

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the death of upper and lower motor neurons. MicroRNAs (miRNAs) are reported to be closely related to the development of ALS. However, the precise functions of miRNAs in the pathogenesis of ALS remain largely unknown. In previous studies, we determined that miRNA-193b-3p was significantly downregulated in patients with sporadic ALS (sALS). Here, we observed that miRNA-193b-3p was downregulated in the SOD1G93A mouse model of ALS and promoted cell death in NSC-34 cells. We further found that miR-193b-3p directly targeted tuberous sclerosis 1 (TSC1) to regulate mechanistic target of rapamycin complex 1 (mTORC1) activity. Downregulation of miR-193b-3p led to TSC1 increase accompanied with mTORC1 inactivation, and vice versa. Moreover, downregulation of miR-193b-3p promoted protective autophagy and cell survival in NSC-34 cells. In contrast, upregulation of miR-193b-3p activated mTORC1 signaling, leading to inhibition of autophagy and promotion of cell death. Taken together, our study suggests that downregulation of miR-193b-3p is required for cell survival by targeting TSC1/mTOR signaling in NSC-34 cells and provides a novel target for improving the clinical therapy of ALS.
Keywords: MicroRNA-193b-3p, TSC1, mTORC1, cell death, autophagy
Introduction
Amyotrophic lateral sclerosis (ALS) is a common fatal motor neuron disease characterized by selective degeneration of upper and lower motor neurons (Boillee et al., 2006; Kiernan et al., 2011; Turner et al., 2011) leading to progressive muscle atrophy and weakness. ALS can be classified into sporadic ALS (sALS) or familial ALS (fALS). Several causative genes including chromosome 9 open reading frame 72 (C9orf72, DeJesus-Hernandez et al., 2011), Cu/Zn-superoxide dismutase (SOD1; Rosen et al., 1993), TAR DNA-binding protein (TARDBP) which encodes TDP43 (Sreedharan et al., 2008), and fused in sarcoma (FUS; Vance et al., 2009) have been shown to be involved in the development of ALS. Approximately 20% of fALS and 5% of sALS cases are caused by mutations in the SOD1 gene (Majoor-Krakauer et al., 2003; Robberecht and Philips, 2013). Aberrant misfolded proteins resulting from SOD1 mutation contribute to increased cellular stress and axon degeneration (Wilcox et al., 2009; Saccon et al., 2013). An animal model with overproduction of pathogenic human SOD1 protein develops late-onset progressive neurodegenerative disease (Saccon et al., 2013). However, the exact mechanisms and pathological processes responsible for the initiation and progression of motor neuron degeneration remain largely unknown.
Excessive cellular stress causes progressive damage to motor neurons (Turner and Atkin, 2006; Barber and Shaw, 2010). Accordingly, the neuronal defensive system must be evoked to protect neurons from death (Novoselov et al., 2013; Wang et al., 2015). Identification and investigation of ALS-relevant molecular alterations may aid in the identification of potential therapeutic targets. For example, SOD1 mutants specifically render vulnerable motor neurons dependent on endogenous neuroprotection signaling involving excitability and mechanistic target of rapamycin (mTOR; Leibinger et al., 2012; Saxena et al., 2013). mTOR signaling senses extracellular stimuli and regulates many biological processes, such as cell growth, energy metabolism and autophagy (Thomson et al., 2009; Laplante and Sabatini, 2012). mTOR is a PI3K-like serine/threonine protein kinase that is evolutionarily conserved in all eukaryotes (Dazert and Hall, 2011; Bordon, 2013). Dysregulation of mTOR signaling has been shown to be closely associated with cancers, metabolic diseases as well as neurodegenerative diseases. mTOR resides in two distinct complexes referred to as mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2; Sarbassov et al., 2005). The tuberous sclerosis tumor suppressor complex (TSC), composed of TSC1 and TSC2, negatively regulates mTORC1 activity (Yang et al., 2016).
Notably, mTORC1 has been identified as a key regulator of autophagy inhibition (Kim et al., 2011). Autophagy could protect cells from death (Hara et al., 2006; Komatsu et al., 2006), especially in the case of motor neurons (Barmada et al., 2014). Autophagy is a highly conserved intracellular pathway involved in the elimination of proteins and organelles by lysosomes. Autophagy is now recognized as an arbiter of neuronal survival and death decisions in neurodegenerative diseases (Banerjee et al., 2010). In ALS, defective autophagy has also been implicated in the accumulation of ubiquitinated TDP-43 inclusions and motor neuron degeneration (Caccamo et al., 2009). Moreover, some studies report that autophagic clearance of mutant SOD1 exert protective effect against motor neuron loss in an ALS mouse model (Crippa et al., 2010). Therefore, autophagy seems to be protective for the survival of motor neurons in ALS. However, the precise role and regulatory factors of autophagy in motor neuron degeneration in ALS remain to be determined.
MicroRNAs (miRNAs) are small, single-stranded, noncoding RNAs that consist of approximately 18–22 nucleotides and can regulate protein expression either by translational inhibition or targeted mRNA cleavage (Bartel, 2009; Guo et al., 2010). Growing evidence suggests that miRNAs play an important role in neurodegenerative diseases, including ALS (Akerblom et al., 2012; Goodall et al., 2013; Zhu et al., 2013; Parisi et al., 2016). Furthermore, mutations of TARDBP and FUS in ALS, both of which are closely related to miRNA processing, give rise to more links between ALS and miRNAs (Morlando et al., 2012; Di Carlo et al., 2013). A single miRNA may have multiple mRNA targets, allowing it to be involved in diverse pathological processes (Filipowicz et al., 2008). However, the exact mechanisms and pathological processes responsible for the initiation and progression of motor neuron degeneration by miRNAs remain largely unknown.
In previous studies, we investigated the miRNA expression profiles of Chinese sALS patients to explore new potential biomarkers for the diagnosis of ALS. We noted that miR-193b-3p was downregulated in sALS patients and provided high diagnostic accuracy for sALS (Chen et al., 2016). Here, we used NSC-34 cells to investigate the fundamental functions of miR-193b-3p in the development of ALS. We found that miR-193b-3p was downregulated in mouse model of ALS and promoted cell death in NSC-34 cells. Our work suggests that downregulation of miR-193b-3p is required for cell survival by targeting TSC1/mTOR signaling to promote autophagy. These findings may inform novel therapeutic targets for ALS.
Materials and Methods
Reagents and Chemicals
For cell culture in vitro, Dulbecco’s Modified Eagle’s Medium (DMEM), trypsin and Fetal Bovine Serum (FBS) were obtained from GIBCO Invitrogen (Carlsbad, CA, USA). The anti-TSC1, TSC2, p70S6K, pp70S6K (Thr389), 4EBP1, p-4EBP1 (Thr37/46) and GAPDH antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). p62 and LC3 antibodies were purchased from Novus Biological Inc. (USA). The mTOR inhibitor Torin1 was purchased from Tocris Bioscience. All other reagents were obtained from Sigma-Aldrich with the highest purity available.
Cell Culture and Treatment
For in vitro experiments, we used the NSC-34 cell line provided by Dr. N.R. Cashman (University of Toronto, Toronto, Canada; Cashman et al., 1992). NSC-34 cells were cultured in DMEM (Gibco) containing 10% (FBS, Gibco), 100 U/mL penicillin, 100 mg/mL streptomycin (Invitrogen) at 37°C in a 95% air/5% CO2 atmosphere at constant humidity. Transfection of miR-193b-3p mimics, inhibitors and scrambled sequences (Ribobio, Guangzhou, China) were carried out when the cell confluent was 80%–90% using RNAiMAX (Invitrogen) according to the manufacturer’s instructions.
SOD1G93A Transgenic Mice
SOD1G93A transgenic mice were purchased from the Jackson Lab (Bar Harbor, ME, USA). Animal care and procedures were performed in accordance with the Laboratory Animal Care Guidelines approved by the Animal Care and Use Committee of Sichuan University West-China Hospital.
Luciferase Reporter Assay
The TSC1 3′UTR was cloned into the XbaI and EcoRI sites of the pMIR-REPORT luciferase vector (Ambion, USA), and the reconstituted plasmid was named pWT. The TSC1 3′UTR mutations were introduced using the Multisite-Quickchange kit (Stratagene, CA, USA) according to the manufacturer’s protocol and cloned into the pMIR-REPORT luciferase vector (Ambion, USA), and the reconstituted plasmid was named pMUT. All inserted or mutated sequences were confirmed by sequencing. NSC-34 cells were transfected with miR-193b-3p mimics and pWT using Lipofectamine RNAiMAX transfection reagent according to the manufacturer’s instructions. miR-193b-3p mimics and pMT, or miRNA negative control (miR-NC) and pWT, or miR-NC and pMT were also transfected into NSC-34 cells as controls. Luciferase activity was measured in cell lysates 48 h after transfection using the Dual-Light® Luminescent Reporter Gene Assay kit (Applied Biosystems, CA, USA).
miRNA Extraction and RT-qPCR Assay
Total miRNA was extracted and collected from cells using the miRNeasy Mini Kit (Qiagen, Germany) according to the manufacturer’s protocol. Isolated miRNAs were reverse-transcribed to complementary DNA (cDNA) using a miScript II reverse transcription kit (Qiagen, Germany) with the standard protocol. Quantitative real-time PCR was carried out with the miScript miRNA PCR Array (Qiagen, Germany) using the SYBR-green-based real-time PCR (RT-PCR) method on a Bio-Rad PCR machine according to the manufacturer’s protocol. The primers for miRNAs were purchased from Ribobio, China. U6 rRNA was used as an internal control. Data analysis was performed using the 2−ΔΔCt method.
RNA Extraction and RT-qPCR Assay
Total RNAs were extracted from cultured cells using Trizol reagent (Invitrogen, USA). cDNA was synthesized from 2 μg of total RNA according to the manufacturer’s instructions (Thermo Fisher Scientific, USA). Quantitative real-time PCR was performed using the Bio-Rad iQ5 system (Bio-Rad, USA), and the relative gene expression was normalized to the internal control GAPDH. Primer sequences for SYBR-green probes of target genes were as follows, and data analysis was performed using the 2−ΔΔCt method.
TSC1: 5′-ATGGCCCAGTTAGCCAACAT-3′ and 5′-CAGAATTGAGGGACTCCTT GAAG-3′;
GAPDH: 5′-AGGTCGGTGTGAACGGATTTG-3′ and 5′-TGTAGACCATGTAGTT GAGGTCA-3′;
Protein Extractions and Western Blots
To extract total proteins, cultured NSC-34 cells were sonicated with lysis buffer (2% SDS with protease and phosphatase inhibitors). The protein concentration of each extract was measured by the BCA Protein Assay kit (Thermo Scientific Pierce). Equal amounts of denatured proteins (~20 μg) from each extract were separated by SDS-PAGE. Proteins were transferred onto PVDF membranes following standard procedures. The membranes were blocked with 5% skimmed milk in TBST (TBS with 0.1% Tween 20, pH 7.6) for 1 h at room temperature on a rocker and then incubated with various antibodies diluted in TBST (1:1000) at 4°C, overnight. The membranes were then washed three times with TBST for 10 min each wash, and the membranes were incubated with appropriate secondary antibodies diluted in TBST (1:10,000 for both the goat anti-rabbit and goat anti-mouse IgG antibodies) for 2 h at room temperature. The membranes were washed three times with TBST at room temperature for 10 min. Proteins were then detected with ECL reagent (Thermo Scientific/Pierce, Rockford, IL, USA), and the membranes were exposed to film (Kodak). Films were scanned, and optical densities were quantified using ImageJ software.
Cell Viability Assay
A Cell Counting Kit-8 (CCK-8; Dojindo, Japan) assay was used to determine NSC-34 cell viability. Cells were seeded in a 96-well plate at a density of approximately 2–4 × 103 cells per well in 200 μl of culture medium and treated as designated. The absorbance was measured in a microplate reader (Gene Company Limited, China) at a wavelength of 450 nm.
Annexin V-FITC/PI Apoptosis Assay
For apoptosis examinations, we used the Annexin V-FITC/PI apoptosis assay. NSC-34 cells were digested into single cell suspensions using EDTA-free trypsin, and cells were stained according to the instructions provided with the Annexin V-FITC/PI Apoptosis Detection kit (KeyGen, Nanjing, China). The cells were analyzed after 20 min by flow cytometry.
GFP-LC3 Puncta Imaging
To assay autophagic status, NSC-34 cells were transfected with GFP-LC3 plasmids and miR-193b-3p mimics, inhibitors or scrambled sequence. Cells were grown on glass coverslips and treated as designated. Cells were fixed with 4% paraformaldehyde and 4% sucrose and permeabilized with 0.1% Triton X-100 for 10 min. Finally, cells were rinsed and mounted on cover glasses with Prolong Gold anti-fade reagent with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, USA) and visualized using an Olympus IX 81 (Olympus, Tokyo, Japan) microscope.
Statistical Analysis
All quantitative results of western blots, real-time PCR and cell assays were presented as the mean and standard error of the mean (SEM) from at least three independent experiments and analyzed by SPSS 22 Package (SPSS, USA). P values were calculated using two-tailed, unpaired Student’s t test or analysis of variance (ANOVA) with an least significant differences (LSD) post-test analysis, and the values 0.05 (*), 0.01 (**) and 0.001 (***) were assumed as the level of significance for the statistic tests carried out.
Results
miR-193b-3p Is Downregulated in the Mouse Model of ALS and Promotes Cell Death in NSC-34 Cells
In a previous study, we demonstrated that miR-193b-3p was downregulated and might be a candidate miRNA in the pathogenesis of ALS (Chen et al., 2016). To confirm the pathogenic role of downregulation of miR-193b-3p in ALS, we further examined the expression of miR-193b-3p in the SOD1G93A ALS mouse model (Ferrante et al., 1997; Chiu et al., 2008). The results showed that the expression of miR-193b-3p was also downregulated in the spinal cord of SOD1G93A ALS mice compared with wild-type controls (~90 days; Figure 1A). We also noted that miR-193b-3p was decreased in the spinal cord of ALS mice from ~90 days to ~150 days (Supplementary Figure S1A). To investigate the role of miR-193b-3p, we transfected NSC-34 hybrid mouse motor neuron-like cells with mimics or inhibitors of miR-193b-3p (Figure 1B). qRT-PCR results showed that the expression of miR-193b-3p was dramatically induced by miR-193b-3p mimics and significantly blocked by its inhibitors (Figure 1C). To investigate the role of miR-193b-3p in cell survival, we detected cell apoptosis in NSC-34 cells by flow cytometry assay. The results showed that miR-193b-3p overexpression induced either early or late apoptosis in NSC-34 cells, while its inhibition exhibited opposite effects (Figures 2A,B). Furthermore, we confirmed this finding by investigating cell viability using the CCK-8 assay. The results showed that cell viability was decreased by miR-193b-3p overexpression and increased by its inhibition (Figure 2C). Therefore, we propose that upregulation of miR-193b-3p could promote cell death in NSC-34 cells.
Figure 1.
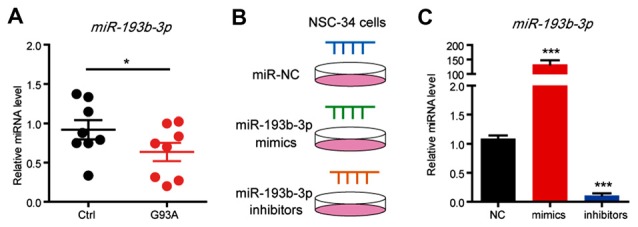
miR-193b-3p is downregulated in the mouse model of amyotrophic lateral sclerosis (ALS). (A) qRT-PCR results show the miRNA levels of miR-193b-3p in the spinal cord of SOD1G93A mutants compared with controls (~90 days). The results were averages of eight pairs of littermate mice. Data represent the mean ± standard error of the mean (SEM). *P < 0.05 vs. controls. (B,C) qRT-PCR results show the miRNA levels of miR-193b-3p in NSC-34 cells transfected with miR-193b-3p mimics, inhibitors or scrambled sequence. The results were averages of four independent experiments. Data represent the mean ± SEM. ***P < 0.001 vs. controls.
Figure 2.
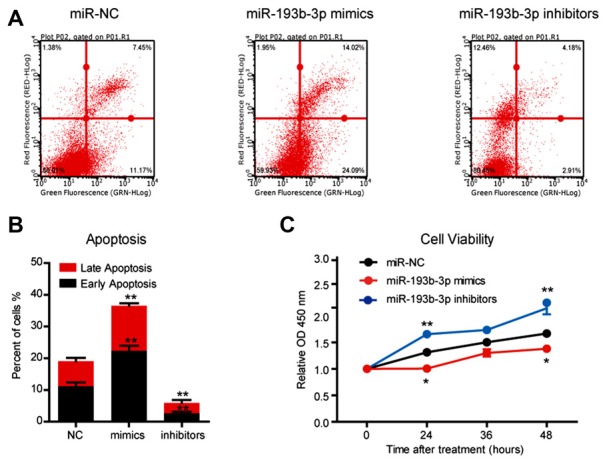
miR-193b-3p promotes cell death in NSC-34 cells. (A,B) The results of flow cytometry assays and quantification of cell apoptosis using FITC/PI staining in NSC-34 cells transfected with miR-193b-3p mimics, inhibitors or scrambled sequence. The results were averages of three independent experiments. Data represent the mean ± SEM. **P < 0.01 vs. controls. (C) The results of Cell Counting Kit-8 (CCK-8) assay indicate cell viability in NSC-34 cells transfected with miR-193b-3p mimics, inhibitors or scrambled sequence. The results were averages of three independent experiments. Data represent the mean ± SEM. *P < 0.5 and **P < 0.01 vs. control.
miR-193b-3p Directly Targets TSC1 and Regulates mTORC1 Activity in NSC-34 Cells
To identify the potential targets of miR-193b-3p in humans, we first screened the targets of miR-193b-3p by sequence analysis. The results showed that TSC1, a well-known regulator of mTORC1 signaling was a potential candidate of miR-193b-3p in NSC-34 cells (Figure 3A). By luciferase reporter assay, we confirmed the targeting sites of miR-193b-3p within TSC1 (Figure 3B). qRT-PCR results showed that the relative expression of TSC1 was dramatically decreased by miR-193b-3p overexpression (mimics) and increased by its inhibition (inhibitors; Figure 3C). We also analyzed other potential targets of miR-193b-3p including Pten, Pfn1 and Prkca (Supplementary Figure S1B,C). To focus on how miR-193b-3p regulated TSC1, we further investigated TSC1 protein expression levels and mTORC1 activity in NSC-34 cells. Western blot results showed that the protein level of TSC1 was dramatically reduced by miR-193b-3p mimics and induced by its inhibitors (Figures 4A,B). Consistently, the indicators of mTORC1 signaling, pp70S6K and p4EBP1, were both increased by miR-193b-3p mimics and decreased by its inhibitors (Figures 4A,C,D). To confirm that miR-193b-3p mimics increased p70S6K and 4EBP1 phosphorylation in mTOR dependent manner, we applied Torin1 (a specific ATP-competitive inhibitor of mTOR) to NSC-34 cells transfected with miR-193b-3p mimics and examined the phosphorylation of p70S6K and 4EBP1. The results showed that Torin1 could block the miR-193b-3p mimic-induced phosphorylation of p70S6K and 4EBP1, suggesting that miR-193b-3p mimics increase p70S6K and 4EBP1 phosphorylation in an mTOR-dependent manner (Supplementary Figure S2A). To further prove that miR-193b-3p targets TSC1 to regulate mTOR signaling, we applied miR-193b-3p inhibitors to TSC1 knockdown NSC-34 cells. The results showed that miR-193b-3p inhibitors could neither increase TSC1 protein nor decrease mTORC1 activity in TSC1 knockdown cells (Supplementary Figure S2B). Therefore, we propose that miR-193b-3p indeed targets TSC1 to control mTOR signaling.
Figure 3.
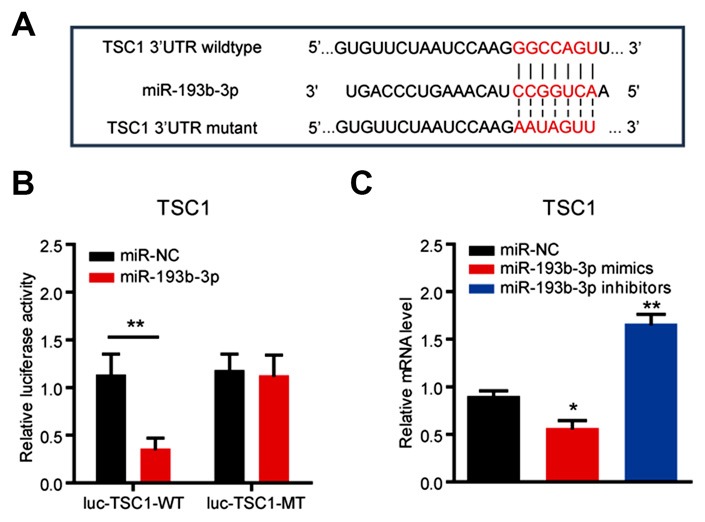
miR-193b-3p directly targets TSC1. (A) Sequence analysis of miR-193b-3p mature miRNA binding with the 3′-UTR of TSC1. (B) Luciferase reporter assay shows a reduction of luciferase activity in NSC-34 cells with wild-type TSC1 3′UTR (luc-TSC1-WT) plasmids. The results were averages of three independent experiments. Data represent the mean ± SEM. **P < 0.01 vs. controls. (C) qRT-PCR results indicate the mRNA levels of TSC1 in NSC-34 cells transfected with miR-193b-3p mimics, inhibitors or scrambled sequence (100 nM, respectively). The results were averages of four independent experiments. Data represent the mean ± SEM. *P < 0.5 and **P < 0.01 vs. controls. miR-NC, miRNA negative control.
Figure 4.

miR-193b-3p activates mTOR complex 1 (mTORC1) signaling by decreasing TSC1 in NSC-34 cells. (A–D) Western blots and quantification to show the protein levels of TSC1/2 and mTORC1 indicators (pp70S6K and p4EBP1) in NSC-34 cells transfected with miR-193b-3p mimics, inhibitors or scrambled sequence. The results were averages of four independent experiments. Data represent the mean ± SEM. *P < 0.5 and **P < 0.01 vs. controls. (E,F) Western blots and quantification to show the protein levels of mTORC1 indicators (pp70S6K and p4EBP1), TSC1/2, p62 and LC3 in the spinal cord of SOD1G93A mutants compared with controls. The results were averages of three pairs of littermate mice. Data represent the mean ± SEM. *P < 0.05 vs. controls.
Next, we examined the activity of mTORC1 signaling in SOD1G93A mutant mice and found that pp70S6K and p4EBP1, two important indicators of mTORC1 signaling, were both decreased in the spinal cord of SOD1G93A mutant mice compared with controls, consistent with the inhibition manipulation of miR-193b-3p (Figures 4E,F). We also noted that autophagy was activated in the spinal cord of SOD1G93A mutant mice, as indicated by the increase of autophagy markers LC3 and p62 (Figure 4E). Taken together, our data revealed that miR-193b-3p was a positive regulator of mTORC1 signaling and directly targeted TSC1 to control mTORC1 activity in NSC-34 cells.
miR-193b-3p Inhibits Autophagy in NSC-34 Cells
The current understanding of TSC1/mTOR in cell survival involves multiple aspects, including cell proliferation, metabolism and autophagy (Matsuzawa et al., 2015). mTOR is widely accepted as a negative regulator of autophagy (Kim et al., 2011). Autophagy is now recognized as protective for neuronal survival in neurodegenerative diseases (Hara et al., 2006; Komatsu et al., 2006; Lee, 2012). To investigate whether miR-193b-3p contributes to cell death through impairing autophagy, we examined the autophagic status under the conditions of miR-193b-3p overexpression (mimics) and inhibition (inhibitors) in NSC-34 cells. The results showed that miR-193b-3p inhibition enhanced autophagy (indicated by decreased p62 and increased ratio of LC3II/I) in NSC-34 cells. We also found that miR-193b-3p mimics increased p62 protein expression and decreased the ratio of LC3II/I in NSC-34 cells (Figures 5A,B, and Supplementary Figure S2C). To confirm the effect of miR-193b-3p on autophagy, we transfected GFP-LC3 into NSC-34 cells and examined whether miR-193b-3p mimics or inhibitors could alter GFP-puncta formation. Images showed that miR-193b-3p inhibitors dramatically increased the formation of GFP-LC3 puncta, whereas miR-193b-3p mimics decreased the formation of GFP-LC3 puncta (Figure 5C). Taken together, these results indicate that miR-193b-3p negatively regulates autophagy in NSC-34 cells.
Figure 5.
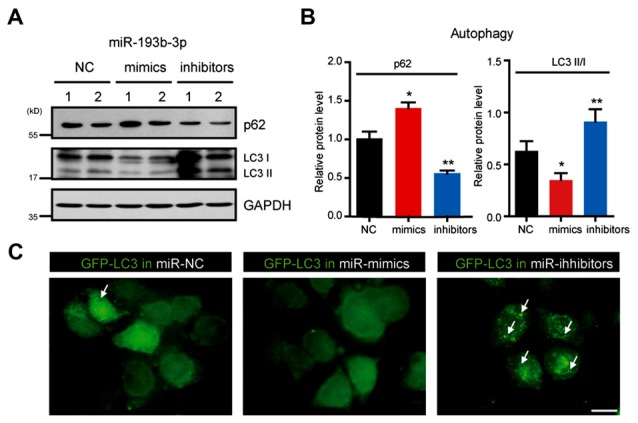
miR-193b-3p inhibits autophagy in NSC-34 cells. (A,B) Western blots and quantification to show the protein levels of p62 and LC3 (II/I) in NSC-34 cells transfected with miR-193b-3p mimics, inhibitors or scrambled sequence. The results were averages of four independent experiments. Data represent the mean ± SEM. *P < 0.5 and **P < 0.01 vs. controls. (C) Representative images of GFP-LC3 puncta (white arrows) in NSC-34 cells transfected with miR-193b-3p mimics, inhibitors or scrambled sequence. Scale bar, 10 μm.
Discussion
Increasing evidence suggests that miRNAs play an important role in the development of ALS. Ours and other previous studies have assessed miRNA expression profiles in ALS patients. Despite these findings, it is still unknown how alterations of specific miRNAs contribute to the development of ALS. In this study, we tried to clarify whether miR-193b-3p, a downregulated miRNA in ALS patients reported in our previous study (Chen et al., 2016), involves in cell survival by targeting TSC1/mTOR signaling in autophagy (Figure 6). Our findings suggest that downregulation of miR-193b-3p is required for cell survival and provide novel molecular mechanisms for the detection and treatment of ALS.
Figure 6.
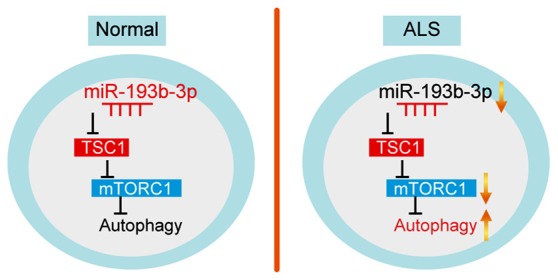
Model. Schematic representation highlighting the role of miR-193b-3p in the development of ALS. miR-193b-3p directly targets TSC1 to regulate mTORC1 activity. Inhibition of miR-193b-3p leads to TSC1 increase and mTORC1 inactivation, resulting in enhanced autophagy.
Interestingly, miRNA-193a-3p and miRNA-193a-5p could directly target and downregulate the ERBB4/PIK3R3/mTOR/ S6K2 signaling pathway (Yu et al., 2015). It has been reported that ERBB4 and S6K2 are the direct targets of miR-193a-3p and that PIK3R3 and mTOR are the direct targets of miR-193a-5p in non-small-cell lung cancer (Yu et al., 2015). In this study, we obtained evidence that miR-193b-3p targets the upstream factor of mTOR (TSC1) to counteract the effect of miR-193a. Therefore, we propose that these effects may be beneficial for the precise regulation of mTOR signaling, and miR-193b-3p directly targets TSC1 and modulates mTOR activity. To clarify the role of miR-193b-3p/TSC1/mTOR axis in the development of ALS, we utilized NSC-34 cells as an in vitro model. NSC-34 is a hybrid motor neuron-like cell line produced by the fusion of neuroblastoma with mouse motor neuron-enriched primary spinal cord cells (Eggett et al., 2000; Matusica et al., 2008; Maier et al., 2013), and it is widely used in studies of ALS in vitro. We found that miR-193b-3p regulates autophagy for cell survival in NSC-34 cells through TSC1/mTOR. These results at least reveal potential roles of miR-193b-3p in cell death of motor neurons in the development of ALS.
Neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and ALS are associated with the permanent loss of neuronal structure and function (Laplante and Sabatini, 2012). Recently, the protective role of mTOR has been noted in neuronal degeneration. Genetic and pharmacological evidence has shown that deletion of TSC1, a negative regulator of mTOR signaling, led to constitutive activation of mTOR, neuroprotective effects and potently enhanced axon regeneration (Park et al., 2008). Moreover, mTOR activity in motor neurons influenced the progression rates of motor dysfunction, muscle denervation and cell death, suggesting that mTOR signaling is required for endogenous neuroprotection to counteract disease progression in fALS (Saxena et al., 2013). However, mTORC1 signaling coordinately activates anabolic processes, such as protein synthesis, while inhibiting the cellular catabolism of autophagy (Chan, 2009). Autophagy could protect cells from death (Hara et al., 2006; Komatsu et al., 2006), especially in motor neurons (Barmada et al., 2014). Accumulating evidence indicates that maintaining a balanced autophagic flux is essential in neuronal physiology. Neurons are highly specialized cells that depend on dynamic cellular processes for their proper function. Once neurons encounter stress, they develop multiple cellular processes to resist these pressures, such as autophagy (Nikoletopoulou et al., 2015). Autophagy is a tightly regulated cellular degradation pathway, which is often defective or hyperactive in neurodegenerative diseases (Laplante and Sabatini, 2012). Currently, accumulating evidence suggests that autophagy is deregulated in neurodegenerative diseases and may play key roles in the etiology of these pathologies (Rubinsztein, 2006). Many studies in cellular and animal models of ALS indicate enhanced autophagy activity in ALS (Morimoto et al., 2007; Sasaki, 2011), in addition to the occurrence of autophagy-mediated clearance of mutant SOD1 and TDP-43 (Nassif et al., 2010). For example, the number of autophagic vacuoles is significantly increased in the motor neurons of the spinal cords of SOD1G93A mice compared with controls (Massey et al., 2006). Therefore, it is predicted that mechanistic target of rapamycin (mTORC1) activity should be downregulated in ALS to meet the demand of increased autophagy. Our findings confirmed that miR-193b-3p is downregulated in ALS patients and mouse models, accompanied by decreased mTORC1 activity. The co-reduction of miR-193b-3p and mTORC1 activity promotes autophagy to protect cells from death.
Autophagy plays an important role in neurodegenerative diseases. However, the contribution of autophagy to the pathology of ALS has not yet been fully elucidated. Although autophagic alteration has been confirmed in ALS patients and experimental models, it remains controversial whether activating autophagy is beneficial or detrimental for motor neuron degeneration. Studies suggest that defects in autophagic flux or specific autophagy-regulatory processes, rather than simple induction of autophagy, may contribute to motor neuron degeneration (Banerjee et al., 2010). In this study, we found that miR-193b-3p targets TSC1 and thus modulates mTOR activity, which negatively regulates autophagy. Inhibition of miR-193b-3p downregulates mTOR activity and activates autophagy, which may protect cells from death. Therefore, we propose that the effect of miR-193b-3p on TSC1/mTOR signaling is fundamentally important to cell survival in ALS development. However, further longitudinal study in ALS patients will be helpful to confirm these results. Nevertheless, manipulating the autophagy process is a complicated dilemma. It is anticipated that more specific autophagic regulators will be discovered and deeper understanding of autophagy biology will be obtained in the near future, which will help decode the mystery of autophagy in ALS pathogenesis and the therapeutic value of autophagy modulators for this devastating disease.
In conclusion, our results suggest that miR-193b-3p directly targets TSC1 to regulate mTORC1 activity. Inhibition of miR-193b-3p leads to increased TSC1 and mTORC1 inactivation. Increased miR-193b-3p activates mTORC1 signaling, inhibits autophagy and thus promotes cell death. Moreover, the downregulation of miR-193b-3p could be a potential biomarker for the detection of ALS development. Taken together, our work supports the hypothesis that miR-193b-3p decrease is required for cell survival by improving autophagy through the TSC1/mTOR pathway and might inform the development of early therapeutic strategies in ALS.
Author Contributions
HS conceived and designed the research. CL collected, analyzed and interpreted the data and drafted the manuscript. CL, YC, QW and BC performed the experiments. HS, YC and XC revised the article critically. All authors approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The present study was supported by the funding of the National Science Fund of China (Grant No. 81371394 and No. 81511140101) and the National Key Research and Development Program of China (No. 2016YFC0901504).
Glossary
Abbreviations
- ALS
amyotrophic lateral sclerosis
- C9orf72
chromosome 9 open reading frame 72
- fALS
familial amyotrophic lateral sclerosis
- FUS
fused in sarcoma
- miRNA
microRNA
- miR-NC
miRNA negative control
- mTOR
mechanistic target of rapamycin
- mTORC1
mTOR complex 1
- mTORC2
mTOR complex 2
- sALS
sporadic amyotrophic lateral sclerosis
- SOD1
superoxide dismutase 1
- TARDBP
TAR DNA-binding protein
- TSC
tuberous sclerosis complex.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnmol.2017.00160/full#supplementary-material
miR-193b-3p is downregulated in the mouse model of ALS, related to Figures 1, 3. (A) qRT-PCR results to show the miRNA levels of miR-193b-3p in the spinal cord of SOD1G93A mutants compared with controls, from 60 days to 150 days. The results were averages of eight pairs of littermate mice. Data represent the mean ± SEM. *P < 0.05 vs. controls. (B) Sequence analysis of miR-193b-3p mature miRNA binding with 3′-UTR of Pten, Pfn1 and Prkca. (C) Luciferase reporter assay to show the reduction of luciferase activity in NSC-34 cells with 3′-UTR of Pten, Pfn1 and Prkca plasmids. The results were averages of three independent experiments. Data represent the mean ± SEM. ***P < 0.001 vs. controls.
miR-193b-3p regulates TSC1/mTOR signaling and autophagy in NSC-34 cells, related to Figures 4, 5. (A) Western blots show the protein levels of mTORC1 indicators (pp70S6K and p4EBP1) in NSC-34 cells by miR-193b-3p mimics with treatment of Torin1 (200 nM for 12 h). (B) Western blots show the protein levels of TSC1 and mTORC1 indicators (pp70S6K), and autophagy markers (p62 and LC3) in NSC-34 cells by miR-193b-3p inhibitors with TSC1 knockdown. (C) Original blots of Figure 5A.
References
- Akerblom M., Sachdeva R., Barde I., Verp S., Gentner B., Trono D., et al. (2012). MicroRNA-124 is a subventricular zone neuronal fate determinant. J. Neurosci. 32, 8879–8889. 10.1523/jneurosci.0558-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R., Beal M. F., Thomas B. (2010). Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci. 33, 541–549. 10.1016/j.tins.2010.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber S. C., Shaw P. J. (2010). Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 48, 629–641. 10.1016/j.freeradbiomed.2009.11.018 [DOI] [PubMed] [Google Scholar]
- Barmada S. J., Serio A., Arjun A., Bilican B., Daub A., Ando D. M., et al. (2014). Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 10, 677–685. 10.1038/nchembio.1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillee S., Yamanaka K., Lobsiger C. S., Copeland N. G., Jenkins N. A., Kassiotis G., et al. (2006). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. 10.1126/science.1123511 [DOI] [PubMed] [Google Scholar]
- Bordon Y. (2013). Infectious disease: hushing mTOR boosts immunity to pathogens. Nat. Rev. Immunol. 13:847. 10.1038/nri3562 [DOI] [PubMed] [Google Scholar]
- Caccamo A., Majumder S., Deng J. J., Bai Y., Thornton F. B., Oddo S. (2009). Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J. Biol. Chem. 284, 27416–27424. 10.1074/jbc.M109.031278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman N. R., Durham H. D., Blusztajn J. K., Oda K., Tabira T., Shaw I. T., et al. (1992). Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 194, 209–221. 10.1002/aja.1001940306 [DOI] [PubMed] [Google Scholar]
- Chan E. Y. (2009). mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci. Signal. 2:pe51. 10.1126/scisignal.284pe51 [DOI] [PubMed] [Google Scholar]
- Chen Y., Wei Q., Chen X., Li C., Cao B., Ou R., et al. (2016). Aberration of miRNAs expression in leukocytes from sporadic amyotrophic lateral sclerosis. Front. Mol. Neurosci. 9:69. 10.3389/fnmol.2016.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu I. M., Chen A., Zheng Y., Kosaras B., Tsiftsoglou S. A., Vartanian T. K., et al. (2008). T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. U S A 105, 17913–17918. 10.1073/pnas.0804610105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crippa V., Sau D., Rusmini P., Boncoraglio A., Onesto E., Bolzoni E., et al. (2010). The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 19, 3440–3456. 10.1093/hmg/ddq257 [DOI] [PubMed] [Google Scholar]
- Dazert E., Hall M. N. (2011). mTOR signaling in disease. Curr. Opin. Cell Biol. 23, 744–755. 10.1016/j.ceb.2011.09.003 [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Carlo V., Grossi E., Laneve P., Morlando M., Dini Modigliani S., Ballarino M., et al. (2013). TDP-43 regulates the microprocessor complex activity during in vitro neuronal differentiation. Mol. Neurobiol. 48, 952–963. 10.1007/s12035-013-8564-x [DOI] [PubMed] [Google Scholar]
- Eggett C. J., Crosier S., Manning P., Cookson M. R., Menzies F. M., McNeil C. J., et al. (2000). Development and characterisation of a glutamate-sensitive motor neurone cell line. J. Neurochem. 74, 1895–1902. 10.1046/j.1471-4159.2000.0741895.x [DOI] [PubMed] [Google Scholar]
- Ferrante R. J., Shinobu L. A., Schulz J. B., Matthews R. T., Thomas C. E., Kowall N. W., et al. (1997). Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann. Neurol. 42, 326–334. 10.1002/ana.410420309 [DOI] [PubMed] [Google Scholar]
- Filipowicz W., Bhattacharyya S. N., Sonenberg N. (2008). Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 9, 102–114. 10.1038/nrg2290 [DOI] [PubMed] [Google Scholar]
- Goodall E. F., Heath P. R., Bandmann O., Kirby J., Shaw P. J. (2013). Neuronal dark matter: the emerging role of microRNAs in neurodegeneration. Front. Cell. Neurosci. 7:178. 10.3389/fncel.2013.00178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H., Ingolia N. T., Weissman J. S., Bartel D. P. (2010). Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840. 10.1038/nature09267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- Kiernan M. C., Vucic S., Cheah B. C., Turner M. R., Eisen A., Hardiman O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. 10.1016/S0140-6736(10)61156-7 [DOI] [PubMed] [Google Scholar]
- Kim J., Kundu M., Viollet B., Guan K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- Laplante M., Sabatini D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-A. (2012). Neuronal autophagy: a housekeeper or a fighter in neuronal cell survival? Exp. Neurobiol. 21, 1–8. 10.5607/en.2012.21.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibinger M., Andreadaki A., Fischer D. (2012). Role of mTOR in neuroprotection and axon regeneration after inflammatory stimulation. Neurobiol. Dis. 46, 314–324. 10.1016/j.nbd.2012.01.004 [DOI] [PubMed] [Google Scholar]
- Maier O., Böhm J., Dahm M., Brück S., Beyer C., Johann S. (2013). Differentiated NSC-34 motoneuron-like cells as experimental model for cholinergic neurodegeneration. Neurochem. Int. 62, 1029–1038. 10.1016/j.neuint.2013.03.008 [DOI] [PubMed] [Google Scholar]
- Majoor-Krakauer D., Willems P. J., Hofman A. (2003). Genetic epidemiology of amyotrophic lateral sclerosis. Clin. Genet. 63, 83–101. 10.1046/j.0009-9163.2002.00001.x [DOI] [PubMed] [Google Scholar]
- Massey A. C., Zhang C., Cuervo A. M. (2006). Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol. 73, 205–235. 10.1016/s0070-2153(05)73007-6 [DOI] [PubMed] [Google Scholar]
- Matsuzawa Y., Oshima S., Takahara M., Maeyashiki C., Nemoto Y., Kobayashi M., et al. (2015). TNFAIP3 promotes survival of CD4 T cells by restricting MTOR and promoting autophagy. Autophagy 11, 1052–1062. 10.1080/15548627.2015.1055439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matusica D., Fenech M. P., Rogers M. L., Rush R. A. (2008). Characterization and use of the NSC-34 cell line for study of neurotrophin receptor trafficking. J. Neurosci. Res. 86, 553–565. 10.1002/jnr.21507 [DOI] [PubMed] [Google Scholar]
- Morimoto N., Nagai M., Ohta Y., Miyazaki K., Kurata T., Morimoto M., et al. (2007). Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1167, 112–117. 10.1016/j.brainres.2007.06.045 [DOI] [PubMed] [Google Scholar]
- Morlando M., Dini Modigliani S., Torrelli G., Rosa A., Di Carlo V., Caffarelli E., et al. (2012). FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 31, 4502–4510. 10.1038/emboj.2012.319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassif M., Matus S., Castillo K., Hetz C. (2010). Amyotrophic lateral sclerosis pathogenesis: a journey through the secretory pathway. Antioxid. Redox Signal. 13, 1955–1989. 10.1089/ars.2009.2991 [DOI] [PubMed] [Google Scholar]
- Nikoletopoulou V., Papandreou M. E., Tavernarakis N. (2015). Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 22, 398–407. 10.1038/cdd.2014.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoselov S. S., Mustill W. J., Gray A. L., Dick J. R., Kanuga N., Kalmar B., et al. (2013). Molecular chaperone mediated late-stage neuroprotection in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. PLoS One 8:e73944. 10.1371/journal.pone.0073944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi C., Napoli G., Amadio S., Spalloni A., Apolloni S., Longone P., et al. (2016). MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ. 23, 531–541. 10.1038/cdd.2015.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K. K., Liu K., Hu Y., Smith P. D., Wang C., Cai B., et al. (2008). Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322, 963–966. 10.1126/science.1161566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W., Philips T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. 10.1038/nrn3430 [DOI] [PubMed] [Google Scholar]
- Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. 10.1038/364362c0 [DOI] [PubMed] [Google Scholar]
- Rubinsztein D. C. (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786. 10.1038/nature05291 [DOI] [PubMed] [Google Scholar]
- Saccon R. A., Bunton-Stasyshyn R. K., Fisher E. M., Fratta P. (2013). Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136, 2342–2358. 10.1093/brain/awt097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov D. D., Ali S. M., Sabatini D. M. (2005). Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 17, 596–603. 10.1016/j.ceb.2005.09.009 [DOI] [PubMed] [Google Scholar]
- Sasaki S. (2011). Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 70, 349–359. 10.1097/NEN.0b013e3182160690 [DOI] [PubMed] [Google Scholar]
- Saxena S., Roselli F., Singh K., Leptien K., Julien J. P., Gros-Louis F., et al. (2013). Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80, 80–96. 10.1016/j.neuron.2013.07.027 [DOI] [PubMed] [Google Scholar]
- Sreedharan J., Blair I. P., Tripathi V. B., Hu X., Vance C., Rogelj B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. 10.1126/science.1154584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson A. W., Turnquist H. R., Raimondi G. (2009). Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 9, 324–337. 10.1038/nri2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner B. J., Atkin J. D. (2006). ER stress and UPR in familial amyotrophic lateral sclerosis. Curr. Mol. Med. 6, 79–86. 10.2174/156652406775574550 [DOI] [PubMed] [Google Scholar]
- Turner M. R., Grosskreutz J., Kassubek J., Abrahams S., Agosta F., Benatar M., et al. (2011). Towards a neuroimaging biomarker for amyotrophic lateral sclerosis. Lancet Neurol. 10, 400–403. 10.1016/S1474-4422(11)70049-7 [DOI] [PubMed] [Google Scholar]
- Vance C., Rogelj B., Hortobágyi T., De Vos K. J., Nishimura A. L., Sreedharan J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. 10.1126/science.1165942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.-Y., Ren M., Jiang H.-Z., Wang J., Jiang H.-Q., Yin X., et al. (2015). Notch pathway is activated in cell culture and mouse models of mutant SOD1-related familial amyotrophic lateral sclerosis, with suppression of its activation as an additional mechanism of neuroprotection for lithium and valproate. Neuroscience 301, 276–288. 10.1016/j.neuroscience.2015.06.002 [DOI] [PubMed] [Google Scholar]
- Wilcox K. C., Zhou L., Jordon J. K., Huang Y., Yu Y., Redler R. L., et al. (2009). Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J. Biol. Chem. 284, 13940–13947. 10.1074/jbc.M809687200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., Chen S., Du J., He J., Wang Y., Li Z., et al. (2016). NK cell development requires Tsc1-dependent negative regulation of IL-15-triggered mTORC1 activation. Nat. Commun. 7:12730. 10.1038/ncomms12730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T., Li J., Yan M., Liu L., Lin H., Zhao F., et al. (2015). MicroRNA-193a-3p and -5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Oncogene 34, 413–423. 10.1038/onc.2013.574 [DOI] [PubMed] [Google Scholar]
- Zhu H., Bhattacharyya B. J., Lin H., Gomez C. M. (2013). Skeletal muscle calpain acts through nitric oxide and neural miRNAs to regulate acetylcholine release in motor nerve terminals. J. Neurosci. 33, 7308–7324. 10.1523/JNEUROSCI.0224-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miR-193b-3p is downregulated in the mouse model of ALS, related to Figures 1, 3. (A) qRT-PCR results to show the miRNA levels of miR-193b-3p in the spinal cord of SOD1G93A mutants compared with controls, from 60 days to 150 days. The results were averages of eight pairs of littermate mice. Data represent the mean ± SEM. *P < 0.05 vs. controls. (B) Sequence analysis of miR-193b-3p mature miRNA binding with 3′-UTR of Pten, Pfn1 and Prkca. (C) Luciferase reporter assay to show the reduction of luciferase activity in NSC-34 cells with 3′-UTR of Pten, Pfn1 and Prkca plasmids. The results were averages of three independent experiments. Data represent the mean ± SEM. ***P < 0.001 vs. controls.
miR-193b-3p regulates TSC1/mTOR signaling and autophagy in NSC-34 cells, related to Figures 4, 5. (A) Western blots show the protein levels of mTORC1 indicators (pp70S6K and p4EBP1) in NSC-34 cells by miR-193b-3p mimics with treatment of Torin1 (200 nM for 12 h). (B) Western blots show the protein levels of TSC1 and mTORC1 indicators (pp70S6K), and autophagy markers (p62 and LC3) in NSC-34 cells by miR-193b-3p inhibitors with TSC1 knockdown. (C) Original blots of Figure 5A.