

Abstract
Objective:
To describe a second hereditary sensory autonomic neuropathy type VI (HSAN-VI) family harboring 2 novel heterozygous mutations in the dystonin (DST) gene and to evaluate their effect on neurons derived from induced pluripotent stem cells (iPSC).
Methods:
The family consisted of 3 affected siblings from nonconsanguineous healthy parents. All members underwent clinical and electrophysiologic evaluation and genetic analysis. Two patients underwent quantitative sensory testing (QST), cardiovascular reflexes, dynamic sweat test, and skin biopsy to evaluate somatic and autonomic cutaneous innervation and to get fibroblast cultures for developing iPSC-derived neurons.
Results:
Onset occurred in the first decade, with painless and progressive mutilating distal ulcerations leading to amputation and joint deformity. Sensation to pain, touch, and vibration was reduced. Autonomic disturbances included hypohidrosis, pupillary abnormalities, and gastrointestinal and sexual dysfunction. Nerve conduction studies showed a severe axonal sensory neuropathy. QST and autonomic functional studies were abnormal. Skin biopsy revealed a lack of sensory and autonomic nerve fibers. Genetic analysis revealed 2 pathogenic mutations in the DST gene affecting exclusively the DST neuronal isoform-a2. Neurons derived from iPSC showed absence or very low levels of DST protein and short and dystrophic neuritis or no projections at all.
Conclusions:
Unlike the previous HSAN-VI family, our description indicates that DST mutations may be associated with a nonlethal and nonsyndromic phenotype. Neuronal loss affects large and small sensory nerve fibers as well as autonomic ones. Induced-PSC findings suggest that dystonin defect might alter proper development of the peripheral nerves. Dystonin-a2 plays a major role in the HSAN-VI phenotype.
Hereditary sensory and autonomic neuropathy type VI (HSAN-VI) was initially described in 2012 in a single family of 3 Ashkenazi infants presenting with severe dysautonomic symptoms, distal contractures, severe psychomotor retardation, and early death.1 These were associated with a homozygous frameshift mutation in the dystonin (DST) gene.1 The report of the first AR mutation in DST paralleled the earlier identification of mutations in the murine DST gene in mouse strains with a neurodegenerative disease called dystonia musculorum (dt).2–4
In the DST gene, 4 different promoters and many alternative splicing events result in numerous isoforms, among which are the 3 major neuronal isoforms known as dystonin-a1, -a2, and -a3, the 3 muscular isoforms named dystonin-b1, -b2, and -b3, and one epithelial isoform named dystonin-e.5–10 The 3 neuronal isoforms derive through the use of alternative transcription start sites (TSS) in the first exons resulting in the specificity of the N-terminal regions that determine cellular localizations and functions.6,11–16
We report the second HSAN-VI family harboring 2 novel heterozygous mutations in the DST gene. We provide clinical, electrophysiologic, functional, and morphologic characterization of this family. We also show the effects of DST mutations in neurons derived from induced pluripotent stem cells (iPSC).
METHODS
The family under study consisted of a healthy sister and 3 affected siblings from nonconsanguineous healthy parents (figure 1A) originating from a small village in Southern Italy. All members underwent clinical and electrophysiologic evaluation and genetic analysis. Two patients underwent quantitative sensory testing (QST), cardiovascular reflexes (CVR), dynamic sweat test, and skin biopsy. Skin samples were taken to study cutaneous innervation, to measure the expression of DST gene, and to obtain fibroblast cultures for developing iPSC-derived neurons.
Figure 1. Pedigree and genetic features.

(A) Pedigree of the family with segregation analysis. (B) Pictures from patients show acromutilation and joint deformities. (C) Schematic diagram of DST gene. The putative promoters for the 3 neuronal isoforms (Dst-a1, -a2, -a3) are indicated by curved arrows. Blue boxes indicate the unique exons of isoform-a2, the red box indicates the unique exon of isoform-a1, and the green box indicates the unique exon of isoform-a3. The blue line represents the transcript of isoform-a2, the red line represents the transcript of isoform-a1, and the green line represents the transcript of isoform-a3. Full arrows show the position of mutations in our family, the empty arrow shows the mutation (exon 83, c.14865delA) previously reported in hereditary sensory and autonomic neuropathy type VI. (D) Chromatograms show the sequence of exon 4 and exon 5 from the affected members of the family.
Standard protocol approvals, registrations, and patient consents.
All participants signed an informed consent approved by the local ethical board of University Federico II.
Quantitative sensory testing.
Six sensory modalities were tested on right dorsum, thigh, leg, and foot as previously described.17
Cardiovascular reflexes.
We evaluated heart rate variability at rest and during deep breathing, Valsalva ratio, 30/15 ratio, and blood pressure response to standing and to sustained handgrip.18
Dynamic sweat test.
Activated sweat gland density per cm2 and sweat volume per gland and per cm2 were calculated analyzing sweat drop imprints on serial frames from digital recording.19
Skin biopsy.
Skin samples from fingertip (III finger), thigh, and leg (union of middle third with the lower third) of patients were used to evaluate somatic and autonomic cutaneous innervation.20
Genetic analysis.
Conventional Sanger sequencing of RAB7 and WNK1 genes was performed in the probands using standard methodologies as previously reported.21 DNA specimens of the family members were further analyzed by whole exome sequencing following standard protocols on the Illumina (San Diego, CA) HiSeq platform using the Agilent (Santa Clara, CA) capture Sure Select Human All Exon V4 design at the Hussman Institute for Human Genomics, Miller School of Medicine, Miami, Florida. Raw data were aligned with BWA and variants were called with GATK as previously described.22 All variant data were then analyzed on the GENESIS platform.23 Pathogenic variants, identified in the DST gene, were confirmed by direct Sanger sequencing (figure 1D) that was also used to determine whether the pathogenic variants cosegregated with disease (figure 1A).
Fibroblast cultures.
Fibroblasts, derived from skin samples obtained from the dorsum (10th dorsal dermatome), were cultured up to 4–5 passages in DMEM with 2 mM l-glutamine, 20% fetal bovine serum (FBS), 100 U/mL penicillin, and streptomycin at 37°C in 5% CO2 atmosphere.
Generation and differentiation of iPSC.
Retroviral vectors expressing Oct4, Sox2, Myc, and Klf4 (all gifts from Shinya Yamanaka; Addgene [Cambridge, MA] plasmid #17,217, #17,218, #17,220, #17,219;24induction of pluripotent stem cells from adult human fibroblasts by defined factors)25 were transfected into Plat-A cells (Cell Biolabs Inc., San Diego, CA) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following instructions. Supernatants of transfected cells containing viruses were mixed at a ratio of 1:1:1:1. Normal and affected dermal fibroblasts were plated on a mouse feeder layer and after 24 hours and 2 rounds of infection were performed with virus mix containing polybrene (8 μg/mL). The medium was replaced after 48 hours with the following human iPSC medium: DMEM/F12 containing 20% knockout serum replacement, 10 ng/mL bFGF, 1 mM l-GIn, 100 mM nonessential aminoacids, 100 mM 2-mercaptoethanol, 1 × penicillin/streptomycin (all from Invitrogen). The medium was changed every other day. Starting from 21 days, iPSC clones were picked to isolate and expand them in human iPSC medium on mouse feeder layer. The stemness of isolated clones was verified for the presence of stemness markers and for the absence of differentiated markers. For differentiation, the human iPSC were pretreated with 10 μM ROCK inhibitor. After 1 hour, the medium was removed and cells were treated with DMEM/F12 containing 1 mg/mL collagenase IV for 5 minutes at 37°C. After 5 minutes, the cells were washed with iPSC medium, scraped, and harvested by centrifugation. Then cells were plated at high density (2–4 × 105 cells per cm2) on Matrigel-coated plates in iPSC medium containing ROCK inhibitor. The following day, the medium was switched to neuronal inducing medium containing DMEM/F12 (Life Technologies, Carlsbad, CA), 25 μg/mL insulin (Sigma, St. Louis, MO), 50 μg/mL transferrin (Sigma), 30 nM sodium selenite (Sigma), 20 nM progesterone (Sigma), 100 nM putrescine (Sigma), and penicillin/streptomycin. After 7 days of differentiation, B27 (Life Technologies) was added and kept for the following 7–15 days, changing the medium every 2–3 days.
Fluorescence microscopy.
For fluorescence analysis, the cells were treated with 4% paraformaldehyde for fixing, with 50 mM NH4Cl for quenching, and with phosphate-buffered saline (PBS) containing 0.2% Triton X-100, 10% FBS, and 1% bovine serum albumin to permeabilize them. The staining was performed with specific antibodies: anti-Nanog (Cell Signaling Technology, Beverly, MA), anti-Oct4 (Santa Cruz Biotechnology, Dallas, TX), anti-β-tubulin (Sigma), and anti-dystonin (catalogue number AB15065; Merck Millipore, Billerica, MA). Secondary antibodies conjugated with Alexa 488 or 546 (Life Technologies) were used. Nuclei were visualized by staining with DAPI. Images were collected using epifluorescence microscopy (DMI 4000B; Leica, Newcastle, UK) equipped with a Plan Fluotar ×20 (NA 0.4) objective lens with a cooled monochrome CCD camera (DFC365 FX, Leica Microsystems) or scanning confocal microscope (LSM 510 META, Carl Zeiss Microimaging, Jena, Germany) equipped with a Plan Neofluar ×20 (NA 0.5) objective lens, by utilizing Argon (488 nm) and HeNe (546 nm) lasers.
Western blotting.
Cells, grown adhesion, were washed with ice-cold PBS and lysed in JS buffer (Hepes pH 7.5 50 mM, NaCl 150 mM, glycerol 1%, Triton X-100 1%, MgCl2 1.5 mM, EGTA 5 mM) including 15 μg/mL of protease inhibitor (Antipain, Pepstatin, and Leupeptin from Sigma). Cell lysates were centrifuged for 20 minutes at 10,000 g. Protein concentration was determined by the Bradford method. Protein extracts were resolved through sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes. Specific primary antibodies, anti-dystonin and anti-GAPDH (Cell Signaling), were used and detected with the appropriate horseradish peroxidase–conjugated secondary antibodies (GE Healthcare, Cleveland, OH).
Extraction of RNA and quantitative PCR.
The extraction of total RNA from biopsies was performed using Trisure (Bioline, London, UK) and following instructions. cDNA was obtained using M-MLV RT kit (New England Biolabs, Ipswich, MA). Quantitative PCR (qPCR) was carried out using the Fast SYBR Green PCR Master mix and QuantStudio 7 Flex PCR system (both from Applied Biosystems, Foster City, CA). The housekeeping GAPDH mRNA was used for normalization; the mRNA levels were calculated using the comparative 2-ΔCt method. The primers used for amplification are as follows:
DST-a1: forward 5′-ACGTGGAGGAGCAGGAGTAT-3′; reverse 5′-TGTCCCGTTCATCTTTGTACCT-3′
DST-a2: forward 5′-GATTCAAGGGGGCTCAGTGC-3′; reverse 5′-ACTTTGTCCCGTTCATCTGCT-3′
Protein gene product 9.5: forward 5′-CAGAGGACACCCTGCTGAAG-3′; reverse 5′-AAGCGGACTTCTCCTTGCTC-3′
GAPDH: forward 5′- GTCGGAGTCAACGGATTTGG-3′; reverse 5′- AAAAGCAGCCCTGGTGACC-3′
RESULTS
Clinical, electrophysiologic, functional, and morphologic features.
The 3 affected brothers since infancy had severely impaired pain sensitivity resulting in poor healing of distal ulcerations, amputations of fingers or toes, and joint deformities (figure 1B). Developmental milestones were reached normally and no signs of mental retardation were observed. On neurologic examination, the sensations to pain, touch, and vibration were reduced and deep tendon reflexes were altered. Muscle weakness was limited to intrinsic foot muscles. All patients complained of autonomic disturbances, including reduced sweating with heat intolerance, pupillary abnormalities (absent light reflexes), chronic diarrhea, and sexual dysfunction (erectile dysfunction) (table 1). Nerve conduction studies showed a severe generalized axonal sensory neuropathy while motor abnormalities (absent/reduced amplitude of compound muscular action potentials) were limited to intrinsic foot muscles (table e-1 at Neurology.org). Needle EMG revealed mild chronic neurogenic changes in distal muscles of the lower limbs.
Table 1.
Clinical features
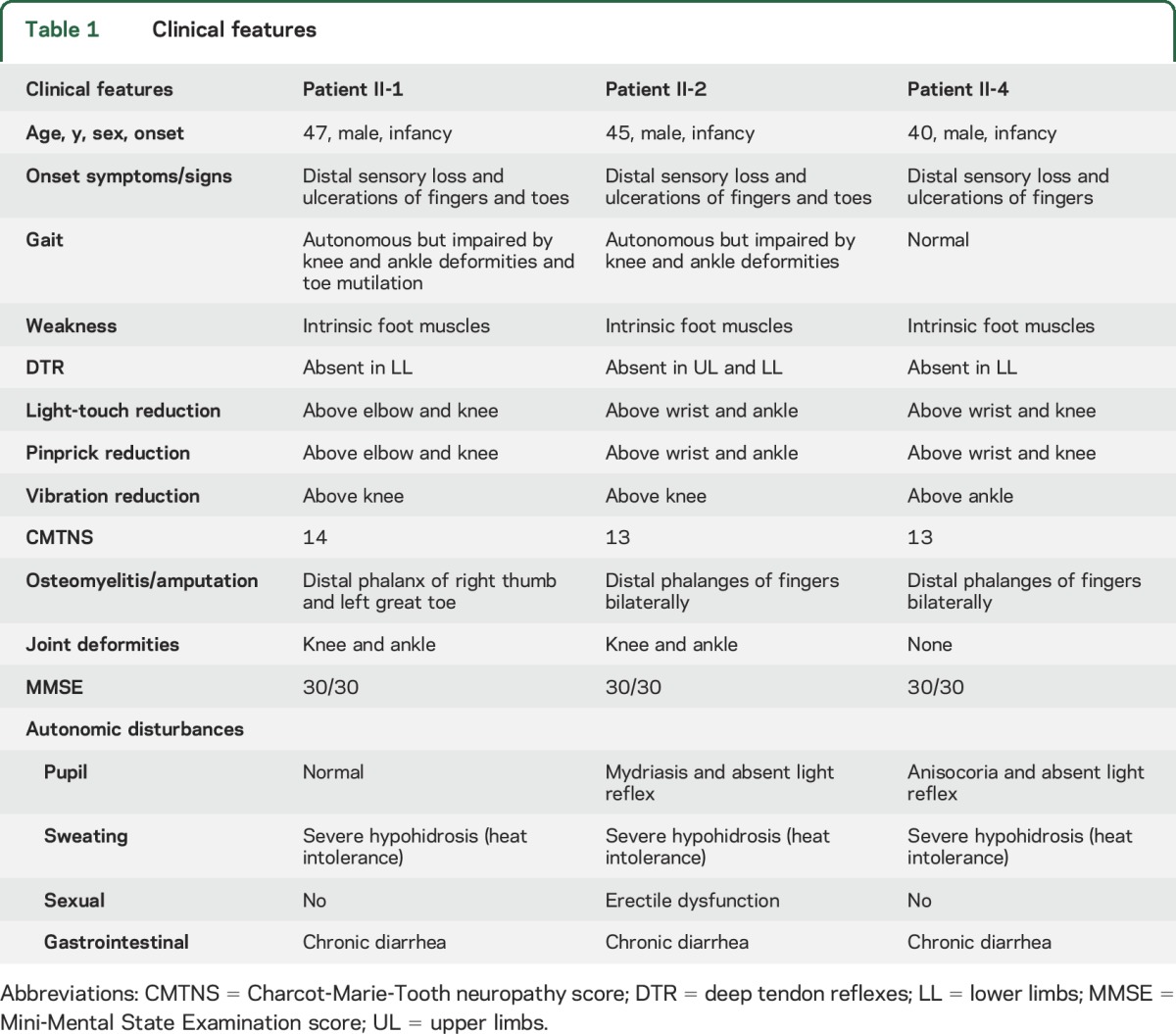
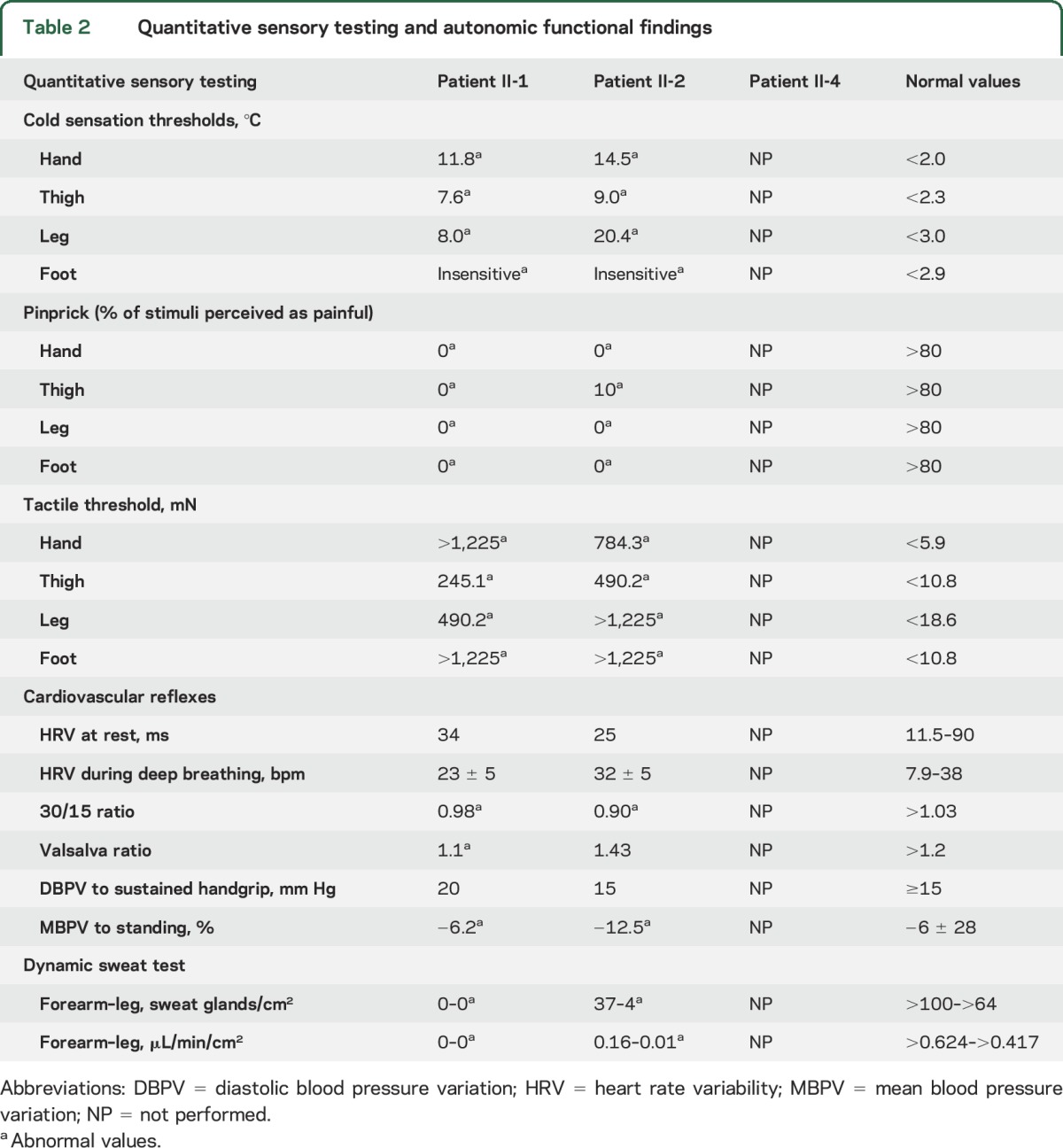
QST was altered for all tested modalities (table 2). In particular, patients were unable to perceive noxious thermal stimuli and innocuous warm stimuli. Very high intensity stimuli were necessary for the perception of cold, touch, and mechanical pain sensory modalities. CVR showed abnormalities of 30/15 ratio and blood pressure response to standing consistent with both sympathetic and parasympathetic system impairment (table 2). Sudomotor function was markedly impaired. Sympathetic skin response was absent in all patients as well as dynamic sweat test, which showed a severe reduction of sweat production in both upper and lower limbs (table 2).
Table 2.
Quantitative sensory testing and autonomic functional findings
The immunohistochemical study of skin innervation showed the lack of myelinated and unmyelinated somatic and autonomic nerve fibers. Notably, somatic innervation study revealed a loss of Meissner corpuscles, myelinated fibers, and epidermal nerve fibers in both proximal and distal sites. Likewise, autonomic innervation studies revealed a complete nerve fiber loss around sweat glands and arteriovenous anastomoses (figure 2). The other members of the family, including the parents (I-1 and I-2) and the sister (II-3), were clinically and electrophysiologically unaffected.
Figure 2. Immunohistochemical study of skin innervation.

Confocal images from fingertip (A, B, G, H) and leg (C, D, E, F) in a control (A, C, E, G) and in patient II-1 (B, D, F, H); and in green nerve fibers (protein gene product 9.5), in red basement membranes and vessels (COLIV), and in blue epidermis and endothelia (ULEX). In patient skin Meissner corpuscles (B compared to A), epidermal nerve fibers (D compared to C), nerves around sweat glands (F compared to E), and arteriovenous anastomosis (H compared to G) are absent. Scale bar = 100 μm.
Genetic analysis.
Whole exome sequencing was performed on 2 affected family members: II-1 and II-2. Filtering for population allele frequency (Exome Variant Server [EVS]: minor allele frequency<0.01) under both homozygous and compound heterozygous models revealed 5 candidate genes: DST, FNDC3B, GNG7, PEG3, and ZIM2. All but DST carried missense variants without loss of function effects and did not reach a threshold of polyphen >0.5. Given the phenotypic overlap with the mouse model for DST, analysis focused on the variants in DST isoform 2 (NM_001144769): c.616C>T, p.R206W (exon 4; chr6:56716204), and c.687+1 G>A, p.K229fs×21 (exon 5; chr6:56600025). Sanger sequencing was performed in DST and confirmed full cosegregation with the phenotype (figure 1A). The p.R206W change was rare (EVS 1/10,299 chromosome), conserved (polyphen 1, phastcons 0.69), and predicted to be damaging by SIFT. The p.K229fs×21 variant constitutes a loss of function frameshift mutation, which disrupts the canonical splice donor site of exon 5 mRNA. Translation is predicted to read into the intron for another 21 residues before encountering a termination codon. Likely, the affected transcript will undergo nonsense mediated decay given its proximal premature stop. The observed base change is very rare (EVS not observed in 10,299 chromosomes; GENESIS not observed in 12,500 chromosomes) and the nucleotide is highly conserved (phastcons 1). Given the complete segregation in 3 affected and 1 unaffected meioses, we calculated the simple probability that the 2 observed variants cosegregate independently by chance using the methods of Jarvik and Browning26 adapted to a recessive trait. We found that probability N equals (1/2)2 (affected meiosis) × 3/4 (unaffected meiosis) = (1/5.3) × (1/5.3) (for 2 independent segregating variants) = 1/∼28. According to ACMG Pathogenicity Classification, N < 1/16 in more than one family supports strong evidence for pathogenicity.27
Neurons derived from iPSC.
To explore the possible alterations of neurons bearing the mutations present in the family under study, we isolated dermal fibroblasts from skin biopsies from 2 patients. In one case, we were able to develop a primary fibroblast culture; these cells were reprogrammed by transducing them with the Yamanaka cocktail.25 Many iPSC clones were isolated and compared with iPSC derived from a healthy control. The DST iPSC appeared identical to the control cells and expressed the normal profile of pluripotency markers (figure 3A). Both normal and DST iPSC were differentiated into neurons. The differentiation of iPSC derived from normal fibroblasts led to the formation of neurons with the expected phenotype, with long neurites and a normal distribution of microtubular structures. On the contrary, the differentiation of DST iPSC resulted in the expression of neuron-specific β3-tubulin, but the phenotype of the β3-tubulin-positive cells was different from that of the neurons derived from normal iPSC. The most striking difference concerned the length of the neurites, which were always shorter than those observed in control cells. Indeed, in most cases, despite the robust expression of the neuron-specific marker β3-tubulin, no clear slender projections were seen protruding from the cell body (figure 3B).
Figure 3. Analysis of dystonin in normal and affected differentiated induced pluripotent stem cells (iPSC).

(A) Normal and affected undifferentiated iPSC were checked for the expression of stemness markers Oct4 and Nanog by immunostaining before differentiation. Scale bar, 100 μm. (B) Normal and affected differentiated iPSC were stained with a specific antibody against β-tubulin revealed by Alexa-488 conjugated secondary antibody. Images were collected by fluorescence microscopy. Scale bar, 50 μm. (C) Confocal images of normal and affected differentiated iPSC stained with β-tubulin (green) and dystonin (red). Scale bar, 10 μm. (D) Western blotting of dystonin in normal and affected differentiated iPSC. GAPDH is used as loading control. Molecular weight markers are indicated.
Then we addressed the expression and the distribution of dystonin in the neurons generated from iPSC. As shown in figure 3C, neurons derived from control iPSC expressed a clearly detectable dystonin signal, mostly located around the nucleus and in a few cases in the proximal part of projections (figure e-1). On the contrary, in the cells obtained from DST iPSC, the dystonin antibody failed to detect any signal, thus indicating that the protein is expressed at low levels, if any, compared to control cells. To further address this point, we prepared total extracts from control and DST neurons and examined them by Western blotting. Considering the very large size of the various dystonin isoforms, the precise identification of the cognate bands in the Western blotting is challenging. However, there are several bands migrating more slowly than the larger molecular marker (higher than 245 kD) that represent good candidates. In the extract from DST neurons these bands were absent or, in some cases, barely detectable (figure 3D).
Dystonin gene expression.
The levels of the mRNAs encoding DST-a1 and -a2 isoforms were measured by qPCR in the RNA isolated from skin biopsies of a patient and a healthy donor. Dystonin-a2 was the most represented isoform in these 2 samples and the amount of this mRNA was similar in the patient and the control specimen (figure e-2).
DISCUSSION
We report the second family with an autosomal recessive defect in the neuronal isoforms of dystonin in humans. Our description broadens the clinical spectrum of HSAN-VI and hence will guide clinical genetic testing going forward. The reported Southern Italian family carried 2 novel compound heterozygous mutations in the proximal and N-terminal part of DST whereas the previous report of HSAN-VI in an Ashkenazi Jewish family described a homozygous mutation in the distal and C-terminal portion of DST. Both our mutations fall within the neuronal isoform-a2 of DST (figure 1C).
The phenotype of our family was milder than in the previous report. Though the clinical picture was dominated by severe sensory and autonomic neuropathy leading to ulceration, amputations, and joint deformities, the neuronal dystonin defect was eventually compatible with adult lifespan. We did not observe neonatal hypotonia or psychomotor retardation. Neurophysiologic and histopathologic data revealed a severe axonal sensory neuropathy and were consistent with a degenerative process primarily affecting sensory neurons. This is in congruence with dt mice, where main pathologic features included severe degeneration of sensory neurons in the dorsal root ganglia (DRG) and at late stages of motor neurons.28–34 These observations fit well with our electrophysiologic data that revealed mild involvement of motor nerve fibers.
Interestingly, in dt mice, sensory degeneration involves large and medium-sized proprioceptive DRG neurons whereas small-caliber nociceptive DRG neurons are relatively spared.35,36 However, skin biopsies obtained from our patients showed a loss of large-caliber fiber as well as a complete loss of small somatic nerve fibers. Consistent with this finding, clinical examination and quantitative sensory testing demonstrated a severe impairment of nociception. Since dt mice typically die around 4 weeks of age, it is conceivable that the full phenotypic representation has not fully developed. Thus the described Italian family, presumably with a residual DST activity, provides an insight into the effects of neuronal dystonin on small nerve fibers in adulthood. Finally, our patients exhibited a widespread involvement of the parasympathetic and sympathetic autonomic nervous system. The spectrum of clinical disturbances included sudomotor, gastrointestinal, genitourinary, and ocular symptoms. Our pathologic findings resembled those observed in footpad skin of young adult dt mice that showed a marked impairment of sweat gland sympathetic innervation.37
Within the nervous system, the 3 major neuronal isoforms (dystonin-a1, -a2, -a3) are characterized by unique N-terminal regions that depend on alternative use of TSS in the first exons.15 The uniqueness of each isoform dictates cellular localization and function.7,15 Our data add interesting insights as to which isoforms are implicated in the pathogenesis of HSAN-VI in humans. The first description of HSAN-VI reported a homozygous frameshift mutation starting at Glu4995 and thus leading to the loss of a functional domain common to all major neuronal dystonin isoforms.1 Instead, in our family the occurrence of both pathogenic mutations in the region of gene that is unique to neuronal isoform-a2 strongly supports the possibility that dystonin-a2 defects represent the key transcript in HSAN-VI. We hypothesize that the milder phenotype in our family is due to a hypomorphic effect of the missense variant p.R206W, limiting the functional consequences of a severe loss of function. However, we cannot exclude that dystonin-a1 may partially compensate for the loss of the isoform-a2. Supporting this assumption is the mouse model dttg4 characterized by a loss of the actin-binding domain that is common to both dystonin-a1 and -a2 isoforms. The experimental restoration of dystonin-a2 expression reduces sensory neuron degeneration and dramatically extends lifespan, though mice do eventually manifest the disease.38 This also holds the promise of a potential therapeutic strategy if future studies can confirm this compensatory effect.
Our analysis of neurons derived from DST iPSC showed evident abnormalities. In comparison to normal iPSC-derived neurons, immunostaining of patient iPSC indicated absence or very low levels of DST protein (figure 3C). This was confirmed by Western blot analysis (figure 3D). Since the levels of mRNA encoding DST-a2 were similar in patients and controls (figure e-2), our results may suggest that mutations present in the patients hamper the expression of a normal amount of protein at post-transcriptional level.
Finally, while neurons derived from normal iPSC have long and branched neurites, those derived from the DST iPSC have short and dystrophic neuritis, or no projection at all (figure 3B). These observations suggest the possibility that phenotypes observed in vivo, i.e., the absence of myelinated fibers and epidermal nerve fibers in the skin, could be the consequence of developmental defects affecting the ability of neurites to reach the epidermis.
Supplementary Material
GLOSSARY
- CVR
cardiovascular reflexes
- DRG
dorsal root ganglia
- EVS
Exome Variant Server
- FBS
fetal bovine serum
- HSAN-VI
hereditary sensory and autonomic neuropathy type VI
- iPSC
induced pluripotent stem cells
- PBS
phosphate-buffered saline
- qPCR
quantitative PCR
- QST
quantitative sensory testing
- TSS
transcription start sites
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
F.M.: conception and design of the study, acquisition and analysis of data, and drafting manuscript and figures. S. Parisi: acquisition and analysis of data and drafting figures. M.N.: acquisition and analysis of data and drafting manuscript and figures. F.T.: acquisition and analysis of data and drafting figures. S Paladino: acquisition and analysis of data. C.P.: acquisition and analysis of data and drafting manuscript. ST.: acquisition and analysis of data and drafting figures. C.N.: acquisition and analysis of data. A.P.R.: acquisition and analysis of data. V.P.: acquisition and analysis of data and drafting figures. F.M.S.: acquisition and analysis of data. M.E.S.: drafting manuscript. T.R.: design of the study, acquisition and analysis of data, and drafting manuscript. S.Z.: acquisition and analysis of data and drafting manuscript. L.S.: conception and design of the study, acquisition and analysis of data, and drafting manuscript.
STUDY FUNDING
This work was supported for S.Z. and M.E.S. by NIH (R01NS075764 and U54NS065712).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Edvardson S, Cinnamon Y, Jalas C, et al. Hereditary sensory autonomic neuropathy caused by a mutation in dystonin. Ann Neurol 2012;71:569–572. [DOI] [PubMed] [Google Scholar]
- 2.Brown A, Bernier G, Mathieu M, Rossant J, Kothary R. The mouse dystonia musculorum gene is a neural isoform of bullous pemphigoid antigen 1. Nat Genet 1995;10:301–306. [DOI] [PubMed] [Google Scholar]
- 3.Duchen LW, Falconer DS, Strich SJ. Dystonia musculorum: a hereditary neuropathy of mice affecting mainly sensory pathways. J Physiol 1963;165:7–9. [Google Scholar]
- 4.Pool M, Boudreau Larivière C, Bernier G, et al. Genetic alterations at the Bpag1 locus in dt mice and their impact on transcript expression. Mamm Genome 2005;16:909–917. [DOI] [PubMed] [Google Scholar]
- 5.Leung CL, Sun D, Zheng M, Knowles DR, Liem RK. Microtubule actin cross-linking factor (MACF): a hybrid of dystonin and dystrophin that can interact with the actin and microtubule cytoskeletons. J Cell Biol 1999;147:1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jefferson JJ, Leung CL, Liem RK. Dissecting the sequence specific functions of alternative N-terminal isoforms of mouse bullous pemphigoid antigen 1. Exp Cell Res 2006;312:2712–2725. [DOI] [PubMed] [Google Scholar]
- 7.Young KG, Kothary R. Dystonin/Bpag1: a link to what? Cell Motil Cytoskeleton 2007;64:897–905. [DOI] [PubMed] [Google Scholar]
- 8.Künzli K, Favre B, Chofflon M, Borradori L. One gene but different proteins and diseases: the complexity of dystonin and bullous pemphigoid antigen 1. Exp Dermatol 2016;25:10–16. [DOI] [PubMed] [Google Scholar]
- 9.Sawamura D, Nomura K, Sugita Y, et al. Bullous pemphigoid antigen (BPAG1): cDNA cloning and mapping of the gene to the short arm of human chromosome 6. Genomics 1990;8:722–726. [DOI] [PubMed] [Google Scholar]
- 10.Guo L, Degenstein L, Dowling J, et al. Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell 1995;81:233–243. [DOI] [PubMed] [Google Scholar]
- 11.Sonnenberg A, Liem RK. Plakins in development and disease. Exp Cell Res 2007;313:2189–2203. [DOI] [PubMed] [Google Scholar]
- 12.Jefferson JJ, Ciatto C, Shapiro L, Liem RK. Structural analysis of the plakin domain of bullous pemphigoid antigen1 (BPAG1) suggests that plakins are members of the spectrin superfamily. J Mol Biol 2007;366:244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bouameur JE, Favre B, Borradori L. Plakins, a versatile family of cytolinkers: roles in skin integrity and in human diseases. J Invest Dermatol 2014;134:885–894. [DOI] [PubMed] [Google Scholar]
- 14.Leung CL, Zheng M, Prater SM, Liem RK. The BPAG1 locus: alternative splicing produces multiple isoforms with distinct cytoskeletal linker domains, including predominant isoforms in neurons and muscles. J Cell Biol 2001;154:691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrier A, Boyer JG, Kothary R. Cellular and molecular biology of neuronal dystonin. Int Rev Cell Mol Biol 2013;300:85–120. [DOI] [PubMed] [Google Scholar]
- 16.Ryan SD, Ferrier A, Kothary R. A novel role for the cytoskeletal linker protein dystonin in the maintenance of microtubule stability and the regulation of ER-Golgi transport. Bioarchitecture 2012;2:2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nolano M, Provitera V, Estraneo A, et al. Sensory deficit in Parkinson's disease: evidence of a cutaneous denervation. Brain 2008;131:1903–1911. [DOI] [PubMed] [Google Scholar]
- 18.Vita G, Princi P, Calabro R, Toscano A, Manna L, Messina C. Cardiovascular reflex tests: assessment of age-adjusted normal range. J Neurol Sci 1986;75:263–274. [DOI] [PubMed] [Google Scholar]
- 19.Provitera V, Nolano M, Caporaso G, Stancanelli A, Santoro L, Kennedy WR. Evaluation of sudomotor function in diabetes using the dynamic sweat test. Neurology 2010;74:50–56. [DOI] [PubMed] [Google Scholar]
- 20.Nolano M, Manganelli F, Provitera V, et al. Small nerve fiber involvement in CMT1A. Neurology 2015;84:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mandich P, Fossa P, Capponi S, et al. Clinical features and molecular modelling of novel MPZ mutations in demyelinating and axonal neuropathies. Eur J Hum Genet 2009;17:1129–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rebelo AP, Abrams AJ, Cottenie E, et al. Cryptic amyloidogenic elements in the 3' UTRs of neurofilament genes trigger axonal neuropathy. Am J Hum Genet 2016;98:597–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez M, Falk MJ, Gai X, Postrel R, Schüle R, Zuchner S. Innovative genomic collaboration using the GENESIS (GEM.app) platform. Hum Mutat 2015;36:950–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park IH, Lerou PH, Zhao R, Huo H, Daley GQ. Generation of human-induced pluripotent stem cells. Nat Protoc 2008;3:1180–1186. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 26.Jarvik GP, Browning BL. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am J Hum Genet 2016;98:1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duchen LW, Strich SJ, Falconer DS. Clinical and pathological studies of an hereditary neuropathy in mice (dystonia musculorum). Brain 1964;87:367–378. [DOI] [PubMed] [Google Scholar]
- 29.Duchen LW. Dystonia musculorum: an inherited disease of the nervous system in the mouse. Adv Neurol 1976;14:353–565. [PubMed] [Google Scholar]
- 30.Al-Ali SY, al-Zuhair AG. Fine structural study of the spinal cord and spinal ganglia in mice afflicted with a hereditary sensory neuropathy, dystonia musculorum. J Submicrosc Cytol Pathol 1989;21:737–748. [PubMed] [Google Scholar]
- 31.Dowling J, Yang Y, Wollmann R, Reichardt LF, Fuchs E. Developmental expression of BPAG1-n: insights into the spastic ataxia and gross neurologic degeneration in dystonia musculorum mice. Dev Biol 1997;187:131–142. [DOI] [PubMed] [Google Scholar]
- 32.Sotelo C, Guenet JL. Pathologic changes in the CNS of dystonia musculorum mutant mouse: an animal model for human spinocerebellar ataxia. Neuroscience 1988;27:403–424. [DOI] [PubMed] [Google Scholar]
- 33.De Repentigny Y, Ferrier A, Ryan SD, Sato T, Kothary R. Motor unit abnormalities in dystonia musculorum mice. PLoS One 2011;6:e21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horie M, Watanabe K, Bepari AK, et al. Disruption of actin-binding domain-containing dystonin protein causes dystonia musculorum in mice. Eur J Neurosci 2014;40:3458–3471. [DOI] [PubMed] [Google Scholar]
- 35.Bernier G, Brown A, Dalpé G, De Repentigny Y, Mathieu M, Kothary R. Dystonin expression in the developing nervous system predominates in the neurons that degenerate in dystonia musculorum mutant mice. Mol Cell Neurosci 1995;6:509–520. [DOI] [PubMed] [Google Scholar]
- 36.Carlsten JA, Kothary R, Wright DE. Glial cell line-derived neurotrophic factor-responsive and neurotrophin-3-responsive neurons require the cytoskeletal linker protein dystonin for postnatal survival. J Comp Neurol 2001;432:155–168. [DOI] [PubMed] [Google Scholar]
- 37.Tseng KW, Peng ML, Wen YC, Liu KJ, Chien CL. Neuronal degeneration in autonomic nervous system of dystonia musculorum mice. J Biomed Sci 2011;18:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferrier A, Sato T, De Repentigny Y, et al. Transgenic expression of neuronal dystonin isoform 2 partially rescues the disease phenotype of the dystonia musculorum mouse model of hereditary sensory autonomic neuropathy VI. Hum Mol Genet 2014;23:2694–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.