

Abstract
APOBEC3G (A3G) belongs to the AID/APOBEC protein family of cytidine deaminases (CDA) that bind to nucleic acids. A3G mutates the HIV genome by deamination of dC to dU, leading to accumulation of virus-inactivating mutations. Binding to cellular RNAs inhibits A3G binding to substrate single-stranded (ss) DNA and CDA activity. Bulk RNA and substrate ssDNA bind to the same three A3G tryptic peptides (amino acids 181–194, 314–320, and 345–374) that form parts of a continuously exposed protein surface extending from the catalytic domain in the C terminus of A3G to its N terminus. We show here that the A3G tyrosines 181 and 315 directly cross-linked ssDNA. Binding experiments showed that a Y315A mutation alone significantly reduced A3G binding to both ssDNA and RNA, whereas Y181A and Y182A mutations only moderately affected A3G nucleic acid binding. Consistent with these findings, the Y315A mutant exhibited little to no deaminase activity in an Escherichia coli DNA mutator reporter, whereas Y181A and Y182A mutants retained ∼50% of wild-type A3G activity. The Y315A mutant also showed a markedly reduced ability to assemble into viral particles and had reduced antiviral activity. In uninfected cells, the impaired RNA-binding capacity of Y315A was evident by a shift of A3G from high-molecular-mass ribonucleoprotein complexes to low-molecular-mass complexes. We conclude that Tyr-315 is essential for coordinating ssDNA interaction with or entry to the deaminase domain and hypothesize that RNA bound to Tyr-315 may be sufficient to competitively inhibit ssDNA deaminase-dependent antiviral activity.
Keywords: cytidine deaminase, DNA binding protein, human immunodeficiency virus (HIV), mass spectrometry (MS), protein cross-linking, RNA, RNA binding protein, APOBEC, HIV
Introduction
APOBEC3G (A3G)5 is a nucleic acid-binding protein that functions in innate immunity by restricting HIV. It belongs to the AID/APOBEC protein family of cytidine deaminases (CDA) known to possess either single-stranded DNA (ssDNA) or RNA-editing activity (1–6). During reverse transcription of HIV, A3G that is packaged within the virion mutates single-stranded regions of the viral genome by deamination of dC to dU, and the accumulated missense and nonsense mutations impair or deactivate the production of functional viruses (7–9). Fluorescence anisotropy experiments showed that A3G has affinity for RNA and ssDNA in the low nanomolar range (10). In order for A3G to serve its antiviral function, it must assemble with nascent viral particles by binding to cellular and viral RNAs (11–18). In contrast, A3G binding to cellular RNAs, particularly to highly abundant Alu and hY1, hY3-hY5 RNAs (19), inhibits A3G assembly with virions by forming cytoplasmic high-molecular-mass ribonucleoprotein (RNP) complexes that are sequestered in P-bodies (20–25).
A3G contains at least two zinc-binding domains, one associated with the catalytically inactive N-terminal CD1 domain and the other within the catalytically active C-terminal CD2 domain (1, 26–28). It has been established that CD1 can bind RNA and mediate A3G incorporation into virions (15, 29, 30), and CD2 contains the catalytic domain (29, 31, 32), but full-length protein was required for oligomerization with RNA and robust CDA activity on ssDNA (31–33).
A3G assembly and disassembly on ssDNA is an ordered process involving A3G dimers and multimers (34, 35). ssDNA binding and CDA activity are inhibited in an RNA dose-dependent manner when A3G binds to RNA (20, 36). Although the structure of full-length A3G has only been approximated by small-angle X-ray scattering (37), structures for the catalytic CD2 and non-catalytic CD1 have been determined (31, 38–41). A3G CD2 co-crystal structures with single deoxynucleotides or oligodeoxynucleotides have been obtained for CD2 of A3G and A3B revealing potential ssDNA coordination residues that lie within and proximal to the catalytic groove (42, 43). In recent studies (10), cross-linking of full-length and catalytically active A3G to deoxyoligonucleotides and mass spectrometry (MS) of tryptic peptides showed A3G interactions with ssDNA within peptides at amino acids (aa) 181–194, 314–320, and 345–374. In three-dimensional reconstruction, these peptides modeled proximally to the catalytic center of CD2 and along a continuously exposed surface on the back of A3G (relative to the catalytic face) toward CD1 in the N terminus (10, 44).
In this study, we used site-directed mutagenesis and functional end-point analysis to show that tyrosines 181 and 315 were responsible for cross-linking to nucleic acids. A novel finding in our studies is that Tyr-315 in the C terminus of A3G is essential for RNA binding. Y315A mutants had a comparable fold as native A3G but did not form high-molecular-mass RNP and had an impaired ability to assembly with HIV virions. Although other studies have presumed RNA only binds to the N terminus of A3G (a single RNA-binding domain model), the data reported here reveal a significant role of Tyr-315 in determining RNA-dependent oligomerization of A3G in RNP critical to viral particle assembly of A3G and cellular cytoplasmic RNP formation. The distribution of RNA-binding peptides within the N- and C-terminal domains of A3G suggests that a dual domain RNA-binding model may more accurately account for the diversity of functional outcomes due to different RNA sequences bound to A3G. We hypothesize that Tyr-315 plays an essential role for both RNA and ssDNA binding whose occupancy may be key to gating entry of ssDNA to the active site and accounts for the observed RNA competitive inhibition of ssDNA binding and deaminase activity (10, 36).
Results
A3G tyrosines 181 and 315 are cross-linked to ssDNA in A3G/BrdU-ssDNA samples
We employed protein/nucleic acid photocross-linking coupled with MS and applied a comparative approach for MS analysis of tryptic peptides as candidate ssDNA-binding sites (44). Three A3G peptides were determined previously as being involved in ssDNA binding: aa 181–194, 314–320, and 345–374 (10). In this study, we took advantage of bromodeoxyuridine (BrdU)-modified ssDNA cross-linked to full-length and native A3G (supplemental Fig. S1) to identify amino acid residues that were cross-linked to nucleotides in ssDNA.
Previously, studies of protein and peptides cross-linked to 5-bromouracil (BU) established that its photoreactivity may be dependent on cross-linking parameters and the peptide microenvironment (45–47). At low-energy excitation, cross-linking may yield the reactive BU triplet state (n,π* state), created by initial single electron transfer from an oxidizable group in an amino acid residue (e.g. cross-linking to the aromatic ring of Trp or Tyr, followed by ion radical coupling and elimination of bromine (HBr) to yield the cross-link). Alternatively, excitation with a higher energy singlet state, a π,π* state, may yield a uracil radical and a bromine atom in competition with intersystem cross-linking to the triplet state. Subsequent reaction of the uracil-bromine atom doubled radical pair may yield cross-linking to a nearby amino acid. These radicals were proposed to have additional reactivities. In the second scenario, excitation of the BU chromophore with UV may result in C–Br bond homolysis in an upper singlet state and single electron transfer from a neighboring group in the triplet state. Even though BrdU (HBr) was often lost after protein cross-linking to nucleic acids (48), it also was possible to observe BrdU modifications in cross-linked peptides. In addition, because bromine was at position C5 in BrdU, it may be stabilized as was suggested for APOBEC3A in respect of edited nucleotide that is engaged in T-shaped π-stacking interaction with Tyr-130, Thr-31, and His-70 (42). Importantly, because of the naturally occurring bromine stable isotopes (79Br and 81Br, typically present at an ∼1:1 ratio), tryptic peptides cross-linked to BrdU-containing ssDNA may be distinguished in MS mass-to-change (m/z) spectra as double peaks of about similar intensity with a 1.998 atomic unit difference for monoisotopic peptides or 0.999 m/z difference for +2 charge peptides, etc. Although the presence of stable 13C, 15N, and 2H isotopes may complicate MS analysis, using BrdU-modified ssDNA may facilitate the identification of cross-linked peptides in MS samples and individual cross-linked residues within the MS/MS tryptic peptide fragmentation data.
A 25-nt ssDNA with a single centrally located BrdU adjacent to the dC for editing was cross-linked to A3G, and the cross-linked product in the reaction was resolved (gel shifted) from uncross-linked protein by SDS-PAGE. Protein subjected to UV irradiation and migrating with the molecular weight of monomeric A3G served as control for maximum MS peptide identification over the primary sequence of A3G. Extensive nuclease treatment of these samples prior to MS analysis ensured that they would only have one nucleotide modification at the cross-linked position. Gel-shifted and control samples were analyzed using Q Exactive (Thermo Fisher Scientific) mass spectrometer, and the MS data were initially searched for differences in peptides identified in the ssDNA cross-linked in A3G sample due to altered m/z compared with those identified in control A3G lacking ssDNA cross-linking. Peptides corresponding to these differences were analyzed for 79Br and 81Br BrdU-modified double peak peptides with Compass Software (IsotopePattern, Bruker Daltonics) and the chemistry of modification set as corresponding to BrdU, C9H11N2O8P1Br. Two sets of peaks, with m/z values in the range of 1111–1113 (charge +4) and 663–665 (charge +3), were identified that showed MS1 peaks matching the predicted m/z values of the A3G-BrdU-modified peptides, aa 181–213, sequence YYILLHIMLGEILRHSMDPPTFTFNFNNEPWVR (one missed trypsin cleavage) (Fig. 1A) and aa 314–326, sequence IYDDQGRCQEGLR (one missed trypsin cleavage) (Fig. 1B), suggesting that these peptides contain cross-linked 79Br/81Br-modified amino acid residues (see details under “Experimental procedures” and in Fig. 1, A and B, legend). MS/MS fragmentation data of these peaks were in agreement with corresponding peptide sequences (Fig. 2, A and B). To identify individual amino acid residues cross-linked to BrdU within A3G peptides determined by Compass IsotopePattern Mascot, searches were performed with parameters set to identify BrdU modifications of residues commonly known to cross-link to nucleic acids: cysteine, tyrosine, and phenylalanine and also tryptophan, histidine, methionine, serine, arginine, and lysine (49). Tyr-181 in tryptic peptide aa 181–194 and Tyr-315 in tryptic peptide aa 314–320 were identified as BrdU-modified residues in cross-linked samples (MS/MS spectra of these peptides with indicated positions of modified tyrosines are shown in Fig. 2, A and B). For example, the m/z value of b11+ ion corresponding to Tyr-181 in peptide aa 181–194 was found to be 548.01, which is much higher than the expected value of m/z 163.06 for tyrosine alone, and the difference between the observed and expected (tyrosine alone) m/z value was equal to the m/z of BrdU. Similarly, an observed m/z value for the b22+ ion, Ile-314, Tyr-315 in peptide aa 314–320, was 331.11 and not 138.57 as if Tyr-315 was not cross-linked to BrdU (note that this peptide has 2+ charge). The supplemental Tables 1 and 2 list all observed ions to complement MS/MS fragmentation data presented in Fig. 2. Both tyrosines at positions 181 and 315 were confirmed to be modified by cross-linked BrdU using BioTools software to manually examine the fragmentation patterns of the corresponding peptide spectra observed during liquid chromatography separation and fragmentation (MS/MS data).
Figure 1.


A, initial identification of 79Br- and 81Br-containing BrdU-cross-linked A3G peptides aa 181–213 that matched predicted MS1 values. Tryptic peptides of A3G/BrdU cross-linked sample were analyzed on Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer and searched for BrdU modifications, including 79Br and 81Br isotope difference with Compass software (Compass IsotopePattern, Bruker Daltonics). Panel a, 79Br- and 81Br-containing peptide peaks with m/z values in a range of 1111–1113, corresponding to the A3G peptide YYILLHIMLGEILRHSMDPPTFTFNFNNEPWVR (aa 181–213, m/z charge +4, one missed trypsin cleavage site) were identified (panel b) in a fraction eluted at 18.96 min followed by (panel c) Compass IsotopePattern analysis and (panel d) MS/MS fragmentation of this peptide that matched a predicted fragmentation pattern. It should be noted that due to the presence in the samples of other stable isotopes besides 79Br and 81Br, including 13C (mostly) 2H, and 15N, the MS1 pattern of each shown A3G peptide analyzed by Xcalibur and Compass is represented by several peaks (typically 2–4, depending on peptide change, with decreasing relative intensity). As evident from Compass IsotopePattern analysis (panel c), we observed eight peptide peaks in an area of 1111–1113 m/z that at the charge 4+ and peak distance ∼0.5 m/z (not counting common 13C-double peaks) may constitute two identical peptides that differ only by ∼2.0 m/z that corresponds to bromine-isotope difference of 79Br and 81Br in BrdU cross-linker. B, initial identification of 79Br- and 81Br-containing BrdU-cross-linked A3G peptides aa 314–326 that matched predicted MS1 values. Tryptic peptides of the A3G/BrdU cross-linked sample were analyzed on Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer and searched for BrdU modifications including 79Br and 81Br isotope difference with Compass software (Compass IsotopePattern, Bruker Daltonics). Panel a, 79Br- and 81Br-containing peptide peaks with m/z values in a range of 663–665, corresponding to the A3G peptide IYDDQGRCQEGLR (aa 314–326, m/z charge +3, one missed trypsin cleavage site) were identified (panel b) in a fraction eluted at 12.26 min followed by (panel c) Compass IsotopePattern analysis and (panel d) MS/MS fragmentation of this peptide that matched a predicted fragmentation pattern. It should be noted that due to the presence in the samples of other stable isotopes besides 79Br and 81Br, including 13C (mostly), 2H, and 15N, the MS1 pattern of each shown A3G peptide analyzed by Xcalibur and Compass is represented by several peaks (typically 2–4, depending on peptide change, with decreasing relative intensity). As evident from Compass IsotopePattern analysis (panel c), we observed six peptide peaks in an area of 663–665 m/z that at the charge 3+ and peak distance ∼0.67 m/z (not counting common 13C-double peaks) may constitute two identical peptides that differ only by ∼2.0 m/z that correspond to BrdU-isotope difference of 79Br and 81Br in BrdU cross-linker.
Figure 2.

Identification of A3G tyrosines 181 (top panel) and 315 (bottom panel) as cross-linked residues to 25-nt BrdU-modified ssDNA. UV light-induced cross-linking was performed with assembled A3G-ssDNA complex, and the cross-linked product was separated onto SDS-polyacrylamide gel and in-gel digested with trypsin. A3G tryptic peptides were analyzed on Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer and searched with Mascot for amino acid residue BrdU modifications. Shown are A3G peptide MS/MS fragmentation spectra: aa 181–194 with BrdU-modified Tyr-181 (A) and aa 314–320 with BrdU-modified Tyr-315 (B). Peptide sequences are shown on the right. Most abundant b and y ion values and their m/z charge are marked on the spectra (no charge is shown for +1 fragment values). The positions of the BrdU-cross-linked fragments on the spectra are indicated with arrows. The complete list of identified b and y ions is presented in supplemental Tables 1 and 2 (1+ (monoisotopic) and 2+ mass/charge values). Abs. Int., absorbance intensity.
A3G tyrosines 181, 182, and 315 are involved in ssDNA binding
To obtain definitive proof that Tyr-181 and Tyr-315 in A3G actually are involved in ssDNA binding, point mutants were created that replaced Tyr-181, Tyr-182, and Tyr-315 to neutral alanine residues. Tyr-182 was included in our studies even though it was not identified by MS as being cross-linked to BrdU because it was adjacent to Tyr-181, and we wanted to evaluate its potential to bind ssDNA when Tyr-181 was mutated. The corresponding mutant constructs were expressed as full-length proteins in Hi-Five insect cells using the Baculovirus protein expression system and purified by nickel-nitrilotriacetic acid affinity resin with a final concentration of 1–3 mg/ml soluble protein (supplemental Fig. S1).
Thermal fluorimetry (thermal shift assays) determines the temperature-dependent ordered unfolding of proteins as detected by SYPRO Orange dye binding to exposed hydrophobic regions and was used here to assess mutant protein folding relative to wild-type (WT) A3G. Purified WT A3G and all three single tyrosine replacement proteins showed near identical dissociation curves as indicated by detection of comparable amounts of SYPRO Orange dye binding across a temperature gradient with only minor difference in melting temperature (supplemental Fig. S2). Such minor differences in melting temperature (not exceeding 2–3 °C) typically are associated with pipetting variations and not with altered protein stability (50).
WT and mutant A3G protein nucleic acid binding was quantified by fluorescence anisotropy (FA) over a range of A3G input (Fig. 3A). The binding assays showed that the tyrosine mutants had diminished affinity for substrate ssDNA (Fig. 3A). Y181A mutant had a kd = 4.18 ± 0.79 (n = 3) for binding to 25 nt of ssDNA that was about 2-fold lower than the kd value observed for WT A3G, 2.18 ± 0.17 (n = 3). Y182A mutant protein had a kd value for ssDNA binding that was similar to Y181A (data not shown). In marked contrast, the Y315A mutant showed substantially lower affinity to ssDNA, kd = 45.3 ± 9.65 (n = 3). The data suggested that Tyr-315 was critical for ssDNA binding in vitro and Tyr-181/Tyr-182 that are more distal from CD2 had less significant roles in coordinating ssDNA binding.
Figure 3.

Binding of A3G tyrosine mutants to AlexaFluor647-labeled nucleic acids is reduced as compared with the WT A3G. Fluorescence anisotropy changes due to A3G binding to AlexaFluor647-labeled 25-nt ssDNA and 25-nt RNA were determined by addition of increasing concentrations of A3G to 2 nm ssDNA (A) or RNA (B) (10). The average normalized change in anisotropy (y axis) of three individual experiments was plotted as a function of protein concentration (x axis), and error bars represent the standard error of the mean (S.E.) of the data. The results were fit to a single-site binding equation accounting for ligand consumption, and A3G dissociation constant (Kd) was determined.
A3G Tyr-315 is critical for the DNA mutator phenotype
We reasoned that because alanine substitutions of A3G Tyr-181, Tyr-182, and Tyr-315 reduced substrate ssDNA binding to A3G, the corresponding A3G mutants might also have reduced deaminase activity. WT and mutant A3Gs were expressed in an Escherichia coli DNA mutator reporter system that quantifies growth of bacterial colonies on plates under rifampicin selection due to mutations in the reporter β-subunit of RNA polymerase gene rpoB that confer rifampicin resistance (51–53). Plasmids containing WT and mutant A3G were transformed into reporter strain, and transformants expressing similar amounts of full-length WT or mutant A3G (supplemental Fig. S3) were plated onto rifampicin selection media.
WT A3G produced on average 25 RifR colonies per 108 cells (Fig. 4). In agreement with the slight reduction in ssDNA binding seen with both single Y181A and Y182A proteins, these mutants had ∼2-fold fewer rifampicin-resistant colonies relative to WT A3G. Bacterial transformants expressing A3G Y181A/Y182A double mutation did not further decrease the number of rifampicin-resistant colonies. The data suggested that although MS analysis identified Tyr-181 as predominantly involved in ssDNA binding, we cannot rule out that Tyr-181 or Tyr-182 could support ssDNA binding and deaminase activity. The activity of single-site mutants versus double-site mutants suggested that simultaneous occupancy of Tyr-181 and Tyr-182 by ssDNA was not required.
Figure 4.
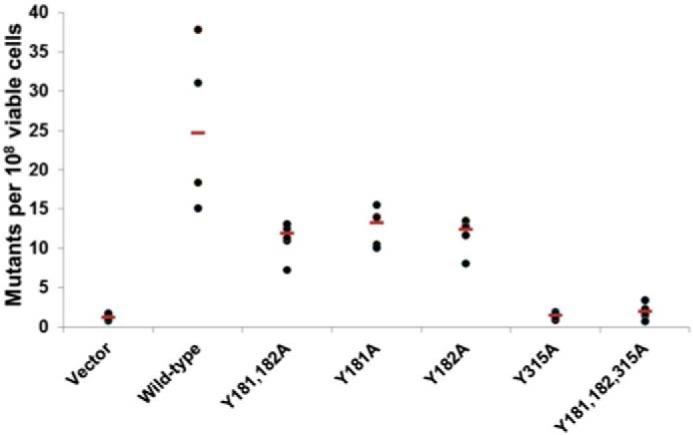
Mutational activity of A3G tyrosine mutants is diminished, especially the Y315A mutant. Mutational activity of A3G mutants due to the expression of cytidine deaminases in E. coli was determined by using a bacterial reverse rifampicin resistance reporter assay (β-subunit of RNA polymerase) (51, 53). Mutational frequency for each WT or mutant A3G strain was calculated as the number of rifampicin-resistant colonies per 108 of viable cells after making correction for the number of colonies on control plates and shown as scatter diagrams with the median value drawn through (red lines). Each experiment was repeated four times. Bacterial strains tested were vector (vector alone); wild type (wild-type A3G); Y181A/Y182A (A3G with double Y181A and Y182A mutations); Y181A (A3G with a single Y181A mutation); Y182A (A3G with a single Y182A mutation); Y315A (A3G with a single Y315A mutation); Y181A/Y182A/Y315A (A3G with a triple Y181A, Y182A, and Y315A mutations).
Few or no rifampicin-resistant colonies were induced by the A3G Y315A mutant that was similar to the number of RifR colonies seen with empty vector control. The data showed that A3G Y315A mutant lost >90% of WT A3G DNA mutagenic activity. Taken together, the DNA mutator assay results suggested that binding of ssDNA to Tyr-315 was essential for A3G deamination of dC to dU in ssDNA substrates, consistent with the highly conserved position of this residue at an entry point for ssDNA relative to catalytic residues in zinc-dependent deaminase domain (1, 42, 54).
Tyr-315 mutation reduces the formation of A3G-containing high-molecular-mass RNPs in mammalian cells
We recently reported that RNA as well as ssDNA binds to A3G tryptic peptides in the CD2 domain (10). Quantitative FA data showed that WT A3G had a slightly higher affinity for the 25-nt RNA than for ssDNA (kd = 1.39 ± 0.14 and kd = 2.18 ± 0.17, (n = 3) respectively) (Fig. 3B). Our FA experiments also showed that the Y181A mutant had about 2.5-fold reduced affinity for RNA binding than WT A3G (similar to the difference in ssDNA binding). In contrast to prior studies that used non-quantitative RT-PCR to assess the role of Tyr-315 in RNA binding (15), the Y315A mutant showed a 10-fold reduction in kd values for RNA as compared with WT A3G (kd = 13.85 ± 1.75 (n = 3), see Fig. 3B). It is noteworthy that the Y315A mutation reduced binding to ssDNA to a greater extent than to RNA (∼20-fold versus ∼10-fold, respectively). We suggest that this may not be solely due to the relative higher affinity of A3G to RNA, but rather it could have been due to the greater number of RNA-binding residues distributed in the N- and C-terminal domains of A3G compared with the three peptides found to bind to ssDNA.
Velocity sedimentation of A3G WT, Y181A, Y182A, and Y315A mutant proteins from the cell extracts of transfected HEK293T cells was evaluated to confirm the effect of these mutations on RNA binding in cells. Cellular A3G typically is assembled in high-molecular-mass RNP complexes that are formed through RNA-bridged protein-protein interactions in cytoplasm processing P-bodies (20–25). A3G ribonucleoproteins are of megadalton molecular mass and therefore sediment to the high-density fractions in glycerol gradients during velocity sedimentation analysis (34, 44, 55). The majority of WT A3G was recovered in high-density fractions and at the bottom of the glycerol gradient (Fig. 5, left panels). Y181A and Y182A mutants sedimented as more evenly distributed complexes and were recovered throughout the gradient. Therefore, relative to WT A3G these two mutants had reduced capacity for RNA-dependent oligomerization into high-molecular-mass complexes. RNase A digestion of cell extracts prior to fractionation markedly reduced the sedimentation of Y181A and Y182A mutants to that comparable with WT A3G following treatment with RNase A (Fig. 5, right panels). Similar sedimentation and RNase A digestion sensitivities were observed for the double Y181A/Y182A mutant (data not shown).
Figure 5.
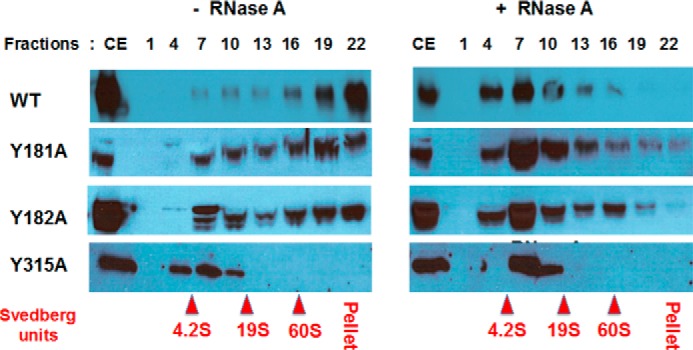
Mutations of A3G tyrosines 181, 182, and especially 315 causes decrease in A3G association with high-molecular-mass ribonucleoprotein complexes. Cell extracts (CE) of transformed with WT and mutant A3G constructs HEK293T cells were not treated (left panel) or treated (right panel) with RNase A and fractionated on 10–50% glycerol density gradient. Each third fraction from the top (lane 1) to the bottom (lane 22) of the gradient was separated onto SDS-PAGE and Western-blotted with anti-A3G antibody. Positions of ovalbumin (42 kDa, 3.5S), BSA (67 kDa, 4.2S), aldolase (160 kDa, 7.4S), catalase (250 kDa, 11.3S), and thyroglobulin (660 kDa, 19S) and in vitro assembled spliceosomes (60S) used as gradient calibration controls are shown in red.
In contrast, Y315A mutant protein showed a significantly altered profile of sedimentation as compared with WT A3G or Y181A and Y182A (Fig. 5, bottom left panels). Y315A sedimented in fractions typically seen only for WT A3G after RNase A digestion. RNase A digestion of Y315A cell extract resulted in relatively minor changes in the sedimentation of the mutant protein. These findings suggest that most if not all of the HEK293T cellular RNA binding to A3G and A3G oligomerization was dependent on the interaction of RNAs with Tyr-315. Moreover, the sedimentation of Y315A without or with RNase digestion to low-molecular-mass regions of the gradient comparable with WT A3G following RNase digestions supported the conclusion from thermal fluorimetry that Y315A was not aggregated. Given the essential role of Tyr-315 in ssDNA deamination, bulk cellular RNA binding to Tyr-315 may account for the inhibition of A3G antiviral activity reported in the literature when A3G becomes sequestered in P-bodies. Although previously it was believed that residues in A3G N-terminal CD1 were primarily responsible for RNA binding, A3G oligomerization, virion incorporation, and sequestration as RNP in P-bodies (21–23), mutations of residues within the N terminus predicted to bind RNA did not all affect RNA binding nor were they able to uniformly affect A3G binding to all RNA sequences (7, 15, 33, 56–58). Our data suggested a novel and unanticipated role for Tyr-315 in RNA binding and support a two RNA binding domain model for A3G (59).
Tyr-315 is required for robust A3G antiviral activity
A3G binding to HIV RNA (11, 13, 16, 21, 60) and host cell 7SL RNA (11, 12, 14, 16, 30, 61, 62) are thought to be essential for assembly of A3G with nascent viral particles (in addition to interactions with HIV Gag protein (60, 63, 64)). We evaluated the importance of tyrosines 181, 182, and 315 for A3G incorporation with viral particles using pseudotyped HIV-1 virus particles produced in HEK293T cells. These studies were conducted with pseudotyped HIV lacking Vif expression to prevent Vif-mediated A3G degradation thus preserving the cellular abundance of A3G necessary for assembly with virions.
The expression of WT and mutant A3G proteins in transiently transfected HEK293T cells was quantified by determining the ratio of the whole-band densitometry for A3G-specific Western blotting signals in cell extracts relative to densitometry for control GAPDH-specific western blotting signals present in each lane (supplemental Fig. S4, top panel, ratio values are shown below each lane/sample). Based on such assessment, A3G protein expression in transfected cells varied no more than 2.5-fold between WT and mutants proteins. Viral particles assembled and released from each transfected cell culture were analyzed for A3G content relative to HIV p24 capsid protein by calculating the ratios of the specific western blotting signal densitometry for these proteins in the same lane (supplemental Fig. S4, bottom panel, ratio values are shown below each lane). Both Y181A and Y182A mutant proteins were detected in total cell extracts and within viral particles in similar relative abundance compared with WT A3G. This suggested that the RNA-binding activity of A3G Y181A and Y182A mutants was sufficient for their assembly with virions and that RNA binding contributed by these residues is not essential for A3G incorporation within virions. In contrast, recovery of the Y315A mutant protein with viral particles was reduced as compared with its abundance in cell extracts (ratios 0.5 and 2.0, respectively). These results suggest that although Tyr-315 was required for RNA binding, the Y315A mutation markedly reduced but did not completely abolish A3G assembly with virions. In fact, the literature suggests that A3G association with the viral particle also depends on its interaction with the nuclear capsid portion of Gag (18, 63, 65).
Antiviral activity of A3G requires its assembly within the viral core in association with HIV genomic RNA placing A3G proximal to reverse transcription complexes and immediate access to ssDNA for CDA activity (20, 66). It is anticipated that A3G mutations that reduce its packaging within virions will preserve the infectivity of those virions. To evaluate the effects of tyrosine replacement mutants on A3G antiviral activity, p24 normalized viral particles were used to infect HeLa-based TZM-bl reporter cells that express luciferase-reporter from a TAT-transactivated HIV LTR promoter.
As anticipated, WT A3G assembled with viral particles markedly suppressed their infectivity (Fig. 6). The infectivity profiles of viral particles containing Y181A and Y182A mutants were similar to those observed for virions containing WT A3G. This finding was consistent with recovery of these A3G mutants with virions and their deaminase activity in the bacterial DNA mutator assay. Virions containing A3G Y315A mutant showed substantial infectivity (greater than 60% of control virions infectivity that lacked A3G). Although Y315A mutant had significantly diminished ssDNA binding and CDA activity, low levels of Y315A were incorporated with virions. Therefore, we cannot rule out some antiviral activity through a deaminase-independent mechanism (67–71), or residual deaminase activity may result from the Y315A proteins assembled with virions.
Figure 6.
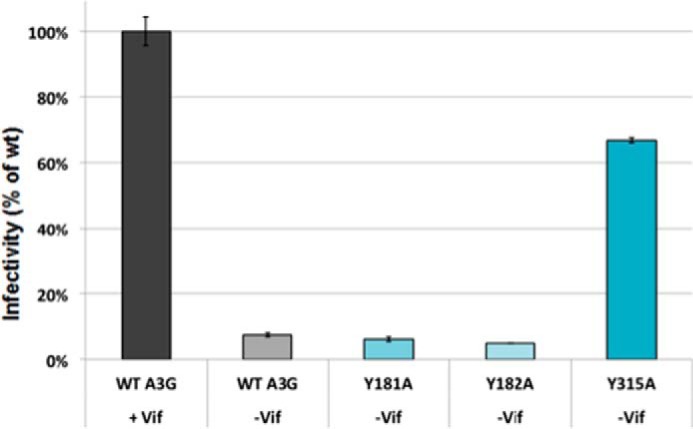
Infectivity assays of A3G WT and tyrosine mutants showed that Tyr-315 mutant lost antiviral function. Infectivity of TZM-bl cells is measured by the RLU from the expression of a stably integrated HIV LTR-driven luciferase gene. TZM-bl reporter cells were infected with 500 ng of p24-normalized pseudotyped virus particles containing WT A3G in presence of Vif and WT and mutant A3G without Vif. SteadyGlo Reagent (Promega) was used to detect the level of luciferase expression and RLU were quantified. The relative percent change in infectivity of WTA3G and mutants without Vif was compared with infectivity with WT A3G in the presence of Vif, which was set as the maximum infectivity (100%); n = 3 with error bars representing standard deviations.
Discussion
The mechanism by which RNA inhibits A3G ssDNA deaminase activity has long been of interest and thought to solely involve RNA-bridged oligomerization of A3G through its N terminus. In this report, site-directed mutagenesis of candidate nucleic acid-binding residues was informed by MS of tryptic peptides from native and full-length A3G cross-linked to ssDNA and by binding assays of the mutant proteins to ssDNA and RNA. We demonstrate that Tyr-315 is essential for ssDNA binding and consequently deaminase activity on ssDNA. RNA binding to the C-terminal CD of A3G, and directly to Tyr-315, is shown for the first time. The data shift the paradigm in the field from a single RNA-binding domain model to a dual RNA-binding domain model (59) and provide a rational explanation for why RNA is a competitive inhibitor of A3G ssDNA binding and deaminase activity on ssDNA (2, 10, 36). MS and FA revealed Tyr-181 and Tyr-182 as novel N-terminal ssDNA-binding residues. In this regard, the predicted coordination of ssDNA resulting from binding to Tyr-181/Tyr-182 provided a rational explanation for why full-length A3G is required for maximal deaminase and ssDNA-binding activity even though the N-terminal CD is catalytically inactive.
We began this study by determining the amino acids in A3G bound to ssDNA. MS data suggested that BrdU containing ssDNA substrates cross-linked to A3G at tyrosines Tyr-181 and Tyr-315. Replacement of each of these tyrosines for alanines did not affect the general protein folding but showed that Tyr-315 was essential for ssDNA binding and deaminase activity. Photo-cross-linking can only occur when the residues in A3G protein have close proximity with nucleotides approaching 0 Å (49). Therefore, we do not rule out that residues other than Tyr-181, Tyr-182, and Tyr-315 also have roles in coordinating nucleic acid binding or in the mechanism for dC to dU deamination.
Crystal structure of the catalytic CD2 domain of A3G and structures of CD2 co-crystallized with mononucleotide or CCC-trinucleotide predicted several different possibilities for substrate ssDNA binding depending on how DNA bends into the corresponding protein groove: “brim” model (31), “kinked” model (38, 40), and “straight” model (39). Interestingly, all these models predicted an involvement of peptide sequence in loop 7, RIYDDGQR, aa 313–320, particularly residues Arg-313, Tyr-315, Asp-316, and Asp-317 in ssDNA binding. Loop 7 of A3 proteins has been proposed to be a nucleotide specificity box (72, 73). A3F structural studies predicted that residues Tyr-307/308, Phe-309, and Trp-310 in loop 7 may form favorable stacking interactions with the deaminase hot spot (5′-TTCA-3′) in ssDNA substrates. These aromatic residues are not conserved across the A3 family and are replaced by YYFQ and YDDQ in A3C and A3G, respectively (74). Mutating the YDDQ box in A3G to the aromatic-rich YYFW of A3F resulted in a change of substrate preference from 5′-CC-3′ to 5′-TC-3′ or 5′-GC-3′, suggesting that mutating these four residues are sufficient in determining the nearest neighbor nucleotide context for deamination (73). Similarly, a D317Y mutation in loop 7 of the C terminus of A3G (Tyr-132 is the homologous position in A3A) was sufficient to alter the local dinucleotide preference for deamination from 5′-CC to 5′-TC (75). The other A3G residues within CD2 predicted to coordinate binding of ssDNA are His-216 (in the kinked model) and His-367 (in the straight model) along with Asn-244, Trp-285, Arg-374, and Arg-376 (38–40).
Arg-374/376 residues located on helix 6 in the C terminus of A3G with amino acid sequence LDEHSQDLSGRLR have been predicted to interact with ssDNA (38, 40). We consistently observed A3G C-terminal peptide aa 345–374 as “missing” in our MS analysis of BrdU ssDNA cross-linked samples as compared with uncross-linked A3G, suggesting that it was chemically modified after cross-linking to ssDNA (and RNA) (10). This peptide is 30 residues long (molecular mass without modification is 3514 Da), so high confidence identification of the modified residue has been problematic due to MS technical limitations. Similarly, Arg-376-containing tryptic peptide is only a dipeptide, LR, and too small for accurate MS detection; therefore, a different approach is required for identification of cross-linked residue in A3G.
While this paper was being reviewed, APOBEC3A (contains only one CD domain) and an A3A loop 1-substituted A3B co-crystal structure with a ssDNA oligodeoxynucleotide were reported (42). The structures suggested that the deaminase hot spot nearest neighbor preference in ssDNA, 5′-TC, was accommodated by bending the ssDNA substrate such that the 5′-T is rotated out of the pocket through hydrogen binding with flexible loop domains, and the deaminated C is positioned deep with the catalytic cleft through interactions with several residues key to zinc coordination and catalysis. It is of interest to note that Tyr-315 is conserved among A3 proteins and was shown positioned in the co-crystal structure with 5′-TTTTCAT-3′ deoxyoligonucleotide where it was predicted to have rotational flexibility and serve in gating ssDNA access to the catalytic site, creating an opened or closed catalytic cleft conformation (42).
Although we found no evidence for BrdU cross-linking to catalytic residues in CD2 known to be involved in zinc ion coordination, these interactions may not have been stable enough for cross-linking or not positioned appropriately for cross-linking to the BrdU nucleotide. It is important to keep in mind that A3G models based on crystal structures of half of the protein may be conditionally accurate and that Tyr-315 becomes an essential high affinity binding site for ssDNA only in the context of full-length A3G, which retains the capability of forming holoenzyme oligomers that are required for robust deaminase activity (34, 76). We cannot rule out that there may have been local misfolding due to tyrosine substitution that affected RNA or DNA binding even though we demonstrated expression of soluble full-length A3G mutant proteins with appropriate sedimentation characteristics (without and with RNA) and with near WT A3G thermo fluorescent dye-binding profiles.
MS also identified candidate RNA-binding sites. Earlier computational modeling of APOBEC structures for exposed residues that may bind to nucleic acids (15, 38, 40, 77) predominantly focused on mutagenesis of the N terminus of A3G. Tyr-315 was evaluated in these studies but found not to be important for RNA binding but important for deaminase activity and viral restriction (15). Non-conservative mutation of Tyr-315 had only a moderate decrease in binding to RNAs co-assembled with A3G in virions such as hY1 RNA and no significant change in hY4 and 7SL RNA binding (15). These analyses were based on fixed cycle number RT-PCR detection of RNAs. We propose these studies may have overestimated how much RNA was bound to Tyr-315 mutants as the template-saturating conditions of RT-PCR are not quantitative. We cannot rule out that RNA primary sequence, RNA size, and RNA secondary structure differences may have contributed to the discrepancy in experimental findings.
Tyr-181 (Tyr-182) in the N terminus of A3G also cross-linked to ssDNA and had a moderate contribution to ssDNA binding. Although these mutations did not impair packaging in viral particles, each resulted in an ∼50% loss of deaminase activity. Earlier A3G site-directed mutagenesis studies suggested that mutations within the CD1 domain moderately reduced CDA activity of the full-length A3G (29) and affected A3G oligomerization required for processive deamination (15, 78). In fact the C-terminal catalytic domain had little or no deaminase activity when expressed without the N terminus (29, 79). The importance of the N terminus for deaminase activity was underscored by the high viral load and decreased CD4+ T-cell phenotype associated with the naturally occurring mutation H186R in A3G (80). Although A3G-H186R retained the ability to incorporate into HIV-1 virions and retained catalytic activity (as did the Y181A mutant), it had a decreased efficiency in mutagenesis due to reduced scanning along ssDNA (78). A3G CD1 W94A/W127A double substitutions altered ssDNA substrate specificity from 5′-CC to 5′-TC (56). W127A substitution also reduced the ability of A3G to scan ssDNA for new deamination sites presumably because of the loss of protein homodimerization (81). Considering these findings Tyr-181/182 may be part of a domain that serves as an ssDNA coordination site for bending ssDNA bound to the catalytic site (42) or facilitates ssDNA-dependent oligomerization of A3G as dimeric ssDNA recognition complexes (34) or higher order holoenzyme complexes (33, 34, 82). Residues in the CD2 ssDNA entry site may coordinate with Tyr-315 in the context of full-length A3G to stabilize ssDNA at the catalytic site and thereby enhance the catalytic efficiency of A3G.
Our data on A3G mutant binding to nucleic acids point to physical evidence for how RNA competitive inhibition of ssDNA binding could be mediated by RNA occupancy and displacement of ssDNA from Tyr-315 within the C terminus of A3G. Previously, we demonstrated through kinetic competition binding assays that RNA directly inhibited A3G CDA activity (36). FA, EMSA, and near equilibrium binding studies showed that A3G assembly and disassembly on ssDNA was an ordered process involving A3G dimers and multimers but that RNA stochastically dissociated A3G dimers and higher-order oligomers from ssDNA (10). Given that Y315A mutation significantly affected binding of both ssDNA and RNA, we suggest that the mechanism of inhibition of ssDNA binding and CDA inactivation by RNA can be explained by direct competitive binding to Tyr-315.
Y315A mutation also had a profound inhibitory effect on A3G assembly with virions in pseudo-type virus-infected cells and the formation of high-molecular-mass RNP in uninfected cells. Y181A and Y182A only moderately reduced the aggregate sedimentation of RNP but were not able to prevent A3G assembly with virions. Interestingly, Pan et al. (83) confirmed the existence of two separate RNA-binding sites, with different affinity to A3G by using atomic force microscopy.
MS analysis of A3G cross-linked to RNA suggested that in addition to peptides in the C terminus, multiple peptides in the N terminus of A3G cross-linked to RNA (10). Mutations of Trp-94 and Trp-127 in A3G significantly reduced binding to several RNAs tested, namely HIV, 7SL, hY1, and hY3 RNAs as determined by A3G immunoprecipitation and RT-PCR (11, 17, 56, 84). S28E, Y124A, and F126L mutants showed alterations in binding to these RNAs (84). A3G W94A/W127A double replacement mutant protein remained localized to P-bodies and exerted DNA mutator activity in bacterial reporter strain but showed no to low HIV restriction despite packaging with virions (56). Molecular modeling suggested that residues Trp-94 and Tyr-124–Trp-127 were involved in protein-protein intermolecular interaction in an A3G homodimer formation during nucleic acid binding (27, 84). A critical distinction of Y315A mutant phenotype contrasting with the W94A/W124A double mutant is the requirement of Tyr-315 for A3G cellular and antiviral functions. Sedimentation in glycerol density gradient (Fig. 5) and thermal shift assays (supplemental Fig. S2) suggested that the phenotype of Y315A was not due to aggregation or misfolding. Further study of the region containing Tyr-181, Tyr-182, and Tyr-315 may provide understanding of how A3G coordinates ssDNA substrates for process deamination and how it is regulated as a host defense factor and potentially targeted therapeutically.
In this study, we have used an AU-rich RNA probe to identify candidate general RNA-binding sites on A3G. An important open question that can now be addressed in future studies will be whether there are residues in either the N or C terminus of A3G that selectively bind to different RNA sequences or secondary structures and how this determines A3G cellular and antiviral functions.
Experimental procedures
Construction of A3G mutants
The N-His6-HA-A3G construct containing the wild-type full-length human A3G cDNA with N-terminal His6 and HA epitope tag was previously cloned in our laboratory into pIRES vector (85). It should be noted that the His6 tag did not affect A3G function because tagged protein binding affinity to nucleic acids and its CDA activity in bacterial test system was not affected. For protein production, the N-terminal His6-A3G DNA fragment was subcloned into EcoRI/XhoI restriction sites of pFastBac1 vector (Thermo Fisher Scientific), and the resultant construct was verified by DNA sequencing. His6-A3G mutant constructs containing Y181A, Y182A, and Y315A replacements in vector pUC57 were obtained from GeneScript, Inc. (Piscataway, NJ), and then subcloned into EcoRI/XhoI restriction sites of pFastBac1 and separately in the same sites of pIRES vectors. In addition, A3G WT and mutant sequences were subcloned into EcoRI/XhoI sites of pTrc99A vector under IPTG-inducible promoter to use in DNA mutator assays.
A3G protein expression and purification
To produce A3G WT and mutant proteins, the corresponding His6-A3G constructs in pFastBac1 were transformed into E. coli DH10Bac helper strain to produce the corresponding bacmids that then transfected into Sf9 insect cells with X-tremeGENE HP DNA transfection reagent (Roche Applied Science). All other procedures with insect cells were performed as described in the Bac-to-Bac manual (Thermo Fisher Scientific), including obtaining the initial viral stock and transducing the High-Five insect cells with recombinant viruses for protein production. A3G protein purification was carried out as described previously (10), frozen in liquid nitrogen, and stored at −80 °C until needed.
Thermo fluorometry (thermal shift assays)
A3G WT and mutant protein thermal shift assays were performed using Stratagene Mx3000P real-time quantitative PCR system instrument (Hauptman-Woodward Medical Research Institute, Buffalo, NY). Reactions were carried out in MicroAmp Optical 96-well reaction plates in 30-μl reactions with 1× PBS buffer containing 5 μm protein (final concentration) and 2 μl of 1:500 diluted SYPRO Orange dye (Sigma). The assays were performed by heating the plates at ∼2 °C/min from 25 to 95 °C and reading the fluorescence of the sample. Protein samples were quantified in triplicate. Protein dissociation curves were plotted from the data using GraphPad Prism 6 software.
ssDNA and RNA for A3G in vitro binding and cross-linking experiments
The following ssDNA oligonucleotides were used: AlexaFluor647®-labeled 25-nt Sub ssDNA, 5′-A647-C6-TTATTTTTAAGGATTTATTTATTTA-3′; 25-nt Sub BrdU-modified ssDNA, 5′-TTATTTCBrdUAAGGATTTATTTATTTA-3′, and the RNA oligonucleotide AlexaFluor647®-labeled 25-nt Sub RNA, 5′-A647-UUAUUUUUAAGGAUUUAUUUAUUUA-3′. These fluorescently labeled or BrdU-modified oligonucleotides used for electrophoretic mobility shift assays (EMSA), FA, and photo-cross-linking experiments were obtained from Integrated DNA Technologies (Coralville, IA).
A3G assembly with nucleic acids and EMSA
Complexes containing A3G bound to nucleic acids were prepared by incubating varying amounts of A3G and varying molar ratios of AlexaFluor647-labeled oligonucleotides in deaminase buffer (1× DB, 40 mm Tris, pH 7.2, 50 mm NaCl, 10 mm MgCl2, 1 mm DTT, 0.1% Triton X-100, 2% glycerol) for 20 min at 37 °C. Complexes assembled in each reaction condition were resolved on a 5% native gel (36) and visualized using a TyphoonTM 9410 Trio Imager (GE Healthcare) by excitation at 633 nm wavelength and measuring fluorescence at 670 nm.
Equilibrium fluorescence anisotropy
Binding of A3G WT and mutant proteins were quantitatively measured by using equilibrium fluorescence anisotropy. AlexaFluor647®-labeled ssDNA or RNA was added to each reaction at a fixed 2 nm concentration and incubated with purified 0–40 nm A3G proteins at 37 °C for 20 min in 1× DB buffer. Fluorescence anisotropy measurements were carried out in Fluoromax-4 fluorometer (Horiba Scientific, Edison, NJ), excitation 647 nm, and emission 670 nm with 5-nm band passes, each sample was done in triplicate with the mean values calculated. The change in anisotropy was calculated by subtracting the average anisotropy of the free 5′-labeled ssDNA or RNA from the average anisotropy of the A3G/nucleic acid assembly reactions and plotted against A3G concentration. The Kd value was calculated through non-linear regression by fitting the data to the following equation, where A3G/NA is the fraction of A3G bound to nucleic acid (NA) and Bmax is the enzyme's capacity to bind substrate: A3G/NA = Bmax ([A3G])/(Kd + [A3G]).
UV-induced cross-linking of A3G to ssDNA and sample preparations for MS
Photo-cross-linking of proteins to nucleic acids is frequently used to map interacting regions/sites (49). The amino acids commonly identified in photo-adducts are cysteine, methionine, serine, arginine, lysine, histidine, phenylalanine, tryptophan, and tyrosine. Pyrimidines, especially uridine, are typically 10 times more effective for cross-linking to amino acids as compared with purines. Photo-cross-linking in combination with high-resolution MS provides an opportunity for direct identification of interacting sites (86). However, often because of chemical heterogeneity in the composition of the cross-linked entity, it has been challenging to identify the amino acid-nucleosides within cross-linked peptides.
A3G was assembled with 25-nt ssDNA containing BrdU at a 4:1 molar ratio (protein/ssDNA) in 1× PBS at 37 °C for 20 min, cooled on ice, and irradiated for 20 min with 302-nm wavelength UV light. The A3G cross-linked products were separated onto 12% SDS-PAGE and visualized after staining with Coomassie SimpleBlue SafeStain (Thermo Fisher Scientific) as proteins bands with slower mobility in the gel as compared with monomeric A3G. A3G cross-linked to RNA showed a marked reduction in mobility causing the bands to be positioned near the top of the resolving gel. Monomeric A3G and A3G cross-linked protein bands were cut out from SDS-polyacrylamide gels, washed three times in 25 mm Tris, pH 7.5, 2 mm EDTA, and processed as described previously (44). Briefly, ssDNA-cross-linked samples were digested with 10 μg of DNase I (Sigma) and 300 units of micrococcal nuclease (Thermo Fisher Scientific) for 4–6 h at 37 °C in presence of Mg2+ and Ca2+. The gel slices were washed three times with 25 mm ammonium bicarbonate (AmBc), dehydrated with solution containing 50% acetonitrile and 25 mm AmBc, and dried out in a vacuum-speed centrifuge. The gel slices were rehydrated with 25 mm AmBc, and then two more rounds of the dehydration-rehydration procedure were performed to remove any residual amounts of nucleases or digested nucleic acids. Samples were then treated with 10 mm DTT at 55 °C for 1 h and iodoacetamide at room temperature in the dark for 45 min. The samples then were washed with 25 mm AmBc, dehydrated with 50% acetonitrile and 25 mm AmBc solution, vacuum-dried, and digested overnight with MS-grade trypsin (G-Biosciences, 0.5 μg/μl) in 25 mm AmBc at 1:20 ratio overnight. The extracted peptides were then purified with C18 resin (Thermo Fisher Scientific) as suggested by the manufacturer and concentrated to 10 μl in a vacuum-speed centrifuge.
Analysis of A3G tryptic peptides by mass spectrometry
MS analysis of tryptic peptides was performed using either an LTQ Orbitrap XL or Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific). Samples were adjusted to 0.1% formic acid in water and vortexed before autosampler loaded onto a home-pulled and home-packed C18 analytical column. Columns were pulled to a tip of ∼10 μm with a Sutter laser puller and packed using a pressure bomb to 10 cm with C18AQ 5 μm of 200 Å media (Michrom), ending with an internal column diameter of 75 μm. Columns were equilibrated to initial run conditions prior to loading the sample on the column. Solvent A was 0.1% formic acid in water, and Solvent B was 0.1% formic acid in acetonitrile. Peptides were eluted with the following chromatographic profile: 0% B for 2 min, ramping to 40% B over 23 min then to 70% B over 1 min, remaining at 70% B for 3 min, and finally returning to initial run conditions.
Data were collected as RAW files and converted to MGF files using Bioworks Browser. Initially, when analyzing peptide spectra we took an advantage of the fact that bromine in BrdU-containing ssDNA is present in two naturally occurring stable isotopes (79Br and 81Br, usually at ∼1:1 ratio); therefore, the cross-linked peptides should produce double MS1 peaks with similar intensity and with about 1.998 atomic units difference for +1 m/z charge peptides, about 0.999 unit for +2 m/z charge peptides, etc. It should be noted that because of the presence of other stable isotopes besides 79Br and 81Br in the samples, including 13C, 2H, and 15N, MS1 peptide pattern typically may show 2–4 peaks depending on peptide change. To identify such double 79Br- and 81Br-containing A3G peptides, we used Compass IsotopePattern software (Bruker Daltonics) and searched MS data for BrdU modification, C9H11N2O8P1Br. Although previous studies showed that BrdU (HBr) is often lost after protein cross-linking to nucleic acids (47, 48), we observed full size BrdU modifications of A3G tryptic peptides.
To identify BrdU-modified residues in A3G tryptic peptides, we used MASCOT software (Matrix Science). Search parameters included the following: trypsin as an enzyme; three missed cleavages; 0.05 Da for MS and MS/MS data; 1 for #13C, 2+; +3; +4 for charge state; decoy search and acceptance criteria of a minimum one peptide greater than identity score; minimum score of 20; and false discovery rate less than 5% (87). To identify individual amino acid residues cross-linked to BrdU within A3G peptides determined by Compass IsotopePattern software, Mascot searches were performed for BrdU modifications at cysteine, tyrosine, phenylalanine, tryptophan, histidine, methionine, serine, arginine, or lysine that are all known as being involved in cross-linking to nucleic acids (49). Also, to facilitate identification of such peptides, we created a custom database that included only A3G protein. To verify the A3G-identified cross-linked peptides, we used BioTools software (Bruker Daltonics) to manually analyze MS/MS fragmentation patterns and to determine individual cross-linked residues within these peptides by matching the observed peak values with anticipated modified residues.
A3G mutational activity (DNA mutator assay)
The DNA mutator assay measures the induction of rifampicin resistance in E. coli that is caused by the accumulation of mutations in the rpoB gene encoding the β-subunit of RNA polymerase (51, 53). Briefly, E. coli strain BW310 Hfr[PO-45] relA1 spoT1 thi-1 ung-1 deficient for uracil DNA glycosylase was transformed with pTrc99A vector alone or plasmids encoding WT or mutant A3G. Parental strain KL16, which expresses normal uracil-DNA glycosylase and can repair deaminase-induced dC-to-dU mutations, was used as a control. Single colonies of each transformation in triplicates were grown overnight in the presence of carbenicillin and 1 mm IPTG, and the aliquots of 10-fold dilutions were plated on carbenicillin- and rifampicin-containing plates. Mutation frequency was calculated as the number of rifampicin-resistant colonies per 108 of viable cells after making corrections for the number of colonies on control plates. Each experiment was repeated four times independently.
Glycerol density gradient fractionation
Glycerol density gradient sedimentation was used to determine the association of A3G with RNA as RNP complexes (34, 44). WT and mutant A3G constructs subcloned in pIRES vector (85) were transfected into HEK293T human cells using X-tremeGENE 9 DNA transfection reagent (Roche Applied Science) with puromycin added the next day to 10-cm dishes for selection. Cells were collected at 60–64 h post-transfection time; cell extracts were prepared in buffer 1× DB, 0.5% Triton X-100 with protease inhibitors (Roche Applied Science), and then cell extracts from each dish were split into two halves with one-half treated with RNase A and T1 for 30 min to ensure maximum digestion of RNA. Cell extracts (500 μg) were centrifuged at 200,000 × g for 10 h at 5 °C through 11-ml linear 10–50% glycerol gradients, and 500-μl fractions were collected from the top of each gradient. The absorbance at 280 and 260 nm was determined for each fraction. The aliquots of each third fraction were separated on 12% SDS-polyacrylamide gels, Western blotted, and probed with anti-A3G (C-terminal) rabbit polyclonal antibody (AIDS Reagent Program (National Institutes of Health), catalog no. 10201) to determine the sedimentation distribution of A3G WT and mutants in the gradient (three separate cell extracts independently). The sedimentation of ovalbumin (42 kDa, 3.5S), BSA (67 kDa, 4.2S), aldolase (160 kDa, 7.4S), catalase (250 kDa, 11.3S), and thyroglobulin (660 kDa, 19S) and in vitro assembled spliceosomes (60S) were sedimented as gradient calibration controls. The densitometric quantification of WT A3G from Western blotting signals for each gradient fraction without and with RNase digestion served to calibrate the gradient for the maximum and minimum RNA-dependent A3G sedimentation.
HIV-1 infectivity assay
WT and each of the A3G mutants (N-terminally HA-tagged) were subcloned into pIRES-P vector (85) under CMV promoter and transfected into HEK293T cells with FuGENE HD transfection reagent (Promega) in 6-well dishes. Cells were transfected with a 5–10-fold range of plasmid concentrations to ensure that we determined the amount of plasmid DNAs to produce the same level of protein expression. It should be noted that HEK293T cells do not naturally express A3G. 48 h post-transfection, the cells were lysed with Reporter Lysis buffer (Promega), and whole-cell extracts together with H9 human T-cell extract as control were probed by Western blotting with anti-A3G (C-terminal) rabbit polyclonal antibody (AIDS Reagent Program (National Institutes of Health), catalog no. 10201). The Western blotting signals were quantified by scanning densitometry to identify the amount of transfecting plasmids that enabled A3G and mutants expression equal to that expressed naturally in H9 cells.
The antiviral activity of A3G mutants was measured by a single-round infectivity assay with pseudotyped HIV-1 produced in HEK293T cells. The HIV proviral vector pDHIV3-GFP encodes for all HIV genes except nef (replaced with enhanced GFP) and env. The pΔVif-DHIV3-GFP proviral vector in addition contains an early stop codon within the Vif gene. Use of this ΔVif virus was necessary so that A3G could be assembled with virions and not subjected to Vif-dependent degradation. HEK293T cells were co-transfected with the pΔVif-DHIV3-GFP, plasmid encoding VSV-G coat protein (pVSV-G) that provides the env gene function in trans position and WT A3G or A3G mutant constructs. Media on the plates were replaced 4 h after transfection, and then media were harvested 20 h later for viral particle isolation. The pseudotyped virus was normalized by p24 ELISA (PerkinElmer Life Sciences), and 500 pg of protein in p24 equivalents were used to infect the reporter TZM-bl cells in 96-well plates in triplicate. In this reporter cell line infectivity is proportional to the expression of a stable firefly LTR-luciferase gene that is driven by the HIV LTR promoter (88). 48 h after infection SteadyGlo Reagent (Promega) was added to each well for 30 min. Luminescence units (RLU) were quantified, and the relative percent change in infectivity due to WT A3G or RNA-binding mutants in cells without Vif was determined from the range established by the infectivity of WT A3G with Vif containing virus set as the maximum infectivity.
A3G WT and mutant protein expression levels in cell extracts were evaluated by Western blottings with HA antibody and normalized by the level of GAPDH protein expression. Viral particles (50 ng of protein in p24 equivalents) were isolated by passing the media through 0.45-micron filters, clearing by 1000 × g centrifugation for 10 min, and then pelleting through a 20% sucrose cushion by ultracentrifugation (Beckman SW 41 rotor, 25,000 rpm, 2 h at 4 °C). The content of A3G in each viral preparation was determined by densitometric scans of Western blottings probed with monoclonal anti-p24 (AIDS Reagent Program (National Institutes of Health), catalog no. 3537) as loading control and anti-HA antibody to detect either WT or mutant A3G proteins.
Statistical analysis
The statistical analyses for nucleic acid binding to A3G and functional end point data were done using GraphPad Prism 6 (GraphPad, La Jolla, CA) programs.
Author contributions
B. P. and H. C. S. designed the research; B. P., R. J., R. P. B., R. G., T. D. S., and R. A. S. performed the research; B. P., R. J., A. E. F., R. P. B., and H. C. S. analyzed the data; and B. P., R. P. B. and H. C. S. wrote the paper.
Supplementary Material
Acknowledgments
We are thankful to Kevin Welle (University of Rochester Medical Center Mass Spectrometry Resource Laboratory) for running MS samples and Elizabeth Snell (Hauptman-Woodward Medical Research Institute, Buffalo, NY) for helping with thermal shift assay experiments.
This work was supported in part by National Institutes of Health Grant NIGMS110568 (to H. C. S.) and by a grant from the University of Rochester Center for AIDS Research (CFAR), a National Institutes of Health-funded program by Grant P30 AI078498. Authors associated with OyaGen, Inc. declare that they are pursuing antagonists of Vif as a means of enabling A3G innate immunity toward therapeutics and cure for HIV as a potential conflict of interest; the pseudotype virus infectivity studies on RNA-binding mutants performed in this paper are of basic science and not of immediate commercial interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S4 and Tables S1–S2.
- A3G
- APOBEC3G
- CD1/2
- catalytic domain 1/2
- CDA
- cytidine deaminase
- FA
- fluorescence anisotropy
- RNP
- ribonucleoprotein
- ssDNA
- single stranded DNA
- nt
- nucleotide
- RLU
- relative luminescence unit
- aa
- amino acid
- AmBc
- ammonium bicarbonate
- BU
- 5-bromouracil
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside.
References
- 1. Salter J. D., Bennett R. P., and Smith H. C. (2016) The APOBEC protein family: united by structure, divergent in function. Trends Biochem. Sci. 41, 578–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith H. C. (2016) RNA binding to APOBEC deaminases; not simply a substrate for C to U editing. RNA Biol. 2016, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harris R. S., and Dudley J. P. (2015) APOBECs and virus restriction. Virology 479, 131–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sharma S., Patnaik S. K., Taggart R. T., and Baysal B. E. (2016) The double-domain cytidine deaminase APOBEC3G is a cellular site-specific RNA editing enzyme. Sci. Rep. 6, 39100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharma S., Patnaik S. K., Taggart R. T., Kannisto E. D., Enriquez S. M., Gollnick P., and Baysal B. E. (2015) APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 6, 6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shlyakhtenko L. S., Lushnikov A. Y., Miyagi A., Li M., Harris R. S., and Lyubchenko Y. L. (2013) Atomic force microscopy studies of APOBEC3G oligomerization and dynamics. J. Struct. Biol. 184, 217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huthoff H., and Malim M. H. (2005) Cytidine deamination and resistance to retroviral infection: towards a structural understanding of the APOBEC proteins. Virology 334, 147–153 [DOI] [PubMed] [Google Scholar]
- 8. Smith H. C. (2011) APOBEC3G: a double agent in defense. Trends Biochem. Sci. 36, 239–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Refsland E. W., and Harris R. S. (2013) The APOBEC3 family of retroelement restriction factors. Curr. Top. Microbiol. Immunol. 371, 1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Polevoda B., McDougall W. M., Tun B. N., Cheung M., Salter J. D., Friedman A. E., and Smith H. C. (2015) RNA binding to APOBEC3G induces the disassembly of functional deaminase complexes by displacing single-stranded DNA substrates. Nucleic Acids Res. 43, 9434–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. York A., Kutluay S. B., Errando M., and Bieniasz P. D. (2016) The RNA binding specificity of human APOBEC3 proteins resembles that of HIV-1 nucleocapsid. PLoS Pathog. 12, e1005833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Apolonia L., Schulz R., Curk T., Rocha P., Swanson C. M., Schaller T., Ule J., and Malim M. H. (2015) Promiscuous RNA binding ensures effective encapsidation of APOBEC3 proteins by HIV-1. PLoS Pathog. 11, e1004609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Friew Y. N., Boyko V., Hu W. S., and Pathak V. K. (2009) Intracellular interactions between APOBEC3G, RNA, and HIV-1 Gag: APOBEC3G multimerization is dependent on its association with RNA. Retrovirology 6, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bogerd H. P., and Cullen B. R. (2008) Single-stranded RNA facilitates nucleocapsid: APOBEC3G complex formation. RNA 14, 1228–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huthoff H., Autore F., Gallois-Montbrun S., Fraternali F., and Malim M. H. (2009) RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 5, e1000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang W., Du J., Yu K., Wang T., Yong X., and Yu X. F. (2010) Association of potent human antiviral cytidine deaminases with 7SL RNA and viral RNP in HIV-1 virions. J. Virol. 84, 12903–12913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khan M. A., Goila-Gaur R., Opi S., Miyagi E., Takeuchi H., Kao S., and Strebel K. (2007) Analysis of the contribution of cellular and viral RNA to the packaging of APOBEC3G into HIV-1 virions. Retrovirology 4, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Svarovskaia E. S., Xu H., Mbisa J. L., Barr R., Gorelick R. J., Ono A., Freed E. O., Hu W. S., and Pathak V. K. (2004) Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J. Biol. Chem. 279, 35822–35828 [DOI] [PubMed] [Google Scholar]
- 19. Chiu Y. L., Witkowska H. E., Hall S. C., Santiago M., Soros V. B., Esnault C., Heidmann T., and Greene W. C. (2006) High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. U.S.A. 103, 15588–15593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Soros V. B., Yonemoto W., and Greene W. C. (2007) Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 3, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kozak S. L., Marin M., Rose K. M., Bystrom C., and Kabat D. (2006) The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J. Biol. Chem. 281, 29105–29119 [DOI] [PubMed] [Google Scholar]
- 22. Gallois-Montbrun S., Kramer B., Swanson C. M., Byers H., Lynham S., Ward M., and Malim M. H. (2007) Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 81, 2165–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wichroski M. J., Robb G. B., and Rana T. M. (2006) Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog. 2, e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kreisberg J. F., Yonemoto W., and Greene W. C. (2006) Endogenous factors enhance HIV infection of tissue naive CD4 T cells by stimulating high molecular mass APOBEC3G complex formation. J. Exp. Med. 203, 865–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stopak K. S., Chiu Y. L., Kropp J., Grant R. M., and Greene W. C. (2007) Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J. Biol. Chem. 282, 3539–3546 [DOI] [PubMed] [Google Scholar]
- 26. Shaban N. M., Shi K., Li M., Aihara H., and Harris R. S. (2016) 1.92 Angstrom zinc-free APOBEC3F catalytic domain crystal structure. J. Mol. Biol. 428, 2307–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shandilya S. M., Nalam M. N., Nalivaika E. A., Gross P. J., Valesano J. C., Shindo K., Li M., Munson M., Royer W. E., Harjes E., Kono T., Matsuo H., Harris R. S., Somasundaran M., and Schiffer C. A. (2010) Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure 18, 28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marx A., Galilee M., and Alian A. (2015) Zinc enhancement of cytidine deaminase activity highlights a potential allosteric role of loop-3 in regulating APOBEC3 enzymes. Sci. Rep. 5, 18191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Navarro F., Bollman B., Chen H., König R., Yu Q., Chiles K., and Landau N. R. (2005) Complementary function of the two catalytic domains of APOBEC3G. Virology 333, 374–386 [DOI] [PubMed] [Google Scholar]
- 30. Wang T., Tian C., Zhang W., Sarkis P. T., and Yu X. F. (2008) Interaction with 7SL RNA but not with HIV-1 genomic RNA or P bodies is required for APOBEC3F virion packaging. J. Mol. Biol. 375, 1098–1112 [DOI] [PubMed] [Google Scholar]
- 31. Chen K. M., Harjes E., Gross P. J., Fahmy A., Lu Y., Shindo K., Harris R. S., and Matsuo H. (2008) Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 452, 116–119 [DOI] [PubMed] [Google Scholar]
- 32. Iwatani Y., Takeuchi H., Strebel K., and Levin J. G. (2006) Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 80, 5992–6002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chelico L., Prochnow C., Erie D. A., Chen X. S., and Goodman M. F. (2010) Structural model for deoxycytidine deamination mechanisms of the HIV-1 inactivation enzyme APOBEC3G. J. Biol. Chem. 285, 16195–16205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McDougall W. M., Okany C., and Smith H. C. (2011) Deaminase activity on single-stranded DNA (ssDNA) occurs in vitro when APOBEC3G cytidine deaminase forms homotetramers and higher-order complexes. J. Biol. Chem. 286, 30655–30661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shlyakhtenko L. S., Lushnikov A. Y., Li M., Lackey L., Harris R. S., and Lyubchenko Y. L. (2011) Atomic force microscopy studies provide direct evidence for dimerization of the HIV restriction factor APOBEC3G. J. Biol. Chem. 286, 3387–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McDougall W. M., and Smith H. C. (2011) Direct evidence that RNA inhibits APOBEC3G ssDNA cytidine deaminase activity. Biochem. Biophys. Res. Commun. 412, 612–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wedekind J. E., Gillilan R., Janda A., Krucinska J., Salter J. D., Bennett R. P., Raina J., and Smith H. C. (2006) Nanostructures of APOBEC3G support a hierarchical assembly model of high molecular mass ribonucleoprotein particles from dimeric subunits. J. Biol. Chem. 281, 38122–38126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Holden L. G., Prochnow C., Chang Y. P., Bransteitter R., Chelico L., Sen U., Stevens R. C., Goodman M. F., and Chen X. S. (2008) Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature 456, 121–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Furukawa A., Nagata T., Matsugami A., Habu Y., Sugiyama R., Hayashi F., Kobayashi N., Yokoyama S., Takaku H., and Katahira M. (2009) Structure and real-time monitoring of the enzymatic reaction of APOBEC3G which is involved in anti-HIV activity. Nucleic Acids Symp. Ser. 2009, 53, 87–88 [DOI] [PubMed] [Google Scholar]
- 40. Lu X., Zhang T., Xu Z., Liu S., Zhao B., Lan W., Wang C., Ding J., and Cao C. (2015) Crystal structure of DNA cytidine deaminase ABOBEC3G catalytic deamination domain suggests a binding mode of full-length enzyme to single-stranded DNA. J. Biol. Chem. 290, 4010–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kouno T., Luengas E. M., Shigematsu M., Shandilya S. M., Zhang J., Chen L., Hara M., Schiffer C. A., Harris R. S., and Matsuo H. (2015) Structure of the Vif-binding domain of the antiviral enzyme APOBEC3G. Nat. Struct. Mol. Biol. 22, 485–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shi K., Carpenter M. A., Banerjee S., Shaban N. M., Kurahashi K., Salamango D. J., McCann J. L., Starrett G. J., Duffy J. V., Demir Ö., Amaro R. E., Harki D. A., Harris R. S., and Aihara H. (2017) Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 24, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xiao X., Li S. X., Yang H., and Chen X. S. (2016) Crystal structures of APOBEC3G N-domain alone and its complex with DNA. Nat. Commun. 7, 12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Polevoda B., McDougall W. M., Bennett R. P., Salter J. D., and Smith H. C. (2016) Structural and functional assessment of APOBEC3G macromolecular complexes. Methods 107, 10–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dietz T. M., and Koch T. H. (1987) Photochemical coupling of 5-bromouracil to tryptophan, tyrosine and histidine, peptide-like derivatives in aqueous fluid solution. Photochem. Photobiol. 46, 971–978 [DOI] [PubMed] [Google Scholar]
- 46. Gott J. M., Willis M. C., Koch T. H., and Uhlenbeck O. C. (1991) A specific, UV-induced RNA-protein cross-link using 5-bromouridine-substituted RNA. Biochemistry 30, 6290–6295 [DOI] [PubMed] [Google Scholar]
- 47. Willis M. C., LeCuyer K. A., Meisenheimer K. M., Uhlenbeck O. C., and Koch T. H. (1994) An RNA-protein contact determined by 5-bromouridine substitution, photocrosslinking and sequencing. Nucleic Acids Res. 22, 4947–4952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kramer K., Huummel P., Hsiao H.-H., Luo X., Wahl M., and Urlaub H. (2011) Mass-spectrometric analysis of proteins cross-linked to 4-thio-uracil- and 5-bromo-uracil-substituted RNA. Int. J. Mass Spectrometry 304, 184–194 [Google Scholar]
- 49. Meisenheimer K. M., and Koch T. H. (1997) Photocross-linking of nucleic acids to associated proteins. Crit. Rev. Biochem. Mol. Biol. 32, 101–140 [DOI] [PubMed] [Google Scholar]
- 50. Boivin S., Kozak S., and Meijers R. (2013) Optimization of protein purification and characterization using Thermofluor screens. Protein Expr. Purif. 91, 192–206 [DOI] [PubMed] [Google Scholar]
- 51. Petersen-Mahrt S. K., Harris R. S., and Neuberger M. S. (2002) AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 418, 99–103 [DOI] [PubMed] [Google Scholar]
- 52. Harris R. S., Petersen-Mahrt S. K., and Neuberger M. S. (2002) RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 10, 1247–1253 [DOI] [PubMed] [Google Scholar]
- 53. Smith H. C. (2007) Measuring editing activity and identifying cytidine-to-uridine mRNA editing factors in cells and biochemical isolates. Methods Enzymol. 424, 389–416 [DOI] [PubMed] [Google Scholar]
- 54. Aydin H., Taylor M. W., and Lee J. E. (2014) Structure-guided analysis of the human APOBEC3-HIV restrictome. Structure 22, 668–684 [DOI] [PubMed] [Google Scholar]
- 55. Ma J., Li X., Xu J., Zhang Q., Liu Z., Jia P., Zhou J., Guo F., You X., Yu L., Zhao L., Jiang J., and Cen S. (2013) The roles of APOBEC3G complexes in the incorporation of APOBEC3G into HIV-1. PLoS ONE 8, e74892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bélanger K., and Langlois M. A. (2015) RNA-binding residues in the N terminus of APOBEC3G influence its DNA sequence specificity and retrovirus restriction efficiency. Virology 483, 141–148 [DOI] [PubMed] [Google Scholar]
- 57. Wang T., Tian C., Zhang W., Luo K., Sarkis P. T., Yu L., Liu B., Yu Y., and Yu X. F. (2007) 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J. Virol. 81, 13112–13124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bach D., Peddi S., Mangeat B., Lakkaraju A., Strub K., and Trono D. (2008) Characterization of APOBEC3G binding to 7SL RNA. Retrovirology 5, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Smith H. C., Bennett R. P., Kizilyer A., McDougall W. M., and Prohaska K. M. (2012) Functions and regulation of the APOBEC family of proteins. Semin. Cell Dev. Biol. 23, 258–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schäfer A., Bogerd H. P., and Cullen B. R. (2004) Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology 328, 163–168 [DOI] [PubMed] [Google Scholar]
- 61. Strebel K., and Khan M. A. (2008) APOBEC3G encapsidation into HIV-1 virions: which RNA is it? Retrovirology 5, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tian C., Wang T., Zhang W., and Yu X. F. (2007) Virion packaging determinants and reverse transcription of SRP RNA in HIV-1 particles. Nucleic Acids Res. 35, 7288–7302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cen S., Guo F., Niu M., Saadatmand J., Deflassieux J., and Kleiman L. (2004) The interaction between HIV-1 Gag and APOBEC3G. J. Biol. Chem. 279, 33177–33184 [DOI] [PubMed] [Google Scholar]
- 64. Alce T. M., and Popik W. (2004) APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J. Biol. Chem. 279, 34083–34086 [DOI] [PubMed] [Google Scholar]
- 65. Luo K., Liu B., Xiao Z., Yu Y., Yu X., Gorelick R., and Yu X. F. (2004) Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J. Virol. 78, 11841–11852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu H., Chertova E., Chen J., Ott D. E., Roser J. D., Hu W. S., and Pathak V. K. (2007) Stoichiometry of the antiviral protein APOBEC3G in HIV-1 virions. Virology 360, 247–256 [DOI] [PubMed] [Google Scholar]
- 67. Iwatani Y., Chan D. S., Wang F., Maynard K. S., Sugiura W., Gronenborn A. M., Rouzina I., Williams M. C., Musier-Forsyth K., and Levin J. G. (2007) Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 35, 7096–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guo F., Cen S., Niu M., Yang Y., Gorelick R. J., and Kleiman L. (2007) The interaction of APOBEC3G with human immunodeficiency virus type 1 nucleocapsid inhibits tRNA3Lys annealing to viral RNA. J. Virol. 81, 11322–11331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Luo K., Wang T., Liu B., Tian C., Xiao Z., Kappes J., and Yu X. F. (2007) Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J. Virol. 81, 7238–7248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Holmes R. K., Koning F. A., Bishop K. N., and Malim M. H. (2007) APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 282, 2587–2595 [DOI] [PubMed] [Google Scholar]
- 71. Li X. Y., Guo F., Zhang L., Kleiman L., and Cen S. (2007) APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J. Biol. Chem. 282, 32065–32074 [DOI] [PubMed] [Google Scholar]
- 72. Dang Y., Abudu A., Son S., Harjes E., Spearman P., Matsuo H., and Zheng Y. H. (2011) Identification of a single amino acid required for APOBEC3 antiretroviral cytidine deaminase activity. J. Virol. 85, 5691–5695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Siu K. K., Sultana A., Azimi F. C., and Lee J. E. (2013) Structural determinants of HIV-1 Vif susceptibility and DNA binding in APOBEC3F. Nat. Commun. 4, 2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Langlois M. A., Beale R. C., Conticello S. G., and Neuberger M. S. (2005) Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 33, 1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rathore A., Carpenter M. A., Demir Ö., Ikeda T., Li M., Shaban N. M., Law E. K., Anokhin D., Brown W. L., Amaro R. E., and Harris R. S. (2013) The local dinucleotide preference of APOBEC3G can be altered from 5′-CC to 5′-TC by a single amino acid substitution. J. Mol. Biol. 425, 4442–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Salter J. D., Krucinska J., Raina J., Smith H. C., and Wedekind J. E. (2009) A hydrodynamic analysis of APOBEC3G reveals a monomer-dimer-tetramer self-association that has implications for anti-HIV function. Biochemistry 48, 10685–10687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Harjes S., Solomon W. C., Li M., Chen K. M., Harjes E., Harris R. S., and Matsuo H. (2013) Impact of H216 on the DNA binding and catalytic activities of the HIV restriction factor APOBEC3G. J. Virol. 87, 7008–7014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Feng Y., and Chelico L. (2011) Intensity of deoxycytidine deamination of HIV-1 proviral DNA by the retroviral restriction factor APOBEC3G is mediated by the noncatalytic domain. J. Biol. Chem. 286, 11415–11426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Li X., Ma J., Zhang Q., Zhou J., Yin X., Zhai C., You X., Yu L., Guo F., Zhao L., Li Z., Zeng Y., and Cen S. (2011) Functional analysis of the two cytidine deaminase domains in APOBEC3G. Virology 414, 130–136 [DOI] [PubMed] [Google Scholar]
- 80. An P., Bleiber G., Duggal P., Nelson G., May M., Mangeat B., Alobwede I., Trono D., Vlahov D., Donfield S., Goedert J. J., Phair J., Buchbinder S., O'Brien S. J., Telenti A., and Winkler C. A. (2004) APOBEC3G genetic variants and their influence on the progression to AIDS. J. Virol. 78, 11070–11076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ara A., Love R. P., and Chelico L. (2014) Different mutagenic potential of HIV-1 restriction factors APOBEC3G and APOBEC3F is determined by distinct single-stranded DNA scanning mechanisms. PLoS Pathog. 10, e1004024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chelico L., Sacho E. J., Erie D. A., and Goodman M. F. (2008) A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J. Biol. Chem. 283, 13780–13791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pan Y., Sun Z., Maiti A., Kanai T., Matsuo H., Li M., Harris R. S., Shlyakhtenko L. S., and Lyubchenko Y. L. (2017) Nanoscale characterization of interaction of APOBEC3G with RNA. Biochemistry 56, 1473–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bulliard Y., Turelli P., Röhrig U. F., Zoete V., Mangeat B., Michielin O., and Trono D. (2009) Functional analysis and structural modeling of human APOBEC3G reveal the role of evolutionarily conserved elements in the inhibition of human immunodeficiency virus type 1 infection and Alu transposition. J. Virol. 83, 12611–12621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hobbs S., Jitrapakdee S., and Wallace J. C. (1998) Development of a bicistronic vector driven by the human polypeptide chain elongation factor 1α promoter for creation of stable mammalian cell lines that express very high levels of recombinant proteins. Biochem. Biophys. Res. Commun. 252, 368–372 [DOI] [PubMed] [Google Scholar]
- 86. Kramer K., Sachsenberg T., Beckmann B. M., Qamar S., Boon K. L., Hentze M. W., Kohlbacher O., and Urlaub H. (2014) Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat. Methods 11, 1064–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lapek J. D. Jr., McGrath J. L., Ricke W. A., and Friedman A. E. (2012) LC/LC-MS/MS of an innovative prostate human epithelial cancer (PHEC) in vitro model system. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 893, 34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Platt E. J., Wehrly K., Kuhmann S. E., Chesebro B., and Kabat D. (1998) Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 72, 2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.