

Abstract
Myotonic dystrophy (DM) is an autosomal dominant neuromuscular disease primarily characterized by myotonia and progressive muscle weakness. The pathogenesis of DM involves microsatellite expansions in noncoding regions of transcripts that result in toxic RNA gain-of-function. Each successive generation of a DM family carries larger repeat expansions, leading to an earlier age of onset with increasing disease severity. At present, diagnosis of DM is challenging and requires special genetic testing to account for somatic mosaicism and meiotic instability. While progress in genetic testing has been made, more rapid, accurate, and cost-effective approaches for measuring repeat lengths are needed to establish clear correlations between repeat size and disease phenotypes.
1. Introduction
DNA repeat expansions are responsible for more than 20 inherited neurological disorders—some of these include Huntington’s disease, fragile X syndrome, spinal and bulbar muscular atrophy, as well as the most common form of familial amyotrophic lateral sclerosis [1]. In multiple repeat diseases, repeat length is correlated to disease severity and age of onset [2], yet molecular pathways that go awry due to expanded repeats can differ. Studies of myotonic dystrophy (dystrophia myotonica, DM) first demonstrated the concept that microsatellite repeats in noncoding regions can be transcribed into pathogenic RNAs [3]. Expansions can occur in the germline, leading to genetic anticipation across multiple generations, and can also occur somatically during various stages of human development with preferences for distinct tissues, ages, genders, and populations [4] [5] [6] [7]. Furthermore, the rate of expansion in somatic cells can vary within the same tissue [8].
2. Overview of Myotonic Dystrophy
Myotonic dystrophy exists in two clinically and molecularly defined forms: myotonic dystrophy type 1 (DM1), also known as Steinert’s disease; and myotonic dystrophy type 2 (DM2), also known as proximal myotonic myopathy, both of which are inherited in an autosomal dominant fashion [9]. DM1 is caused by a CTG expansion in the 3′ untranslated region of the dystrophia myotonica protein kinase (DMPK) gene on chromosome 19q13 [10] [11], while DM2 is caused by a CCTG expansion located within intron 1 of the cellular nucleic-acid-binding protein (CNBP, formerly ZNF9) gene on chromosome 3q21 [12].
A healthy individual with normal DMPK alleles has 5 to 37 repeats (35 has also commonly been used as an upper threshold for normal repeat length [13]) [14]. DM1 patients who have repeats between 38 and 50 are said to have a “pre-mutation” allele and can be asymptomatic throughout their lifetime. However, they are at increased risk of having children with larger repeats [15]. Penetrance tends to grow as repeat length increases, but extreme variability in penetrance of specific symptoms exists in the patient population [14]. Somatic mosaicism and intergenerational instability are biased towards expansion in DM1 [4], although contraction can rarely occur. It is estimated that a decrease in the CTG repeat size during transmission from parents to child is about 6.4%, most frequently during paternal transmissions [16]. Children of DM1 parents typically inherit repeat lengths considerably larger than those present in the transmitting parent, the phenomenon known as “anticipation,” where disease severity increases and age of onset decreases in successive generations. Up to 5% of DM1 patients have interrupted repeats, in which the CTG repeat tract contains GGC, CCG, or CTC repeats [17] [18]. Some of these interruptions have been associated with stabilization of the CTG repeat tract length [19].
The repeat expansion of DM2 in intron 1 of CNBP is found within the context of a complex (TG)n(TCTG)n(CCTG)n sequence. While non-pathogenic alleles contain up to 26 repeats, the range of repeats in patients is extremely broad, with measurements from 75 to 11,000 units (on average 5,000) [12]. Unlike DM1, the size of the repeat DNA expansion in DM2 does not correlate with age of onset or disease severity [20]. This is further supported by the observation that individuals homozygous for repeat expansions have clinical features indistinguishable from that of their heterozygous siblings [21]. Phenotypes and anticipation in DM2 are almost always milder than DM1, and DM2 lacks the congenital form [22].
The combined prevalence of DM1 and DM2 is approximately 1 in 8,000 (12.5 per 100,000), but this is likely an underestimate because of difficulty in clinical identification of minimally affected individuals [7]. Although DM2 is generally rarer than DM1, recent epidemiological data in Germany and Finland suggest that DM2 occurs more frequently than previously observed [23]. Similarly, the prevalence of DM1 can vary widely: in Taiwan, approximately 0.5 in 100,000 people are affected; while in the United Kingdom, the number can range from 7.1 to 10.6 in 100,000 [24]. Different factors could play a role in such variations: for instance, a founder effect is assumed to have increased the prevalence of DM1 to 1 in 500 in the Saguenay-Lac-Saint-Jean region of Northeastern Quebec [25].
Despite these key differences, DM1 and DM2 share several hallmark clinical features such as myotonia, cataracts, and cardiac conduction defects [26]. The fact that two independent mutations cause the similar disease pathology has led to the RNA toxicity hypothesis where the expanded repeat-containing RNAs form ribonuclear foci that sequester and disrupt the normal activities of RNA binding proteins belonging to the MBNL and CELF families [27] (for more details, see Ashizawa’s review on “RNA foci” and Thornton’s review on “DM: approaches to therapy” in the same issue, as well as [28]). In this review, we discuss mechanisms of repeat expansion, approaches for measuring repeat lengths, and the relationships between repeat length and phenotypes in DM.
3. Mechanisms of Repeat Expansion in Mitotic and Post-Mitotic Tissues
Several molecular mechanisms for repeat instability have been proposed, mainly in the context of DNA replication, recombination, transcription and/or repair (Box 1). Most of these mechanisms involve folding of microsatellite repeats into an unusual secondary structure, kinetically trapping the otherwise unstable DNA repeats [29]. In the case of DM, (CTG)n(CAG)n and (CCTG)n(CAGG)n repeats form hairpin-like secondary structures, which are stabilized by both Watson-Crick (WC) and non-WC base pairs [30] [31].
Box 1. Mechanisms of Repeat Expansion and Contraction.
The hairpin-like structures are formed during an out-of-register realignment of the complementary repetitive strands during DNA replication, recombination, transcription, and/or repair. Flanking DNA is shown in black. Parental repeat and complementary strands are shown in red and orange, respectively. The newly synthesized repeat strand is shown in blue. (a) A model of repeat instability generated during replication fork stalling and restart. Formation of a hairpin in the lagging-strand promotes the stalling of a DNA polymerase. To overcome this obstacle, fork reversal promotes unwinding of newly synthesized strands (as well as reannealing of parental strands), exposing a structure-prone 3′ repetitive run. A hairpin structure is formed and retained during leading strand synthesis, which leads to expansion. Repeat contraction can occur if the DNA polymerase skips the hairpin on the lagging-strand template. (b) Recombination model of repeat instability. Cleavage of stable DNA hairpin during replication generates single-stranded 3′ repetitive fragments. Invasion of these fragments with or without repeats can lead to expansion or contraction via recombination. (c) Transcription model of repeat instability. Unwinding of repetitive DNA strands during transcription can generate template strand (TS) or non-template strand (NTS) slip-outs. RNA Polymerase II (RNAPII) can stall at the either slip-out and initiate the transcription-coupled repair (TCR). If RNAPII stalls at the NTS hairpin, TCR cuts the portion of TS and copies the slip-out during repair. Conversely, RNAPII can stall at the distal end of the TS slip-out where TCR can excise near the 5′ end of the NTS slip-out, subsequently removing the repeats by 3′-end cleavage. (d) A DNA-repair-dependent repeat expansion model. DNA damage induces a small gap in the structure-prone repetitive strand. Hairpin formation prevents FEN1 endonuclease from loading onto a repetitive flap. The binding of mismatch repair proteins, MSH2 and MSH3, stabilizes the hairpin leading to expansion upon completion of repair.
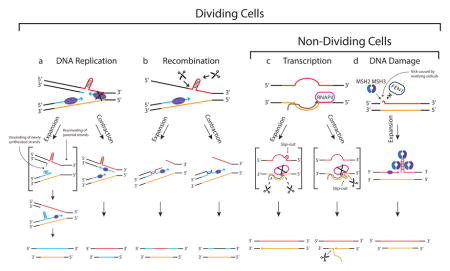
Proof of principle studies from yeast have demonstrated that repeat instability can be based on replication fork stalling and restart [32] [33], ruling out the classic model of strand slippage for DM [34]. In this model, the formation of a stable secondary structure during lagging strand synthesis could stall a DNA polymerase, slowing down the overall replication fork progression as the lagging and leading strand syntheses are coordinated. To minimize the stalling of replication fork, DNA polymerase can skip an Okazaki fragment to resume lagging strand synthesis (contraction pathway) or promote fork reversal (for more details on fork reversal mechanism, please see the review by Neelsen and Lopes [•35]) to generate a structure-prone single-stranded repeat extension at the 3′ end of the leading strand. If the 3′ repetitive hairpin persists when the replication restarts, repeats can expand. Recombination can also account for repeat instability in mitotically dividing cells. In bacteria, longer repeats increase the rate of recombination [36] [37] while in yeast, CTG repeats cause chromosomal breakage [38]. As Mirkin described in his review [39], one possible mechanism for recombination-based instability is that repeats promote the double-strand breaks in DNA causing the invasion of fragments into sister chromatids.
The transcription and repair models of repeat instability can likewise account for expansion in both mitotic and post-mitotic cells. During transcription, the formation of slip-outs on either strand can stall RNA polymerase II, facilitating the transcription-coupled repair. Depending on the location of the excision, subsequent patch repair could lead to expansion or contraction [40]. Similarly, studies on transgenic mouse models of Huntington’s disease and DM have demonstrated that loss of MSH2/MSH3 mismatch repair proteins can decrease the frequency of repeat expansion [41] [42] [43]. This discovery has led to a theory that the MSH2/MSH3 complex can stabilize the secondary structure and prevent the flip removal by FEN1, leading to expansion during DNA repair. Although this theory is highly supported in a yeast model [44], it is less clear in a mouse model where repeat instability was unaltered in Fen1-knockout mice [45].
As disease symptoms in DM are most prominent in post-mitotic tissues such as the heart, skeletal muscle, and central nervous system (CNS), it is thought that DNA repair-dependent mechanisms, and potentially transcription-coupled nucleotide excision repair, may drive repeat instability in these tissues. For more details on tissue-specific DNA repair mechanisms of repeat instability, see the review by Dion [•46].
4. Diagnosis and Laboratory Methods to Measure Repeat Lengths
A wide variety of DM symptoms can bring patients to the clinic, including myotonia, muscle weakness, cardiac arrhythmias, hypersomnia, gastrointestinal (GI) tract issues, and cataracts. Prior to the discovery of the genetic basis for DM, muscle biopsies were commonly used as a diagnostic tool, but typically, a definitive diagnosis is now made via genetic testing (Figure 1). If clinical features suggest DM1 but DM1 genetic testing is negative, DM2 testing is performed.
Figure 1. Schematic of genetic testing used to diagnose DM.

DM is a multisystemic disorder that primarily affects skeletal muscle, heart, brain, eyes, CNS, and GI tract. Electromyography (EMG) is used to assess myotonia in suspected individuals. Genetic testing is performed using the patient’s DNA sample, typically from the blood. There are two distinct laboratory procedures for genetic testing: SP-PCR and TP-PCR. SP-PCR requires single-molecule dilutions of extracted DNA, which are individually PCR amplified and probed with Southern blot hybridization. This method readily amplifies repeats from individual molecules, avoiding the amplification bias for the smaller allele commonly observed in conventional PCR. Similarly, TP-PCR is an improved method for detecting larger repeats. During the early amplification cycles, 5′ fluorescently labeled primer P1 and repeat specific primer P3 with 5′ tail sequence (in yellow) generate multiple products. The primer P4, which shares the 5′ tail sequence, subsequently amplifies the products from the previous amplification cycles. A 10:1 ratio of P4 to P3 ensures that P3 is exhausted in the early amplification cycles. Flanking and repeat DNA are shown in black and red, respectively.
A number of approaches are taken to measure repeat lengths. A PCR across the DM1 locus is usually first conducted to determine whether there are two different short alleles, or a short allele and a longer allele of <150 repeats. Extremely long repeats are challenging to amplify by PCR. Therefore, the presence of a single short PCR product does not rule out DM1 [11]. In such cases, other methods such as triplet-repeat primed PCR (TP-PCR), small-pool PCR (SP-PCR) and/or Southern blots, on either PCR products or restriction-digested genomic DNA, are performed.
Triplet-repeat primed PCR
Warner et al. [47] introduced a robust and reliable method to identify (but not the size of) an expanded allele using TP-PCR. This technique is typically cheaper and faster than Southern blotting for diagnostic purposes. However, TP-PCR can lead up to 9% false positive results due to sequence interruptions between repeats [48] [49]. More recently, an improved bi-directionally labeled TP-PCR was developed by Radvansky et al. [50], in which TP-PCR is performed in a reverse direction with two individual fluorescently-labeled flanking primers. This method can detect expansions carrying sequence interruptions both in DM1 and DM2. In the context of DM2, quadruplet-repeat primed PCR is also commonly performed to size the CCTG repeat tract [20].
Small-pool PCR
Conventional PCR and Southern blot approaches typically show expanded alleles as a smear rather than a discrete band on a gel, due to somatic instability [51]. SP-PCR, in which small amounts of input DNA up to 2 genomic equivalents are separately PCR-amplified and detected by Southern, showed that these smears are indeed CTG tracts of discrete lengths from different nuclei [4]. This method allows for assay of the entire repeat length distribution, as well as estimation of the progenitor allele length [52].
Finally, an approach combining rolling circle amplification and Southern blotting to identify expanded repeats in DM1 and DM2 with low DNA input requirements [53] and a method which utilizes a special PCR enzyme mix and machine to screen normal DMPK alleles in less than 15 minutes have been described [•54].
5. Repeat Range, Penetrance, Age of Onset, and Relationship to Phenotypes
DM1 patients can be subdivided into several categories, based on clinical features, age of onset, and disease severity. As larger patient cohorts are studied and more precise methods applied to measure clinical symptoms, the granularity of these categories has increased. Traditionally, DM1 has been divided into late-onset, adult, and congenital forms. However, in a recent study by De Antonio et al. [••55], separate categories of congenital, infantile, juvenile, adult and late-onset forms of DM1 are described, with respective mean repeat lengths of ~1000, 800, 600, 400, and 200 according to registry records (Table 1). Across all these forms, symptoms span a broad range of tissues, including skeletal muscle, heart, CNS, as well as the GI tract, eyes, reproductive tract, endocrine system, and immune system. Although in general many symptoms occur earlier when repeats are longer, there is extreme variation across individuals. For example, while many adult patients first present myotonia, the first symptom affecting other adults with DM1 may be hypersomnolence, subcapsular/iridescent “Christmas tree” cataracts, or atrial fibrillation.
Table 1.
Summary of Repeat Range, Penetrance, Age of Onset, and Phenotypes
| Phenotype | Most prominent clinical symptoms | Repeat range | Age of Onset | Life Span | Penetrance and Anticipation (Yes/No) |
|---|---|---|---|---|---|
| Pre-mutation | None | 38–49 | - | Normal | At increased risk of penetrance Uncertain |
| Late-onset DM1 | Cataracts, hypersomnia, myotonia | 100–600 | >40 years | Normal | Full Penetrance Yes |
| Adult DM1 | Myotonia, cardiac arrhythmias, hypersomnia, gastrointestinal difficulties, muscle weakness and wasting, cataracts, male hypogonadism, insulin resistance, cognitive challenges, left ventricular dysfunctions | 250–750 | 20–40 years | Shortened | Full Penetrance Yes |
| Juvenile DM1 | Similar symptoms as adult DM1 but more severe | 400–800 | 10–20 years | Shortened | Full Penetrance Yes |
| Infantile DM1 | Similar symptoms as congenital DM1 but less severe | 500–1100 | 1 mo. – 10 years | Shortened | Full Penetrance Yes |
| Congenital DM1 | Developmental defects, hypotonia, respiratory insufficiency, cardiac defects, severe cognitive challenges, facial dysmorphism, dysphagia | 750–1400 | Birth | At increased risk of infant mortality or shortened | Full Penetrance Yes |
| DM2 | Proximal muscle weakness and wasting, cognitive challenges, cardiac arrhythmias, myalgic pain, hypertension | 100–10,000 | Adult | Normal | Uncertain |
There has been much discussion regarding whether to draw distinctions between the congenital, infantile, and juvenile forms of DM1; each is successively milder, with the congenital exhibiting profound developmental defects, including hypotonia, respiratory insufficiency, cardiac defects, severe muscle weakness, cognitive challenges, and facial dysmorphism. Congenitally affected fetuses are associated with excess amniotic fluid and decreased movement [56] [57], and occur almost exclusively from maternal transmissions (although there are exceptions [58]). The infantile and juvenile forms of DM1 are less severe than congenital, but still exhibit many of the same features, in particular, cognitive challenges [59] [60]. Severe myotonia is also much more prominent in the juvenile form [55].
Core features of adult and late-onset DM1 are cataracts, myotonia, GI problems, muscle weakness, and cardiac arrhythmias. Cardiac conduction defects with arrhythmias contribute to the shortened life span of adult DM1 patients [61] [62]. Mild intellectual deficits can be found in both of adult and late-onset patients [•63], but clinical depression and personality disorders are more common in adult DM1 [64] [65]. Nocturnal apnoeic episodes and excessive daytime somnolence also have significant repercussions on the quality of adult DM1 patient’s life [66] [67].
Repeat lengths in DM2 have not been observed to correlate with specific phenotypes, such as proximal muscle weakness (often the quadriceps), cardiac arrhythmias, and cognitive decline, but this is likely in part due to the relative paucity of studies focused on measuring these correlations. The repeat length in DM2 can be extremely long, presenting technical challenges.
6. Concluding Remarks
As the molecular basis of DM continues to unfold, it is clear that RNA toxicity has a major role in disease pathology. Based on this framework, one would expect that repeat length and expression levels correlate with disease severity and age of onset. However, heterogeneity across tissues, somatic instability, and the relative technical difficulty of accurately measuring repeat length distributions present challenges in establishing these correlations, and importantly, identifying additional genetic modifiers that may protect or exacerbate particular disease symptoms. New sequencing, such as those that have been applied in the other repeat diseases (PacBio single-molecule, real-time sequencing for Fragile X [••68]), may allow for more rapid, cost-effective, and accurate measurements of long repeat lengths at a single-nucleotide resolution, and enhance our overall understanding of expansion repeat diseases.
Highlights.
Distinct mechanisms for repeat expansion in mitotic and post-mitotic tissues of DM patients.
Overview of the diagnostic methodologies for measuring repeat lengths in DM
Relationship of DM repeat range with penetrance, age of onset, and clinical symptoms
Acknowledgments
E.T.W. is supported by (NIH DP5 OD 017865). A.K. is supported by grants from the US National Institute of Health (R01HL126845), March of Dimes (5-FY14-112), and the Center for Advanced Study at the University of Illinois. The authors apologize to those whose work could not be cited because of lack of space.
Footnotes
8. Competing interests statement
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
• • of outstanding interest
- 1.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.La Spada AR. Trinucleotide repeat instability: genetic features and molecular mechanisms. Brain Pathol. 1997;7:943–963. doi: 10.1111/j.1750-3639.1997.tb00895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 4.Monckton DG, Wong LJ, Ashizawa T, Caskey CT. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum Mol Genet. 1995;4:1–8. doi: 10.1093/hmg/4.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Cleary JD, Tome S, Lopez Castel A, Panigrahi GB, Foiry L, Hagerman KA, Sroka H, Chitayat D, Gourdon G, Pearson CE. Tissue- and age-specific DNA replication patterns at the CTG/CAG-expanded human myotonic dystrophy type 1 locus. Nat Struct Mol Biol. 2010;17:1079–1087. doi: 10.1038/nsmb.1876. [DOI] [PubMed] [Google Scholar]
- 6.Dogan C, De Antonio M, Hamroun D, Varet H, Fabbro M, Rougier F, Amarof K, Arne Bes MC, Bedat-Millet AL, Behin A, et al. Gender as a Modifying Factor Influencing Myotonic Dystrophy Type 1 Phenotype Severity and Mortality: A Nationwide Multiple Databases Cross-Sectional Observational Study. PLoS One. 2016;11:e0148264. doi: 10.1371/journal.pone.0148264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suominen T, Bachinski LL, Auvinen S, Hackman P, Baggerly KA, Angelini C, Peltonen L, Krahe R, Udd B. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet. 2011;19:776–782. doi: 10.1038/ejhg.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 9.Harper PS, Brook JD, Newman E. Myotonic dystrophy. 3. London: Saunders; 2001. [Google Scholar]
- 10.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 11.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O’Hoy K, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 12.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 13.New nomenclature and DNA testing guidelines for myotonic dystrophy type 1 (DM1) The International Myotonic Dystrophy Consortium (IDMC) Neurology. 2000;54:1218–1221. doi: 10.1212/wnl.54.6.1218. [DOI] [PubMed] [Google Scholar]
- 14.Turner C, Hilton-Jones D. The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry. 2010;81:358–367. doi: 10.1136/jnnp.2008.158261. [DOI] [PubMed] [Google Scholar]
- 15.Martorell L, Monckton DG, Sanchez A, Lopez De Munain A, Baiget M. Frequency and stability of the myotonic dystrophy type 1 premutation. Neurology. 2001;56:328–335. doi: 10.1212/wnl.56.3.328. [DOI] [PubMed] [Google Scholar]
- 16.Ashizawa T, Anvret M, Baiget M, Barcelo JM, Brunner H, Cobo AM, Dallapiccola B, Fenwick RG, Jr, Grandell U, Harley H, et al. Characteristics of intergenerational contractions of the CTG repeat in myotonic dystrophy. Am J Hum Genet. 1994;54:414–423. [PMC free article] [PubMed] [Google Scholar]
- 17.Musova Z, Mazanec R, Krepelova A, Ehler E, Vales J, Jaklova R, Prochazka T, Koukal P, Marikova T, Kraus J, et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am J Med Genet A. 2009;149A:1365–1374. doi: 10.1002/ajmg.a.32987. [DOI] [PubMed] [Google Scholar]
- 18.Braida C, Stefanatos RK, Adam B, Mahajan N, Smeets HJ, Niel F, Goizet C, Arveiler B, Koenig M, Lagier-Tourenne C, et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum Mol Genet. 2010;19:1399–1412. doi: 10.1093/hmg/ddq015. [DOI] [PubMed] [Google Scholar]
- 19.Rolfsmeier ML, Lahue RS. Stabilizing effects of interruptions on trinucleotide repeat expansions in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:173–180. doi: 10.1128/mcb.20.1.173-180.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, Schneider C, Koch MC, Beilman GJ, Harrison AR, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 2003;60:657–664. doi: 10.1212/01.wnl.0000054481.84978.f9. [DOI] [PubMed] [Google Scholar]
- 21.Schoser BG, Kress W, Walter MC, Halliger-Keller B, Lochmuller H, Ricker K. Homozygosity for CCTG mutation in myotonic dystrophy type 2. Brain. 2004;127:1868–1877. doi: 10.1093/brain/awh210. [DOI] [PubMed] [Google Scholar]
- 22.Schneider C, Ziegler A, Ricker K, Grimm T, Kress W, Reimers CD, Meinck H, Reiners K, Toyka KV. Proximal myotonic myopathy: evidence for anticipation in families with linkage to chromosome 3q. Neurology. 2000;55:383–388. doi: 10.1212/wnl.55.3.383. [DOI] [PubMed] [Google Scholar]
- 23.Udd B, Meola G, Krahe R, Thornton C, Ranum LP, Bassez G, Kress W, Schoser B, Moxley R. 140th ENMC International Workshop: Myotonic Dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on management. Neuromuscul Disord. 2006;16:403–413. doi: 10.1016/j.nmd.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 24.Theadom A, Rodrigues M, Roxburgh R, Balalla S, Higgins C, Bhattacharjee R, Jones K, Krishnamurthi R, Feigin V. Prevalence of muscular dystrophies: a systematic literature review. Neuroepidemiology. 2014;43:259–268. doi: 10.1159/000369343. [DOI] [PubMed] [Google Scholar]
- 25.Yotova V, Labuda D, Zietkiewicz E, Gehl D, Lovell A, Lefebvre JF, Bourgeois S, Lemieux-Blanchard E, Labuda M, Vezina H, et al. Anatomy of a founder effect: myotonic dystrophy in Northeastern Quebec. Hum Genet. 2005;117:177–187. doi: 10.1007/s00439-005-1298-8. [DOI] [PubMed] [Google Scholar]
- 26.Cho DH, Tapscott SJ. Myotonic dystrophy: emerging mechanisms for DM1 and DM2. Biochim Biophys Acta. 2007;1772:195–204. doi: 10.1016/j.bbadis.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 27.Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol. 2007;20:572–576. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- 28.Chau A, Kalsotra A. Developmental insights into the pathology of and therapeutic strategies for DM1: Back to the basics. Dev Dyn. 2015;244:377–390. doi: 10.1002/dvdy.24240. [DOI] [PubMed] [Google Scholar]
- 29.McMurray CT. DNA secondary structure: a common and causative factor for expansion in human disease. Proc Natl Acad Sci U S A. 1999;96:1823–1825. doi: 10.1073/pnas.96.5.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gacy AM, Goellner G, Juranic N, Macura S, McMurray CT. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell. 1995;81:533–540. doi: 10.1016/0092-8674(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 31.Dere R, Napierala M, Ranum LP, Wells RD. Hairpin structure-forming propensity of the (CCTG. CAGG) tetranucleotide repeats contributes to the genetic instability associated with myotonic dystrophy type 2. J Biol Chem. 2004;279:41715–41726. doi: 10.1074/jbc.M406415200. [DOI] [PubMed] [Google Scholar]
- 32.Pelletier R, Krasilnikova MM, Samadashwily GM, Lahue R, Mirkin SM. Replication and expansion of trinucleotide repeats in yeast. Mol Cell Biol. 2003;23:1349–1357. doi: 10.1128/MCB.23.4.1349-1357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krasilnikova MM, Mirkin SM. Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Mol Cell Biol. 2004;24:2286–2295. doi: 10.1128/MCB.24.6.2286-2295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petruska J, Hartenstine MJ, Goodman MF. Analysis of strand slippage in DNA polymerase expansions of CAG/CTG triplet repeats associated with neurodegenerative disease. J Biol Chem. 1998;273:5204–5210. doi: 10.1074/jbc.273.9.5204. [DOI] [PubMed] [Google Scholar]
- 35•.Neelsen KJ, Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol. 2015;16:207–220. doi: 10.1038/nrm3935. This review provides a detailed summary of in vivo mechanisms and the consequences of fork reversal during replication. [DOI] [PubMed] [Google Scholar]
- 36.Jakupciak JP, Wells RD. Genetic instabilities in (CTG. CAG) repeats occur by recombination. J Biol Chem. 1999;274:23468–23479. doi: 10.1074/jbc.274.33.23468. [DOI] [PubMed] [Google Scholar]
- 37.Dere R, Wells RD. DM2 CCTG*CAGG repeats are crossover hotspots that are more prone to expansions than the DM1 CTG*CAG repeats in Escherichia coli. J Mol Biol. 2006;360:21–36. doi: 10.1016/j.jmb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 38.Freudenreich CH, Kantrow SM, Zakian VA. Expansion and length-dependent fragility of CTG repeats in yeast. Science. 1998;279:853–856. doi: 10.1126/science.279.5352.853. [DOI] [PubMed] [Google Scholar]
- 39.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 40.Salinas-Rios V, Belotserkovskii BP, Hanawalt PC. DNA slip-outs cause RNA polymerase II arrest in vitro: potential implications for genetic instability. Nucleic Acids Res. 2011;39:7444–7454. doi: 10.1093/nar/gkr429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pearson CE, Ewel A, Acharya S, Fishel RA, Sinden RR. Human MSH2 binds to trinucleotide repeat DNA structures associated with neurodegenerative diseases. Hum Mol Genet. 1997;6:1117–1123. doi: 10.1093/hmg/6.7.1117. [DOI] [PubMed] [Google Scholar]
- 42.Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet. 1999;23:471–473. doi: 10.1038/70598. [DOI] [PubMed] [Google Scholar]
- 43.van den Broek WJ, Nelen MR, Wansink DG, Coerwinkel MM, te Riele H, Groenen PJ, Wieringa B. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet. 2002;11:191–198. doi: 10.1093/hmg/11.2.191. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y, Zhang H, Veeraraghavan J, Bambara RA, Freudenreich CH. Saccharomyces cerevisiae flap endonuclease 1 uses flap equilibration to maintain triplet repeat stability. Mol Cell Biol. 2004;24:4049–4064. doi: 10.1128/MCB.24.9.4049-4064.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van den Broek WJ, Nelen MR, van der Heijden GW, Wansink DG, Wieringa B. Fen1 does not control somatic hypermutability of the (CTG)(n)*(CAG)(n) repeat in a knock-in mouse model for DM1. FEBS Lett. 2006;580:5208–5214. doi: 10.1016/j.febslet.2006.08.059. [DOI] [PubMed] [Google Scholar]
- 46•.Dion V. Tissue specificity in DNA repair: lessons from trinucleotide repeat instability. Trends Genet. 2014;30:220–229. doi: 10.1016/j.tig.2014.04.005. This review provides a detailed summary of tissue-specific DNA repair mechanisms responsible for trinucleotide repeat instability. [DOI] [PubMed] [Google Scholar]
- 47.Warner JP, Barron LH, Goudie D, Kelly K, Dow D, Fitzpatrick DR, Brock DJ. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J Med Genet. 1996;33:1022–1026. doi: 10.1136/jmg.33.12.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kakourou G, Dhanjal S, Mamas T, Serhal P, Delhanty JD, SenGupta SB. Modification of the triplet repeat primed polymerase chain reaction method for detection of the CTG repeat expansion in myotonic dystrophy type 1: application in preimplantation genetic diagnosis. Fertil Steril. 2010;94:1674–1679. doi: 10.1016/j.fertnstert.2009.10.050. [DOI] [PubMed] [Google Scholar]
- 49.Radvansky J, Ficek A, Minarik G, Palffy R, Kadasi L. Effect of unexpected sequence interruptions to conventional PCR and repeat primed PCR in myotonic dystrophy type 1 testing. Diagn Mol Pathol. 2011;20:48–51. doi: 10.1097/PDM.0b013e3181efe290. [DOI] [PubMed] [Google Scholar]
- 50.Radvansky J, Ficek A, Kadasi L. Upgrading molecular diagnostics of myotonic dystrophies: multiplexing for simultaneous characterization of the DMPK and ZNF9 repeat motifs. Mol Cell Probes. 2011;25:182–185. doi: 10.1016/j.mcp.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 51.Prior TW American College of Medical Genetics Laboratory Quality Assurance C. Technical standards and guidelines for myotonic dystrophy type 1 testing. Genet Med. 2009;11:552–555. doi: 10.1097/GIM.0b013e3181abce0f. [DOI] [PubMed] [Google Scholar]
- 52.Higham CF, Monckton DG. Modelling and inference reveal nonlinear length-dependent suppression of somatic instability for small disease associated alleles in myotonic dystrophy type 1 and Huntington disease. J R Soc Interface. 2013;10:20130605. doi: 10.1098/rsif.2013.0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakamori M, Sobczak K, Moxley RT, 3rd, Thornton CA. Scaled-down genetic analysis of myotonic dystrophy type 1 and type 2. Neuromuscul Disord. 2009;19:759–762. doi: 10.1016/j.nmd.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54•.Connelly CM, Porter LR, TerMaat JR. PCR amplification of a triple-repeat genetic target directly from whole blood in 15 minutes as a proof-of-principle PCR study for direct sample analysis for a clinically relevant target. BMC Med Genet. 2014;15:130. doi: 10.1186/s12881-014-0130-5. This article provides a rapid PCR screening method directly from patient’s blood samples to identify the presence of normal DMPK alleles, which can function as a negative DM1 screen if both alleles of normal size are clearly detected. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55••.De Antonio M, Dogan C, Hamroun D, Mati M, Zerrouki S, Eymard B, Katsahian S, Bassez G French Myotonic Dystrophy Clinical N. Unravelling the myotonic dystrophy type 1 clinical spectrum: A systematic registry-based study with implications for disease classification. Rev Neurol (Paris) 2016;172:572–580. doi: 10.1016/j.neurol.2016.08.003. A large cohort of DM1 patients was studied from the DM-Scope registry and classified into five subtypes (including two new subtypes: infantile and juvenile) based on their distribution of CTG expansion size and age of major symptom onset. [DOI] [PubMed] [Google Scholar]
- 56.Zaki M, Boyd PA, Impey L, Roberts A, Chamberlain P. Congenital myotonic dystrophy: prenatal ultrasound findings and pregnancy outcome. Ultrasound Obstet Gynecol. 2007;29:284–288. doi: 10.1002/uog.3859. [DOI] [PubMed] [Google Scholar]
- 57.Esplin MS, Hallam S, Farrington PF, Nelson L, Byrne J, Ward K. Myotonic dystrophy is a significant cause of idiopathic polyhydramnios. Am J Obstet Gynecol. 1998;179:974–977. doi: 10.1016/s0002-9378(98)70200-5. [DOI] [PubMed] [Google Scholar]
- 58.Zeesman S, Carson N, Whelan DT. Paternal transmission of the congenital form of myotonic dystrophy type 1: a new case and review of the literature. Am J Med Genet. 2002;107:222–226. doi: 10.1002/ajmg.10141. [DOI] [PubMed] [Google Scholar]
- 59.Echenne B, Bassez G. Congenital and infantile myotonic dystrophy. Handb Clin Neurol. 2013;113:1387–1393. doi: 10.1016/B978-0-444-59565-2.00009-5. [DOI] [PubMed] [Google Scholar]
- 60.Douniol M, Jacquette A, Guile JM, Tanguy ML, Angeard N, Heron D, Plaza M, Cohen D. Psychiatric and cognitive phenotype in children and adolescents with myotonic dystrophy. Eur Child Adolesc Psychiatry. 2009;18:705–715. doi: 10.1007/s00787-009-0037-4. [DOI] [PubMed] [Google Scholar]
- 61.Melacini P, Villanova C, Menegazzo E, Novelli G, Danieli G, Rizzoli G, Fasoli G, Angelini C, Buja G, Miorelli M, et al. Correlation between cardiac involvement and CTG trinucleotide repeat length in myotonic dystrophy. J Am Coll Cardiol. 1995;25:239–245. doi: 10.1016/0735-1097(94)00351-p. [DOI] [PubMed] [Google Scholar]
- 62.Groh WJ, Lowe MR, Zipes DP. Severity of cardiac conduction involvement and arrhythmias in myotonic dystrophy type 1 correlates with age and CTG repeat length. J Cardiovasc Electrophysiol. 2002;13:444–448. doi: 10.1046/j.1540-8167.2002.00444.x. [DOI] [PubMed] [Google Scholar]
- 63•.Jean S, Richer L, Laberge L, Mathieu J. Comparisons of intellectual capacities between mild and classic adult-onset phenotypes of myotonic dystrophy type 1 (DM1) Orphanet J Rare Dis. 2014;9:186. doi: 10.1186/s13023-014-0186-5. This clinical study provides evidence that intellectual impairment in classic adult-onset DM1 patients are also common in mild late-onset DM1 patients characterized by low general intellectual functioning in both verbal and nonverbal abilities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winblad S, Jensen C, Mansson JE, Samuelsson L, Lindberg C. Depression in Myotonic Dystrophy type 1: clinical and neuronal correlates. Behav Brain Funct. 2010;6:25. doi: 10.1186/1744-9081-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winblad S, Lindberg C, Hansen S. Temperament and character in patients with classical myotonic dystrophy type 1 (DM-1) Neuromuscul Disord. 2005;15:287–292. doi: 10.1016/j.nmd.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 66.Hansotia P, Frens D. Hypersomnia associated with alveolar hypoventilation in myotonic dystrophy. Neurology. 1981;31:1336–1337. doi: 10.1212/wnl.31.10.1336. [DOI] [PubMed] [Google Scholar]
- 67.Cirignotta F, Mondini S, Zucconi M, Barrot-Cortes E, Sturani C, Schiavina M, Coccagna G, Lugaresi E. Sleep-related breathing impairment in myotonic dystrophy. J Neurol. 1987;235:80–85. doi: 10.1007/BF00718014. [DOI] [PubMed] [Google Scholar]
- 68••.Loomis EW, Eid JS, Peluso P, Yin J, Hickey L, Rank D, McCalmon S, Hagerman RJ, Tassone F, Hagerman PJ. Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome Res. 2013;23:121–128. doi: 10.1101/gr.141705.112. This study demonstrates that the SMRT sequencing can generate real-time fluorescent pulse data to help sequence FMR1 alleles containing large CGG repeats. [DOI] [PMC free article] [PubMed] [Google Scholar]