

Abstract
Recent advances in metabolomic and genome mining approaches have uncovered a poorly understood metabolome that originates solely or in part from bacterial enzyme sources. Whether living on exposed surfaces or within our intestinal tract, our microbial inhabitants produce a remarkably diverse set of natural products and small molecule metabolites that can impact human health and disease. Highlighted here, the gut microbe-derived metabolite trimethylamine N-oxide has been causally linked to the development of cardiovascular diseases. Recent studies reveal drugging this pathway can inhibit atherosclerosis development in mice. Building on this example, we discuss challenges and untapped potential of targeting bacterial enzymology for improvements in human health.
Keywords: atherosclerosis, cardiovascular disease, drug discovery, metabolomics, microbiome
Introduction
Many centuries have passed since the seminal discovery of microorganisms by Hooke and van Leeuwenhoek (1) and the subsequent game-changing postulates put forth by Koch (2) for demonstration of a causal role for microorganisms in infectious diseases. Recently, there has been a rapid advancement in our appreciation of the undeniable association between commensal microorganisms (bacteria, archaea, viruses, and other eukaryotes) and both physiological processes critical to our health and human disease susceptibility (3–5). Despite this, with the exception of antibiotics and vaccines, there has been an unfortunate gap in the development of therapies that leverage this newfound appreciation by selectively targeting microorganisms for improvement of health in the human host. This is because we are still only in our infancy of understanding the mechanisms by which human resident microorganisms impact normal host physiology and human disease.
Recent research at the microbiome-host interface has been dominated by genomic or metagenomic sequencing approaches, which have allowed a near comprehensive cataloguing of what types of microorganisms are present in different compartments of the human body. At the same time, advances in the fields of mass spectrometry and computational biology have allowed for the identification of natural products and metabolites that originate solely or in part from bacterial sources (Fig. 1). In addition to the growing list of bacterially derived metabolites, it has long been understood that the host immune system can selectively recognize microbe-associated molecular patterns (MAMPs)3 via cognate pattern-recognition receptors on host immune cells (6, 7). Even though we now understand what types of microbes are present, some of the metabolites they produce, and what MAMP repertoire they possess, there are very few examples of where this information has been leveraged into human-relevant therapeutics. Hence, the purpose of this review is to highlight the potential for targeting microbial enzymology for improvement of human health. Here, we discuss the current state of knowledge surrounding drug discovery in the microbiome space, with particular focus on promising developments in the field of cardiovascular disease (CVD). We also critically discuss the untapped potential of transitioning away from currently favored non-selective microbiome therapeutic approaches (prebiotics, probiotics, antibiotics, fecal transplantation, etc.) to instead utilize non-lethal small molecules to manipulate microorganism-host interactions. Discussion here is intended to highlight the few known examples of microbiome-targeted small molecule therapeutics and to provide a framework for future investigation in this area.
Figure 1.

Small molecule metabolites originating from the human microbiome. Diverse bacterial ecosystems present in the oral cavity, upper and lower gastrointestinal tract, skin surface, lungs, and almost every exposed orifice studied possess the enzymatic machinery to generate chemically diverse and biologically active metabolites that impact host health and disease. Collectively, human microbiota represent a major contributor to the chemical diversity in the human metaorganism, and many known bacterial metabolites have dedicated host receptor systems that allow for microbe-host cross-talk that modulates human health and disease.
Challenges and opportunities in microbiome-targeted drug discovery
The advent of high throughput sequencing, either by classic 16S ribosomal RNA taxonomic approaches or by more recent metagenomic deep sequencing, has allowed for the cataloguing of microorganisms that are present in a sample without the need of culturing (8, 9). Large international microbiome research consortia have leveraged such methodology to reveal that healthy humans are inhabited by a multitude of different species of gut bacteria, and microbial diversity can be dynamically altered by a number of variables, including age, host genetics, diet, and antibiotic exposure (5, 10–12). More importantly, enhanced proportion or absence of certain microbial taxa is associated with a number of human diseases, including obesity (13–17), diabetes (18–20), alcoholic and non-alcoholic fatty liver disease (21–24), cancer (25–27), osteoporosis (28, 29), hypertension (30, 31), autism (32, 33), autoimmune diseases (20, 34, 35), CVD (36, 37), and likely many others. Undoubtedly, high throughput sequencing approaches have uncovered a number of compelling microorganism-disease links. However, how to move from association to causation remains a daunting challenge.
A major limitation that persists with culture-independent taxonomic sequencing approaches is that many disease associations are made at the broad level (e.g. phylum, class, order, or genus) and not at the species or strain level. Moreover, there are many examples where operational taxonomic units within a taxon negatively associate, whereas some others positively associate, with disease phenotypes. This makes disease associations above the species level extremely difficult to interpret because many taxa are lumped together in a microbe-disease association. In addition to this lumping issue, DNA-based metagenomic approaches have inherent biases that limit the ability to fully understand druggable microbial function. Current limitations in DNA-based metagenomics are based on variable amounts of DNA isolated from the source sample, DNA fragmentation biases, PCR amplification biases, and sequencing and read mapping bias (8). Given these limitations with genomic sequencing methods, and the fact that many microbial gene products are only expressed under certain conditions, additional approaches are desperately needed.
To identify robust microbiome drug targets, it will be most informative to look beyond what taxa or genes are present and instead to investigate what genes are being expressed and, most importantly, what the gene products are doing to impact host disease. Although methodology is still immature at this point, there are shotgun sequencing approaches under development to quantify bacterial mRNA expression (metatranscriptomics) and protein expression (metaproteomics). At this point, metatranscriptomic methods are still in their infancy, and given that bacterial transcriptional profiles can change within minutes, sample collection and stabilization are a major hurdle. Another clear caveat of both metatranscriptomics and metaproteomics is limit of detection thresholds. For instance, low abundant gene products in low abundant bacteria will likely be missed, which is less of a limitation with current metagenomic methods. Once properly matured, metatranscriptomic and metaproteomic methods have the potential to strengthen metagenomic approaches because they provide a readout one step closer to the desired functional readouts of bacteria. Another approach to move beyond DNA sequencing approaches that only provide a catalogue of what taxa and genes are present, and not their function, is to employ the complementary approach of metabolomics. Understanding the chemical secretome (natural products, metabolites, and small molecules) originating from our microbial inhabitants is a powerful complementary goal because these molecules are the most likely causative links between our microbiome and human disease. Ultimately, combining metagenomic, metatranscriptomic, metaproteomic, and metabolomic platforms are necessary for identifying the disease-causing products from the microbiome and their impact on the human host.
More than any other approach, annotation of the microbial metabolite secretome (Fig. 1) provides the highest likelihood of identifying human relevant drug targets and biomarkers of disease. This is because microbe-derived metabolites can more readily cross host epithelial barriers than microbes themselves, making them the most likely agents to impact host cells, and contribute to disease in organs lacking a resident microbiome. Unfortunately, we are still only scratching the surface in the area of metaorganismal metabolomics. Identification of microorganism-derived metabolites typically couples mass spectrometry or nuclear magnetic resonance (NMR) detection with antibiotics or germ-free experimental conditions. Using such approaches, several recent metabolomics studies have identified disease-associated microbial metabolites (30, 32, 38, 39). However, our current annotation of the microbial metabolome is vastly incomplete due to the fact that less than 2% of chemical data generated by mass spectrometry can be annotated (40, 41). The remaining unidentified metabolites that are altered under germ-free or antibiotic-suppressed states represent a treasure trove of potentially biologically active compounds that have the potential to be attractive drug targets. It should also be noted that the above estimates likely vastly underestimate the magnitude of the metabolome, because volatile and poorly ionizing molecular species are typically not even detected by typical mass spectrometry and NMR approaches. Moreover, most studies have thus far focused on fasting samples, with a staggering number of metabolites possible in the postprandial metabolome. To overcome current annotation limitations in analyte structural identification in the typical metabolomics analyses, the field is making rapid advances in database expansion and development, along with dissemination of platforms that allow for more comprehensive curation of mass spectrometry data (42, 43). As discovery-based metabolomic studies progress, it is important to understand that production of microbial secretory products is quite dynamic and in most cases substrate-driven. For example, several well known bacterial metabolites such as short chain fatty acids, secondary bile acids, and trimethylamine N-oxide (TMAO) are produced in greater amounts after ingestion of a meal (44–46). In fact, gut microbes represent a filter of our greatest environmental exposure, our diet. Therefore, it is extremely important to consider diet and substrate availability as a major determinant of the microbial metabolome. Unfortunately, almost all germ-free metabolomics studies have been done by feeding rodent chow, which is not standardized in its composition from batch to batch and by no means represents a human relevant dietary milieu. Also, it is important to recognize that careful evaluation of the gut microbial metabolome requires defined dietary conditions where substrates are present at physiologically relevant levels. Ultimately, the identification of disease-causing microbial metabolites, and the host receptors that sense them, is a requisite step in selectively targeting pathways in which microbial products contribute to disease susceptibility or progression in humans. Although there are very few examples where disease-causing microbial metabolites/pathways have been pursued as drug targets, recent progress in the CVD field provides a framework for future microbiome-centered drug discovery.
An early success story of drug discovery in the gut microbial enzymology space: Small molecule inhibition of TMAO production
Within the last 5 years, the gut microbial metabolite TMAO has quickly gained traction as both a strong biomarker for human CVD risk, as well as a contributory participant in the pathogenesis of atherosclerosis (39, 47–65), thrombosis (66), heart failure (67–70), and chronic kidney disease (71–76). In fact, the TMAO pathway is uniquely positioned as one of the first bona fide gut microbiome-centered drug targets with several active discovery programs underway. In support of the notion that drugging of microbial enzymes is a tractable approach, we highlight the gut microbial TMAO pathway because it is one of the first examples where small molecules have been successfully used as a non-lethal microbial pathway inhibitor to attenuate disease progression in preclinical models (56). The only other known example as of the writing of this review are the seminal studies by Redinbo and colleagues employing selective inhibition of microbial β-glucuronidase to attenuate gastrointestinal toxicity of select cancer drugs and non-steroidal anti-inflammatory agents (77, 78). This latter topic is the focus of another Minireview in this series by Pellock and Redinbo (101), so it will not be further discussed in this review.
Interest in TMAO as a drug target is bolstered by a wealth of recent independent human studies, which have amply demonstrated that elevated circulating TMAO levels are associated with increased risk of CVD in a variety of cohorts (39, 47–65). Speaking to its causative role in CVD pathogenesis, experimental elevation of TMAO by dietary provision worsens atherosclerosis, thrombosis, and adverse cardiac remodeling in mice (39, 47, 66, 70, 79, 80). The TMAO pathway is a diet-driven pathway where nutrients enriched in a Western diet (phosphatidylcholine, choline, l-carnitine, etc.) are metabolized by distinct gut microbial enzyme complexes (CutC/D, CntA/B, and YeaW/X) to generate the primary gut microbial metabolite trimethylamine (TMA). TMA is subsequently metabolized by host enzymes in the liver called flavin-containing monooxygenases to produce TMAO (Fig. 2). In addition to microbe-derived TMA and TMAO, there are a number of potential dietary sources of TMA and TMAO, such as some cold water-dwelling fish or seafood. Several recent reviews have highlighted the clinical relevance and therapeutic potential of the TMAO pathway in CVD (81, 82). Briefly, the link between TMAO and CVD risk was first discovered via unbiased and untargeted metabolomic screening of human plasma in stable cardiac patients undergoing elective coronary angiography (39). Many follow-up studies with independent cohorts have supported the link between TMAO levels and CVD risk in humans (58–65) and have also confirmed that TMAO feeding worsens cardiovascular phenotypes in mice (70, 79, 80). However, it is important to note that a few studies report not finding an association between circulating TMAO and CVD risks, although these have tended to be either small or to have low numbers of cardiovascular events (83–85). Fulfilling Koch's postulate of disease susceptibility, microbial transplantation studies employing delivery of microbial communities with a high (versus low) capacity to generate TMA and TMAO results in enhancement of atherosclerosis and thrombosis in mice (57, 66). Reductions in the TMAO levels in animal models, either via genetic approaches or small molecule inhibition of microbial TMA lyase enzyme activity, have both resulted in abrogation of choline diet-dependent enhancement in atherosclerosis and other CVD-related phenotypes (51–53, 56). Collectively, the gut microbe-derived metabolite TMAO represents a strong prognostic biomarker and causative agent in atherosclerosis and thrombotic vascular disease. The question that remains is how best to therapeutically intervene on this metaorganismal nutrient metabolism pathway.
Figure 2.
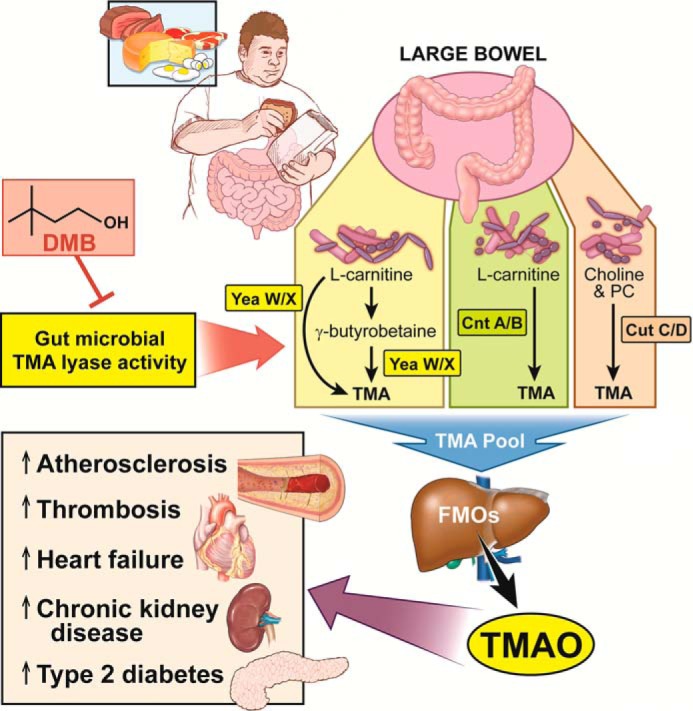
Drugging the gut microbial TMAO pathway for the treatment of prevention of cardiometabolic disease. Dietary consumption of choline, phosphatidylcholine (PC), carnitine, γ-butyrobetaine, and likely other methylamine-containing source nutrient gut microbes provides substrate for gut bacterial production of TMA through the collective actions of several TMA lyase enzymes. Bacteria expressing the YeaW/X enzyme complex can sequentially convert l-carnitine to γ-butyrobetaine and then γ-butyrobetaine to TMA. TMA can be generated from another l-carnitine TMA lyase enzyme complex known as CntA/B. Choline and phosphatidylcholine can be used by the CutC/D enzyme complex to generate a substantial pool of TMA. Once generated from these distinct sources, TMA enters the portal circulation where it is ultimately delivered to the host liver. The host flavin-containing monooxygenase (FMO) family of enzymes, especially FMO3, can then convert TMA to TMAO. TMAO can then promote atherosclerosis, thrombosis, heart failure, kidney disease, and insulin resistance via tissue- or cell type-specific reprogramming. Inhibition and choline and l-carnitine TMA lyase activity by DMB can blunt atherosclerosis progression in mice. The TMAO pathway represents one of the first pathways where small molecule inhibitors targeting microbial enzymes can benefit host disease.
Recent proof-of-concept preclinical studies demonstrate that small molecule inhibition of gut microbial TMA lyase enzymes can be a therapeutically tractable approach for attenuating atherosclerosis (56). By screening structural analogues of choline, a compound (3,3-dimethyl-1-butanol, DMB) was identified to serve as a tool drug to test whether selective inhibition in choline-dependent production of TMA has beneficial effects in murine models of atherosclerosis (56). Importantly, this inhibitor was effective at suppressing TMA production both in cultured human commensal strains and in vivo in mice. Provision of DMB in apolipoprotein E (apoE) knock-out mice on a choline- or carnitine-supplemented diet was shown to lower systemic levels of TMAO with associated reductions in endogenous macrophage foam cell formation and aortic root atherosclerotic lesion development (56). This study for the first time showed that small molecule inhibition of a bacterial enzyme activity can retard or prevent atherosclerotic CVD. It thus provides a framework for analogous drug discovery in other microbe-associated diseases.
Although the primary indication for TMA lyase inhibitors would likely be cardiovascular-related, such drugs may also hold promise in several other disease states. For example, elevated TMAO levels have also been linked to both development of chronic kidney disease (CKD) and adverse outcomes in humans (72–76). It has long been known that TMAO is actively excreted into the urine, and diminished renal function impedes the proper elimination of TMAO (86). A metabolomics study in the Framingham population revealed elevated choline and TMAO levels are seen in subjects with normal renal function who are at risk for incident development of CKD (76). In several large human studies, TMAO has been found to be elevated in people suffering with CKD and who are at risk for cardiovascular disease and its adverse events (71–76). Interestingly, animal model studies have shown chronic feeding with either dietary choline or TMAO can result in progressive renal functional impairment and tubulointerstitial fibrosis in mice, with accompanying activation of the phospho-Smad3 pro-fibrotic pathway (71). In addition to kidney disease, TMAO has also been linked to insulin resistance and type 2 diabetes mellitus (T2DM) by several independent groups (87, 88). A seminal study by Biddinger and co-workers (53) recently demonstrated that mice with selective hepatic insulin resistance have elevated levels of circulating TMAO. This study demonstrated that male mice lacking the insulin receptor in the liver (LIRKO mice) have a profound up-regulation of the TMAO-producing enzyme FMO3 (flavin monooxygenase 3) in the liver (53). Furthermore, antisense oligonucleotide-mediated inhibition of FMO3 in LIRKO mice was shown to inhibit TMAO production and to protect against the hyperlipidemic and proatherogenic phenotype of these mice (53). In agreement, an independent study showed that dietary supplementation with TMAO can exacerbate glucose intolerance in high fat fed mice (89). Although mechanisms underlying the link between the TMAO pathway and both T2DM and CKD are not known, there is mounting evidence that TMAO may be a contributory factor in the pathogenesis of these common diseases, as well as the CVD-related morbidity and mortality accompanying these disorders. In addition, recent studies suggest TMAO levels may similarly be associated with the presence and severity of non-alcoholic fatty liver disease (18, 23, 24). TMA lyase inhibition may thus hold promise in the prevention or treatment of these common chronic metabolic disorders that are also frequently associated with atherothrombotic disease risks.
Another patient population that could immediately potentially benefit from TMA lyase inhibitors would be those suffering from trimethylaminuria (86). Subjects with trimethylaminuria typically adopt stringent dietary restrictions in animal products in an effort to limit TMA elevation, although choline is an abundant component of bile (in the form of phosphatidylcholine), and the epithelial cells lining the alimentary tract rapidly turn over, shedding membrane particles into the gut lumen. There thus are ample sources of endogenous TMA precursors even in the most ardent vegan diet, and dietary efforts to limit TMAO production has limits to its potential benefits. When given in combination with a low choline or a low carnitine diet, TMA lyase inhibitors would be predicted to rapidly ameliorate fish odor symptoms in those with primary mutations in FMO3 (86). Collectively, the TMAO pathway represents one of the first examples where non-lethal drugs targeting microbial enzymes can provide protection against host disease.
Using the metaorganismal TMAO pathway as an example, we now have a methodological framework to identify diseases where “drugging the microbiome” with non-lethal microbial enzyme inhibitors may be of potential benefit. First, we believe it is important that the initial discovery process include human clinical investigations. These are critical in serving as a lens with which to identify microbe-derived metabolites that reproducibly show strong and independent associations with the presence and severity of human disease. It is also extremely important to recognize that untargeted metabolomics studies are semi-quantitative due to the limitations in inclusion of appropriate internal standards before candidate analytes are revealed. It is therefore critical that hypotheses generating untargeted metabolomics studies be subsequently independently validated using robust analytical approaches (e.g. stable isotope dilution mass spectrometry-based) across multiple non-overlapping human cohorts to establish whether the levels of the metabolite can predict future risk of disease, as has been done for TMAO (47–49, 58–63). Once clinical relevance is established, mechanistic studies to reveal whether the observed associations are causally linked to the disease process are critical. Testing of the microbial requirement for generation of the metabolite and whether it is directly linked to disease causation can be established by feeding or directly administering the nutrient precursors or the metabolite itself in germ-free or conventionally reared mice (39, 47, 50, 70, 79, 80). Concurrent with these studies, investigation of the breadth of specific nutrient precursors that can give rise to the microbe-generated metabolite(s) provides insights into the global meta-organismal pathway, its dietary precursors, and substrates to use in both microbial enzyme discovery efforts and drug development efforts. Thus, once disease association and causation are established, it is key to identify relevant microbial communities, participants, and enzymes that are the predominant sources of the metabolite in question using a combination of reference genome mining and microbial biochemical approaches (90, 91). In parallel, it is imperative to identify microbe host co-metabolites involved in metaorganismal pathways (such as conversion of TMA into TMAO) and whether host receptors exist to sense the metabolites of interest.
With each new gut microbial-derived metabolite identified that reveals mechanistic connection to disease, there may thus exist within the host many additional points of potential therapeutic intervention beyond the microbial enzyme source. Examples can include host enzymes involved in biotransformation of the initial gut microbial metabolite or host receptors or transporters in the pathway. Using the TMAO pathway as an example, there is one known host receptor for TMA identified, TAAR5 (trace-amine associated receptor 5) (92, 93). TAAR5 is a G-protein-coupled receptor that mediates olfactory recognition specifically of TMA and not TMAO (92, 93). Unfortunately, the receptor that recognizes TMAO in mammals remains unknown at present but is an obvious potential therapeutic target once identified. It is also possible that TMAO exerts its biological effects by acting as a key osmolyte and protein stabilizer, especially in the renal medulla or papilla (94–96). It is important to realize that the human genome presents us with ∼20,000 protein-coding drug targets. However, current estimates based on large microbiome sequencing consortia estimate that there are likely at least 2 orders of magnitude more bacterial genes in human commensals (97). Combining both the human and microbial genome repertoire, there is almost unlimited potential for new drug targets. Although the exact numbers of microbial metabolites are not known, bacteria within the Streptomycetaceae family have been estimated to synthesize over a million small molecules themselves (98). When taking into account the entire human gut microbiome community, the potential chemical diversity is quite staggering.
Concluding remarks
The concept that microorganisms can cause human disease is not new. Beyond infectious diseases, a role for gut microbes in health and disease likewise is not new. For example, over a century ago the Russian zoologist Eli Metchnikoff was awarded the Nobel Prize for his pioneering work in phagocytosis, and in part for his suggestion that human disease originates from “poisoning of the body from bacterial by-products.” Unfortunately, Dr. Metchnikoff would be disappointed to know that in 2016 almost all major efforts in drug discovery have instead focused only on host targets. It is hard to fathom that such large-scale efforts have been undertaken for discovery of antibiotics to ameliorate pathobionts (99), whereas drugs that target microbial metabolites have not been pursued. There is an exciting time ahead for drug discovery, where we target either the gene products of gut microorganisms or associated host enzymes and receptors along discovered metaorganismal pathways linked to disease susceptibility. As we embark on this new frontier, it is essential that we move beyond approaches that merely profile lists of which microorganisms are more or less abundant and instead focus on which microbial genes are being expressed, and more importantly, what bioactive chemicals are being secreted that exert effects on the host and impact human disease. Processes and insights learned during antibiotic drug discovery will no doubt be important in non-lethal targeting of microbial pathways, such as improved understanding of lateral gene transfer and its potential impact on gut microbe metabolite-targeted therapeutics. Furthermore, improved understanding of how existing and new small molecule therapeutics affect microbial ecology is another area for investigation. Using mice to model human microbiome-disease pathways no doubt has some limitations because of obvious microbial community structure differences (10–12) and known differences in innate immune pathways (100). Nonetheless, by starting with exploratory studies seeking to reveal strong associations between a metabolite of interest and human disease presence and severity, more focused and potentially clinically relevant preclinical animal model studies can be designed.
Although non-selective approaches that broadly alter microbial community structure such as prebiotics, probiotics, and fecal microbial transplantation may have some promise, their rational scientific development is remarkably challenging. Moving toward selective small molecule non-lethal therapeutics that target defined microbial pathways or host participants along discovered causal metaorganismal pathways represents a scientifically manageable approach. This transition is similar to the movement made in the cancer field from broadly cytotoxic chemotherapeutic regimens to carefully targeted small molecule and antibody-based therapeutics. Using small molecules to inhibit gut microbial enzymes for amelioration of disease is tractable (56). In fact, drugging gut microbiome enzymology potentially represents an ideal scenario if such small molecules can be designed to avoid systemic absorption or exposure, thereby minimizing the potential for host side effects and avoiding development of unanticipated adverse side effects from altering the microbial community. As such small molecule drug discovery efforts advance, it will be extremely important to understand the effects of such drugs on microbial ecology and whether resistance to small molecule therapeutics will arise with chronic exposure. Drug discovery advances in the microbiome space are being aggressively pursued and will require a multidisciplinary approach. Initial discovery efforts require access to well annotated clinical repositories with deep phenotype, expertise in untargeted metabolomics, and compound identification to unambiguously associate metabolite levels with human disease, microbial enzymology, and clinical chemistry, coupled with complementary metagenomics, metatranscriptomics, and bioinformatics to facilitate both microbial and host enzyme and transporter/receptor participation. Such studies are daunting because of the diverse skill sets needed to make meaningful connections. However, it is these types of multidisciplinary studies that form the basis for therapies for the next generation where we selectively yet non-lethally target enzyme pathways within the microorganisms that inhabit us to make the human part of us thrive.
Acknowledgments
We apologize for excluding many important recent advances in microbiome-related drug discovery due to space constraints. We sincerely thank David Schumick for providing the illustrations included in this work.
This work was supported by National Institutes of Health Grants R01 HL122283 (to J. M. B.), P50 AA024333 (to J. M. B.), R01 HL103866 (to S. L. H.), P01 HL076491 (to S. L. H.), R01DK10600 (to S. L. H.), and R01HL126827 (to S. L. H.) from NHLBI and Office of Dietary Supplements. This is the third article in the Host-Microbiome metabolic interplay Minireview series. Dr. Hazen reports being listed as co-inventor on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics. Dr. Hazen reports having been paid as a consultant for the following companies: Esperion, Proctor & Gamble, and Takeda. Dr. Hazen reports receiving research funds from Astra Zeneca, Pfizer, Proctor & Gamble, Roche Diagnostics, and Takeda. Dr. Hazen reports having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics and therapeutics for the following companies: Cleveland Heart Laboratory, Esperion, Frantz Biomarkers, LLC, and Siemens. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MAMP
- microbe-associated molecular pattern
- CKD
- chronic kidney disease
- CVD
- cardiovascular disease
- TMA
- trimethylamine
- TMAO
- trimethylamine-N-oxide
- T2DM
- type 2 diabetes mellitus
- DMB
- 3,3-dimethyl-1-butanol.
References
- 1. Gest H. (2004) The discovery of microorganisms by Robert Hooke and Antoni Van Leeuwenhoek, fellows of the Royal Society. Notes Rec. R. Soc. Lond. 58, 187–201 [DOI] [PubMed] [Google Scholar]
- 2. Koch R. (1880) Investigations into the Etiology of Traumatic Infectious Diseases. (Watson Cheyne W., ed), The New Syndenham Society, West, Newman and Co., London [Google Scholar]
- 3. Sonnenburg J. L., and Bäckhed F. (2016) Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clemente J. C., Ursell L. K., Parfrey L. W., and Knight R. (2012) The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lozupone C. A., Stombaugh J. I., Gordon J. I., Jansson J. K., and Knight R. (2012) Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Medzhitov R. (2001) Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1, 135–145 [DOI] [PubMed] [Google Scholar]
- 7. Meylan E., Tschopp J., and Karin M. (2006) Intracellular pattern recognition receptors in the host response. Nature 442, 39–44 [DOI] [PubMed] [Google Scholar]
- 8. Nayfach S., and Pollard K. S. (2016) Toward accurate and quantitative comparative metagenomics. Cell 166, 1103–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jovel J., Patterson J., Wang W., Hotte N., O'Keefe S., Mitchel T., Perry T., Kao D., Mason A. L., Madsen K. L., and Wong G. K. (2016) Characterization of the gut microbiome using 16S or shotgun metagenomics. Front. Microbiol. 7, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qin J., Li R., Raes J., Arumugam M., Burgdorf K. S., Manichanh C., Nielsen T., Pons N., Levenez F., Yamada T., Mende D. R., Li J., Xu J., Li S., Li D., et al. (2010) A human gut microbiome gene catalogue established by metagenomic sequencing. Nature 464, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arumugam M., Raes J., Pelletier E., Le Paslier D., Yamada T., Mende D. R., Fernandes G. R., Tap J., Bruls T., Batto J. M., Bertalan M., Borruel N., Casellas F., Fernandez L., Gautier L., et al. (2011) Enterotypes of the human gut microbiome. Nature 473, 174–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Human Microbiome Project Consortium (2012) Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bäckhed F., Ding H., Wang T., Hooper L. V., Koh G. Y., Nagy A., Semenkovich C. F., and Gordon J. I. (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U.S.A. 101, 15718–15723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Turnbaugh P. J., Ley R. E., Mahowald M. A., Magrini V., Mardis E. R., and Gordon J. I. (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 [DOI] [PubMed] [Google Scholar]
- 15. Bäckhed F., Manchester J. K., Semenkovich C. F., and Gordon J. I. (2007) Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. U.S.A. 104, 979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Turnbaugh P. J., Hamady M., Yatsunenko T., Cantarel B. L., Duncan A., Ley R. E., Sogin M. L., Jones W. J., Roe B. A., Affourtit J. P., Egholm M., Henrissat B., Heath A. C., Knight R., and Gordon J. I. (2009) A core gut microbiome in obese and lean twins. Nature 457, 480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ley R. E., Bäckhed F., Turnbaugh P., Lozupone C. A., Knight R. D., and Gordon J. I. (2005) Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. U.S.A. 102, 11070–11075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dumas M. E., Barton R. H., Toye A., Cloarec O., Blancher C., Rothwell A., Fearnside J., Tatoud R., Blanc V., Lindon J. C., Mitchell S. C., Holmes E., McCarthy M. I., Scott J., Gauguier D., and Nicholson J. K. (2006) Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. U.S.A. 103, 1251–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cani P. D., Amar J., Iglesias M. A., Poggi M., Knauf C., Bastelica D., Neyrinck A. M., Fava F., Tuohy K. M., Chabo C., Waget A., Delmée E., Cousin B., Sulpice T., Chamontin B., et al. (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56, 1761–1772 [DOI] [PubMed] [Google Scholar]
- 20. Wen L., Ley R. E., Volchkov P. Y., Stranges P. B., Avanesyan L., Stonebraker A. C., Hu C., Wong F. S., Szot G. L., Bluestone J. A., Gordon J. I., and Chervonsky A. V. (2008) Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455, 1109–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan A. W., Fouts D. E., Brandl J., Stärkel P., Torralba M., Schott E., Tsukamoto H., Nelson K. E., Brenner D. A., and Schnabl B. (2011) Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 53, 96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bajaj J. S., Heuman D. M., Hylemon P. B., Sanyal A. J., White M. B., Monteith P., Noble N. A., Unser A. B., Daita K., Fisher A. R., Sikaroodi M., and Gillevet P. M. (2014) Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 60, 940–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Henao-Mejia J., Elinav E., Jin C., Hao L., Mehal W. Z., Strowig T., Thaiss C. A., Kau A. L., Eisenbarth S. C., Jurczak M. J., Camporez J. P., Shulman G. I., Gordon J. I., Hoffman H. M., and Flavell R. A. (2012) Inflammsome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Y. M., Liu Y., Zhou R. F., Chen X. L., Wang C., Tan X. Y., Wang L. J., Zheng R. D., Zhang H. W., Ling W. H., and Zhu H. L. (2016) Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 6, 19076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arthur J. C., Perez-Chanona E., Mühlbauer M., Tomkovich S., Uronis J. M., Fan T. J., Campbell B. J., Abujamel T., Dogan B., Rogers A. B., Rhodes J. M., Stintzi A., Simpson K. W., Hansen J. J., Keku T. O., Fodor A. A., and Jobin C. (2012) Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yoshimoto S., Loo T. M., Atarashi K., Kanda H., Sato S., Oyadomari S., Iwakura Y., Oshima K., Morita H., Hattori M., Hattori M., Honda K., Ishikawa Y., Hara E., and Ohtani N. (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101 [DOI] [PubMed] [Google Scholar]
- 27. Schulz M. D., Atay C., Heringer J., Romrig F. K., Schwitalla S., Aydin B., Ziegler P. K., Varga J., Reindl W., Pommerenke C., Salinas-Riester G., Böck A., Alpert C., Blaut M., Polson S. C., et al. (2014) High fat diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 514, 508–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sjögren K., Engdahl C., Henning P., Lerner U. H., Tremaroli V., Lagerquist M. K., Bäckhed F., and Ohlsson C. (2012) The gut microbiota regulates bone mass in mice. J. Bone Miner. Res. 27, 1357–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li J. Y., Chassaing B., Tyagi A. M., Vaccaro C., Luo T., Adams J., Darby T. M., Weitzmann M. N., Mulle J. G., Gewirtz A. T., Jones R. M., and Pacifici R. (2016) Sex steroid deficiency-associated bone loss is microbiota dependent and prevented by probiotics. J. Clin. Invest. 126, 2049–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pluznick J. L., Protzko R. J., Gevorgyan H., Peterlin Z., Sipos A., Han J., Brunet I., Wan L. X., Rey F., Wang T., Firestein S. J., Yanagisawa M., Gordon J. I., Eichmann A., Peti-Peterdi J., and Caplan M. J. (2013) Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. U.S.A. 110, 4410–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Durgan D. J., Ganesh B. P., Cope J. L., Ajami N. J., Phillips S. C., Petrosino J. F., Hollister E. B., and Bryan R. M. Jr. (2016) Role of the gut microbiome in obstructive sleep apnea-induced hypertension. Hypertension 67, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hsiao E. Y., McBride S. W., Hsien S., Sharon G., Hyde E. R., McCue T., Codelli J. A., Chow J., Reisman S. E., Petrosino J. F., Patterson P. H., and Mazmanian S. K. (2013) Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buffington S. A., Di Prisco G. V., Auchtung T. A., Ajami N. J., Petrosino J. F., and Costa-Mattioli M. (2016) Microbial reconstitution reverses maternal diet-induced social and synaptic deficits in offspring. Cell 165, 1762–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee Y. K., Menezes J. S., Umesaki Y., and Mazmanian S. K. (2011) Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 108, 4615–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berer K., Mues M., Koutrolos M., Rasbi Z. A., Boziki M., Johner C., Wekerle H., and Krishnamoorthy G. (2011) Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479, 538–541 [DOI] [PubMed] [Google Scholar]
- 36. Koren O., Spor A., Felin J., Fåk F., Stombaugh J., Tremaroli V., Behre C. J., Knight R., Fagerberg B., Ley R. E., and Bäckhed F. (2011) Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 108, 4592–4598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karlsson F. H., Fåk F., Nookaew I., Tremaroli V., Fagerberg B., Petranovic D., Bäckhed F., and Nielsen J. (2012) Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 3, 1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rothhammer V., Mascanfroni I. D., Bunse L., Takenaka M. C., Kenison J. E., Mayo L., Chao C. C., Patel B., Yan R., Blain M., Alvarez J. I., Kébir H., Anandasabapathy N., Izquierdo G., Jung S., et al. (2016) Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22, 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Z., Klipfell E., Bennett B. J., Koeth R., Levison B. S., Dugar B., Feldstein A. E., Britt E. B., Fu X., Chung Y. M., Wu Y., Schauer P., Smith J. D., Allayee H., Tang W. H., et al. (2011) Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. da Silva R. R., Dorrestein P. C., and Quinn R. A. (2015) Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. U.S.A. 112, 12549–12550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Johnson S. R., and Lange B. M. (2015) Open-access metabolomics databases for natural product research: present capabilities and future potential. Front. Bioeng. Biotechnol. 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wishart D. S. (2016) Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 15, 473–484 [DOI] [PubMed] [Google Scholar]
- 43. Bingol K., Bruschweiler-Li L., Li D., Zhang B., Xie M., and Brüschweiler R. (2016) Emerging new strategies for successful metabolite identification in metabolomics. Bioanalysis 8, 557–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fleming S. E., Fitch M. D., and Chansler M. W. (1989) High-fiber diets influence on characteristics of ceal digesta including short-chain fatty acid concentrations and pH. Am. J. Clin. Nutr. 50, 93–99 [DOI] [PubMed] [Google Scholar]
- 45. Northfield T. C., and McColl I. (1973) Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut 14, 513–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boutagy N. E., Neilson A. P., Osterberg K. L., Smithson A. T., Englund T. R., Davy B. M., Hulver M. W., and Davy K. P. (2015) Short-term high-fat diet increases postprandial trimethylamine-N-oxide in humans. Nutr. Res. 35, 858–864 [DOI] [PubMed] [Google Scholar]
- 47. Koeth R. A., Wang Z., Levison B. S., Buffa J. A., Org E., Sheehy B. T., Britt E. B., Fu X., Wu Y., Li L., Smith J. D., DiDonato J. A., Chen J., Li H., Wu G. D., et al. (2013) Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19, 576–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tang W. H., Wang Z., Levison B. S., Koeth R. A., Britt E. B., Fu X., Wu Y., and Hazen S. L. (2013) Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 368, 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Z., Tang W. H., Buffa J. A., Fu X., Britt E. B., Koeth R. A., Levison B. S., Fan Y., Wu Y., and Hazen S. L. (2014) Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur. Heart J. 35, 904–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Koeth R. A., Levison B. S., Culley M. K., Buffa J. A., Wang Z., Gregory J. C., Org E., Wu Y., Li L., Smith J. D., Tang W. H., DiDonato J. A., Lusis A. J., and Hazen S. L. (2014) γ-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of l-carnitine to TMAO. Cell Metab. 20, 799–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shih D. M., Wang Z., Lee R., Meng Y., Che N., Charugundla S., Qi H., Wu J., Pan C., Brown J. M., Vallim T., Bennett B. J., Graham M., Hazen S. L., and Lusis A. J. (2015) Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J. Lipid Res. 56, 22–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Warrier M., Shih D. M., Burrows A. C., Ferguson D., Gromovsky A. D., Brown A. L., Marshall S., McDaniel A., Schugar R. C., Wang Z., et al. (2015) The TMAO-generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep. 10, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miao J., Ling A. V., Manthena P. V., Gearing M. E., Graham M. J., Crooke R. M., Croce K. J., Esquejo R. M., Clish C. B., Morbid Obesity Study Group, Vicent D., and Biddinger S. B. (2015) Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat. Commun. 6, 6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Senthong V., Li X. S., Hudec T., Coughlin J., Wu Y., Levison B., Wang Z., Hazen S. L., and Tang W. H. (2016) Plasma trimethylamine N-oxide, a gut microbe-generated phosphatidylcholine metabolite, is associated with atherosclerotic burden. J. Am. Coll. Cardiol. 67, 2620–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Senthong V., Wang Z., Li X. S., Fan Y., Wu Y., Tang W. H., and Hazen S. L. (2016) Intestinal microbiota-generated metabolite trimethylamine N-oxide and 5-year mortality risk in stable coronary artery disease: the contributory role of intestinal microbiota in a COURAGE-like patient cohort. J. Am. Heart Assoc. 5, e002816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang Z., Roberts A. B., Buffa J. A., Levison B. S., Zhu W., Org E., Gu X., Huang Y., Zamanian-Daryoush M., Culley M. K., DiDonato A. J., Fu X., Hazen J. E., Krajcik D., DiDonato J. A., et al. (2015) Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 163, 1585–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gregory J. C., Buffa J. A., Org E., Wang Z., Levison B. S., Zhu W., Wagner M. A., Bennett B. J., Li L., DiDonato J. A., Lusis A. J., and Hazen S. L. (2015) Transmission of atherosclerosis susceptibility with gut microbial transplantation. J. Biol. Chem. 290, 5647–5660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shafi T., Powe N. R., Meyer T. W., Hwang S., Hai X., Melamed M. L., Banerjee T., Coresh J., and Hostetter T. H. (2017) Trimethylamine N-oxide and cardiovascular events in hemodialysis patients. J. Am. Soc. Nephrol. 28, 321–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zheng Y., Li Y., Rimm E. B., Hu F. B., Albert C. M., Rexrode K. M., Manson J. E., and Qi L. (2016) Dietary phosphatidylcholine and risk of all-cause and cardiovascular-specific mortality among United States women and men. Am. J. Clin. Nutr. 104, 173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Randrianarisoa E., Lehn-Stefan A., Wang X., Hoene M., Peter A., Heinzmann S. S., Zhao X., Königsrainer I., Königsrainer A., Balletshofer B., Machann J., Schick F., Fritsche A., Häring H. U., Xu G., Lehmann R., and Stefan N. (2016) Relationship of serum trimethylamine N-oxide (TMAO) levels with early atherosclerosis in humans. Sci. Rep. 6, 26745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Skagen K., Trøseid M., Ueland T., Holm S., Abbas A., Gregersen I., Kummen M., Bjerkeli V., Reier-Nilsen F., Russell D., Svardal A., Karlsen T. H., Aukrust P., Berge R. K., Hov J. E., et al. (2016) The carnitine-butyrobetaine-trimethylamine-oxide pathway and its association with cardiovascular mortality in patients with carotid atherosclerosis. Atherosclerosis 247, 64–69 [DOI] [PubMed] [Google Scholar]
- 62. Mente A., Chalcraft K., Ak H., Davis A. D., Lonn E., Miller R., Potter M. A., Yusuf S., Anand S. S., and McQueen M. J. (2015) The relationship between trimethylamine-N-oxide and prevalent cardiovascular disease in a multiethnic population living in Canada. Can. J. Cardiol. 31, 1189–1194 [DOI] [PubMed] [Google Scholar]
- 63. Miller P. E., Haberlen S. A., Brown T. T., Margolick J. B., DiDonato J. A., Hazen S. L., Witt M. D., Kingsley L. A., Palella F. J. Jr., Budoff M., Jacobson L. P., Post W. S., and Sears C. L. (2016) Brief Report: Intestinal microbiota-produced trimethylamine-N-oxide and its association with coronary stenosis and HIV serostatus. J. Acquir. Immune. Defic. Syndr. 72, 114–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rohrmann S., Linseisen J., Allenspach M., von Eckardstein A., and Müller D. (2016) Plasma concentration of trimethylamine-N-oxide are directly associated with dairy food consumption and low-grade inflammation in a German adult population. J. Nutr. 146, 283–289 [DOI] [PubMed] [Google Scholar]
- 65. Chen M. L., Yi L., Zhang Y., Zhou X., Ran L., Yang J., Zhu J. D., Zhang Q. Y., and Mi M. T. (2016) Resveratrol attenuates trimethylamine-N-oxide (TMAO)-induced atherosclerosis by regulating TMAO synthesis and bile acid metabolism via remodeling of the gut microbiota. MBio 7, e02210–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhu W., Gregory J. C., Org E., Buffa J. A., Gupta N., Wang Z., Li L., Fu X., Wu Y., Mehrabian M., Sartor R. B., McIntyre T. M., Silverstein R. L., Tang W. H., DiDonato J. A., et al. (2016) Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 165, 111–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tang W. H., Wang Z., Shrestha K., Borowski A. G., Wu Y., Troughton R. W., Klein A. L., and Hazen S. L. (2015) Intestinal microbiota-dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J. Card. Fail. 21, 91–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tang W. H., Wang Z., Fan Y., Levison B., Hazen J. E., Donahue L. M., Wu Y., and Hazen S. L. (2014) Prognostic value of elevated levels of intestinal microbe-generated metabolite, trimethylamine-N-oxide, in patients with heart failure: Refining the gut hypothesis. J. Am. Coll. Cardiol. 64, 1908–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Trøseid M., Ueland T., Hov J. R., Svardal A., Gregersen I., Dahl C. P., Aakhus S., Gude E., Bjørndal B., Halvorsen B., Karlsen T. H., Aukrust P., Gullestad L., Berge R. K., and Yndestad A. (2015) Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J. Intern. Med. 277, 717–726 [DOI] [PubMed] [Google Scholar]
- 70. Organ C. L., Otsuka H., Bhushan S., Wang Z., Bradley J., Trivedi R., Polhemus D. J., Tang W. H., Wu Y., Hazen S. L., and Lefer D. J. (2016) Choline diet and its gut microbe-derive metabolite, trimethylamine N-oxide, exacerbate pressure overload-induced heart failure. Circ. Heart Fail. 9, e002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tang W. H., Wang Z., Kennedy D. J., Wu Y., Buffa J. A., Agatisa-Boyle B., Li X. S., Levison B. S., and Hazen S. L. (2015) Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 116, 448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim R. B., Morse B. L., Djurdjev O., Tang M., Muirhead N., Barrett B., Holmes D. T., Madore F., Clase C. M., Rigatto C., Levin A., CanPREDDICT Investigators (2016) Advanced chronic kidney disease populations have elevated trimethylamine N-oxide levels associated with increased cardiovascular events. Kidney Int. 89, 1144–1152 [DOI] [PubMed] [Google Scholar]
- 73. Missailidis C., Hällqvist J., Qureshi A. R., Barany P., Heimbürger O., Lindholm B., Stenvinkel P., and Bergman P. (2016) Serum trimethylamine-N-oxide is strongly related to renal function and predicts outcome in chronic kidney disease. PLoS ONE 11, e0141738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mafune A., Iwamoto T., Tsutsumi Y., Nakashima A., Yamamoto I., Yokoyama K., Yokoo T., and Urashima M. (2016) Associations among serum trimethylamine-N-oxide (TMAO) levels, kidney function and infarcted coronary artery number in patients undergoing cardiovascular surgery: a cross-sectional study. Clin. Exp. Nephrol. 20, 731–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stubbs J. R., House J. A., Ocque A. J., Zhang S., Johnson C., Kimber C., Schmidt K., Gupta A., Wetmore J. B., Nolin T. D., Spertus J. A., and Yu A. S. (2016) Serum trimethylamine-N-oxide is elevated in CKD and correlates with coronary atherosclerosis burden. J. Am. Soc. Nephrol. 27, 305–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rhee E. P., Clish C. B., Ghorbani A., Larson M. G., Elmariah S., McCabe E., Yang Q., Cheng S., Pierce K., Deik A., Souza A. L., Farrell L., Domos C., Yeh R. W., Palacios I., et al. (2013) A combined epidemiologic and metabolomic approach improves CKD prediction. J. Am. Soc. Nephrol. 24, 1330–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wallace B. D., Wang H., Lane K. T., Scott J. E., Orans J., Koo J. S., Venkatesh M., Jobin C., Yeh L. A., Mani S., and Redinbo M. R. (2010) Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330, 831–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. LoGuidice A., Wallace B. D., Bendel L., Redinbo M. R., and Boelsterli U. A. (2012) Pharmacologic targeting of bacterial β-glucuronidase alleviates nonsteroidal anti-inflammatory drug-induced enteropathy in mice. J. Pharmacol. Exp. Ther. 341, 447–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ufnal M., Jazwiec R., Dadlez M., Drapala A., Sikora M., and Skrzypecki J. (2014) Trimethylamine-N-oxide: a carnitine metabolite that prolongs the hypertensive effect of angiotensin II in rats. Can. J. Cardiol. 30, 1700–1705 [DOI] [PubMed] [Google Scholar]
- 80. Seldin M. M., Meng Y., Qi H., Zhu W., Wang Z., Hazen S. L., Lusis A. J., and Shih D. M. (2016) Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J. Am. Heart Assoc. 5, e002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Brown J. M., and Hazen S. L. (2015) The gut microbial endocrine organ: bacterially derived signals driving cardiometabolic disease. Annu. Rev. Med. 66, 343–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tang W. H., and Hazen S. L. (2014) The contributory role of gut microbiota in cardiovascular disease. J. Clin. Invest. 124, 4204–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yin J., Liao S. X., He Y., Wang S., Xia G. H., Liu F. T., Zhu J. J., You C., Chen Q., Zhou L., Pan S. Y., and Zhou H. W. (2015) Dysbiosis of gut microbiota with reduced trimethylamine-N-oxide level in patients with large-artery atherosclerosis stroke or transient ischemic attack. J. Am. Heart Assoc. 4, e002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Collins H. L., Drazul-Schrader D., Sulpizio A. C., Koster P. D., Williamson Y., Adelman S. J., Owen K., Sanli T., and Bellamine A. (2016) l-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE(−/−) transgenic mice expressing CETP. Atherosclerosis 244, 29–37 [DOI] [PubMed] [Google Scholar]
- 85. Meyer K. A., Benton T. Z., Bennett B. J., Jacobs D. R. Jr, Lloyd-Jones D. M., Gross M. D., Carr J. J., Gordon-Larsen P., and Zeisel S. H. (2016) Microbiota-dependent metabolite trimethylamine N-oxide and coronary artery calcium in the coronary artery risk development in young adults study (CARDIA). J. Am. Heart Assoc. 5, e003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yamazaki H., and Shimizu M. (2013) Survey of variants of human flavin-containing monooxygenase 3 (FMO3) and their drug oxidation activities. Biochem. Pharmacol. 85, 1588–1593 [DOI] [PubMed] [Google Scholar]
- 87. Dambrova M., Latkovskis G., Kuka J., Strele I., Konrade I., Grinberga S., Hartmane D., Pugovics O., Erglis A., and Liepinsh E. (2016) Diabetes is associated with higher trimethylamine N-oxide plasma levels. Exp. Clin. Endocrinol. Diabetes 124, 251–256 [DOI] [PubMed] [Google Scholar]
- 88. Lever M., George P. M., Slow S., Bellamy D., Young J. M., Ho M., McEntyre C. J., Elmslie J. L., Atkinson W., Molyneux S. L., Troughton R. W., Frampton C. M., Richards A. M., and Chambers S. T. (2014) Betaine and trimethylamine-N-oxide as predictors of cardiovascular outcomes show different patterns in diabetes mellitus: an observational study. PLoS ONE 9, e114969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gao X., Liu X., Xu J., Xue C., Xue Y., and Wang Y. (2014) Dietary trimethylamine N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J. Biosci. Bioeng. 118, 476–481 [DOI] [PubMed] [Google Scholar]
- 90. Falony G., Vieira-Silva S., and Raes J. (2015) Microbiology meets big data: the case of gut microbe-derived trimethylamine. Annu. Rev. Microbiol. 69, 305–321 [DOI] [PubMed] [Google Scholar]
- 91. Romano K. A., Vivas E. I., Amador-Noguez D., and Rey F. E. (2015) Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio 6, e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Li Q., Korzan W. J., Ferrero D. M., Chang R. B., Roy D. S., Buchi M., Lemon J. K., Kaur A. W., Stowers L., Fendt M., and Liberles S. D. (2013) Synchronous evolution of an odor biosynthesis pathway and behavioral response. Curr. Biol. 23, 11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wallrabenstein I., Kuklan J., Weber L., Zborala S., Werner M., Altmüller J., Becker C., Schmidt A., Hatt H., Hummel T., and Gisselmann G. (2013) Human trace amine-associated receptor TAAR5 can be activated by trimethylamine. PLoS ONE 8, e54950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Law R. O. (1992) Efflux and accumulation of amino nitrogen in relation to the volume of rat renal inner medullary cells exposed to media of variable osmolality. Biochim. Biophys. Acta 1133, 268–274 [DOI] [PubMed] [Google Scholar]
- 95. Yancey P. H., Clark M. E., Hand S. C., Bowlus R. D., and Somero G. N. (1982) Living with water stress: evolution of osmolyte systems. Science 217, 1214–1222 [DOI] [PubMed] [Google Scholar]
- 96. Lin T. Y., and Timasheff S. N. (1994) Why do some organisms use a urea-methylamine mixture as osmolyte? Thermodynamic compensation of urea and trimethylamine N-oxide interactions with protein. Biochemistry 33, 12695–12701 [DOI] [PubMed] [Google Scholar]
- 97. Grice E. A., and Segre J. A. (2012) The human microbiome: our second genome. Annu. Rev. Genomics Hum. Genet. 13, 151–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Davies J. E. (2004) Streptomycetes and beyond. Microbiol. Aust. 25, 8–10 [Google Scholar]
- 99. Cooper M. A. (2015) A community-based approach to new antibiotic discovery. Nat. Rev. Drug Discov. 14, 587–588 [DOI] [PubMed] [Google Scholar]
- 100. Zschaler J., Schlorke D., and Arnhold J. (2014) Differences in innate immune response between man and mouse. Crit. Rev. Immunol. 34, 433–454 [PubMed] [Google Scholar]
- 101. Pellock S. J., and Redinbo M. R. (2017) Glucuronides in the gut: Sugar-driven symbioses between microbe and host. J. Biol. Chem. 292, 8569–8576 [DOI] [PMC free article] [PubMed] [Google Scholar]