

Abstract
Mammals and their gut microbial communities share extensive and tightly coordinated co-metabolism of dietary substrates. A large number of microbial metabolites have been detected in host circulation and tissues and, in many cases, are linked to host metabolic, developmental, and immunological states. The presence of these metabolites in host tissues intersects with regulation of the host's epigenetic machinery. Although it is established that the host's epigenetic machinery is sensitive to levels of endogenous metabolites, the roles for microbial metabolites in epigenetic regulation are just beginning to be elucidated. This review focuses on eukaryotic chromatin regulation by endogenous and gut microbial metabolites and how these regulatory events may impact host developmental and metabolic phenotypes.
Keywords: chromatin, DNA demethylation, DNA methylation, epigenetics, histone acetylation, histone deacetylase (HDAC), histone methylation, metabolism, microbiome
Introduction
The eukaryotic genome exists in a largely static state, yet gene expression patterns are remarkably plastic in response to environmental stimuli. This adaptability is governed by epigenetic mechanisms that alter chromatin structure through a combination of covalent post-translational modifications (PTMs)4 of histone proteins, histone variant deposition, DNA methylation, and noncoding RNAs. Furthermore, these programming events can result in either transient or more long-term, even transgenerational, effects. For example, histone acetylation has been demonstrated to have a half-life of 53–87 minutes, depending on the specific lysine residue being modified, which is considerably faster than a typical mammalian cell cycle (1). In contrast, parental and early life nutritional status has been shown to elicit persistent effects on the DNA methylomes of offspring. Natural variation in dietary intake of methyl-donor nutrients in rural Gambian mothers, which associated with seasonal differences in maternal plasma biomarker concentrations, enabled prediction of infant DNA methylation patterns based on the season at the time of conception (2). In mice, suboptimal nutrition during fetal development affected the DNA methylome of male F1 offspring and was transmitted through the paternal line to F2 offspring (3). Thus, environmental factors can “program” chromatin states and drive both short- and long-term effects.
The molecular machinery responsible for depositing and removing histone and DNA modifications is known to be sensitive to the availability of small molecule metabolites, many of which serve as co-substrates in these enzyme-catalyzed transformations. For example, histone acetyltransferases (HATs) require sufficient availability of acetyl coenzyme A (acetyl-CoA), a key metabolite at the intersection of catabolic and anabolic metabolism and the major acetyl donor in cells (Fig. 1) (4). In this manner, acetyl-CoA and numerous other endogenous metabolites exert known regulatory roles on histone- and DNA-modifying enzymes.
Figure 1.
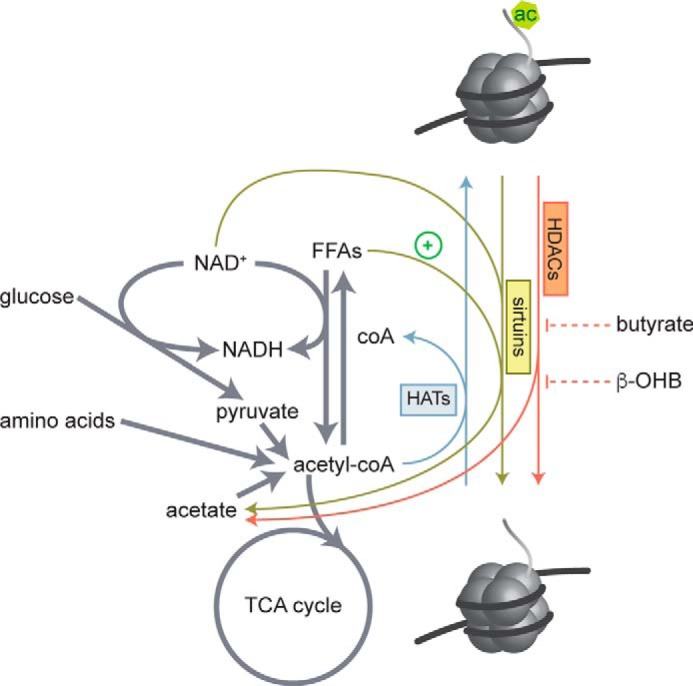
Regulation of histone acetylation by metabolites. HAT enzymes use acetyl-CoA as a necessary co-substrate for histone acetylation and produce CoA. Acetyl-CoA pools are fed by oxidation of free fatty acids (FFAs), glucose, and degradation of amino acids. HDAC enzymes hydrolyze acetyl groups from histone lysine residues and produce acetate. The class III HDACs, sirtuins, require NAD+ as a necessary co-substrate and produce NADH and acetate. Sirt6 is also activated by long chain free fatty acids. Class I, IIa, IIb, and IV sirtuins do not require NAD+ but are inhibited by butyrate and the ketone body β-hydroxybutyrate (β-OHB).
Here we will refer to endogenous metabolites as those generated by the mammalian host (e.g. mouse and human). A major source of metabolic diversity is encoded in the genomes of microbes that colonize the gut of mammals. These communities have co-evolved with their hosts (5) and interact with dietary components and host-derived molecules to produce a myriad of metabolites that are measurable in host circulation and tissues and can modulate physiology and behavior (6, 7). For example, butyrate, a major product of gut microbial fermentation of undigested complex carbohydrates, has been known as a histone deacetylase inhibitor since the 1970s (8). The relationship between SCFAs and a number of other microbial metabolites with host chromatin is detailed in Table 1. In light of these relationships, the gut microbiota may be a key regulator of host metabolo-epigenetic events. The gut microbiota has also been implicated in a number of host metabolic and immunological etiologies (9, 10).
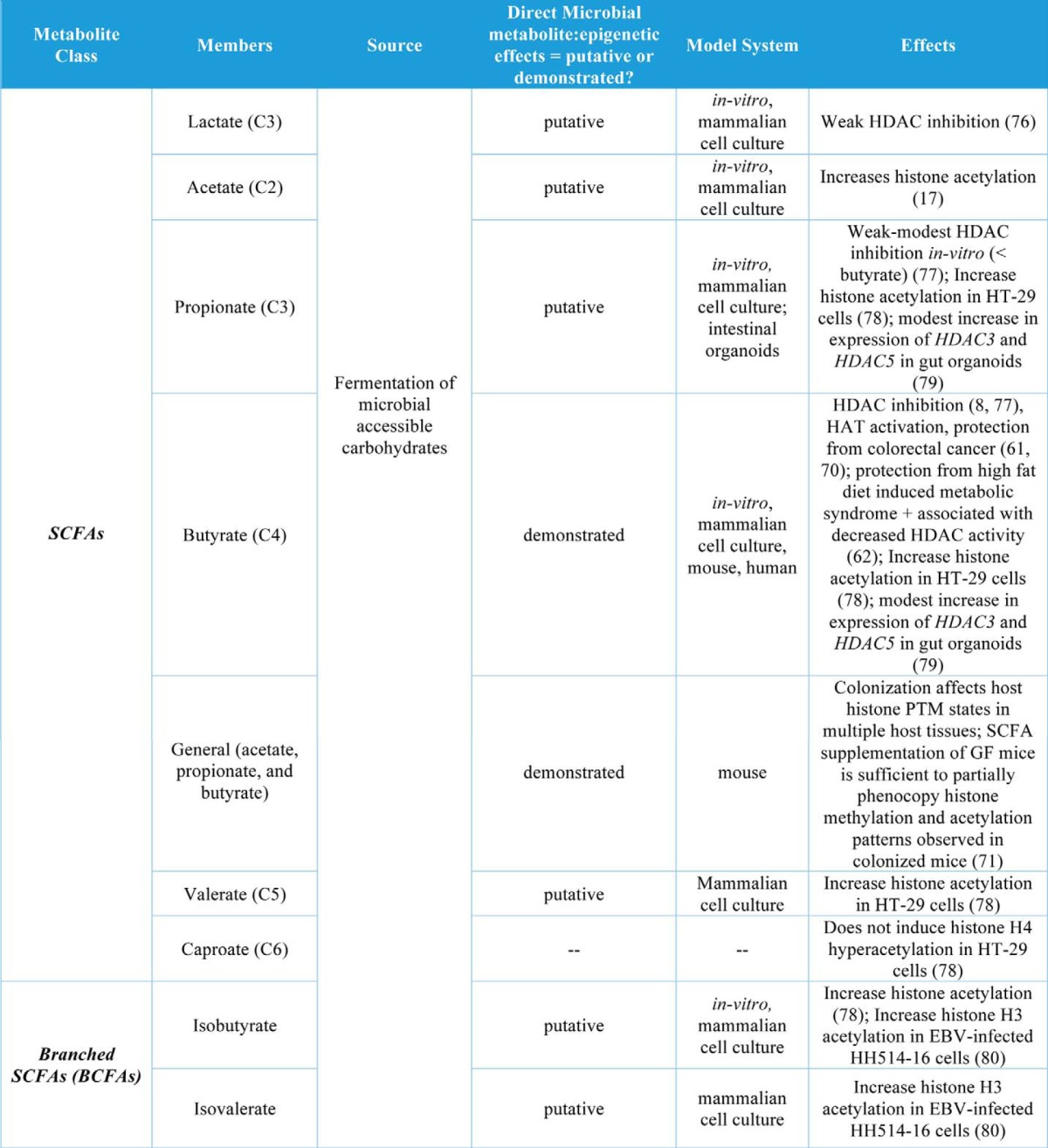
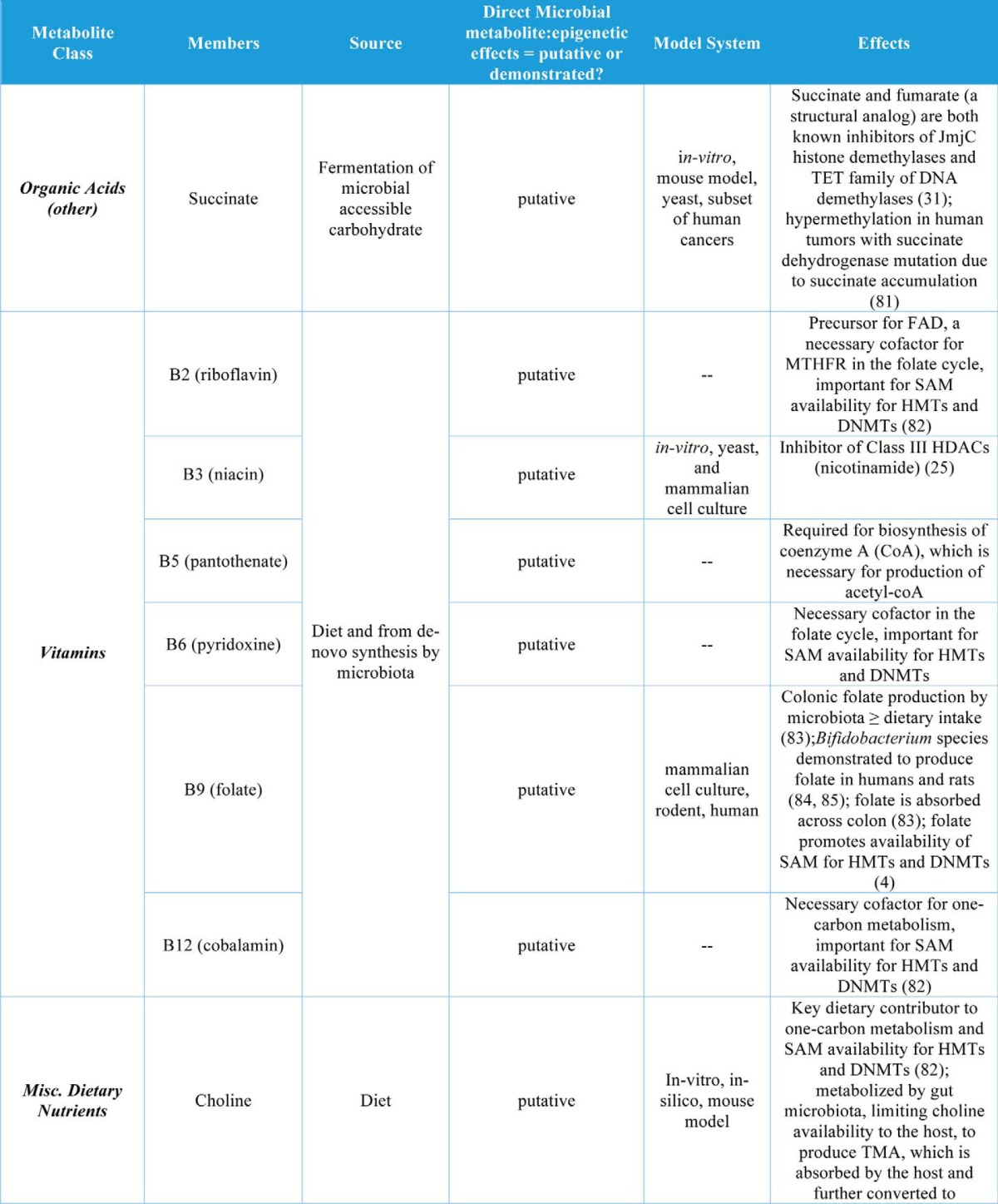
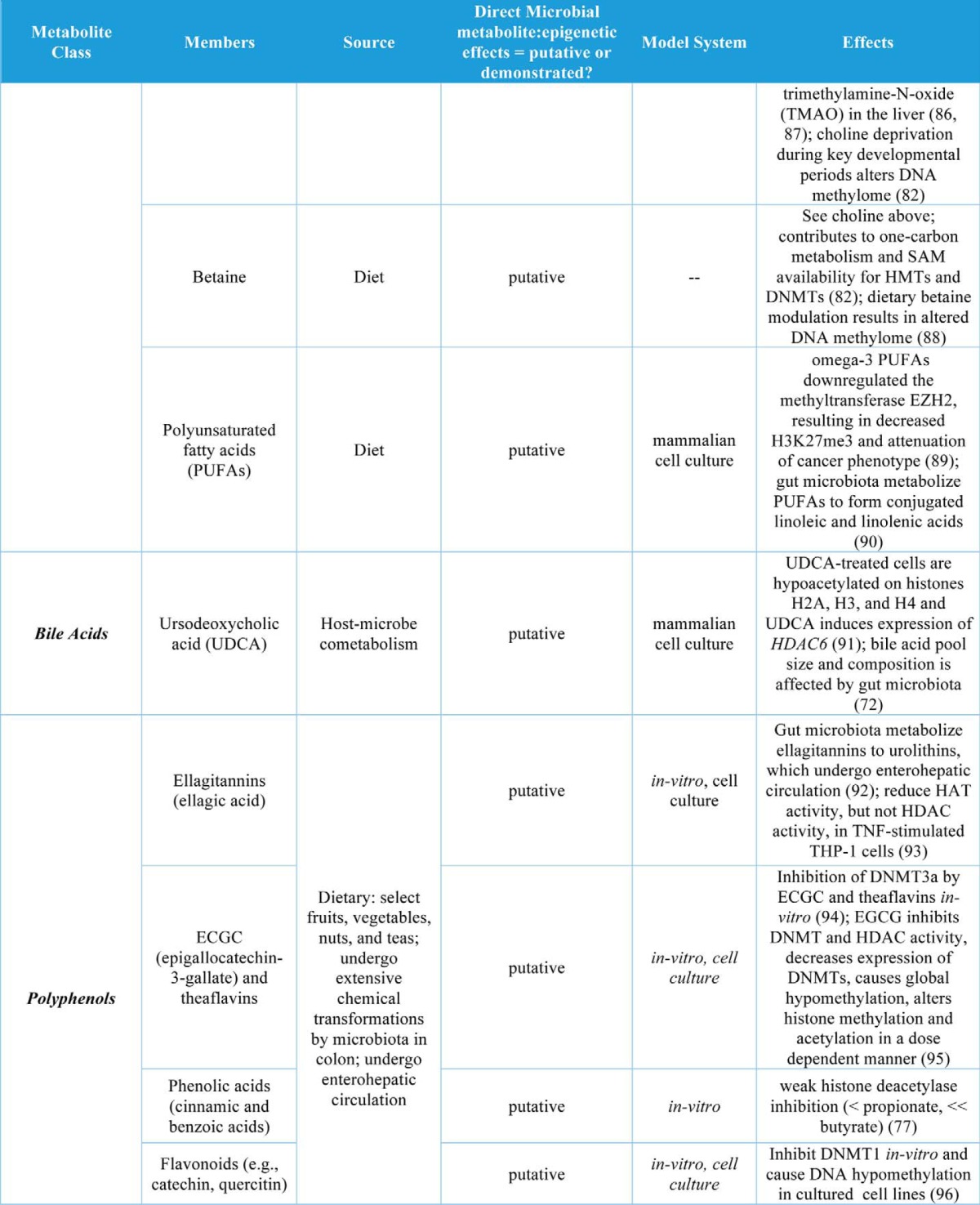
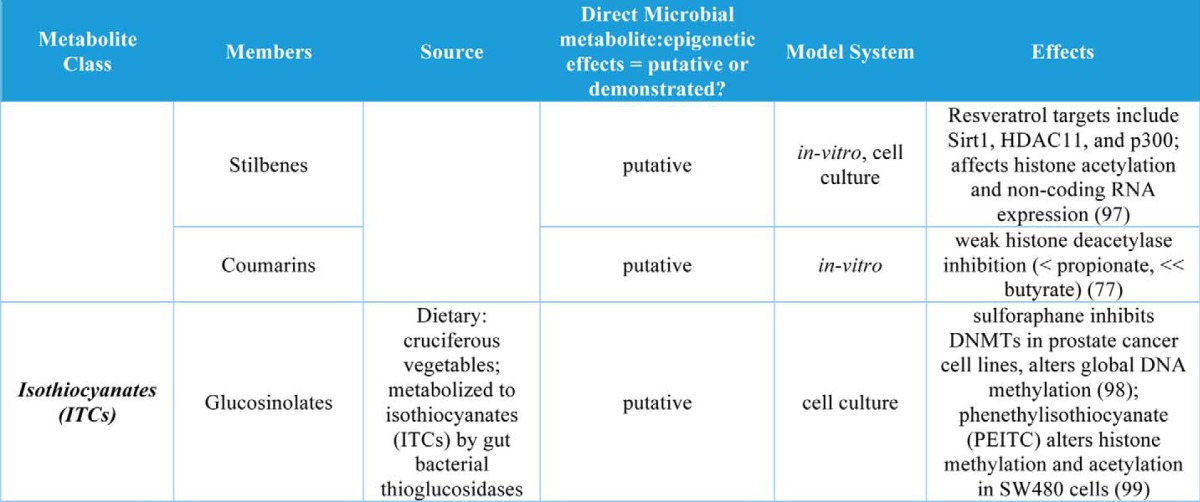
Table 1.
Gut microbial metabolites and their roles in regulation of chromatin states
Gut microbial communities and their hosts communicate via chemical signals in the form of small molecule metabolites and signaling molecules like LPS and peptides (7). Given its sensitivity to metabolite availability, a significant portion of this chemical communication may take place at the level of the host epigenome. Thus, the microbiota may not only exert transient effects on host phenotypes, but also “program” lasting and even multigenerational outcomes. Here we focus on recent literature surrounding metabolic regulation of host chromatin states and how this intersects with what is currently known about the gut microbiota, its co-metabolism of substrates with the host, and their chemical communication with one another. Although we could not exhaustively cover the literature for the vast fields of epigenetics and gut microbiota-host interactions, we provide references where it is possible to direct readers to more extensive coverage of specific topics.
Histone PTM states: Regulation by endogenous metabolites
The eukaryotic genome is compressed by a factor of >10,000 into the highly structured and organized nucleoprotein complex known as chromatin. The fundamental unit of chromatin is the nucleosome, which is composed of a hetero-octamer of core histone proteins (two copies each of histone H2A, H2B, H3, and H4) wrapped by ∼146 bp of double-stranded genomic DNA. Histones are small, highly basic, and globular proteins with flexible N-terminal tails. The N-terminal tails are subject to a multitude of covalent PTMs, the most abundant and well-studied of which are acetylation, methylation, and phosphorylation (4, 11). The modification state of histone proteins affects chromatin structure and thereby any process requiring physical access to the DNA itself. This includes transcription and DNA repair, recombination, and replication.
Histone acetylation is generally associated with open chromatin states and active transcription. Acetylation leads to charge neutralization of lysine residues, affecting electrostatic interactions between DNA and residues within histone octamers. Acetylated residues also serve as binding sites for other factors that can play a role in transcriptional activation, including histone-modifying and chromatin-remodeling enzymes, as well as transcription factors (12). Histone methylation is associated with both transcriptional activation and silencing, depending on both the location and the degree of methylation of a particular lysine residue on the histone tail. For example, trimethylation at histone H3 lysine 4 (H3K4me3) is found at active or poised promoters (13), whereas H3K4me1 is typically associated with enhancers (14). In contrast, H3K27me3 is located in areas of closed chromatin or transcriptionally silenced genes (15). Similar to histone acetylation, methylated residues also serve as binding sites for a number of regulatory factors (16). Thus, histone PTMs create what has been termed the “histone code,” which consists of combinatorial histone PTM states that serve as both a signal integration platform for diverse environmental signals and landing platform for other effectors.
Histone-modifying enzymes are sensitive to levels of endogenous small molecule metabolites, with some serving as co-substrates while others act as inhibitors. The Km or Ki values of many of these enzymes for their substrates or inhibitors, respectively, are often higher than measured or calculated levels of key metabolites, opening the possibility that fluctuations in these metabolites may regulate enzyme activities. The relationship between metabolism and histone-modifying enzymes and their kinetic parameters has been reviewed thoroughly in Ref. 4. Here we focus on histone acetylation and methylation, as the most common and well-studied histone PTMs in relation to metabolism. The interplay between endogenous metabolites and histone and DNA modification is depicted in Figs. 1–3.
Figure 2.
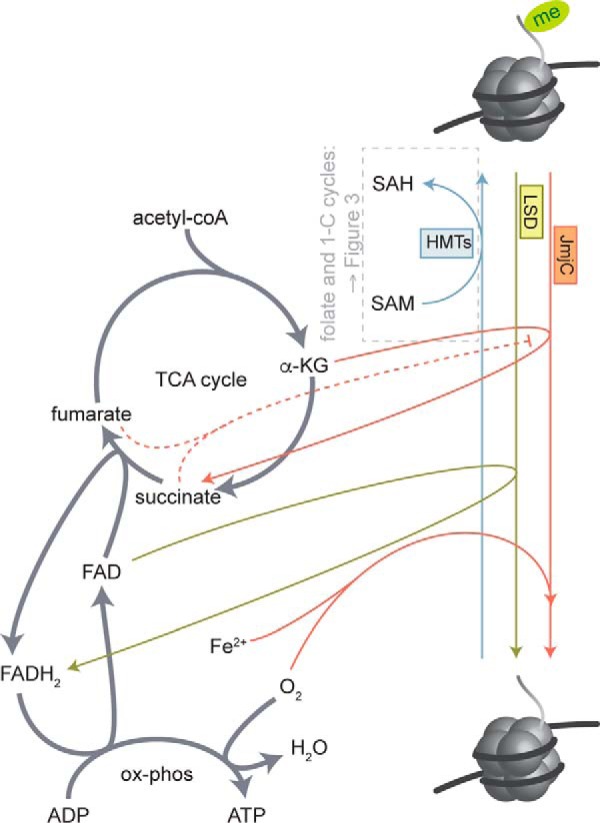
Regulation of histone methylation by central carbon and one-carbon metabolites. HMT enzymes require SAM as a methyl group donor and produce SAH. The relationship between folate and one-carbon metabolism and HMT and DNMT activity is further detailed in Fig. 3. Histone demethylases are regulated by central carbon metabolites and carry out a redox reaction to remove methyl groups from histone lysine residues, producing formaldehyde. The LSD family of demethylases require FAD as an electron acceptor, producing FADH2, whereas the JmjC family uses α-KG as a co-substrate and requires both oxygen and iron. The TCA cycle intermediates succinate and fumarate inhibit JmjC family demethylases.
Figure 3.

Regulation of HMTs and DNMTs by SAM availability via folate and one-carbon metabolism. Dietary contributors are denoted by green arrows. Active one-carbon groups are generated via amino acid- and vitamin-dependent reactions in the folate cycle. These one-carbon groups are then used by methionine synthase (MTR) to generate methionine from homocysteine. Methionine is then adenylated to form SAM via methionine adenosyltransferase (MAT). SAM is used as a methyl donor by both HMTs and DNMTs, producing SAH. SAH is then converted to homocysteine, which can be converted back to methionine via a reaction that uses carbons from choline and produces dimethylglycine (DMG). MTHFR, methylenetetrahydrofolate reductase; SHMT, serine hydroxymethyltransferase; THF, tetrahydrofolate.
Histone acetylation is the result of dynamic balance between the activities of HATs and histone deacetylases (HDACs). HATs catalyze the transfer of an acetyl group from acetyl-CoA onto the ϵ-amino group of lysine residues, releasing coenzyme A (CoA). Notably, coenzyme A acts as a competitive inhibitor of HATs. Acetyl-CoA also serves as a hub for central carbon metabolism with roles in catabolic, anabolic, and energy-producing pathways. Given its dual role as a necessary substrate for HAT enzymes and a central metabolite, acetyl-CoA is a rheostat that communicates cellular metabolic states to chromatin, ultimately regulating transcriptional programming. Cellular concentrations of acetyl-CoA are reported to be 2–20 μm, which is above the Km value for the HATs GCN5 and P/CAF but near the Km value of p300 (4).
The subcellular compartmentalization of metabolic reactions is important to consider in the context of metabolite-driven regulation of histone PTMs. Acetyl-CoA is produced by a number of cytosolic and mitochondrial reactions. It can be made directly from acetate by acetyl-CoA synthetase 1 and 2 (AceCS1 and -2) in the cytosol and mitochondria, respectively. In mitochondria, acetyl-CoA is also produced via β-oxidation of fatty acids and oxidative decarboxylation of pyruvate by the pyruvate dehydrogenase complex (PDC). Mitochondrial acetyl-CoA condenses with oxaloacetate (OAA) to form citrate, which can be shuttled into the cytosol and converted back into acetyl-CoA and OAA by ATP citrate lyase (ACLY). ACLY has been demonstrated to be essential for histone acetylation in response to glucose in mammalian cells; however, supplementation with 1–5 mm acetate partially rescued histone acetylation in the setting of ACLY knockdown (17). Interestingly, both ACLY and PDC have been reported to localize to the nucleus in mammalian cells in response to growth stimuli and in concordance with increased histone acetylation and acetyl-CoA pools (demonstrated for PDC only) (17, 18). AceCS1 has also been demonstrated to be present in the nucleus (17), although its role in histone acetylation in cultured mammalian cells appears to be secondary to ACLY. The fact that these enzymes can translocate to the nucleus suggests that, beyond subcellular compartmentalization, metabolite availability may also be regulated at the level of subnuclear microenvironments and perhaps direct channeling from one enzyme to the other. Additional evidence for this link between cellular metabolism and histone acetylation comes from studies in yeast, where yeast metabolic cycles are associated with histone acetylation and regulation of growth-related genes (19).
Mammalian HDACs are organized into four classes, depending on their homology to yeast orthologues and their factor dependence: class I, IIa, IIb, and IV are all zinc-dependent deacetylases and are generally inhibited by hydroxamic acid inhibitors, including TSA (trichostatin A) and SAHA (suberoylanilide hydroxamic acid, Vorinostat), which chelate the active-site zinc. These metal-dependent HDACs catalyze the hydrolysis of acetyl groups from acetyl-lysine residues, producing acetate and a deacetylated substrate. The class III HDACs, also known as sirtuins, are structurally distinct from other classes of HDAC. Sirtuins require NAD+ as a necessary co-substrate and produce nicotinamide, O-acetyl-ADP-ribose, and the deacetylated substrate. There are seven mammalian sirtuins (Sirt1–7), which share a conserved NAD+-binding site and catalytic domain but diverge in their biological roles due to differences in subcellular localization, tissue expression, and substrate specificity (20). Of the sirtuins, Sirt1 and Sirt6 are localized to the nucleus, whereas Sirt7 is found in the nucleolus, and Sirt2–5 are either mitochondrial or cytosolic. The discovery that yeast orthologue Sir2 (silent information regulator 2) was regulated by NAD+ availability was one of the first reports of a small molecule metabolite regulating chromatin modifications (21). More recently, the histone H3 Lys-9 and Lys-56 deacetylase Sirt6, which has inherently low deacetylase activity in vitro, was reported to be activated by long-chain free fatty acids (20); however, whether these long-chain free fatty acids have a role in vivo remains to be determined.
NAD+/NADH is one of the most important redox coenzymes found in living cells. It plays a role in both catabolic and oxidative pathways, including glycolysis, the TCA cycle, and oxidation of fatty acids. In addition to sirtuins, two other nuclear enzymes may be significant consumers of NAD+: poly(ADP-ribose) polymerase (PARP) and CD38. PARP-1 is activated in response to genotoxic stress and is known to induce a caspase-independent form of cell death termed “parthanatos” (22), which was thought to be caused by excessive consumption of NAD+ and bioenergetic collapse. However, it has recently been shown that the resulting bioenergetic collapse is not dependent upon NAD+ depletion, but rather is due to inhibition of hexokinase and subsequent glycolytic defects by poly(ADP-ribose), a product of the PARP-1 reaction (23). Although precise measurement of subcellular NAD+ has been limited by technical challenges and the fact that the majority of NAD+ is protein-bound, nuclear NAD+ has been estimated to be ∼70–109 μm, which is approximately at or below the Km value of yeast Sir2 (∼100 μm) and mammalian Sirt1 (∼150–170 μm) but not Sirt6 (Kd = 27 μm), which can bind NAD+ in the absence of a peptide substrate, suggesting it exists in a poised state (4, 24). NAD+ and its role in metabolic signaling have been thoroughly reviewed in Ref. 25.
Histone methylation is balanced by the activities of histone methyltransferases (HMTs) and histone demethylases. Regulation of histone methylation by central carbon and one-carbon metabolites is depicted in Figs. 2 and 3, respectively. Histone methyltransferases fall into one of three major families of enzymes, all of which catalyze the addition of a methyl group to the ϵ-amino group of lysine residues or the guanidinyl group of arginine residues: 1) SET domain-containing enzymes; 2) DOT1-like enzymes, which methylate lysine residues; and 3) the protein arginine N-methyltransferase family of enzymes that methylate arginines. Although these enzymes have different target specificities, mechanisms, and kinetic properties, all known histone methyltransferases use S-adenosylmethionine (SAM, also known as AdoMet) as a donor and release S-adenosylhomocysteine (SAH, also known as AdoHcy) as a product. Methylation has also been reported to occur on histidine, cysteine, aspartate, and glutamate residues, although these modifications are much more rare and the biological significance remains to be determined. Methylation of lysine residues is the predominant form of histone methylation and exists in mono-, di-, and tri-methyl forms. As such, we will limit our focus here to methylation of lysine residues and its regulation by small molecule metabolites. Histone methylation and biological roles have been thoroughly reviewed in Ref. 16.
HMTs are regulated by the availability of the methyl donor SAM. This essential co-substrate is synthesized via the one-carbon cycle (also known as the methionine cycle), which utilizes methyl groups derived from dietary folate in the folate cycle (Fig. 3). These two cycles intersect at the vitamin B12-dependent enzyme 5-methyltetrahydrofolate homocysteine methyltransferase (methionine synthase, MTR), where a one-carbon unit from the folate cycle is used to convert homocysteine to methionine. Methionine adenosyltransferase then catalyzes the formation of SAM from methionine and ATP. This reaction is conserved across all domains of life (26). SAM can then be used as a methyl donor by both HMTs and DNA methyltransferases (DNMTs). There are a number of dietary contributors for these pathways, including serine, glycine, choline, betaine, B-vitamins, methionine, and folate (27). SAM availability is also regulated by a number of other factors, including dietary fat intake, alcohol consumption, and oxidative stress (4). In yeast, folate and methionine restriction reduced histone H3K4 di- and trimethylation, which is deposited by the Set1 HMT, and associated gene expression, but H3K79 methylation, which is deposited by Dot1, was not significantly altered (28). To test whether this was due to differences in enzyme sensitivity to nutrient restriction, Sadhu et al. (28) subjected a strain of yeast expressing hypomorphic Dot1 to folate restriction. These G401A mutants have decreased Dot1 activity relative to the wild type, and the mutation is predicted to be near the SAM-binding site. Although wild-type Dot1 was not affected by folate restriction, the hypomorphic mutant was affected. This suggests that HMTs have varying sensitivities to nutrient restriction that are likely due to differences in Km.
In mammalian cells, SAM concentrations are reported to be 10–100 μm. This is slightly above the measured Km values for both SET domain-containing and non-SET domain-containing HMTs; however, SAH also competitively inhibits SAM binding to HMTs. SAH concentrations are reported to be 0.1–20 μm, which is within the Ki value of both SET and non-SET domain-containing HMTs (4). Thus, it is possible that the ratio of SAM/SAH, which differs by cell type and environmental conditions, is a biologically relevant measure of enzyme activity.
Histone demethylases are also closely coupled to cellular metabolic state (Fig. 2). There are two main classes: the FAD-dependent lysine-specific demethylase (LSD) family demethylases and the α-ketoglutarate (α-KG)-dependent JmjC family demethylases. Using different oxidizing agents, both families carry out a redox reaction to remove the methyl group from histone lysine residues, producing formaldehyde. The LSD family of demethylases uses FAD as an electron acceptor, generating FADH2 (29), whereas the iron-dependent JmjC family uses oxygen and α-KG and generates CO2 and succinate (30). Interestingly, mutation of fumarate hydratase and succinate dehydrogenase in a subset of human cancers leads to accumulation of fumarate and succinate, respectively, both of which have been demonstrated to inhibit the α-KG-dependent JmjC family of histone demethylases, causes aberrant histone and DNA methylation (31, 32).
Iron availability has also recently been demonstrated to affect histone PTM states in mouse myoblast cells, wherein pharmacological iron chelation resulted in reversible increases in histone methylation at JmjC target sites (33). Finally, hypoxia, which is a hallmark of a number of inflammatory conditions and tumor microenvironments, has also been shown to affect histone methylation via inhibition of oxygen-dependent JmjC family demethylases (34, 35). Thus, in a manner similar to acetyl-CoA, these key central carbon metabolites serve as TCA cycle intermediates (α-KG, succinate, fumarate, and FAD), play roles in other oxidative processes such as β-oxidation of fatty acids and oxidative phosphorylation (FAD) and amino acid metabolism (α-KG), and signal cellular metabolic status to chromatin.
Cross-talk between DNA methylation, histone modification, and metabolites
DNA methylation occurs mainly at CpG residues in the genome, and ∼60–80% of the mammalian genome is methylated; however, in regions of active chromatin only ∼10% of CG sites are methylated (36, 37). DNA methylation is associated with repressed transcription and closed chromatin. Some cross-talk between histone modification and DNA methylation has been established, particularly between H3K9 methylation and DNA methylation in the fungi Neurospora crassa, although it remains unclear which is the causative event (38, 39). Regardless, DNMTs have the same requirements for the methyl donor SAM as HMTs and thus are also regulated by nutrient availability and cellular metabolic state, as shown in Fig. 3. For example, both in utero and early life adversity have been shown to affect DNA methylation, in some cases affecting multiple generations (2, 3). Recently, fumarate has also been shown to drive the epithelial to mesenchymal transition, a key step in tumor invasion and metastasis, by inhibiting TET (ten eleven translocation)-mediated demethylation of an anti-metastatic miRNA cluster (32). Thus, similar to histone methylation, both one-carbon and central metabolites can chemically signal to DNA methylation machinery.
Histone PTM states: Regulation by gut microbial metabolites
The gut microbiota produces a large number and variety of bioactive metabolites (6, 7), including both demonstrated and putative regulators of host chromatin as follows: SCFAs, vitamins, bile acids, and compounds derived from metabolism of dietary components, including polyphenols, isothiocyanates, and choline (Table 1). Gut microbial community composition affects metabolic outcomes; for example, the number of genes within a gut microbiome (richness) correlates with metabolic biomarkers (40). Furthermore, dietary intervention has been shown to improve low gene richness and subsequent clinical phenotypes (41). In a small human cohort study, consumption of either an entirely animal- or plant-based diet resulted in alterations in microbial diversity within 1 day of consumption of the altered diet, and consumption of the animal-based diet increased the abundance and activity of Bilophila wadsworthia (42), which has been associated with inflammatory bowel disease (43). Dietary additives common to Westernized human diets cause gut dysbiosis and contribute to metabolic syndrome (44) and gut inflammation (45). Although host genetics have been shown to play a role in shaping gut microbial community composition and metabolism, the effects of diet and environment have been shown to exert broader effects (46, 47).
Although the gut microbiota is necessary for proper immune system and brain development (48, 49), several studies have shown that it contributes to a number of etiologies, including metabolic syndrome and diabetes mellitus (50, 51), obesity and adiposity (52, 53), cardiovascular disease (54, 55), non-alcoholic fatty liver disease (56), inflammatory bowel disease (57), and colon cancer (58). Furthermore, changes in microbiota composition caused by antibiotic exposure early in life affect the gut microbiota and elicit long-lasting effects on host metabolic outcomes (59, 60). Notably, the gut microbiota is also associated with therapeutic effects (61, 62).
There are a number of interesting and putative connections between microbial-host metabolic axes and chromatin regulatory events; however, most of these studies have provided only indirect evidence or used cell culture-based models rather than whole organisms. For example, it is well established that early life adversity affects DNA methylation (63). This is a critical time in life when the microbiome is assembled (64, 65). Interestingly, adverse events during this key developmental period (either in utero or during early life) have been shown to impact both chromatin and the gut microbiota; however, with the exception of microbial production of butyrate in the setting of colon cancer (61), these two have not yet been directly linked. Furthermore, natural seasonal variation in nutrient availability has been linked to alterations in both chromatin states and the microbiota (2, 66). Similar effects have also been separately reported for high fat diet feeding on chromatin and the microbiota (67, 68). Even more intriguingly, Sonnenburg et al. (69) recently showed that diet-microbiota interactions may reprogram transgenerational susceptibility to metabolic disease. Although consumption of a diet low in microbial accessible carbohydrates (MACs) induces largely reversible effects on the gut microbiota within a single generation, continued feeding of a low MACs diet results in loss of microbial taxa that are at increased risk for extinction with each subsequent generation. Thus, key links exist between microbiota-dependent transgenerational effects and potential metabolic effects associated with consumption of a typical Westernized diet (which is low in MACs). Although these effects were not linked to chromatin states, this study presents the intriguing possibility that perhaps transgenerational inheritance in response to nutrient availability is mediated via gut microbiota-host epigenetic responses.
A large number of microbial metabolites have been measured in host circulation and other tissue compartments largely via NMR and mass spectrometry (6, 7, 67). These studies highlight the extensive co-metabolism that occurs between the gut microbiota and host. Table 1 details demonstrated and putative interactions between host epigenetic machinery, gut microbial metabolites, and host-gut microbial co-metabolites. Of note, the SCFAs acetate, propionate, and butyrate are the only examples for which a direct link between microbial metabolite production and host epigenetic programming has been made in a whole organism. Donohoe et al. (61, 70) demonstrate a key link between gut microbial fermentation of dietary fiber and both normal maintenance of healthy colonic epithelium and attenuation of colon cancer via butyrate-mediated effects on histone acetylation and gene expression. Recently, we have also demonstrated that the gut microbiota affects global histone methylation and acetylation and that these effects can be partially mimicked in GF mice that are supplemented with a mixture of acetate, propionate, and butyrate (71). To our knowledge, all other relevant studies have either used purely in vitro methods or involved treatment of cell culture-based models with known microbial metabolites. Thus, there remains enormous potential for discovery, which links commonly available foodstuffs to epigenetic programming in health and disease.
In addition to butyrate, other organic acids (C1, C2, C3, and C5 and branched SCFAs) have been demonstrated to increase histone acetylation, inhibit HDACs, or increase expression of HDACs in cell culture models (Table 1). The organic acid succinate has also been demonstrated to inhibit both histone and DNA demethylases (Table 1). Another major group of co-metabolites are B-vitamins, which are both derived from diet and synthesized de novo by gut bacteria. Vitamins B2, B6, B9, and B12 all play roles in SAM availability and thus may affect histone and DNA methylation, whereas vitamins B3 and B5 may affect histone acetylation via sirtuin inhibition or HAT activation, respectively (Table 1). Other dietary nutrients, including choline, betaine, and polyunsaturated fatty acids (PUFAs), may play roles in histone methylation (Table 1).
Bile acids are regulators of gut microbial community composition and are also regulated by the gut microbiota via microbial production of secondary bile acids that mediate both bile acid pool size and composition (72). The human secondary bile acid ursodeoxycholic acid (a primary bile acid in mice) also induces expression of HDAC6 and decreases global histone acetylation in cultured cells (Table 1). Finally, two classes of phytonutrients that are metabolized by the gut microbiota to bioactive compounds have putative roles in host epigenetic regulation. Both polyphenol metabolites and glucosinolates are derived from plants (select fruits, vegetables, nuts, and teas) and are metabolized by the gut microbiota to form bioactive compounds that may regulate host chromatin at the level of methylation and acetylation of histones as well as DNA methylation (Table 1).
Although gut microbial derivatives of dietary isothiocyanates and polyphenols are potential regulators of host epigenetic machinery, their bioavailability is somewhat limited, and thus future studies will need to determine the relevance of these metabolites in this setting. Interestingly, organic acid production in the distal gut is associated with a decrease in pH (73). This is particularly intriguing within the context of work by McBrian et al. (74), wherein histones are globally deacetylated as extracellular pH decreases and hydrolyzed histone acetate anions are exported with protons as a means to regulate intracellular pH. This suggests that organic acid production in the colon may promote decreased histone acetylation; however, this is at odds with reports of SCFA-driven increases in histone acetylation. Perhaps organic acid-driven effects on pH and on HDACs/HATs are cell type- and tissue-specific.
Conclusions
Much of the extensive chemical communication that occurs between the gut microbiota and host may occur through chromatin-mediated mechanisms. A number of microbial metabolites exert physiological effects via cellular signaling pathways and can even exert effects via multitissue signaling, as demonstrated for glucose homeostasis via gut-brain neural circuits (75). These signaling effects need not be mutually exclusive from chromatin effects, however, emphasizing the importance of elucidating chromatin effects in response to the multitude of gut microbial metabolites. Furthermore, as both proteomic and metabolomic methodologies continue to improve, the identification of novel metabolite-epigenome interactions will drive further exploration of this exciting interaction between the host and its microbial symbionts, undoubtedly yielding key insights into how the microbiota modulates the health of the host.
Acknowledgments
We thank the members of the Epigenetics Theme at the Wisconsin Institute for Discovery, members of the UW-Madison Microbiota and Health Interest Group, and others on and around campus, including Dr. Hannah Carey and Emily Meier for their continued support and inspiring discussions. We apologize for any omission of relevant publications due to space limitations.
This is the sixth article in the Host-Microbiome Metabolic Interplay Minireview series. J. M. D. consults for BioTechne and FORGE Life Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PTM
- post-translational modification
- HAT
- histone acetyltransferase
- HDAC
- histone deacetylase
- HMT
- histone methyltransferase
- SAM
- S-adenosylmethionine
- SAH
- S-adenosylhomocysteine
- LSD
- lysine-specific demethylase
- α-KG
- α-ketoglutarate
- DNMT
- DNA methyltransferase
- MAC
- microbial accessible carbohydrate
- SCFA
- short-chain fatty acid
- PDC
- pyruvate dehydrogenase complex
- OAA
- oxaloacetate
- ACLY
- ATP citrate lyase
- TCA
- tricarboxylic acid.
References
- 1. Evertts A. G., Zee B. M., Dimaggio P. A., Gonzales-Cope M., Coller H. A., and Garcia B. A. (2013) Quantitative dynamics of the link between cellular metabolism and histone acetylation. J. Biol. Chem. 288, 12142–12151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dominguez-Salas P., Moore S. E., Baker M. S., Bergen A. W., Cox S. E., Dyer R. A., Fulford A. J., Guan Y., Laritsky E., Silver M. J., Swan G. E., Zeisel S. H., Innis S. M., Waterland R. A., Prentice A. M., and Hennig B. J. (2014) Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat. Commun. 5, 3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Radford E. J., Ito M., Shi H., Corish J. A., Yamazawa K., Isganaitis E., Seisenberger S., Hore T. A., Reik W., Erkek S., Peters A. H., Patti M.-E., and Ferguson-Smith A. C. (2014) In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science 345, 1255903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fan J., Krautkramer K. A., Feldman J. L., and Denu J. M. (2015) Metabolic regulation of histone post-translational modifications. ACS Chem. Biol. 10, 95–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ley R. E., Hamady M., Lozupone C., Turnbaugh P. J., Ramey R. R., Bircher J. S., Schlegel M. L., Tucker T. A., Schrenzel M. D., Knight R., and Gordon J. I. (2008) Evolution of mammals and their gut microbes. Science 320, 1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wikoff W. R., Anfora A. T., Liu J., Schultz P. G., Lesley S. A., Peters E. C., and Siuzdak G. (2009) Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. U.S.A. 106, 3698–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nicholson J. K., Holmes E., Kinross J., Burcelin R., Gibson G., Jia W., and Pettersson S. (2012) Host-gut microbiota metabolic interactions. Science 336, 1262–1267 [DOI] [PubMed] [Google Scholar]
- 8. Riggs M. G., Whittaker R. G., Neumann J. R., and Ingram V. M. (1977) n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 268, 462–464 [DOI] [PubMed] [Google Scholar]
- 9. Sonnenburg J. L., and Bäckhed F. (2016) Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Belkaid Y., and Hand T. W. (2014) Role of the microbiota in immunity and inflammation. Cell 157, 121–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao Y., and Garcia B. A. (2015) Comprehensive catalog of currently documented histone modifications. Cold Spring Harb. Perspect. Biol. 7, a025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson D. G., and Dent S. Y. (2013) Chromatin: receiver and quarterback for cellular signals. Cell 152, 685–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Santos-Rosa H., Schneider R., Bannister A. J., Sherriff J., Bernstein B. E., Emre N. C., Schreiber S. L., Mellor J., and Kouzarides T. (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419, 407–411 [DOI] [PubMed] [Google Scholar]
- 14. Heintzman N. D., Stuart R. K., Hon G., Fu Y., Ching C. W., Hawkins R. D., Barrera L. O., Van Calcar S., Qu C., Ching K. A., Wang W., Weng Z., Green R. D., Crawford G. E., and Ren B. (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318 [DOI] [PubMed] [Google Scholar]
- 15. Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R. S., and Zhang Y. (2002) Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 16. Greer E. L., and Shi Y. (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wellen K. E., Hatzivassiliou G., Sachdeva U. M., Bui T. V., Cross J. R., and Thompson C. B. (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sutendra G., Kinnaird A., Dromparis P., Paulin R., Stenson T. H., Haromy A., Hashimoto K., Zhang N., Flaim E., and Michelakis E. D. (2014) A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 158, 84–97 [DOI] [PubMed] [Google Scholar]
- 19. Cai L., Sutter B. M., Li B., and Tu B. P. (2011) Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 42, 426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feldman J. L., Baeza J., and Denu J. M. (2013) Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 288, 31350–31356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Imai S., Armstrong C. M., Kaeberlein M., and Guarente L. (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 [DOI] [PubMed] [Google Scholar]
- 22. Fatokun A. A., Dawson V. L., and Dawson T. M. (2014) Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 171, 2000–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andrabi S. A., Umanah G. K., Chang C., Stevens D. A., Karuppagounder S. S., Gagné J.-P., Poirier G. G., Dawson V. L., and Dawson T. M. (2014) Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. U.S.A. 111, 10209–10214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cambronne X. A., Stewart M. L., Kim D., Jones-Brunette A. M., Morgan R. K., Farrens D. L., Cohen M. S., and Goodman R. H. (2016) Biosensor reveals multiple sources for mitochondrial NAD+. Science 352, 1474–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Houtkooper R. H., Cantó C., Wanders R. J., and Auwerx J. (2010) The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 31, 194–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thomas D., and Surdin-Kerjan Y. (1991) The synthesis of the two S-adenosyl-methionine synthetases is differently regulated in Saccharomyces cerevisiae. Mol. Gen. Genet. 226, 224–232 [DOI] [PubMed] [Google Scholar]
- 27. Ducker G. S., and Rabinowitz J. D. (2017) One-carbon metabolism in health and disease. Cell Metab. 25, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sadhu M. J., Guan Q., Li F., Sales-Lee J., Iavarone A. T., Hammond M. C., Cande W. Z., and Rine J. (2013) Nutritional control of epigenetic processes in yeast and human cells. Genetics 195, 831–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Forneris F., Binda C., Vanoni M. A., Mattevi A., and Battaglioli E. (2005) Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 579, 2203–2207 [DOI] [PubMed] [Google Scholar]
- 30. Cascella B., and Mirica L. M. (2012) Kinetic analysis of iron-dependent histone demethylases: α-ketoglutarate substrate inhibition and potential relevance to the regulation of histone demethylation in cancer cells. Biochemistry 51, 8699–8701 [DOI] [PubMed] [Google Scholar]
- 31. Xiao M., Yang H., Xu W., Ma S., Lin H., Zhu H., Liu L., Liu Y., Yang C., Xu Y., Zhao S., Ye D., Xiong Y., and Guan K. L. (2012) Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 26, 1326–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sciacovelli M., Gonçalves E., Johnson T. I., Zecchini V. R., da Costa A. S., Gaude E., Drubbel A. V., Theobald S. J., Abbo S. R., Tran M. G., Rajeeve V., Cardaci S., Foster S., Yun H., Cutillas P., et al. (2016) Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rensvold J. W., Krautkramer K. A., Dowell J. A., Denu J. M., and Pagliarini D. J. (2016) Iron deprivation induces transcriptional regulation of mitochondrial biogenesis. J. Biol. Chem. 291, 20827–20837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou X., Sun H., Chen H., Zavadil J., Kluz T., Arita A., and Costa M. (2010) Hypoxia induces trimethylated H3 lysine 4 by inhibition of JARID1A demethylase. Cancer Res. 70, 4214–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen H., Yan Y., Davidson T. L., Shinkai Y., and Costa M. (2006) Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer Res. 66, 9009–9016 [DOI] [PubMed] [Google Scholar]
- 36. Stadler M. B., Murr R., Burger L., Ivanek R., Lienert F., Schöler A., van Nimwegen E., Wirbelauer C., Oakeley E. J., Gaidatzis D., Tiwari V. K., and Schübeler D. (2011) DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495 [DOI] [PubMed] [Google Scholar]
- 37. Lister R., Pelizzola M., Dowen R. H., Hawkins R. D., Hon G., Tonti-Filippini J., Nery J. R., Lee L., Ye Z., Ngo Q.-M., Edsall L., Antosiewicz-Bourget J., Stewart R., Ruotti V., Millar A. H., Thomson J. A., Ren B., and Ecker J. R. (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Du J., and Patel D. J. (2014) Structural biology-based insights into combinatorial readout and crosstalk among epigenetic marks. Biochim. Biophys. Acta. 1839, 719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fuks F. (2005) DNA methylation and histone modifications: teaming up to silence genes. Curr. Opin. Genet. Dev. 15, 490–495 [DOI] [PubMed] [Google Scholar]
- 40. Le Chatelier E., Nielsen T., Qin J., Prifti E., Hildebrand F., Falony G., Almeida M., Arumugam M., Batto J.-M., Kennedy S., Leonard P., Li J., Burgdorf K., Grarup N., Jørgensen T., et al. (2013) Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546 [DOI] [PubMed] [Google Scholar]
- 41. Cotillard A., Kennedy S. P., Kong L. C., Prifti E., Pons N., Le Chatelier E., Almeida M., Quinquis B., Levenez F., Galleron N., Gougis S., Rizkalla S., Batto J.-M., Renault P., ANRMicroObes consortium, et al. (2013) Dietary intervention impact on gut microbial gene richness. Nature 500, 585–588 [DOI] [PubMed] [Google Scholar]
- 42. David L. A., Maurice C. F., Carmody R. N., Gootenberg D. B., Button J. E., Wolfe B. E., Ling A. V., Devlin A. S., Varma Y., Fischbach M. A., Biddinger S. B., Dutton R. J., and Turnbaugh P. J. (2014) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Devkota S., Wang Y., Musch M. W., Leone V., Fehlner-Peach H., Nadimpalli A., Antonopoulos D. A., Jabri B., and Chang E. B. (2012) Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487, 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suez J., Korem T., Zeevi D., Zilberman-Schapira G., Thaiss C. A., Maza O., Israeli D., Zmora N., Gilad S., Weinberger A., Kuperman Y., Harmelin A., Kolodkin-Gal I., Shapiro H., Halpern Z., et al. (2014) Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 514, 181–186 [DOI] [PubMed] [Google Scholar]
- 45. Chassaing B., Koren O., Goodrich J. K., Poole A. C., Srinivasan S., Ley R. E., and Gewirtz A. T. (2015) Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519, 92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ussar S., Griffin N. W., Bezy O., Fujisaka S., Vienberg S., Softic S., Deng L., Bry L., Gordon J. I., and Kahn C. R. (2015) Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab. 22, 516–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Carmody R. N., Gerber G. K., Luevano J. M. Jr, Gatti D. M., Somes L., Svenson K. L., and Turnbaugh P. J. (2015) Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17, 72–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rooks M. G., and Garrett W. S. (2016) Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16, 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Goyal M. S., Venkatesh S., Milbrandt J., Gordon J. I., and Raichle M. E. (2015) Feeding the brain and nurturing the mind: linking nutrition and the gut microbiota to brain development. Proc. Natl. Acad. Sci. U.S.A. 112, 14105–14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Perry R. J., Peng L., Barry N. A., Cline G. W., Zhang D., Cardone R. L., Petersen K. F., Kibbey R. G., Goodman A. L., and Shulman G. I. (2016) Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature 534, 213–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Qin J., Li Y., Cai Z., Li S., Zhu J., Zhang F., Liang S., Zhang W., Guan Y., Shen D., Peng Y., Zhang D., Jie Z., Wu W., Qin Y., et al. (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60 [DOI] [PubMed] [Google Scholar]
- 52. Caesar R., Tremaroli V., Kovatcheva-Datchary P., Cani P. D., and Bäckhed F. (2015) Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab. 22, 658–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bäckhed F., Manchester J. K., Semenkovich C. F., and Gordon J. I. (2007) Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. U.S.A. 104, 979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Karlsson F. H., Fåk F., Nookaew I., Tremaroli V., Fagerberg B., Petranovic D., Bäckhed F., and Nielsen J. (2012) Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 3, 1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Z., Klipfell E., Bennett B. J., Koeth R., Levison B. S., Dugar B., Feldstein A. E., Britt E. B., Fu X., Chung Y.-M., Wu Y., Schauer P., Smith J. D., Allayee H., Tang W. H., et al. (2011) Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Leung C., Rivera L., Furness J. B., and Angus P. W. (2016) The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 13, 412–425 [DOI] [PubMed] [Google Scholar]
- 57. Sheehan D., Moran C., and Shanahan F. (2015) The microbiota in inflammatory bowel disease. J. Gastroenterol. 50, 495–507 [DOI] [PubMed] [Google Scholar]
- 58. Belcheva A., Irrazabal T., Robertson S. J., Streutker C., Maughan H., Rubino S., Moriyama E. H., Copeland J. K., Kumar S., Green B., Geddes K., Pezo R. C., Navarre W. W., Milosevic M., Wilson B. C., et al. (2014) Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 158, 288–299 [DOI] [PubMed] [Google Scholar]
- 59. Blaser M. J. (2016) Antibiotic use and its consequences for the normal microbiome. Science 352, 544–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cox L. M., Yamanishi S., Sohn J., Alekseyenko A. V., Leung J. M., Cho I., Kim S. G., Li H., Gao Z., Mahana D., Zárate Rodriguez J. G., Rogers A. B., Robine N., Loke P., and Blaser M. J. (2014) Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158, 705–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Donohoe D. R., Holley D., Collins L. B., Montgomery S. A., Whitmore A. C., Hillhouse A., Curry K. P., Renner S. W., Greenwalt A., Ryan E. P., Godfrey V., Heise M. T., Threadgill D. S., Han A., Swenberg J. A., Threadgill D. W., and Bultman S. J. (2014) A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 4, 1387–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gao Z., Yin J., Zhang J., Ward R. E., Martin R. J., Lefevre M., Cefalu W. T., and Ye J. (2009) Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Majnik A. V., and Lane R. H. (2015) The relationship between early-life environment, the epigenome and the microbiota. Epigenomics 7, 1173–1184 [DOI] [PubMed] [Google Scholar]
- 64. Yassour M., Vatanen T., Siljander H., Hämäläinen A.-M., Härkönen T., Ryhänen S. J., Franzosa E. A., Vlamakis H., Huttenhower C., Gevers D., Lander E. S., Knip M., DIABIMMUNE Study Group, and Xavier R. J. (2016) Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 8, 343ra81–343ra81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bokulich N. A., Chung J., Battaglia T., Henderson N., Jay M., Li H., D Lieber A., Wu F., Perez-Perez G. I., Chen Y., Schweizer W., Zheng X., Contreras M., Dominguez-Bello M. G., and Blaser M. J. (2016) Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 8, 343ra82–343ra82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maurice C. F., Knowles S. C., Ladau J., Pollard K. S., Fenton A., Pedersen A. B., and Turnbaugh P. J. (2015) Marked seasonal variation in the wild mouse gut microbiota. ISME J. 9, 2423–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Daniel H., Gholami A. M., Berry D., Desmarchelier C., Hahne H., Loh G., Mondot S., Lepage P., Rothballer M., Walker A., Böhm C., Wenning M., Wagner M., Blaut M., Schmitt-Kopplin P., Kuster B., Haller D., and Clavel T. (2014) High-fat diet alters gut microbiota physiology in mice. ISME J. 8, 295–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ng S.-F., Lin R. C., Laybutt D. R., Barres R., Owens J. A., and Morris M. J. (2010) Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 467, 963–966 [DOI] [PubMed] [Google Scholar]
- 69. Sonnenburg E. D., Smits S. A., Tikhonov M., Higginbottom S. K., Wingreen N. S., and Sonnenburg J. L. (2016) Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Donohoe D. R., Collins L. B., Wali A., Bigler R., Sun W., and Bultman S. J. (2012) The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 48, 612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Krautkramer K. A., Kreznar J. H., Romano K. A., Vivas E. I., Barrett-Wilt G. A., Rabaglia M. E., Keller M. P., Attie A. D., Rey F. E., and Denu J. M. (2016) Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol. Cell 64, 982–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ridlon J. M., Kang D. J., Hylemon P. B., and Bajaj J. S. (2014) Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 30, 332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. den Besten G, van Eunen K., Groen A. K., Venema K., Reijngoud D.-J., and Bakker B. M. (2013) The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 54, 2325–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McBrian M. A., Behbahan I. S., Ferrari R., Su T., Huang T.-W., Li K., Hong C. S., Christofk H. R., Vogelauer M., Seligson D. B., and Kurdistani S. K. (2013) Histone acetylation regulates intracellular pH. Mol. Cell 49, 310–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. De Vadder F., Kovatcheva-Datchary P., Goncalves D., Vinera J., Zitoun C., Duchampt A., Bäckhed F., and Mithieux G. (2014) Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156, 84–96 [DOI] [PubMed] [Google Scholar]
- 76. Latham T., Mackay L., Sproul D., Karim M., Culley J., Harrison D. J., Hayward L., Langridge-Smith P., Gilbert N., and Ramsahoye B. H. (2012) Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 40, 4794–4803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Waldecker M., Kautenburger T., Daumann H., Busch C., and Schrenk D. (2008) Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 19, 587–593 [DOI] [PubMed] [Google Scholar]
- 78. Hinnebusch B. F., Meng S., Wu J. T., Archer S. Y., and Hodin R. A. (2002) The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J. Nutr. 132, 1012–1017 [DOI] [PubMed] [Google Scholar]
- 79. Lukovac S., Belzer C., Pellis L., Keijser B. J., de Vos W. M., Montijn R. C., and Roeselers G. (2014) Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. MBio. 5, e01438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gorres K. L., Daigle D., Mohanram S., and Miller G. (2014) Activation and repression of Epstein-Barr virus and Kaposi's sarcoma-associated herpesvirus lytic cycles by short- and medium-chain fatty acids. J. Virol. 88, 8028–8044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Letouzé E., Martinelli C., Loriot C., Burnichon N., Abermil N., Ottolenghi C., Janin M., Menara M., Nguyen A. T., Benit P., Buffet A., Marcaillou C., Bertherat J., Amar L., Rustin P., De Reyniès A., Gimenez-Roqueplo A.-P., and Favier J. (2013) SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23, 739–752 [DOI] [PubMed] [Google Scholar]
- 82. Kaelin W. G. Jr., and McKnight S. L. (2013) Influence of metabolism on epigenetics and disease. Cell 153, 56–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Aufreiter S., Gregory J. F. 3rd, Pfeiffer C. M., Fazili Z., Kim Y. I., Marcon N., Kamalaporn P., Pencharz P. B., and O'Connor D. L. (2009) Folate is absorbed across the colon of adults: evidence from cecal infusion of 13C-labeled [6S]-5-formyltetrahydrofolic acid. Am. J. Clin. Nutr. 90, 116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pompei A., Cordisco L., Amaretti A., Zanoni S., Raimondi S., Matteuzzi D., and Rossi M. (2007) Administration of folate-producing bifidobacteria enhances folate status in Wistar rats. J. Nutr. 137, 2742–2746 [DOI] [PubMed] [Google Scholar]
- 85. Strozzi G. P., and Mogna L. (2008) Quantification of folic acid in human feces after administration of Bifidobacterium probiotic strains. J. Clin. Gastroenterol. 42, S179–S184 [DOI] [PubMed] [Google Scholar]
- 86. Craciun S., and Balskus E. P. (2012) Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. U.S.A. 109, 21307–21312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Romano K. A., Vivas E. I., Amador-Noguez D., and Rey F. E. (2015) Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio. 6, e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Anderson O. S., Sant K. E., and Dolinoy D. C. (2012) Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 23, 853–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dimri M., Bommi P. V., Sahasrabuddhe A. A., Khandekar J. D., and Dimri G. P. (2010) Dietary ω-3 polyunsaturated fatty acids suppress expression of EZH2 in breast cancer cells. Carcinogenesis 31, 489–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Druart C., Neyrinck A. M., Vlaeminck B., Fievez V., Cani P. D., and Delzenne N. M. (2014) Role of the lower and upper intestine in the production and absorption of gut microbiota-derived PUFA metabolites. PLoS ONE 9, e87560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Akare S., Jean-Louis S., Chen W., Wood D. J., Powell A. A., and Martinez J. D. (2006) Ursodeoxycholic acid modulates histone acetylation and induces differentiation and senescence. Int. J. Cancer 119, 2958–2969 [DOI] [PubMed] [Google Scholar]
- 92. García-Villalba R., Beltrán D., Espín J. C., Selma M. V., and Tomás-Barberán F. A. (2013) Time course production of urolithins from ellagic acid by human gut microbiota. J. Agric. Food Chem. 61, 8797–8806 [DOI] [PubMed] [Google Scholar]
- 93. Kiss A. K., Granica S., Stolarczyk M., and Melzig M. F. (2012) Epigenetic modulation of mechanisms involved in inflammation: Influence of selected polyphenolic substances on histone acetylation state. Food Chem. 131, 1015–1020 [Google Scholar]
- 94. Rajavelu A., Tulyasheva Z., Jaiswal R., Jeltsch A., and Kuhnert N. (2011) The inhibition of the mammalian DNA methyltransferase 3a (Dnmt3a) by dietary black tea and coffee polyphenols. BMC Biochem. 12, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nandakumar V., Vaid M., and Katiyar S. K. (2011) (−)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 32, 537–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lee W. J., Shim J.-Y., and Zhu B. T. (2005) Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol. 68, 1018–1030 [DOI] [PubMed] [Google Scholar]
- 97. Vahid F., Zand H., Nosrat-Mirshekarlou E., Najafi R., and Hekmatdoost A. (2015) The role dietary of bioactive compounds on the regulation of histone acetylases and deacetylases: A review. Gene 562, 8–15 [DOI] [PubMed] [Google Scholar]
- 98. Wong C. P., Hsu A., Buchanan A., Palomera-Sanchez Z., Beaver L. M., Houseman E. A., Williams D. E., Dashwood R. H., and Ho E. (2014) Effects of sulforaphane and 3,3′-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS ONE 9, e86787–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Liu Y., Chakravarty S., and Dey M. (2013) Phenethylisothiocyanate alters site- and promoter-specific histone tail modifications in cancer cells. PLoS ONE 8, e64535–13 [DOI] [PMC free article] [PubMed] [Google Scholar]