Res. 2009;88:848–858. doi: 10.1016/j.exer.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519–2526. doi: 10.1158/1078-0432.CCR-07-2223. [DOI] [PubMed] [Google Scholar]
- Mehta-Shah N, Younes A. Novel targeted therapies in diffuse large B-cell lymphoma. Semin Hematol. 2015;52:126–137. doi: 10.1053/j.seminhematol.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Meirovitz A, Hermano E, Lerner I, Zcharia E, Pisano C, Peretz T, Elkin M. Role of heparanase in radiation-enhanced invasiveness of pancreatic carcinoma. Cancer Res. 2011;71:2772–2780. doi: 10.1158/0008-5472.CAN-10-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades CS, Rouleau C, Echart C, Menon K, Teicher B, Distaso M, Palumbo A, Boccadoro M, Anderson KC, Iacobelli M, Richardson PG. Preclinical studies in support of defibrotide for the treatment of multiple myeloma and other neoplasias. Clin Cancer Res. 2009;15:1210–1221. doi: 10.1158/1078-0432.CCR-08-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizgerd JP, Kubo H, Kutkoski GJ, Bhagwan SD, Scharffetter-Kochanek K, Beaudet AL, Doerschuk CM. Neutrophil emigration in the skin, lungs, and peritoneum: different requirements for CD11/CD18 revealed by CD18-deficient mice. J Exp Med. 1997;186:1357–1364. doi: 10.1084/jem.186.8.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris A, Wang B, Waern I, Venkatasamy R, Page C, Schmidt EP, Wernersson S, Li JP, Spina D. The role of heparanase in pulmonary cell recruitment in response to an allergic but not non-allergic stimulus. PloS one. 2015;10:e0127032. doi: 10.1371/journal.pone.0127032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murch SH, MacDonald TT, Walker-Smith JA, Levin M, Lionetti P, Klein NJ. Disruption of sulphated glycosaminoglycans in intestinal inflammation. Lancet. 1993;341:711–714. doi: 10.1016/0140-6736(93)90485-y. [DOI] [PubMed] [Google Scholar]
- Nadav L, Eldor A, Yacoby-Zeevi O, Zamir E, Pecker I, Ilan N, Geiger B, Vlodavsky I, Katz BZ. Activation, processing and trafficking of extracellular heparanase by primary human fibroblasts. J Cell Sci. 2002;115:2179–2187. doi: 10.1242/jcs.115.10.2179. [DOI] [PubMed] [Google Scholar]
- Nadir Y, Brenner B, Fux L, Shafat I, Attias J, Vlodavsky I. Heparanase enhances the generation of activated factor X in the presence of tissue factor and activated factor VII. Haematologica. 2010;95:1927–1934. doi: 10.3324/haematol.2010.023713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadir Y, Brenner B, Zetser A, Ilan N, Shafat I, Zcharia E, Goldshmidt O, Vlodavsky I. Heparanase induces tissue factor expression in vascular endothelial and cancer cells. J Thromb Haemost. 2006;4:2443–2451. doi: 10.1111/j.1538-7836.2006.02212.x. [DOI] [PubMed] [Google Scholar]
- Naggi A, Casu B, Perez M, Torri G, Cassinelli G, Penco S, Pisano C, Giannini G, Ishai-Michaeli R, Vlodavsky I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J Biol Chem. 2005;280:12103–12113. doi: 10.1074/jbc.M414217200. [DOI] [PubMed] [Google Scholar]
- Naomoto Y, Takaoka M, Okawa T, Nobuhisa T, Gunduz M, Tanaka N. The role of heparanase in gastrointestinal cancer. Oncol Rep. 2005;14:3–8. [PubMed] [Google Scholar]
- Naparstek Y, Cohen IR, Fuks Z, Vlodavsky I. Activated T lymphocytes produce a matrix-degrading heparan sulphate endoglycosidase. Nature. 1984;310:241–244. doi: 10.1038/310241a0. [DOI] [PubMed] [Google Scholar]
- Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129:2475–81. doi: 10.1242/jcs.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardella C, Lahm A, Pallaoro M, Brunetti M, Vannini A, Steinkuhler C. Mechanism of activation of human heparanase investigated by protein engineering. Biochemistry. 2004;43:1862–1873. doi: 10.1021/bi030203a. [DOI] [PubMed] [Google Scholar]
- Nobuhisa T, Naomoto Y, Takaoka M, Tabuchi Y, Ookawa K, Kitamoto D, Gunduz E, Gunduz M, Nagatsuka H, Haisa M, Matsuoka J, Nakajima M, Tanaka N. Emergence of nuclear heparanase induces differentiation of human mammary cancer cells. Biochem Biophys Res Commun. 2005;331:175–180. doi: 10.1016/j.bbrc.2005.03.129. [DOI] [PubMed] [Google Scholar]
- Novick D, Barak S, Ilan N, Vlodavsky I. Heparanase interacts with resistin and augments its activity. PloS one. 2014;9:e85944. doi: 10.1371/journal.pone.0085944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogishima T, Shiina H, Breault JE, Tabatabai L, Bassett WW, Enokida H, Li LC, Kawakami T, Urakami S, Ribeiro-Filho LA, Terashima M, Fujime M, Igawa M, Dahiya R. Increased heparanase expression is caused by promoter hypomethylation and up-regulation of transcriptional factor early growth response-1 in human prostate cancer. Clin Cancer Res. 2005;11:1028–1036. [PubMed] [Google Scholar]
- Ogren S, Lindahl U. Cleavage of macromolecular heparin by an enzyme from mouse mastocytoma. J Biol Chem. 1975;250:2690–2697. [PubMed] [Google Scholar]
- Okada Y, Yamada S, Toyoshima M, Dong J, Nakajima M, Sugahara K. Structural recognition by recombinant human heparanase that plays critical roles in tumor metastasis. Hierarchical sulfate groups with different effects and the essential target disulfated trisaccharide sequence. J Biol Chem. 2002;277:42488–42495. doi: 10.1074/jbc.M206510200. [DOI] [PubMed] [Google Scholar]
- Ostapoff KT, Awasthi N, Cenik BK, Hinz S, Dredge K, Schwarz RE, Brekken RA. PG545, an angiogenesis and heparanase inhibitor, reduces primary tumor growth and metastasis in experimental pancreatic cancer. Mol Cancer Ther. 2013;12:1190–1201. doi: 10.1158/1535-7163.MCT-12-1123. [DOI] [PubMed] [Google Scholar]
- Osterholm C, Folkersen L, Lengquist M, Ponten F, Renne T, Li J, Hedin U. Increased expression of heparanase in symptomatic carotid atherosclerosis. Atherosclerosis. 2013;226:67–73. doi: 10.1016/j.atherosclerosis.2012.09.030. [DOI] [PubMed] [Google Scholar]
- Ostrovsky O, Shimoni A, Baryakh P, Morgulis Y, Mayorov M, Beider K, Shteingauz A, Ilan N, Vlodavsky I, Nagler A. Modification of heparanase gene expression in response to conditioning and LPS treatment: strong correlation to rs4693608 SNP. J Leuk Biol. 2014;95:677–688. doi: 10.1189/jlb.0313147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, Goud B, Benaroch P, Hacohen N, Fukuda M, Desnos C, Seabra MC, Darchen F, Amigorena S, Moita LF, Thery C. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12(sup):19–30. 11–13. doi: 10.1038/ncb2000. [DOI] [PubMed] [Google Scholar]
- Pala D, Rivara S, Mor M, Milazzo FM, Roscilli G, Pavoni E, Giannini G. Kinetic analysis and molecular modeling of the inhibition mechanism of roneparstat (SST0001) on human heparanase. Glycobiology. 2016;26:640–654. doi: 10.1093/glycob/cww003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo A, Larocca A, Genuardi M, Kotwica K, Gay F, Rossi D, Benevolo G, Magarotto V, Cavallo F, Bringhen S, Rus C, Masini L, Iacobelli M, Gaidano G, Mitsiades C, Anderson K, Boccadoro M, Richardson P. Melphalan, prednisone, thalidomide and defibrotide in relapsed/refractory multiple myeloma: results of a multicenter phase I/II trial. Haematologica. 2010;95:1144–1149. doi: 10.3324/haematol.2009.017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. 2006;6:633–643. doi: 10.1038/nri1918. [DOI] [PubMed] [Google Scholar]
- Parish CR, Freeman C, Hulett MD. Heparanase: a key enzyme involved in cell invasion. Biochim Biophys Acta. 2001;1471:M99–108. doi: 10.1016/s0304-419x(01)00017-8. [DOI] [PubMed] [Google Scholar]
- Parish CR, Freeman C, Ziolkowski AF, He YQ, Sutcliffe EL, Zafar A, Rao S, Simeonovic CJ. Unexpected new roles for heparanase in Type 1 diabetes and immune gene regulation. Matrix Biol. 2013;32:228–233. doi: 10.1016/j.matbio.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Parish CR, Hindmarsh EJ, Bartlett MR, Staykova MA, Cowden WB, Willenborg DO. Treatment of central nervous system inflammation with inhibitors of basement membrane degradation. Immunol Cell Biol. 1998;76:104–113. doi: 10.1046/j.1440-1711.1998.00722.x. [DOI] [PubMed] [Google Scholar]
- Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan J, Martins VR, Skog J, Kaplan RN, Brady MS, Wolchok JD, Chapman PB, Kang Y, Bromberg J, Lyden D. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol. 2011;21:139–146. doi: 10.1016/j.semcancer.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Peterson S, Liu J. Deciphering mode of action of heparanase using structurally defined oligosaccharides. J Biol Chem. 2012;287:34836–34843. doi: 10.1074/jbc.M112.390161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SB, Liu J. Unraveling the specificity of heparanase utilizing synthetic substrates. J Biol Chem. 2010;285:14504–14513. doi: 10.1074/jbc.M110.104166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovich E, Feigelson SW, Stoler-Barak L, Hatzav M, Solomon A, Bar-Shai A, Ilan N, Li JP, Engelhardt B, Vlodavsky I, Alon R. Lung ICAM-1 and ICAM-2 support spontaneous intravascular effector lymphocyte entrapment but are not required for neutrophil entrapment or emigration inside endotoxin-inflamed lungs. FASEB J. 2016;30:1767–1778. doi: 10.1096/fj.201500046. [DOI] [PubMed] [Google Scholar]
- Picardo SL, Maher SG, O’Sullivan JN, Reynolds JV. Barrett’s to oesophageal cancer sequence: a model of inflammatory-driven upper gastrointestinal cancer. Dig Surg. 2012;29:251–260. doi: 10.1159/000341498. [DOI] [PubMed] [Google Scholar]
- Pikas DS, Li J-P, Vlodavsky I, Lindahl U. Substrate Specificity of Heparanases from Human Hepatoma and Platelets. J Biol Chem. 1998;273:18770–18777. doi: 10.1074/jbc.273.30.18770. [DOI] [PubMed] [Google Scholar]
- Pisano C, Vlodavsky I, Ilan N, Zunino F. The potential of heparanase as a therapeutic target in cancer. Biochem pharmacol. 2014;89:12–19. doi: 10.1016/j.bcp.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planer D, Metzger S, Zcharia E, Wexler ID, Vlodavsky I, Chajek-Shaul T. Role of heparanase on hepatic uptake of intestinal derived lipoprotein and fatty streak formation in mice. PloS one. 2011;6:e18370. doi: 10.1371/journal.pone.0018370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon IK, Goodall KJ, Phipps S, Chow JD, Pagler EB, Andrews DM, Conlan CL, Ryan GF, White JA, Wong MK, Horan C, Matthaei KI, Smyth MJ, Hulett MD. Mice deficient in heparanase exhibit impaired dendritic cell migration and reduced airway inflammation. Eur J Immunol. 2014;44:1016–1030. doi: 10.1002/eji.201343645. [DOI] [PubMed] [Google Scholar]
- Proskuryakov SY, Gabai VL. Mechanisms of tumor cell necrosis. Curr Pharm Des. 2010;16:56–68. doi: 10.2174/138161210789941793. [DOI] [PubMed] [Google Scholar]
- Purushothaman A, Bandari SK, Liu J, Mobley JA, Brown EE, Sanderson RD. Fibronectin on the Surface of Myeloma Cell-derived Exosomes Mediates Exosome-Cell Interactions. J Biol Chem. 2016;291:1652–1663. doi: 10.1074/jbc.M115.686295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman A, Chen L, Yang Y, Sanderson RD. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem. 2008;283:32628–32636. doi: 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman A, Uyama T, Kobayashi F, Yamada S, Sugahara K, Rapraeger AC, Sanderson RD. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood. 2010;115:2449–2457. doi: 10.1182/blood-2009-07-234757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani VC, Pruett PS, Thompson CA, Delucas LD, Sanderson RD. Heparan sulfate chains of syndecan-1 regulate ectodomain shedding. J Biol Chem. 2012;287:9952–9961. doi: 10.1074/jbc.M111.330803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani VC, Purushothaman A, Stewart MD, Thompson CA, Vlodavsky I, Au JL, Sanderson RD. The heparanase/syndecan-1 axis in cancer: mechanisms and therapies. FEBS J. 2013;280:2294–2306. doi: 10.1111/febs.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani VC, Vlodavsky I, Ng M, Zhang Y, Barbieri P, Noseda A, Sanderson RD. Chemotherapy induces expression and release of heparanase leading to changes associated with an aggressive tumor phenotype. Matrix Biol. 2016 doi: 10.1016/j.matbio.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani VC, Yang Y, Ren Y, Nan L, Sanderson RD. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J Biol Chem. 2011;286:6490–6499. doi: 10.1074/jbc.M110.183277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani VC, Zhan F, He J, Barbieri P, Noseda A, Tricot G, Sanderson RD. Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget. 2015;7:1598–1607. doi: 10.18632/oncotarget.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz A, Ilan N, Vlodavsky I, Li JP, Johansson S. Characterization of Heparanase-induced Phosphatidylinositol 3-Kinase-AKT Activation and Its Integrin Dependence. J Biol Chem. 2013;288:12366–12375. doi: 10.1074/jbc.M112.435172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie JP, Ramani VC, Ren Y, Naggi A, Torri G, Casu B, Penco S, Pisano C, Carminati P, Tortoreto M, Zunino F, Vlodavsky I, Sanderson RD, Yang Y. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin Cancer Res. 2011;17:1382–1393. doi: 10.1158/1078-0432.CCR-10-2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivara S, Milazzo FM, Giannini G. Heparanase: a rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Med Chem. 2016;8:647–680. doi: 10.4155/fmc-2016-0012. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32:955–963. doi: 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roucourt B, Meeussen S, Bao J, Zimmermann P, David G. Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 2015a;25:412–428. doi: 10.1038/cr.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roucourt B, Meeussen S, Bao J, Zimmermann P, David G. Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 2015b;25:412–428. doi: 10.1038/cr.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson RD, Iozzo RV. Targeting heparanase for cancer therapy at the tumor-matrix interface. Matrix Biol. 2012;31:283–284. doi: 10.1016/j.matbio.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Sanderson RD, Yang Y. Syndecan-1: a dynamic regulator of the myeloma microenvironment. Clin Exp Metastasis. 2008;25:149–159. doi: 10.1007/s10585-007-9125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki N, Higashi N, Taka T, Nakajima M, Irimura T. Cell surface localization of heparanase on macrophages regulates degradation of extracellular matrix heparan sulfate. J Immunol. 2004;172:3830–3835. doi: 10.4049/jimmunol.172.6.3830. [DOI] [PubMed] [Google Scholar]
- Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, Zemans RL, Bowman JC, Koyanagi DE, Yunt ZX, Smith LP, Cheng SS, Overdier KH, Thompson KR, Geraci MW, Douglas IS, Pearse DB, Tuder RM. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18:1217–1223. doi: 10.1038/nm.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz M, Munzel PA, Braeuning A. Non-melanoma skin cancer in mouse and man. Archives Toxicol. 2013;87:783–798. doi: 10.1007/s00204-012-0998-9. [DOI] [PubMed] [Google Scholar]
- Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer. 2014;14:517–534. doi: 10.1038/nrc3774. [DOI] [PubMed] [Google Scholar]
- Secchi MF, Masola V, Zaza G, Lupo A, Gambaro G, Onisto M. Recent data concerning heparanase: focus on fibrosis, inflammation and cancer. Biomol Concepts. 2015;6:415–421. doi: 10.1515/bmc-2015-0021. [DOI] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafat I, Ben-Arush MW, Issakov J, Meller I, Naroditsky I, Tortoreto M, Cassinelli G, Lanzi C, Pisano C, Ilan N, Vlodavsky I, Zunino F. Pre-clinical and clinical significance of heparanase in Ewing’s sarcoma. J Cell Mol Med. 2011;15:1857–1864. doi: 10.1111/j.1582-4934.2010.01190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain KH, Dalton WS, Tao J. The tumor microenvironment shapes hallmarks of mature B-cell malignancies. Oncogene. 2015;34:4673–4682. doi: 10.1038/onc.2014.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteingauz A, Boyango I, Naroditsky I, Hammond E, Gruber M, Doweck I, Ilan N, Vlodavsky I. Heparanase Enhances Tumor Growth and Chemoresistance by Promoting Autophagy. Cancer Res. 2015;75:3946–3957. doi: 10.1158/0008-5472.CAN-15-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteingauz A, Ilan N, Vlodavsky I. Processing of heparanase is mediated by syndecan-1 cytoplasmic domain and involves syntenin and alpha-actinin. J Cell Mol Life Sci (CMLS) 2014;71:4457–4470. doi: 10.1007/s00018-014-1629-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteper PJ, Zcharia E, Ashhab Y, Peretz T, Vlodavsky I, Ben-Yehuda D. Role of promoter methylation in regulation of the mammalian heparanase gene. Oncogene. 2003;22:7737–7749. doi: 10.1038/sj.onc.1207056. [DOI] [PubMed] [Google Scholar]
- Simizu S, Ishida K, Osada H. Heparanase as a molecular target of cancer chemotherapy. Cancer Sci. 2004;95:553–558. doi: 10.1111/j.1349-7006.2004.tb02485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Raposo G. Exosomes–vesicular carriers for intercellular communication. Curr Opi Cell Biol. 2009;21:575–581. doi: 10.1016/j.ceb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Sonoda R, Naomoto Y, Shirakawa Y, Fujiwara Y, Yamatsuji T, Noma K, Tanabe S, Takaoka M, Gunduz M, Tsujigiwa H, Nagatsuka H, Ohara N, Yoshino T, Takubo K, Vieth M, Tanaka N. Preferential up-regulation of heparanase and cyclooxygenase-2 in carcinogenesis of Barrett’s oesophagus and intestinal-type gastric carcinoma. Histopathol. 2010;57:90–100. doi: 10.1111/j.1365-2559.2010.03594.x. [DOI] [PubMed] [Google Scholar]
- Stoler-Barak L, Petrovich E, Aychek T, Gurevich I, Tal O, Hatzav M, Ilan N, Feigelson SW, Shakhar G, Vlodavsky I, Alon R. Heparanase of murine effector lymphocytes and neutrophils is not required for their diapedesis into sites of inflammation. FASEB J. 2015;29:2010–2021. doi: 10.1096/fj.14-265447. [DOI] [PubMed] [Google Scholar]
- Tayel A, Abd El Galil KH, Ebrahim MA, Ibrahim AS, El-Gayar AM, Al-Gayyar MM. Suramin inhibits hepatic tissue damage in hepatocellular carcinoma through deactivation of heparanase enzyme. Eur J Pharmacol. 2014;728:151–160. doi: 10.1016/j.ejphar.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Thompson CA, Purushothaman A, Ramani VC, Vlodavsky I, Sanderson RD. Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J Biol Chem. 2013;288:10093–10099. doi: 10.1074/jbc.C112.444562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpl R, Brown JC. Supramolecular assembly of basement membranes. Bioessays. 1996;18:123–132. doi: 10.1002/bies.950180208. [DOI] [PubMed] [Google Scholar]
- Tkach M, Théry C. Communication by extracellular vesicles: Where we are and where we need to go. 2016 doi: 10.1016/j.cell.2016.01.043. [DOI] [PubMed] [Google Scholar]
- Tkachenko E, Rhodes JM, Simons M. Syndecans: new kids on the signaling block. Cir Res. 2005;96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- Udo Hacker KNANP. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- van den Hoven MJ, Rops AL, Bakker MA, Aten J, Rutjes N, Roestenberg P, Goldschmeding R, Zcharia E, Vlodavsky I, van der Vlag J, Berden JH. Increased expression of heparanase in overt diabetic nephropathy. Kidney Int. 2006;70:2100–2108. doi: 10.1038/sj.ki.5001985. [DOI] [PubMed] [Google Scholar]
- van den Hoven MJ, Rops AL, Vlodavsky I, Levidiotis V, Berden JH, van der Vlag J. Heparanase in glomerular diseases. Kidney Int. 2007;72:543–548. doi: 10.1038/sj.ki.5002337. [DOI] [PubMed] [Google Scholar]
- Vincent Rajkumar S. Multiple myeloma: 2014 Update on diagnosis, risk-stratification, and management. Am J Hematol. 2014;89:999–1009. doi: 10.1002/ajh.23810. [DOI] [PubMed] [Google Scholar]
- Vlodavsky I, Bar-Shavit R, Ishai-Michaeli R, Bashkin P, Fuks Z. Extracellular sequestration and release of fibroblast growth factor: a regulatory mechanism? Trends Biochem Sci. 1991;16:268–271. doi: 10.1016/0968-0004(91)90102-2. [DOI] [PubMed] [Google Scholar]
- Vlodavsky I, Beckhove P, Lerner I, Pisano C, Meirovitz A, Ilan N, Elkin M. Significance of heparanase in cancer and inflammation. Cancer Microenviron. 2012;5:115–132. doi: 10.1007/s12307-011-0082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky I, Blich M, Li JP, Sanderson RD, Ilan N. Involvement of heparanase in atherosclerosis and other vessel wall pathologies. Matrix Biol. 2013;32:241–251. doi: 10.1016/j.matbio.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky I, Eldor A, Haimovitz-Friedman A, Matzner Y, Ishai-Michaeli R, Lider O, Naparstek Y, Cohen IR, Fuks Z. Expression of heparanase by platelets and circulating cells of the immune system: possible involvement in diapedesis and extravasation. Invasion & Metastasis. 1992;12:112–127. [PubMed] [Google Scholar]
- Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–347. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky I, Friedmann Y, Elkin M, Aingorn H, Atzmon R, Ishai-Michaeli R, Bitan M, Pappo O, Peretz T, Michal I, Spector L, Pecker I. Mammalian heparanase: gene cloning, expression and function in tumor progression and metastasis. Nat Med. 1999;5:793–802. doi: 10.1038/10518. [DOI] [PubMed] [Google Scholar]
- Vreys V, David G. Mammalian heparanase: what is the message? J Cell Mol Med. 2007;11:427–452. doi: 10.1111/j.1582-4934.2007.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreys V, Delande N, Zhang Z, Coomans C, Roebroek A, Durr J, David G. Cellular Uptake of Mammalian Heparanase Precursor Involves Low Density Lipoprotein Receptor-related Proteins, Mannose 6-Phosphate Receptors, and Heparan Sulfate Proteoglycans. J Biol Chem. 2005;280:33141–33148. doi: 10.1074/jbc.M503007200. [DOI] [PubMed] [Google Scholar]
- Wang F, Wan A, Rodrigues B. The function of heparanase in diabetes and its complications. Can J Diabetes. 2013;37:332–338. doi: 10.1016/j.jcjd.2013.05.008. [DOI] [PubMed] [Google Scholar]
- Wang GZ, Tang XD, Lu MH, Gao JH, Liang GP, Li N, Li CZ, Wu YY, Chen L, Cao YL, Fang DC, Yang SM. Multiple antigenic peptides of human heparanase elicit a much more potent immune response against tumors. Cancer Prev Res (Phila) 2011;4:1285–1295. doi: 10.1158/1940-6207.CAPR-11-0083. [DOI] [PubMed] [Google Scholar]
- Wang Z, Xu H, Jiang L, Zhou X, Lu C, Zhang X. Positive association of heparanase expression with tumor invasion and lymphatic metastasis in gastric carcinoma. Mod Pathol. 2005;18:205–211. doi: 10.1038/modpathol.3800282. [DOI] [PubMed] [Google Scholar]
- Waterman M, Ben-Izhak O, Eliakim R, Groisman G, Vlodavsky I, Ilan N. Heparanase upregulation by colonic epithelium in inflammatory bowel disease. Mod Pathol. 2007;20:8–14. doi: 10.1038/modpathol.3800710. [DOI] [PubMed] [Google Scholar]
- Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121–167. doi: 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- Weissmann M, Arvatz G, Horowitz N, Feld S, Naroditsky I, Zhang Y, Ng M, Hammond E, Nevo E, Vlodavsky I, Ilan N. Heparanase-neutralizing antibodies attenuate lymphoma tumor growth and metastasis. Proc Nat Acad Sci USA. 2016;113:704–709. doi: 10.1073/pnas.1519453113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JC, Laloo AE, Singh S, Ferro V. 1H NMR spectroscopic studies establish that heparanase is a retaining glycosidase. Biochem Biophys Res Commun. 2014;443:185–188. doi: 10.1016/j.bbrc.2013.11.079. [DOI] [PubMed] [Google Scholar]
- Winterhoff B, Freyer L, Hammond E, Giri S, Mondal S, Roy D, Teoman A, Mullany SA, Hoffmann R, von Bismarck A, Chien J, Block MS, Millward M, Bampton D, Dredge K, Shridhar V. PG545 enhances anti-cancer activity of chemotherapy in ovarian models and increases surrogate biomarkers such as VEGF in preclinical and clinical plasma samples. Eur J Cancer. 2015;51:879–892. doi: 10.1016/j.ejca.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtuszkiewicz A, Schuurhuis GJ, Kessler FL, Piersma SR, Knol JC, Pham TV, Jansen G, Musters RJ, van Meerloo J, Assaraf YG, Kaspers GJ, Zweegman S, Cloos J, Jimenez CR. Exosomes secreted by apoptosis-resistant acute myeloid leukemia (AML) blasts harbor regulatory network proteins potentially involved in antagonism of apoptosis. Mol Cell Proteomics. 2016;15:1281–98. doi: 10.1074/mcp.M115.052944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthen GS, Schwab B, 3rd, Elson EL, Downey GP. Mechanics of stimulated neutrophils: cell stiffening induces retention in capillaries. Science. 1989;245:183–186. doi: 10.1126/science.2749255. [DOI] [PubMed] [Google Scholar]
- Wu L, Viola CM, Brzozowski AM, Davies GJ. Structural characterization of human heparanase reveals insights into substrate recognition. Nat Struct Mol Biol. 2015;22:1016–1022. doi: 10.1038/nsmb.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Pan C, Meng K, Zhao L, Du L, Liu Q, Lin R. Hypoxia activates heparanase expression in an NF-kappaB dependent manner. Oncol Rep. 2010;23:255–261. [PubMed] [Google Scholar]
- Wu WK, Coffelt SB, Cho CH, Wang XJ, Lee CW, Chan FK, Yu J, Sung JJ. The autophagic paradox in cancer therapy. Oncogene. 2012;31:939–953. doi: 10.1038/onc.2011.295. [DOI] [PubMed] [Google Scholar]
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- Xiong Z, Lu MH, Fan YH, Cao YL, Hu CJ, Wu YY, Wang SM, Luo G, Fang DC, Li C, Yang SM. Downregulation of heparanase by RNA interference inhibits invasion and tumorigenesis of hepatocellular cancer cells in vitro and in vivo. Int J Oncol. 2012;40:1601–1609. doi: 10.3892/ijo.2012.1338. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ren Y, Ramani VC, Nan L, Suva LJ, Sanderson RD. Heparanase enhances local and systemic osteolysis in multiple myeloma by upregulating the expression and secretion of RANKL. Cancer Res. 2010;70:8329–8338. doi: 10.1158/0008-5472.CAN-10-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young E. The anti-inflammatory effects of heparin and related compounds. Thromb Res. 2008;122:743–752. doi: 10.1016/j.thromres.2006.10.026. [DOI] [PubMed] [Google Scholar]
- Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. 2010;14:2592–2603. doi: 10.1111/j.1582-4934.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Wang L, Chen S. Dual character of Toll-like receptor signaling: pro-tumorigenic effects and anti-tumor functions. Biochim Biophys Acta. 2013;1835:144–154. doi: 10.1016/j.bbcan.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Zcharia E, Philp D, Edovitsky E, Aingorn H, Metzger S, Kleinman HK, Vlodavsky I, Elkin M. Heparanase Regulates Murine Hair Growth. Am J Pathol. 2005a;166:999–1008. doi: 10.1016/S0002-9440(10)62321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zcharia E, Zilka R, Yaar A, Yacoby-Zeevi O, Zetser A, Metzger S, Sarid R, Naggi A, Casu B, Ilan N, Vlodavsky I, Abramovitch R. Heparanase accelerates wound angiogenesis and wound healing in mouse and rat models. Faseb J. 2005b;19:211–221. doi: 10.1096/fj.04-1970com. [DOI] [PubMed] [Google Scholar]
- Zetser A, Bashenko Y, Edovitsky E, Levy-Adam F, Vlodavsky I, Ilan N. Heparanase induces vascular endothelial growth factor expression: correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006;66:1455–1463. doi: 10.1158/0008-5472.CAN-05-1811. [DOI] [PubMed] [Google Scholar]
- Zetser A, Levy-Adam F, Kaplan V, Gingis-Velitski S, Bashenko Y, Schubert S, Flugelman MY, Vlodavsky I, Ilan N. Processing and activation of latent heparanase occurs in lysosomes. J Cell Sci. 2004;117:2249–2258. doi: 10.1242/jcs.01068. [DOI] [PubMed] [Google Scholar]
- Zhang HG, Grizzle WE. Exosomes and cancer: a newly described pathway of immune suppression. Clin Cancer Res. 2011;17:959–964. doi: 10.1158/1078-0432.CCR-10-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang J, Han X, Zhao Z, Du L, Yu T, Wang H. Overexpression of heparanase multiple antigenic peptide 2 is associated with poor prognosis in gastric cancer: Potential for therapy. Oncology lett. 2012a;4:178–182. doi: 10.3892/ol.2012.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ngo JA, Wetzel MD, Marchetti D. Heparanase mediates a novel mechanism in lapatinib-resistant brain metastatic breast cancer. Neoplasia. 2015;17:101–113. doi: 10.1016/j.neo.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wang B, Li JP. Implications of heparan sulfate and heparanase in neuroinflammation. Matrix Biol. 2014;35:174–181. doi: 10.1016/j.matbio.2013.12.009. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wang B, O’Callaghan P, Hjertstrom E, Jia J, Gong F, Zcharia E, Nilsson LN, Lannfelt L, Vlodavsky I, Lindahl U, Li JP. Heparanase overexpression impairs inflammatory response and macrophage-mediated clearance of amyloid-beta in murine brain. Acta Neuropathol. 2012b;124:465–478. doi: 10.1007/s00401-012-0997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ryan DS, Bower KS, Ilan N, Vlodavsky I, Laurie GW. Focus on molecules: heparanase. Exp Eye Res. 2010;91:476–477. doi: 10.1016/j.exer.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YF, Tang XD, Gao JH, Fang DC, Yang SM. Heparanase: a universal immunotherapeutic target in human cancers. Drug Discov Today. 2011;16:412–417. doi: 10.1016/j.drudis.2011.02.015. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Yang J, Qian J, Qiu P, Hanabuchi S, Lu Y, Wang Z, Liu Z, Li H, He J, Lin P, Weber D, Davis RE, Kwak L, Cai Z, Yi Q. PSGL-1/selectin and ICAM-1/CD18 interactions are involved in macrophage-induced drug resistance in myeloma. Leukemia. 2013;27:702–710. doi: 10.1038/leu.2012.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitomirsky B, Assaraf YG. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget. 2015;20(6):1143–56. doi: 10.18632/oncotarget.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitomirsky B, Assaraf YG. Lysosomes as mediators of drug resistance in cancer. Drug Resist Updat. 2016;24:23–33. doi: 10.1016/j.drup.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Zhou H, Roy S, Cochran E, Zouaoui R, Chu CL, Duffner J, Zhao G, Smith S, Galcheva-Gargova Z, Karlgren J, Dussault N, Kwan RY, Moy E, Barnes M, Long A, Honan C, Qi YW, Shriver Z, Ganguly T, Schultes B, Venkataraman G, Kishimoto TK. M402, a novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PloS one. 2011;6:e21106. doi: 10.1371/journal.pone.0021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Yang Y, Xia J, Wang H, Salama ME, Xiong W, Xu H, Shetty S, Chen T, Zeng Z, Shi L, Zangari M, Miles R, Bearss D, Tricot G, Zhan F. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell. 2013;23:48–62. doi: 10.1016/j.ccr.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziolkowski AF, Popp SK, Freeman C, Parish CR, Simeonovic CJ. Heparan sulfate and heparanase play key roles in mouse beta cell survival and autoimmune diabetes. J Clin Invest. 2012;122:132–141. doi: 10.1172/JCI46177. [DOI] [PMC free article] [PubMed] [Google Scholar]