

Abstract
Carbapenemase-producing Enterobacteriaceae are an emerging threat to hospitals worldwide, and antibiotic exposure is a risk factor for developing fecal carriage that may lead to nosocomial infection. Here, we review how antibiotics reduce colonization resistance against Enterobacteriaceae to pinpoint possible control points for curbing their spread. Recent work identifies host-derived respiratory electron acceptors as a critical resource driving a post-antibiotic expansion of Enterobacteriaceae within the large bowel. By providing a conceptual framework for colonization resistance against Enterobacteriaceae, these mechanistic insights point to the metabolism of epithelial cells as a possible target for intervention strategies.
Keywords: antibiotic resistance, antibiotics, Escherichia coli (E. coli), Klebsiella pneumonia, microbiome, Salmonella enterica
Introduction
The lumen of the large intestine is host to a large microbial community, the gut microbiota, which is dominated by obligate anaerobic bacteria belonging to the classes Clostridia (phylum Firmicutes) and Bacteroidia (phylum Bacteroidetes). The concept that the gut microbiota confer “colonization resistance” against members of the family Enterobacteriaceae (phylum Proteobacteria) dates back to the 1950s and 60s, when streptomycin treatment of mice was shown to greatly increase susceptibility to oral infection with pathogenic Salmonella enterica (1–3) or commensal Escherichia coli (4, 5). Antimicrobial-resistant Enterobacteriaceae can expand in the human gut during antibiotic therapy, as shown by challenge of trimoxazole-treated healthy volunteers with a trimoxazole-resistant strain of Klebsiella pneumoniae (6). The relative abundance of endogenous E. coli within the murine gut microbiota increases 1 day after cessation of treatment with cefoperazone, metronidazole, clindamycin, ampicillin, or vancomycin (7). Epidemiological evidence suggests that antibiotics cause a prolonged disruption of the colonic microbiota, thereby decreasing colonization resistance for some time after cessation of treatment. For example, a history of antibiotic usage is a risk factor for developing gastroenteritis with antimicrobial-sensitive S. enterica isolates (8).
Over the last decade, carbapenemase-producing Enterobacteriaceae (CPE),2 such as K. pneumoniae and E. coli, have emerged as a major infectious disease threat to hospitals worldwide, with mortality from invasive nosocomial CPE infections reaching up to 40% (9, 10). One of the risk factors for developing fecal carriage and nosocomial CPE infection is a previous exposure to broad-spectrum antibiotics (11–15), suggesting that an antibiotic-mediated disruption of colonization resistance facilitates the spread of CPE within the hospital. Thus, the development of approaches for maintaining colonization resistance during or following antibiotic therapy might hold the key for limiting communicability of nosocomial CPE infections. Here, we attempt to synthesize recent insights into the mechanisms underlying colonization resistance against Enterobacteriaceae into a coherent conceptual framework to facilitate the development of strategies for curbing their spread by limiting unwanted side effects of antibiotic therapy.
Proposed mechanisms for colonization resistance
Understanding colonization resistance against Enterobacteriaceae is not a trivial task, because the gut microbiota are highly complex and vary greatly between individuals (16). Furthermore, at least three principal mechanisms for disruption of colonization resistance against Enterobacteriaceae have been proposed: gut inflammation, depletion of microbiota-derived inhibitory products, and increased nutrient availability (17–20). In the following we will discuss the relevance of each of these mechanisms for understanding an antibiotic-mediated disruption of colonization resistance against Enterobacteriaceae.
Gut inflammation
Severe intestinal inflammation is a driver of dysbiosis characterized by an expansion of Enterobacteriaceae within the gut-associated microbial community (21, 22). Mechanisms driving this expansion include resistance of Enterobacteriaceae against antimicrobial host defenses induced during inflammation (23, 24) and the generation of respiratory electron acceptors generated as a by-product of the host inflammatory response, which favors growth of facultative anaerobic bacteria (25, 26). Antibiotic treatment does not cause severe gut inflammation, although mild inflammatory changes may be observed on occasion (27, 28). To understand how a history of antibiotic exposure lowers colonization resistance against Enterobacteriaceae, severe gut inflammation can be eliminated from the list of possible mechanisms. However, it remains possible that some of the mechanisms driving an expansion of Enterobacteriaceae during severe inflammation are also operational during an antibiotic-induced mild increase in the inflammatory tone of the gut mucosa.
Depletion of microbiota-derived inhibitory products
The gut microbiota break down complex carbohydrates in the large bowel to fermentation products, the most abundant of which are the short-chain fatty acids butyrate, propionate, and acetate. An antibiotic-mediated depletion of the gut microbiota reduces short-chain fatty acid concentrations in the large intestine of mice (3, 29) and in human feces (30). Because short-chain fatty acids can impede growth of E. coli or S. enterica in the test tube, the presence of these metabolites is proposed to confer colonization resistance against Enterobacteriaceae by “metabolic exclusion” (20). However, this concept does not take into account that microbiota-derived short-chain fatty acids have a profound effect on host cell physiology, which might elevate colonization resistance indirectly through alternative mechanisms (31). Furthermore, Freter and co-workers (32) pointed out in the 1980s that metabolic exclusion cannot explain how Enterobacteriaceae are maintained at a low abundance in healthy individuals. For an inhibitor to maintain Enterobacteriaceae at a constant low level, it would have to be present continuously at precisely the concentration needed to check population growth. Any increase in the inhibitor concentration would lead to an elimination of Enterobacteriaceae from the microbial community, and any decrease would result in their expansion until another resource becomes limiting. Thus, metabolic exclusion by a microbiota-derived inhibitor does not provide a robust theoretical framework for colonization resistance.
Increased nutrient availability
Most microbes present within our gut-associated microbial community have resided in this environment for decades (33). Mathematical models explaining such a stable co-existence of multiple bacterial species suggest that each member within the microbial community must be able to grow faster on a few limiting resources than all other members and that the availability of these resources controls its abundance within the community (34). This concept is known as the “nutrient-niche hypothesis” and suggests that a low abundance of Enterobacteriaceae within the gut microbiota is maintained through a limited availability of critical resources. A disruption of the gut microbiota by antibiotic treatment somehow increases the availability of these limiting resources, thereby triggering an expansion of Enterobacteriaceae. Thus, the nutrient-niche hypothesis raises the following two questions essential for understanding colonization resistance. What are the resources that limit an expansion of Enterobacteriaceae? How does antibiotic treatment elevate their availability? In the following we will attempt to address these two key gaps in knowledge.
What resources control Enterobacteriaceae abundance?
The ability to utilize complex dietary and host polysaccharides depends on the carbohydrate-digestive capacity of the gut microbiota. The members within the gut microbiota that possess the largest carbohydrate substrate range belong to the class Bacteroidia, as suggested by the presence of genes encoding a diverse array of glycoside hydrolases and polysaccharide lyases (35). Bacteroidia prioritize the consumption of sugars liberated from dietary carbohydrates by their glycoside hydrolases and polysaccharide lyases but turn to utilization of host mucus glycans when dietary polysaccharides are absent (36). Streptomycin treatment of mice increases the abundance of microbiota-liberated monosaccharides, such as sialic acid and fucose, and inactivation of genes required for the utilization of these sugars reduces a post-antibiotic expansion of S. enterica in the gut lumen (37). These data suggest that by depleting sugar-consuming bacteria the antibiotic treatment increases the availability of microbiota-liberated monosaccharides, which in turn supports growth of S. enterica. In addition, antibiotic treatment can lead to an increased abundance of sugar-oxidation products, such as glucarate or galactarate, and utilization of these carbon sources drives a post-antibiotic expansion of E. coli and S. enterica (38).
To explain why an increased availability of monosaccharides specifically favors Enterobacteriaceae, the nutrient-niche hypothesis would predict that Enterobacteriaceae must be able to grow faster on this resource than any other member of the gut microbiota (34). The energy yield S. enterica or E. coli obtained by catabolizing hexoses through the Embden-Meyerhof-Parnas pathway followed by mixed acid fermentation is not superior to that generated by obligate anaerobic bacteria competing for available monosaccharides (39). Thus, it is not obvious why sugars would favor growth of Enterobacteriaceae over that of Clostridia or Bacteroidia.
One critical resource that Enterobacteriaceae can use better than obligate anaerobic Clostridia or Bacteroidia is oxygen (O2), which can only be respired by facultative anaerobic bacteria. An elevated availability of oxygen can increase the abundance of facultative anaerobic Enterobacteriaceae within the gut-associated microbial community, as suggested by an expansion of this family near the ileostomy of small bowel transplant patients (40). Enterobacteriaceae also expand within the gut-associated microbial community when electron acceptors for anaerobic respiration become available. This represents one of the mechanisms increasing the abundance of Enterobacteriaceae during severe gut inflammation (22). An elevated mucosal synthesis of inducible nitric-oxide synthase (iNOS) triggered during genetically or chemically induced colitis in mice leads to the production of nitric oxide (NO), which reacts to form nitrate (NO3−) in the gut lumen, thereby driving an uncontrolled expansion of commensal E. coli by nitrate respiration (26).
The ability to utilize exogenous respiratory electron acceptors enables Enterobacteriaceae to gain an edge over Clostridia or Bacteroidia, because respiration generates more energy from catabolism of carbon sources than fermentation. Furthermore, respiration enables Enterobacteriaceae to consume non-fermentable substrates in the inflamed gut, thereby allowing these facultative anaerobic bacteria to sidestep the competition with fermenting Clostridia and Bacteroidia (41). Thus, consistent with the nutrient-niche hypothesis, Enterobacteriaceae can grow faster on carbon sources using respiration than the fermenting Clostridia and Bacteroidia. Furthermore, a limited availability of exogenous electron acceptors in the large intestine helps explain how these facultative anaerobic bacteria are maintained at a low abundance within the gut-associated microbial community. However, these considerations beg the question whether and by which mechanism antibiotic treatment might increase the abundance of respiratory electron acceptors in the absence of overt gut inflammation.
How antibiotic treatment creates resources for Enterobacteriaceae
The colon functions in water absorption by absorbing sodium (Na+) to generate an osmotic gradient. Active Na+ transport mediated by the Na+K+-ATPase in the basolateral membrane of enterocytes in the colon (colonocytes) requires ATP, which is generated by oxidizing microbiota-derived butyrate to carbon dioxide (CO2) in their mitochondria (Fig. 1) (42, 43). Burning microbiota-derived butyrate to generate energy consumes oxygen, thereby rendering surface colonocytes hypoxic (<1% oxygen) (44).
Figure 1.
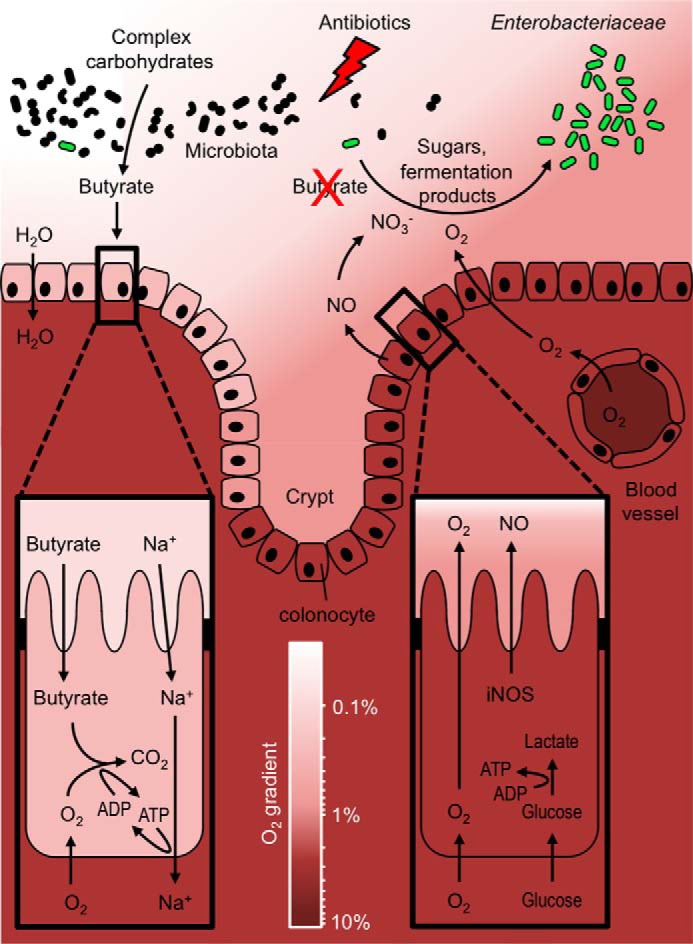
Antibiotics lower colonization resistance against Enterobacteriaceae. The gut microbiota breaks down complex carbohydrates into a variety of fermentation products, such as butyrate. Butyrate serves as the main energy source for colonocytes, which oxidize this short-chain fatty acid to produce ATP for the absorption of sodium (Na+). This metabolism consumes oxygen (O2) thereby rendering the colonic surface hypoxic (<1% oxygen). Antibiotics deplete butyrate-producing bacteria, which forces colonocytes to switch their metabolism to a fermentation of glucose to lactate, thereby increasing the abundance of nitrate (NO3−) and oxygen in the gut lumen. Facultative anaerobic bacteria can use these host-derived electron acceptors to grow better on carbon sources than obligate anaerobic bacteria, which drives a luminal expansion of Enterobacteriaceae.
Streptomycin treatment depletes butyrate-producing bacteria, as indicated by a marked drop in butyrate concentrations in the cecum and colon of mice (3, 29, 45, 46). Butyrate production by the gut microbiota proceeds through the lysine pathway, the glutarate pathway, the 4-aminobutyrate pathway, or the acetyl-CoA pathway (47). The majority of bacteria encoding these pathways are members of the class Clostridia, but genes for butyrate production can also be present in members of the Bacteroidia families Rikenellaceae and Porphyromonadaceae (47, 48). Attempts to restore colonization resistance after streptomycin treatment suggest that transfer of spore-forming Clostridia is most effective in preventing a post-antibiotic expansion of commensal E. coli in the large bowel of mice (49).
The antibiotic-induced depletion of microbiota-derived butyrate forces colonocytes to obtain energy through the fermentation of glucose to lactate (Fig. 1) (50). This energy metabolism does not consume oxygen, thus rendering the colonic surface normoxic (51), which is between 3 and 10% oxygen (52). Elevated oxygenation of surface colonocytes is predicted to increase diffusion of oxygen across the brush border into the gut lumen (53), which might explain why streptomycin treatment increases the redox potential in the murine cecum to that of an aerobic broth culture (3). Oral treatment with streptomycin reduces butyrate concentrations in the large intestine leading to an increase in oxygen availability, which drives a cytochrome bd-II oxidase-dependent aerobic luminal expansion of S. enterica (46). Increasing the colonic butyrate concentration through tributyrin supplementation restores colonocyte hypoxia (51), thereby abrogating an aerobic post-antibiotic expansion of S. enterica in the colon (46). Aerobic respiration also contributes to an E. coli expansion in the colon of streptomycin-treated mice (54).
The shift from fatty acid respiration to glucose fermentation is common in host cells, characterizing, for example, the transition from the prohealing M2 polarization state to the proinflammatory M1 polarization state of macrophages. The synthesis of iNOS is a marker for the fermentation-based metabolic program of M1 macrophages (55). Butyrate reduces glucose fermentation in colonocytes (50) and inhibits iNOS synthesis in this cell type (56). Thus, butyrate-induced changes in the metabolic program of colonocytes might explain why streptomycin treatment induces iNOS synthesis in the large intestine of mice, which increases the availability of host-derived nitrate in the large intestine (27). Nitrate respiration contributes to an expansion of commensal E. coli in the colon of streptomycin-treated mice (27, 57) and synergizes with aerobic respiration to drive a post-antibiotic expansion of S. enterica (Fig. 1) (46).
In summary, antibiotic treatment generates a respiratory nutrient niche that supports an uncontrolled expansion of Enterobacteriaceae within the gut-associated microbial community. Pathogenic and commensal Enterobacteriaceae compete for occupancy of this nutrient niche (23, 58–61), and a better understanding of the mechanisms that control the outcome of this competition may facilitate the development of second-generation probiotics to prevent colonization with potentially harmful members of this family, such as CPE. Alternatively, approaches to maintain epithelial homeostasis could be used to limit the generation of a respiratory nutrient-niche after antibiotic treatment, thereby curbing a post-antibiotic expansion of Enterobacteriaceae.
Conclusions
The picture emerging from these studies is that exogenous respiratory electron acceptors are a limiting resource that controls the abundance of Enterobacteriaceae within the gut-associated microbial community. Antibiotic treatment depletes butyrate-producing bacteria, thereby changing host cell metabolism to elevate iNOS synthesis and increase epithelial oxygenation. Through these mechanisms, antibiotics raise the concentration of host-derived respiratory electron acceptors in the lumen of the large intestine. The resulting respiratory nutrient niche drives an expansion of Enterobacteriaceae, because these facultative anaerobic bacteria can use respiration to catabolize available carbon sources better than obligate anaerobic Clostridia and Bacteroidia (Fig. 1). This conceptual framework incorporates all players previously implicated in colonization resistance, although it changes data interpretation in some instances. For example, the nutrient-niche hypothesis suggests that short-chain fatty acids confer colonization resistance by influencing host cell physiology, not through metabolic exclusion.
After filling a few key gaps in knowledge, it becomes clear that the nutrient-niche hypothesis explains most aspects of colonization resistance, which is of great appeal because it helps reduce the complexity of the problem. Putting the available information into a coherent conceptual framework will help guide the development of treatment strategies designed to alleviate unwanted side effects of antibiotic therapy by pinpointing critical steps, such as colonocyte energy metabolism, that could become targets for intervention. Curbing the nosocomial spread of CPE is a national priority, because these infections have limited treatment options and are associated with high mortality rates. Given the lack of new antibiotics in the pipeline, the identification of intervention strategies to prevent a post-antibiotic expansion of CPE thus holds great potential for limiting communicability of these opportunistic pathogens.
This is the fifth article in the Host-Microbiome Metabolic Interplay Minireview series. M. X. B. and A. J. B. filed invention report number 0577501-16-0038 at iEdison.gov for a treatment to prevent post-antibiotic expansion of Enterobacteriaceae.
- CPE
- carbapenemase-producing Enterobacteriaceae
- iNOS
- inducible nitric-oxide synthase.
References
- 1. Bohnhoff M., Drake B. L., and Miller C. P. (1954) Effect of streptomycin on susceptibility of intestinal tract to experimental Salmonella infection. Proc. Soc. Exp. Biol. Med. 86, 132–137 [DOI] [PubMed] [Google Scholar]
- 2. Bohnhoff M., and Miller C. P. (1962) Enhanced susceptibility to Salmonella infection in streptomycin-treated mice. J. Infect. Dis. 111, 117–127 [DOI] [PubMed] [Google Scholar]
- 3. Meynell G. G. (1963) Antibacterial mechanisms of the mouse gut. II. The role of Eh and volatile fatty acids in the normal gut. Br. J. Exp. Pathol. 44, 209–219 [PMC free article] [PubMed] [Google Scholar]
- 4. Saito K. (1961) Studies on the habitation of pathogenic Escherichia coli in the intestinal tract of mice. I. Comparative experiments on the habitation of each type of resistant pathogenic Escherichia coli under an administration of streptomycin. Paediatr. Jpn. 65, 385–393 [PubMed] [Google Scholar]
- 5. Saito K. (1961) Studies on the habitation of pathogenic Escherichia coli in the intestinal tract of mice. II. Experimental inoculation of type 055 Escherichia coli after long-term administration of streptomycin. Paediatr. Jpn. 65, 394–399 [PubMed] [Google Scholar]
- 6. Vollaard E. J., Clasener H. A., and Janssen A. J. (1992) Co-trimoxazole impairs colonization resistance in healthy volunteers. J. Antimicrob. Chemother. 30, 685–691 [DOI] [PubMed] [Google Scholar]
- 7. Schubert A. M., Sinani H., and Schloss P. D. (2015) Antibiotic-induced alterations of the murine gut microbiota and subsequent effects on colonization resistance against Clostridium difficile. MBio 6, e00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pavia A. T., Shipman L. D., Wells J. G., Puhr N. D., Smith J. D., McKinley T. W., and Tauxe R. V. (1990) Epidemiologic evidence that prior antimicrobial exposure decreases resistance to infection by antimicrobial-sensitive Salmonella. J. Infect. Dis. 161, 255–260 [DOI] [PubMed] [Google Scholar]
- 9. Tängdén T., and Giske C. G. (2015) Global dissemination of extensively drug-resistant carbapenemase-producing Enterobacteriaceae: clinical perspectives on detection, treatment and infection control. J. Intern. Med. 277, 501–512 [DOI] [PubMed] [Google Scholar]
- 10. Doi Y., and Paterson D. L. (2015) Carbapenemase-producing Enterobacteriaceae. Semin. Respir. Crit. Care Med. 36, 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao S. Y., Zhang J., Zhang Y. L., Wang Y. C., Xiao S. Z., Gu F. F., Guo X. K., Ni Y. X., and Han L. Z. (2016) Epidemiology and risk factors for faecal extended-spectrum β-lactamase-producing Enterobacteriaceae (ESBL-E) carriage derived from residents of seven nursing homes in western Shanghai, China. Epidemiol. Infect. 144, 695–702 [DOI] [PubMed] [Google Scholar]
- 12. Lowe C. F., Kus J. V., Salt N., Callery S., Louie L., Khan M. A., Vearncombe M., and Simor A. E. (2013) Nosocomial transmission of New Delhi metallo-β-lactamase-1-producing Klebsiella pneumoniae in Toronto, Canada. Infect. Control Hosp. Epidemiol. 34, 49–55 [DOI] [PubMed] [Google Scholar]
- 13. Razazi K., Derde L. P., Verachten M., Legrand P., Lesprit P., and Brun-Buisson C. (2012) Clinical impact and risk factors for colonization with extended-spectrum β-lactamase-producing bacteria in the intensive care unit. Intensive Care Med. 38, 1769–1778 [DOI] [PubMed] [Google Scholar]
- 14. Shanthi M., and Sekar U. (2010) Extended spectrum β-lactamase producing Escherichia coli and Klebsiella pneumoniae: risk factors for infection and impact of resistance on outcomes. J. Assoc. Physicians India 58, 41–44 [PubMed] [Google Scholar]
- 15. Daikos G. L., Vryonis E., Psichogiou M., Tzouvelekis L. S., Liatis S., Petrikkos P., Kosmidis C., Tassios P. T., Bamias G., and Skoutelis A. (2010) Risk factors for bloodstream infection with Klebsiella pneumoniae producing VIM-1 metallo-β-lactamase. J. Antimicrob. Chemother. 65, 784–788 [DOI] [PubMed] [Google Scholar]
- 16. Tap J., Mondot S., Levenez F., Pelletier E., Caron C., Furet J. P., Ugarte E., Muñoz-Tamayo R., Paslier D. L., Nalin R., Dore J., and Leclerc M. (2009) Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 11, 2574–2584 [DOI] [PubMed] [Google Scholar]
- 17. Stecher B. (2015) The roles of inflammation, nutrient availability and the commensal microbiota in enteric pathogen infection. Microbiol. Spectr. 3, 10.1128/microbiolspec.MBP-0008-2014 [DOI] [PubMed] [Google Scholar]
- 18. Yurist-Doutsch S., Arrieta M. C., Vogt S. L., and Finlay B. B. (2014) Gastrointestinal microbiota-mediated control of enteric pathogens. Annu. Rev. Genet. 48, 361–382 [DOI] [PubMed] [Google Scholar]
- 19. Stecher B., Berry D., and Loy A. (2013) Colonization resistance and microbial ecophysiology: using gnotobiotic mouse models and single-cell technology to explore the intestinal jungle. FEMS Microbiol. Rev. 37, 793–829 [DOI] [PubMed] [Google Scholar]
- 20. Lawley T. D., and Walker A. W. (2013) Intestinal colonization resistance. Immunology 138, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shin N. R., Whon T. W., and Bae J. W. (2015) Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 33, 496–503 [DOI] [PubMed] [Google Scholar]
- 22. Winter S. E., Lopez C. A., and Bäumler A. J. (2013) The dynamics of gut-associated microbial communities during inflammation. EMBO Rep. 14, 319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raffatellu M., George M. D., Akiyama Y., Hornsby M. J., Nuccio S. P., Paixao T. A., Butler B. P., Chu H., Santos R. L., Berger T., Mak T. W., Tsolis R. M., Bevins C. L., Solnick J. V., Dandekar S., and Bäumler A. J. (2009) Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe 5, 476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu J. Z., Jellbauer S., Poe A. J., Ton V., Pesciaroli M., Kehl-Fie T. E., Restrepo N. A., Hosking M. P., Edwards R. A., Battistoni A., Pasquali P., Lane T. E., Chazin W. J., Vogl T., Roth J., et al. (2012) Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe 11, 227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winter S. E., Thiennimitr P., Winter M. G., Butler B. P., Huseby D. L., Crawford R. W., Russell J. M., Bevins C. L., Adams L. G., Tsolis R. M., Roth J. R., and Bäumler A. J. (2010) Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Winter S. E., Winter M. G., Xavier M. N., Thiennimitr P., Poon V., Keestra A. M., Laughlin R. C., Gomez G., Wu J., Lawhon S. D., Popova I. E., Parikh S. J., Adams L. G., Tsolis R. M., Stewart V. J., and Bäumler A. J. (2013) Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spees A. M., Wangdi T., Lopez C. A., Kingsbury D. D., Xavier M. N., Winter S. E., Tsolis R. M., and Bäumler A. J. (2013) Streptomycin-induced inflammation enhances Escherichia coli gut colonization through nitrate respiration. MBio 4, e00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wlodarska M., Willing B., Keeney K. M., Menendez A., Bergstrom K. S., Gill N., Russell S. L., Vallance B. A., and Finlay B. B. (2011) Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infect. Immun. 79, 1536–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Garner C. D., Antonopoulos D. A., Wagner B., Duhamel G. E., Keresztes I., Ross D. A., Young V. B., and Altier C. (2009) Perturbation of the small intestine microbial ecology by streptomycin alters pathology in a Salmonella enterica serovar typhimurium murine model of infection. Infect. Immun. 77, 2691–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Høverstad T., Carlstedt-Duke B., Lingaas E., Norin E., Saxerholt H., Steinbakk M., and Midtvedt T. (1986) Influence of oral intake of seven different antibiotics on faecal short-chain fatty acid excretion in healthy subjects. Scand. J. Gastroenterol. 21, 997–1003 [DOI] [PubMed] [Google Scholar]
- 31. Spees A. M., Lopez C. A., Kingsbury D. D., Winter S. E., and Bäumler A. J. (2013) Colonization resistance: battle of the bugs or Menage a Trois with the host? PLoS Pathog. 9, e1003730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Freter R., Brickner H., Botney M., Cleven D., and Aranki A. (1983) Mechanisms that control bacterial populations in continuous-flow culture models of mouse large intestinal flora. Infect. Immun. 39, 676–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Faith J. J., Guruge J. L., Charbonneau M., Subramanian S., Seedorf H., Goodman A. L., Clemente J. C., Knight R., Heath A. C., Leibel R. L., Rosenbaum M., and Gordon J. I. (2013) The long-term stability of the human gut microbiota. Science 341, 1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Freter R., Brickner H., Fekete J., Vickerman M. M., and Carey K. E. (1983) Survival and implantation of Escherichia coli in the intestinal tract. Infect. Immun. 39, 686–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. El Kaoutari A., Armougom F., Gordon J. I., Raoult D., and Henrissat B. (2013) The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 11, 497–504 [DOI] [PubMed] [Google Scholar]
- 36. Sonnenburg J. L., Xu J., Leip D. D., Chen C. H., Westover B. P., Weatherford J., Buhler J. D., and Gordon J. I. (2005) Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307, 1955–1959 [DOI] [PubMed] [Google Scholar]
- 37. Ng K. M., Ferreyra J. A., Higginbottom S. K., Lynch J. B., Kashyap P. C., Gopinath S., Naidu N., Choudhury B., Weimer B. C., Monack D. M., and Sonnenburg J. L. (2013) Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Faber F., Tran L., Byndloss M. X., Lopez C. A., Velazquez E. M., Kerrinnes T., Nuccio S. P., Wangdi T., Fiehn O., Tsolis R. M., and Bäumler A. J. (2016) Host-mediated sugar oxidation promotes post-antibiotic pathogen expansion. Nature 534, 697–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rivera-Chávez F., and Bäumler A. J. (2015) The pyromaniac inside you: Salmonella metabolism in the host gut. Annu Rev. Microbiol. 69, 31–48 [DOI] [PubMed] [Google Scholar]
- 40. Hartman A. L., Lough D. M., Barupal D. K., Fiehn O., Fishbein T., Zasloff M., and Eisen J. A. (2009) Human gut microbiome adopts an alternative state following small bowel transplantation. Proc. Natl. Acad. Sci. U.S.A. 106, 17187–17192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thiennimitr P., Winter S. E., Winter M. G., Xavier M. N., Tolstikov V., Huseby D. L., Sterzenbach T., Tsolis R. M., Roth J. R., and Bäumler A. J. (2011) Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. U.S.A. 108, 17480–17485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Velázquez O. C., Lederer H. M., and Rombeau J. L. (1997) Butyrate and the colonocyte. Production, absorption, metabolism, and therapeutic implications. Adv. Exp. Med. Biol. 427, 123–134 [PubMed] [Google Scholar]
- 43. Sandle G. I. (1998) Salt and water absorption in the human colon: a modern appraisal. Gut 43, 294–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Furuta G. T., Turner J. R., Taylor C. T., Hershberg R. M., Comerford K., Narravula S., Podolsky D. K., and Colgan S. P. (2001) Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J. Exp. Med. 193, 1027–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smith P. M., Howitt M. R., Panikov N., Michaud M., Gallini C. A., Bohlooly-Y M., Glickman J. N., and Garrett W. S. (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rivera-Chávez F., Zhang L. F., Faber F., Lopez C. A., Byndloss M. X., Olsan E. E., Xu G., Velazquez E. M., Lebrilla C. B., Winter S. E., and Bäumler A. J. (2016) Depletion of butyrate-producing clostridia from the gut microbiota drives an aerobic luminal expansion of Salmonella. Cell Host Microbe 19, 443–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vital M., Howe A. C., and Tiedje J. M. (2014) Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. MBio 5, e00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Louis P., and Flint H. J. (2009) Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 294, 1–8 [DOI] [PubMed] [Google Scholar]
- 49. Itoh K., and Freter R. (1989) Control of Escherichia coli populations by a combination of indigenous clostridia and lactobacilli in gnotobiotic mice and continuous-flow cultures. Infect. Immun. 57, 559–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Donohoe D. R., Wali A., Brylawski B. P., and Bultman S. J. (2012) Microbial regulation of glucose metabolism and cell-cycle progression in mammalian colonocytes. PLoS one 7, e46589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kelly C. J., Zheng L., Campbell E. L., Saeedi B., Scholz C. C., Bayless A. J., Wilson K. E., Glover L. E., Kominsky D. J., Magnuson A., Weir T. L., Ehrentraut S. F., Pickel C., Kuhn K. A., Lanis J. M., et al. (2015) Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17, 662–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carreau A., El Hafny-Rahbi B., Matejuk A., Grillon C., and Kieda C. (2011) Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med 15, 1239–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Espey M. G. (2013) Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota. Free Radic. Biol. Med. 55, 130–140 [DOI] [PubMed] [Google Scholar]
- 54. Jones S. A., Chowdhury F. Z., Fabich A. J., Anderson A., Schreiner D. M., House A. L., Autieri S. M., Leatham M. P., Lins J. J., Jorgensen M., Cohen P. S., and Conway T. (2007) Respiration of Escherichia coli in the mouse intestine. Infect. Immun. 75, 4891–4899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Weigert A., and Brüne B. (2008) Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide 19, 95–102 [DOI] [PubMed] [Google Scholar]
- 56. Zimmerman M. A., Singh N., Martin P. M., Thangaraju M., Ganapathy V., Waller J. L., Shi H., Robertson K. D., Munn D. H., and Liu K. (2012) Butyrate suppresses colonic inflammation through HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T cells. Am. J. Physiol. Gastrointest Liver Physiol 302, G1405–G1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jones S. A., Gibson T., Maltby R. C., Chowdhury F. Z., Stewart V., Cohen P. S., and Conway T. (2011) Anaerobic respiration of Escherichia coli in the mouse intestine. Infect. Immun. 79, 4218–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Deriu E., Liu J. Z., Pezeshki M., Edwards R. A., Ochoa R. J., Contreras H., Libby S. J., Fang F. C., and Raffatellu M. (2013) Probiotic bacteria reduce Salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe 14, 26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nedialkova L. P., Denzler R., Koeppel M. B., Diehl M., Ring D., Wille T., Gerlach R. G., and Stecher B. (2014) Inflammation fuels colicin Ib-dependent competition of Salmonella serovar typhimurium and E. coli in enterobacterial blooms. PLoS Pathog. 10, e1003844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ali M. M., Newsom D. L., González J. F., Sabag-Daigle A., Stahl C., Steidley B., Dubena J., Dyszel J. L., Smith J. N., Dieye Y., Arsenescu R., Boyaka P. N., Krakowka S., Romeo T., Behrman E. J., et al. (2014) Fructose-asparagine is a primary nutrient during growth of Salmonella in the inflamed intestine. PLoS Pathog. 10, e1004209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Behnsen J., Jellbauer S., Wong C. P., Edwards R. A., George M. D., Ouyang W., and Raffatellu M. (2014) The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity 40, 262–273 [DOI] [PMC free article] [PubMed] [Google Scholar]