

Abstract
MF6p/FhHDM-1 is a small protein secreted by the parasitic flatworm (trematode) Fasciola hepatica that belongs to a broad family of heme-binding proteins (MF6p/helminth defense molecules (HDMs)). MF6p/HDMs are of interest for understanding heme homeostasis in trematodes and as potential targets for the development of new flukicides. Moreover, interest in these molecules has also increased because of their immunomodulatory properties. Here we have extended our previous findings on the mechanism of MF6p/HDM-heme interactions and mapped the protein regions required for heme binding and for other biological functions. Our data revealed that MF6p/FhHDM-1 forms high-molecular-weight complexes when associated with heme and that these complexes are reorganized by a stacking procedure to form fibril-like and granular nanostructures. Furthermore, we showed that MF6p/FhHDM-1 is a transitory heme-binding protein as protein·heme complexes can be disrupted by contact with an apoprotein (e.g. apomyoglobin) with higher affinity for heme. We also demonstrated that (i) the heme-binding region is located in the MF6p/FhHDM-1 C-terminal moiety, which also inhibits the peroxidase-like activity of heme, and (ii) MF6p/HDMs from other trematodes, such as Opisthorchis viverrini and Paragonimus westermani, also bind heme. Finally, we observed that the N-terminal, but not the C-terminal, moiety of MF6p/HDMs has a predicted structural analogy with cell-penetrating peptides and that both the entire protein and the peptide corresponding to the N-terminal moiety of MF6p/FhHDM-1 interact in vitro with cell membranes in hemin-preconditioned erythrocytes. Our findings suggest that MF6p/HDMs can transport heme in trematodes and thereby shield the parasite from the harmful effects of heme.
Keywords: cell-penetrating peptide (CPP), heme, homeostasis, oligomerization, parasite, Fasciola, fluke, MF6p/FhHDM-1, MF6p/HDM, trematode
Introduction
Trematodiasis (or trematodosis) refers to a large group of important parasitic diseases caused by infection with trematodes, also known as flatworms or flukes. Considering only the more pathogenic foodborne liver flukes (Clonorchis sinensis, Opisthorchis spp., and Fasciola spp.) and foodborne lung flukes (Paragonimus spp.), more than one billion people are estimated to be at risk of infection (1). Although human infections by trematodes are particularly abundant in Southeast Asia and the Western Pacific region, those caused by Fasciola (i.e. Fasciola hepatica and Fasciola gigantica) and Paragonimus species have a much wider geographic distribution (1, 2).
Given the medical relevance of trematodiasis, considerable effort is being made to obtain genomic and transcriptomic data on foodborne trematodes (for available data, see for example Genomes OnLine Database and WormBase ParaSite). Use of these data, together with bioinformatics tools, has made it possible to identify relevant molecules of possible interest as targets for the development of new antiparasite drugs and vaccines. Unfortunately, most of the reported parasite proteins have been identified by sequence homology, and their physiological function in the parasites has not been demonstrated experimentally (3–5). Moreover, predicting the function of identified proteins is very difficult when they do not contain domains or exhibit substantial homology to any known protein from other well characterized organisms.
Previous studies (6), including a study by our research group (7), have identified and cloned a small protein (molecular mass, 7.8 kDa) that is abundant in somatic and secretory antigens (SAs)3 of F. hepatica and that is recognized by the MF6 monoclonal antibody (mAb). This protein was initially erroneously identified as Fasciola hemoglobin (8), and it was later renamed FhHDM-1 on the basis of its homology with cathelicidin-like host defense peptides (6, 9). However, after recognizing its heme-binding nature, we renamed the protein MF6p/FhHDM-1 (7).
The physiological role of MF6p/FhHDM-1 in the parasite has not yet been convincingly demonstrated; however, because of its ability to bind to lipopolysaccharide (LPS) and to prevent LPS-induced inflammation in mice (6) as well as its ability to suppress antigen processing and presentation in macrophages (9), it has been suggested that the protein may favor parasite survival by suppressing the host immune response (10). Nevertheless, we previously suggested that the protein may play a role in heme homeostasis on the basis of its heme-binding nature and the fact that it accumulates in several internal fluke tissues, mainly parenchymal cells, vitellaria, testicular tubules, and the basal lamina under the tegument (7). To gain further insight into the mode of action of MF6p/HDM proteins, we have carried out a detailed study of the binding characteristics of these proteins in relation to heme and LPS. From the results of the analysis of the Fasciola MF6p/FhHDM-1 and protein orthologs from related trematodes, we conclude that the MF6p/HDM family of proteins (Fig. 1A) are transitory heme-binding proteins probably involved in heme scavenging, storage, and transport in trematodes.
Figure 1.

Sequence alignment of the MF6p/HDM family of proteins. A, aligned sequences of complete mature orthologous proteins present in F. hepatica and related trematodes. The 28–29 regions considered for bioinformatics analyses are underlined in red (N-terminal fragment) and blue (C-terminal fragment). B, aligned sequences of the synthetic peptides used in the present study. Amino acid residues in common with F. hepatica are shaded in green, and residues shared among trematodes other than F. hepatica are shaded in gray. GB, GenBankTM.
Results
The Fasciola MF6p/FhHDM-1 protein promotes the formation of high-molecular-mass protein·hemin complexes
Despite its low molecular mass (7,828 Da), native MF6p/FhHDM-1 (nMF6p/FhHDM-1) has previously been observed to form high-molecular-mass complexes in F. hepatica SAs (6–8). However, the nature of this phenomenon remained unknown until now. Demonstration that MF6p/FhHDM-1 is a heme-binding protein (7) led us to suspect that heme molecules may be involved in the formation of these complexes. To investigate the mechanism of multimerization, we determined the molecular mass of the species formed after mixing a synthetic form of MF6p/FhHDM-1 (sMF6p/FhHDM-1; Fig. 1B) with hemin at different molar ratios. The experiment was performed in an FPLC chromatographic system equipped with a Superdex 75 HR size-exclusion chromatography (SEC) column with hemin (ferric protoporphyrin IX) diluted in TBS buffer (50 mm Tris, 150 mm NaCl, pH 7.3) supplemented with 25 mm caffeine (TBS-C). Caffeine was added to the TBS buffer to prevent aggregation of hemin in the aqueous medium (11, 12). This strategy is probably more effective than the use of non-ionic detergents, which have also been shown to maintain hemin in its monomeric form (13), as they form micelles and may cause dissociation of protein complexes or break hydrophobic interactions between the proteins and heme (14, 15). As caffeine absorbs at 280 nm, a wavelength typically used to measure the optical absorption of proteins, only hemin absorbance was monitored (395 nm). A comparison of the chromatograms obtained for the heme-containing species present in natural F. hepatica SAs and those formed after mixing sMF6p/FhHDM-1 with hemin at different molar ratios prior to loading is shown in Fig. 2. Regarding the elution profile of natural SAs (red line), the second peak, which appeared at an elution volume of 8.36 ml, corresponds to the main fraction of high-molecular-mass nMF6p/FhHDM-1·hemin complexes (≥150 kDa), whereas the peak at 12.21 ml corresponds to other heme-containing species in the range of 25–28 kDa, the same as Fasciola cathepsins (7). The remaining chromatograms correspond to the elution profiles of sMF6p/FhHDM-1·hemin complexes. The sMF6p/FhHDM-1 mixed with hemin at a molar ratio of 0.5:1 (blue line) eluted as a main peak at 10.23 ml, although a shoulder of higher molecular mass (arrow) was also observed. However, when the protein to hemin ratio was increased to 1:1 (black line) and 2:1 (green line), the high-molecular-mass complexes increased proportionally (arrows). The molecular mass of some of the complexes was equivalent to that of the native protein. To rule out any possible effects caused by the presence of caffeine in the running buffer and to study the propensity of sMF6p/FhHDM-1 to form such high-molecular-mass complexes, we first injected a 0.2-ml sample of 0.06 mm hemin in PBS into the column. As expected (16), hemin interacted with resin beads, and no relevant product was observed in the eluate when either PBS or 0.5 m NaOH was used as running buffer (data not shown). Nevertheless, when 0.2 ml of 0.12 mm sMF6p/FhHDM-1 in PBS was injected into the column, the hemin (Fig. 3A, red line) trapped by the column was eluted together with the protein (Fig. 3A, black line) as a main peak at 11.38 ml and with additional species of higher molecular mass (overlapping peaks at 9.97 and 8.56 ml). This was again within the range of the native protein eluted in PBS (Fig. 3B; elution volume, 8.98 ml). Injection of sMF6p/FhHDM-1 into a clean column eluted as a single peak at 11.23 ml (Fig. 3C) corresponding to an apparent molecular mass of 35–40 kDa, which is higher than the true molecular mass of the protein (7.8 kDa), and coinciding with the main peak obtained for the sMF6p/FhHDM-1·hemin complexes observed in Fig. 3A. Together, these data suggest that the sMF6p/FhHDM-1 protein forms oligomers that associate with hemin to form protein·hemin complexes of higher molecular mass.
Figure 2.
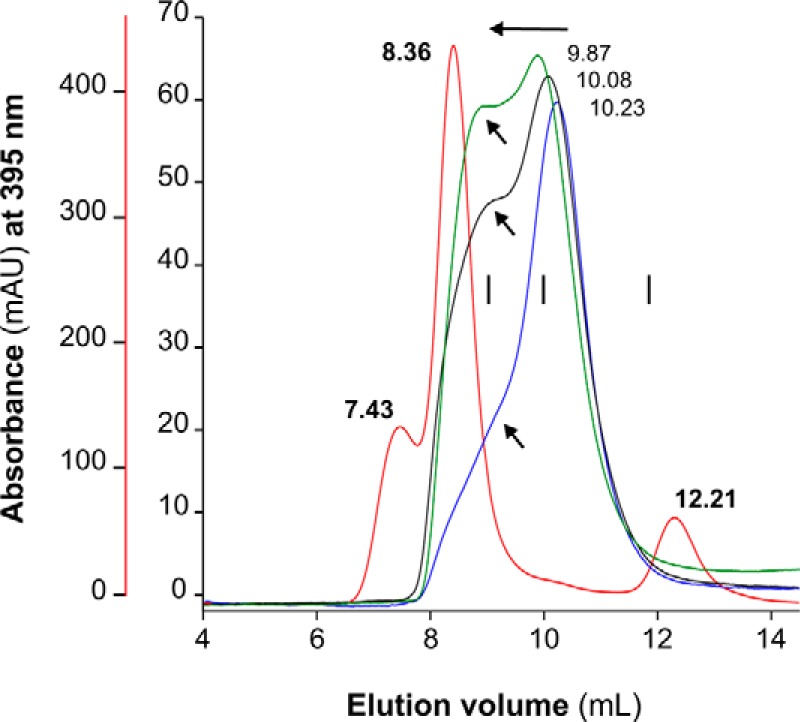
Fractionation of sMF6p/FhHDM-1·hemin complexes by SEC. Elution profiles obtained by SEC (Superdex 75 HR 10/30) of 0.06 mm hemin samples preincubated with sMF6p/FhHDM-1 at protein to hemin molar ratios of 0.5:1 (blue), 1:1 (black), and 2:1 (green) in TBS-C. A sample of F. hepatica SAs diluted in TBS-C (red) was also analyzed for comparison (red vertical axis). Absorbance at a wavelength of 395 nm is shown for heme, and the elution volume of the major peaks is indicated above each peak. The following internal standards are indicated (vertical bars): albumin (67 kDa), ovalbumin (43 kDa), and chymotrypsin (25 kDa). Arrows indicate the tendency of sMF6p/FhHDM-1·hemin to form higher-molecular-mass complexes as the molar ratio of protein to hemin increased. Some of these complexes were eluted at a retention volume equivalent to nMF6p/FhHDM-1·heme complexes present in SAs (8.36 ml). mAU, milliabsorbance units.
Figure 3.
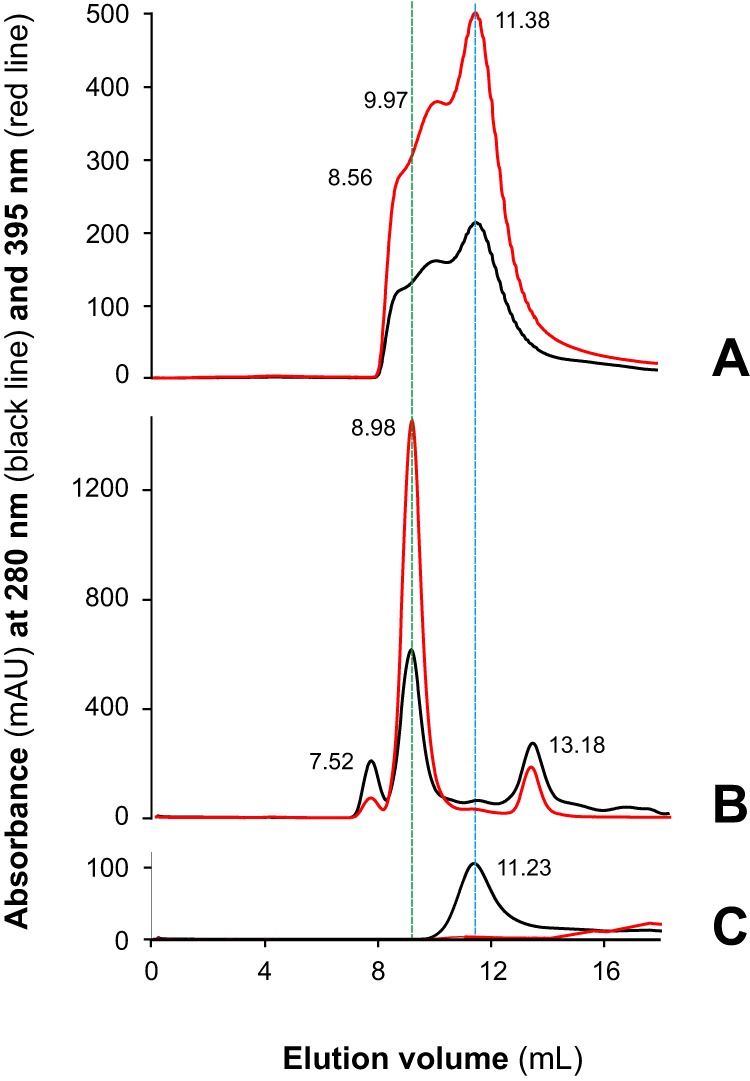
The sMF6p/FhHDM-1 protein tends to oligomerize and form high-molecular-mass complexes with hemin. Shown are elution profiles obtained by SEC (Superdex 75 HR 10/30) of sMF6p/FhHDM-1 (0.2-ml sample at 0.12 mm in PBS) applied to a column to which hemin (0.2-ml sample at 0.06 mm in PBS) was previously adsorbed (A), F. hepatica SAs diluted in PBS (B), and a sample of sMF6p/FhHDM-1 applied to a clean column in the absence of hemin (C). Absorbance at wavelengths of 280 (black) and 395 nm (red) are shown for protein and hemin, respectively. The elution volume of the major peaks is indicated above each peak. Hemin trapped by the column beads in A was eluted with sMF6p/FhHDM-1, forming complexes of several molecular masses between that of nMF6p/FhHDM-1·heme complexes (elution volume, 8.98 ml; B), and sMF6p/FhHDM-1 (elution volume, 11.23 ml; C), which forms oligomers of 35–40 kDa. mAU, milliabsorbance units.
The interaction between native Fasciola MF6p/FhHDM-1 and hemin leads to the formation of filiform and granular nanostructures
To obtain more information about the characteristics of the interaction between MF6p/FhHDM-1 and heme that leads to the formation of high-molecular-mass complexes, we investigated the presence of visible structures under cryogenic transmission electron microscopy (cryo-TEM). This technique is frequently used to determine the 3D structure of protein complexes and viruses (17) and to obtain structural information at the nanometric level about self-assembled structures in the native aqueous environment (18). We used samples of nMF6p/FhHDM-1 protein purified from SAs by affinity chromatography with mAb MF6, neutralized with Tris, and dialyzed against distilled water. Native MF6p/FhHDM-1·hemin complexes present in SAs form variable fibril-like structures of 50–200 nm in length (Fig. 4, A–D, yellow arrows) and that may occasionally circularize. Small (5–20 nm in diameter) electron-dense granule-like structures were also observed in some fields (Fig. 4, B and C, white asterisks).
Figure 4.
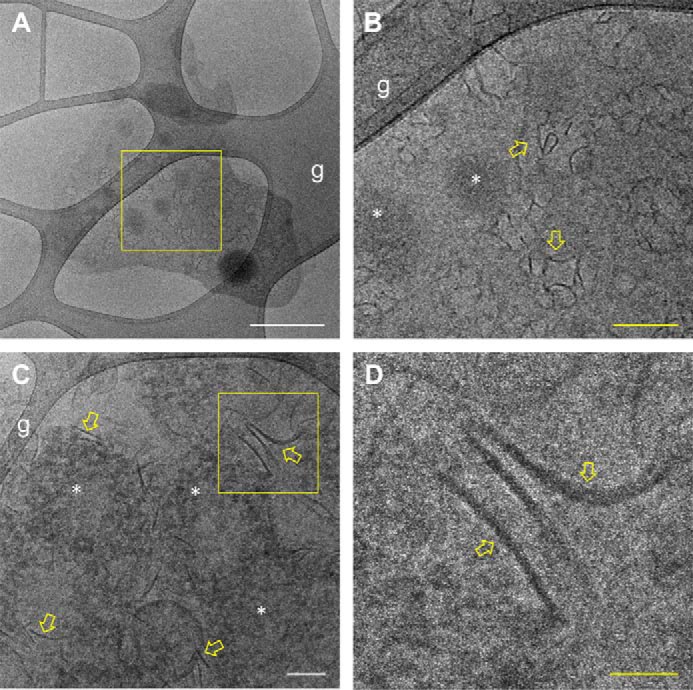
Cryo-TEM analysis of the structure of nMF6p/FhHDM-1·hemin complexes present in F. hepatica SAs. A sample of a solution of immunopurified nMF6p/FhHDM-1 prepared in distilled water was analyzed by cryo-TEM. A, Grid 1, low-magnification cryo-TEM micrograph showing an overall view of nMF6p/FhHDM-1·hemin complexes extended over the lacey carbon grid (g). White scale bar, 400 nm. B, a higher magnification of the square region marked in yellow in A showing the presence of fibril-like nanostructures (yellow arrows) and electron-dense granular structures of 5–20 nm in diameter (white asterisks). Yellow scale bar, 100 nm. C, Grid 2, intermediate-magnification micrograph from another grid showing abundant fibril-like (yellow arrows) and granular nanostructures (white asterisks). White scale bar, 100 nm. D, a higher magnification of the square region marked in yellow in B showing the fibril-like nanostructures in greater detail. Yellow scale bar, 50 nm.
The C-terminal region of the MF6p/FhHDM-1 protein is the main site of interaction with heme as observed by UV/visible spectroscopy
To determine which region of the MF6p/FhHDM-1 protein is the main site of heme binding, we compared the spectrophotometric alterations observed at wavelengths in the range of 300–700 nm after hemin was mixed with the complete sMF6p/FhHDM-1 protein or peptides corresponding to the N- and C-terminal regions as well as with other truncated peptides derived from the latter and with the C-terminal region of MF6p/FhHDM-1 orthologs present in the trematodes Opisthorchis viverrini, C. sinensis, and Paragonimus westermani (Fig. 1B). For this purpose, we first tested the optimal conditions for minimizing hemin aggregation, which involved the addition of a non-ionic detergent (Tween 20) or caffeine to the incubation buffer (TBS). The UV/visible absorption spectra obtained for hemin diluted in TBS alone or containing 0.1% Tween 20 or 25 mm caffeine are shown in Fig. 5A. Comparison of the three spectra showed that addition of caffeine produced the highest absorbance and narrowest Soret band (B band), thus confirming the advantage of using caffeine rather than Tween 20 to retain hemin in its monomeric form. In these experiments, we therefore incubated hemin with the peptides in TBS-C. We also observed that the presence of caffeine produced a significant bathochromic displacement of the Soret peak of hemin spectra from 380 (hemin in TBS) to 401 nm as well as a slight alteration of the weaker heme Q bands, which are typically observed in the range of 500–750 nm (19). The absorption spectra of hemin incubated alone or with the cationic, non-heme-binding molecules used as controls, i.e. lysozyme (LSZ) and polymyxin B (PMX), which were tested at 1:1 and 2:1 protein/peptide to hemin molar ratios, are shown in Fig. 5B. Although the addition of PMX or LSZ to the hemin solution in TBS-C produced a slight increase and decrease, respectively, in the absorbance of the Soret band, no bathochromic (red shift) or hypsochromic (blue shift) displacement occurred with any of the molecules tested.
Figure 5.

Comparison of hemin spectra in several solvent systems. A, UV/visible absorption spectra of hemin (12 μm) in TBS (pH 7.3), TBS-T, or TBS-C. B, UV/visible absorption spectra of hemin (12 μm) alone, mixed with LSZ, or with PMX at protein to hemin molar ratios of 2:1 and 1:1 in TBS-C. Addition of LSZ or PMX did not shift the absorbance maxima of hemin spectra. AU, absorbance units.
In contrast to LSZ and PMX, the Fasciola sMF6p/FhHDM-1 protein caused hypsochromic displacement of the hemin Soret peak plus an increase in the intensity of this band and the emergence of a prominent shoulder (see red arrow) in the spectrum at around 350 nm (Fig. 6A). These changes became more noticeable as the molar ratio of protein to hemin increased (λmax shift from 401 to 389 nm at a ratio of 5:1). In addition to the changes observed in the Soret peak, the interaction between the sMF6p/FhHDM-1 protein and hemin also produced a slight bathochromic displacement in the region of the Q bands (Fig. 6A, inset). Considered together, these data are indicative of a formation of a complex between sMF6p/FhHDM-1 and hemin.
Figure 6.

The C-terminal region of the MF6p/HDM family of proteins is mainly responsible for the alterations observed in the UV/visible absorption spectra of hemin. Shown are UV/visible absorption spectra of hemin (12 μm) prepared in TBS-C alone (black) or incubated with one of the following: sMF6p/FhHDM-1 (sFhMF6p) at protein to hemin molar ratios of 0.5:1, 1:1, 2:1 and 5:1 (A); sMF6p/FhHDM-1, sFhMF6a, sFhMF6c, or BSA at a protein to hemin molar ratio of 2:1 (B); sFhMF6c, two shorter derivatives (sFhMF6c1 and sFhMF6c2), or an N-acetylated and C-amidated derivative of sFhMF6c (sFhMF6cm) at a protein to hemin molar ratio of 2:1 (C); sFhMF6c or the C-terminal region of orthologous proteins sCsMF6c, sOvMF6c, and sPwMF6c (D). The direction of spectral changes of protein plus hemin samples relative to hemin alone is indicated by black arrows. Insets in A and B depict a magnification of the Q bands. A blue shift of the Soret band (λmax, 401–389 nm), a slight red shift of the Q bands, and the emergence of an apparent shoulder close to 350 nm (red arrow) were observed in A as the molar ratio of sMF6p/FhHDM-1 to hemin increased. Although less intense (see B–D), these spectral changes were also reproduced by F. hepatica sFhMF6c (λmax, 393 nm), by the orthologous peptides sOvMF6c (λmax, 392 nm) and sPwMF6c (λmax, 393 nm), and to a lesser extent by sFhMF6c2 (λmax, 398 nm) and sCsMF6c (λmax, 398 nm). Incubation of hemin with sFhMF6a, sFhMF6c1, or BSA did not shift the absorbance maxima of hemin spectra. AU, absorbance units.
Comparison of the activity of the peptides corresponding to the N- (sFhMF6a) and C-terminal (sFhMF6c) regions with that of the parent protein sMF6p/FhHDM-1, all tested at a peptide to hemin ratio of 2:1 (Fig. 6B), revealed that sFhMF6c was able to produce a blue shift displacement of the hemin Soret peak (λmax shift from 401 to 393 nm) together with a red shift in the Q band region (inset) although slightly lower than that of the parent protein. By contrast, the changes induced by sFhMF6a and BSA (control) were limited to changes in the intensity of the Soret band. Thus, the findings suggest that the C terminus is responsible for the heme-binding activity of sMF6p/FhHDM-1.
To delimit the region of interaction with heme, we assayed two truncated peptides derived from the sFhMF6c sequence (sFhMF6c1 and sFhMF6c2; see Fig. 1B). We also assayed an N-acetylated, C-amidated sFhMF6c-derived peptide (sFhMF6cm) to rule out any possible charge effect (20). The sFhMF6cm peptide produced the same blue shift of the hemin Soret peak as sFhMF6c, whereas the truncated fragments did not produce this shift (Fig. 6C). Nevertheless, unlike sFhMF6c1, the peptide sFhMF6c2 broadened the Soret band with the same intensity as the sFhMF6c and sFhMF6cm peptides (see red oblique arrow) and produced a slight blue shift of the hemin Soret peak (λmax shift from 401 to 398 nm).
Finally, we compared the sFhMF6c (positive control) and the carboxyl region of MF6p/FhHDM-1 orthologs from the trematodes C. sinensis, O. viverrini, and P. westermani (Fig. 6D). Interestingly, the ability to interact with hemin (reflected by the blue shift together with the broadening of the Soret band of the hemin spectrum) was not restricted to sFhMF6c (λmax, 393 nm) as this activity was also observed for the C-terminal peptides derived from O. viverrini (λmax, 392 nm) and P. westermani (λmax, 393 nm) and to a lesser extent for the peptide derived from C. sinensis (λmax, 398 nm).
The C-terminal region of the sMF6p/FhHDM-1 protein is required to inhibit the peroxidase-like activity of hemin
In a previous study, we reported that the sMF6p/FhHDM-1 protein was able to inhibit the peroxidase-like activity of hemin as revealed by co-incubation with 3,3′,5,5′-tetramethylbenzidine (TMB) and H2O2 (7). In the present study, we tested whether this inhibitory activity can be also reproduced by the peptides corresponding to the N- (sFhMF6a) or C-terminal (sFhMF6c and sFhMF6cm) regions of sMF6p/FhHDM-1. Both C-terminal peptides (sFhMF6c and sFhMF6cm) were able to inhibit the peroxidase-like activity of hemin, although their activity was lower than that of the parent protein (Fig. 7). On the contrary, the N-terminal peptide did not display any inhibitory activity.
Figure 7.
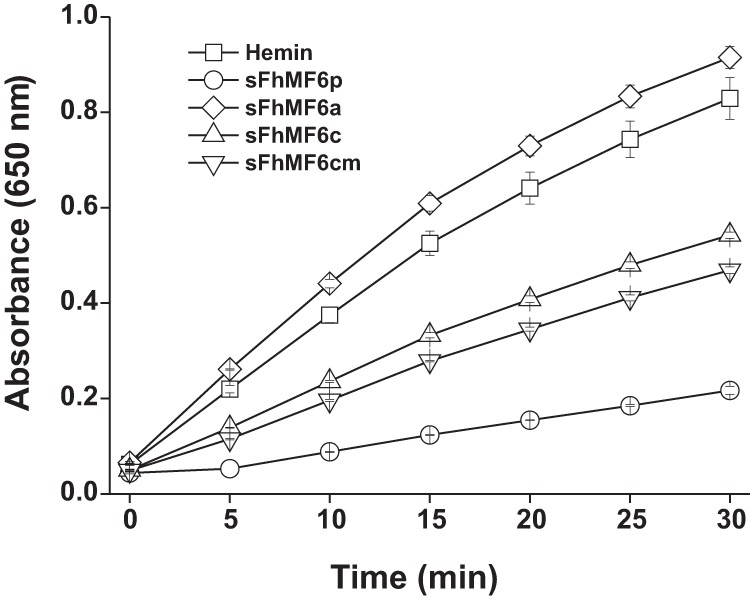
The C-terminal region of sMF6p/FhHDM-1 is responsible for the inhibitory effect on hemin peroxidase-like activity. Hemin alone or preincubated with sMF6p/FhHDM-1 (sFhMF6p), sFhMF6a, sFhMF6c, and sFhMF6cm (N-acetylated and C-amidated sFhMF6c) at a protein to hemin ratio of 5:1 were mixed with a commercial solution of TMB (1 mm TMB, 3 mm H2O2) adjusted to pH 6, and TMB oxidation was monitored at 650 nm for 30 min. The sMF6p/FhHDM-1 protein showed the highest level of inhibitory activity followed by both sFhMF6c and sFhMF6cm, whereas sFhMF6a had no effect on the peroxidase-like activity of hemin. The data points show the mean value ±S.D. (error bars) for duplicate wells.
Apomyoglobin (apoMYO) can sequester heme from sMF6p/FhHDM-1·hemin complexes, restoring holoMYO
We have previously reported that (i) Fasciola sMF6p/FhHDM-1 binds hemin with a dissociation constant (Kd) of ≈10−6 m and (ii) the protein is broadly distributed in the parasite tissues, including parenchymal cells and the fluid of testicular tubules, whereas it is absent from the caeca (7). These findings led us to postulate that the protein may play a role in heme homeostasis by sequestering heme to prevent heme-induced oxidation of parasite tissues and thus possibly serving as a heme donor for the synthesis of heme proteins. To test the hypothesis that sMF6p/FhHDM-1 can bind and donate heme to heme proteins with higher affinity for heme, we performed a hemin transfer experiment in which sMF6p/FhHDM-1·hemin complexes were incubated with horse apoMYO to restore the holo form of MYO. The reaction was monitored spectrophotometrically by observing changes in the UV/visible spectrum of sMF6p/FhHDM-1·hemin complexes after addition of increasing concentrations of apoMYO or BSA (as control) to the mixture. The results of these experiments are shown in Fig. 8, A (for apoMYO) and B (for BSA). The addition of apoMYO to sMF6p/FhHDM-1·hemin complexes caused a bathochromic change in the visible spectrum from 389 to 407 nm at the maximal concentration of apoMYO tested. By contrast, addition of BSA to the sample containing the sMF6p/FhHDM-1·hemin complexes produced an increase in the intensity of the Soret band but no displacement of this band. These results suggest that the Fasciola sMF6p/FhHDM-1 protein binds hemin with an affinity intermediate between those reported for BSA (Kd ≈ 2 × 10−8 m) (21) and for MYO (Kd ≈ 10−13 m) (22). Thus, the Kd of the Fasciola sMF6p/FhHDM-1 protein for hemin is probably at least 2 orders of magnitude lower than we reported previously (7). This discrepancy may be at least partly explained by the different buffers used to maintain hemin its monomeric form in these studies.
Figure 8.
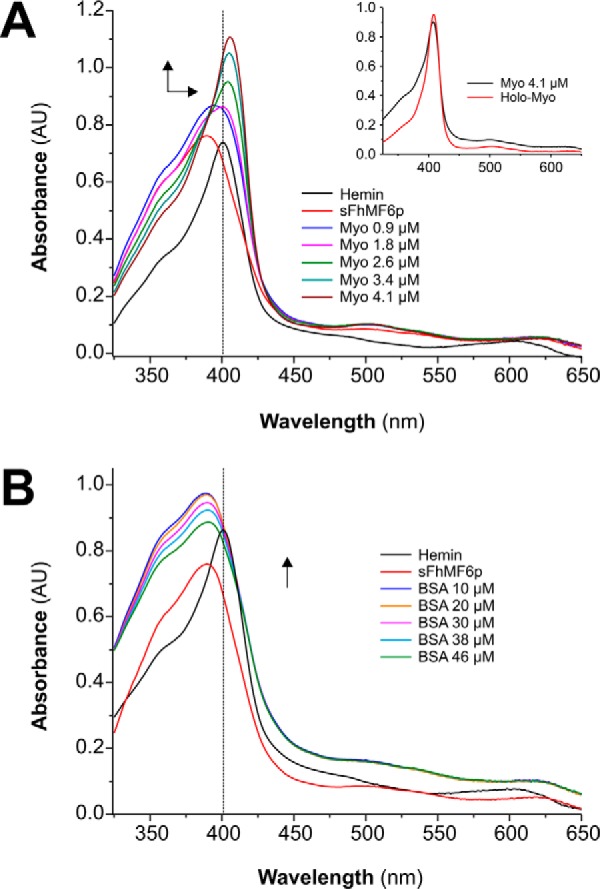
sMF6p/FhHDM-1 transfers hemin to apoMYO but not to BSA. A, 20 μm hemin was incubated with 40 μm sMF6p/FhHDM-1 in TBS containing 25 mm caffeine. Both UV/visible absorption spectra prior to (black) and after (red) addition of sMF6p/FhHDM-1 (sFhMF6p) were measured. The sample was then titrated with increasing amounts of apoMYO (0.9, 1.8, 2.6, 3.4, and 4.1 μm final concentrations), and hemin transfer to apoMYO was followed by the increase in the absorbance at 407 nm. The inset shows the UV/visible spectra of MYO prior to hemin extraction (holoMYO) and after hemin reconstitution (MYO). B, another sample of hemin preincubated with sMF6p/FhHDM-1 was titrated with increasing amounts of BSA (10, 20, 30, 38, and 46 μm final concentrations), and the UV/visible absorption spectra were measured. Arrows represent the direction of spectral changes upon addition of apoMYO or BSA. Spectra were not corrected for dilution. AU, absorbance units.
nMF6p/FhHDM-1·hemin complexes present in Fasciola SAs can be disrupted by apoMYO
Given that apoMYO displays a greater affinity than MF6p/FhHDM-1 for hemin, we tested whether the former is able to disrupt the high-molecular-mass nMF6p/FhHDM-1·hemin complexes present in Fasciola SAs (see Fig. 3B; elution volume, 8.98 ml). For this experiment, we fractionated a sample of Fasciola SAs by SEC on a Superdex 75 HR column in the presence or absence of an excess of apoMYO (Fig. 9). Note that the elution volumes obtained do not exactly match those in Fig. 3B as the SAs analyzed belonged to a different batch. In the absence of apoMYO, most of the heme-containing proteins present in SAs (mainly nMF6p/FhHDM-1 forming complexes of high molecular mass) were eluted at 8.19 ml (Fig. 9A). Nevertheless, incubation of SAs with apoMYO (Fig. 9B) resulted in the collapse of the high-molecular-mass complexes as the corresponding peak decreased drastically (peak II) for both hemin (red chromatogram) and protein (blue chromatogram) signals, whereas the peak at ∼12 ml (peak IV) increased, and a new peak of intermediate molecular mass emerged at an elution volume of 10.37 ml (peak III). Most of the hemin signal appeared in peak IV, although a relevant fraction was also present in peak III. Both peaks are consistent with the chromatogram obtained for native holoMYO (Fig. 9C) and apoMYO (Fig. 9D). By means of MALDI-TOF analysis of the proteins in Fig. 9B, we detected (as expected) nMF6p/FhHDM-1 in peak II, reconstituted holoMYO in peak III, and a mixture of MYO and nMF6p/FhHDM-1 in peak IV. The displacement of hemin and nMF6p/FhHDM-1 to peaks of lower molecular mass after incubation with apoMYO confirms that hemin is the cohesive element in the formation of species of high molecular mass with nMF6p/FhHDM-1.
Figure 9.

Disruption of Fasciola nMF6p/FhHDM-1·hemin complexes of high molecular mass by apoMYO. Size-exclusion chromatography analyses were performed on an FPLC system equipped with a Superdex 75 HR 10/30 column and continuous monitoring at 282 (blue line) and 395 nm (red line). Elution volumes are indicated above each peak fraction. A and B, elution profiles of F. hepatica SAs subjected to SEC alone (A) or mixed with an excess of apoMYO (B). After mixing the SAs with apoMYO, most of the heme signal observed at 8.11 ml in A was eluted at 12.07 ml (B). MALDI-TOF MS confirmed the presence of nMF6p/FhHDM-1 in peak II, MYO in peak III, and a mixture of nMF6p/FhHDM-1 and MYO in peak IV. C and D, elution profiles of myoglobin prior to (C) and after (D) acidification and MEK extraction to obtain apoMYO. Most heme signal (red line) disappeared after MEK treatment, and the protein (black line) eluted as a main peak in both chromatograms (elution volume, 12.7 and 11.88 ml, respectively). A small peak (elution volume, 10.2 ml), probably corresponding to apoMYO aggregates, is also observed in D. mAU, milliabsorbance units.
The peptides corresponding to the C-terminal region of several molecules belonging to the MF6p/HDM family of proteins interact differently with LPS
In addition to the heme-binding nature of the MF6p/FhHDM-1 protein (7), previous studies have reported that this protein and its C-terminal region are able to bind LPS (6). In the present study, we investigated whether this is a generally occurring phenomenon and whether the LPS-binding region overlaps that of heme. For this purpose, we used a recently developed competitive ELISA (23), which uses biotinylated PMX immobilized on deglycosylated avidin to capture LPS. For the study, we compared the LPS-binding ability of PMX (positive control for inhibition) with that of the Fasciola sMF6p/FhHDM-1 protein (either native or synthetic), the N-terminal region (sFhMF6a), the C-terminal region (sFhMF6c), two shorter fragments derived from the sFhMF6c region (sFhMF6c1 and sFhMF6c2), and the C-terminal regions of the three MF6p/FhHDM-1 orthologs from C. sinensis, O. viverrini, and P. westermani. We also considered two types of Escherichia coli serotypes of LPS (O111:B4 and O55:B5) as targets for LPS binding. Our results showed that, in addition to PMX (positive control), the native and synthetic MF6p/FhHDM-1 proteins as well as the C-terminal portion (sFhMF6c) were able to inhibit the binding of O111:B4 (Fig. 10A) and O55:B5 (Fig. 10B) LPS serotypes to biotinylated PMX at 10 μg/ml. However, the remaining peptides, including the sFhMF6c-truncated fragments sFhMF6c1 and sFhMF6c2 (not shown), did not display any inhibitory activity at this concentration or exerted a significant disaggregating effect. Moreover, when the concentrations of peptides/proteins were lowered to 1 μg/ml, PMX again displayed significant inhibition, but the remaining molecules either showed, as above, no inhibition or a significant disaggregating activity. Overall, these data indicate that the integrity of the C-terminal region of the MF6p/FhHDM-1 is required to interact with LPS but also that this interaction is not universal as similar peptides derived from MF6p/FhHDM-1 orthologs from other trematodes did not exert the same inhibitory effect.
Figure 10.
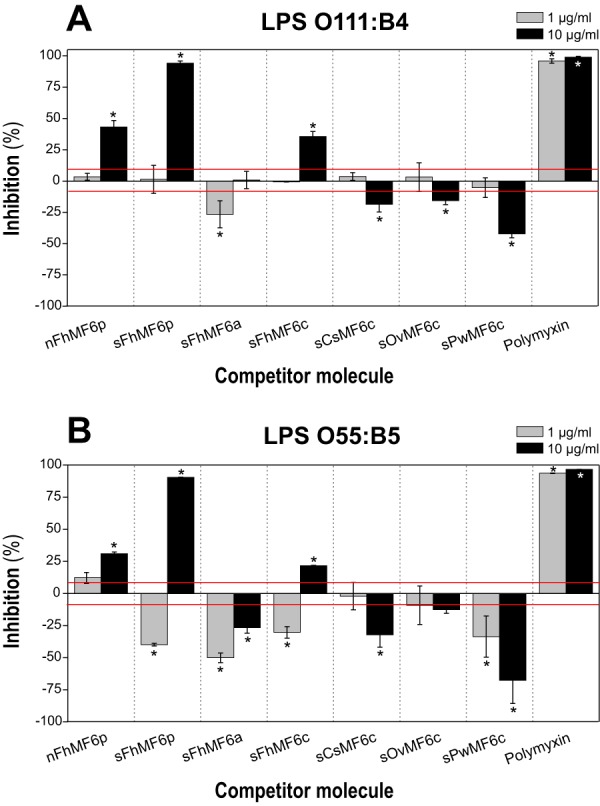
Competitive displacement of LPS binding to PMX. Samples of FITC-LPS from E. coli serotypes O111:B4 (A) and O55:B5 (B) were preincubated at concentrations of 1.25 and 2.5 μg/ml, respectively, in PBS-EDTA-BSA with 10 and 1 μg/ml concentrations of the following proteins/peptides: whole native (nFhMF6p) and synthetic (sFhMF6p); the N- (sFhMF6a) and C-terminal (sFhMF6c) regions of Fasciola MF6p/FhHDM-1; the C-terminal region of orthologous proteins sCsMF6c, sOvMF6c, and sPwMF6c; and PMX. A 100-μl volume of each sample was then added to the wells containing biotinylated PMX captured by deglycosylated avidin. The results are expressed as percentage of inhibition of LPS binding to PMX by the target protein/peptide and are the mean values ±S.D. (error bars) for duplicate wells. The average A (492 nm) obtained for control wells (without inhibitor) was 1.06 ± 0.09 and 0.81 ± 0.05 for serotypes O111:B4 and O55:B5, respectively. The red line indicates the ±S.D. percentage values of control wells. Differences in bars marked with an asterisk were significant at p < 0.05.
The N- and C-terminal regions of sMF6p/FhHDM-1 show different biological effects on red blood cells (RBCs) sensitized with hemin
Taking into account the heme-binding properties of MF6p/FhHDM-1 and the fact that free hemin is able to disturb the bilayer structure of cells (24), we investigated whether MF6p/FhHDM-1 or its derivatives can alter the hemolytic activity of hemin on RBCs. We included ovalbumin (OVA) and LSZ as controls. The study was carried out with RBCs from rabbit by using two different experimental approaches: (i) RBCs (0.5%) were first sensitized with hemin (20 μm) and then incubated with the proteins or peptides (40 μm), and (ii) RBCs were incubated with hemin previously mixed with the corresponding proteins or peptides at the same concentrations as above. We tested the effect of hemin and proteins alone on RBCs in control assays. As expected, hemolysis did not occur when RBCs were incubated with the proteins or peptides alone (Fig. 11A, column 6). We also observed that incubating RBCs with hemin alone did not immediately produce hemolysis but that a significant amount of hemoglobin was released after incubation of the plates for 6 h at room temperature (Fig. 11A, well A7). However, when we first sensitized the RBCs with hemin and subsequently added sMF6p/FhHDM-1 or its N-terminal peptide (sFhMF6a), total hemolysis occurred almost immediately (Fig. 11A, wells 1A and 1B). By contrast, incubation of hemin-sensitized RBCs with the C-terminal portion of MF6p/FhHDM-1 or with the protein OVA or LSZ only led to a pseudoagglutination phenomenon during the incubation period (Fig. 11A, wells C1–E1). In contrast, when the RBCs were incubated with hemin previously mixed with the proteins or peptides, the hemolytic activity of sMF6p/FhHDM-1 and that of the sFhMF6a peptide disappeared along with the hemin-derived hemolysis. Under these conditions, a pseudoagglutination phenomenon occurred with all peptides and proteins except the complete sMF6p/FhHDM-1 protein (Fig. 11A, column 2). We also observed that LSZ was able to precipitate the hemin present in the TBS buffer (Fig. 11A, well E5).
Figure 11.

The sMF6p/FhHDM-1 protein and the peptide sFhMF6a derived from its N-terminal region are able to hemolyze RBCs preconditioned with hemin. A, hemolysis assays showing the effect of the sMF6p/FhHDM-1 protein or derived peptides and hemin on RBCs. The assay was carried out in U-shaped wells of microtiter plates and photographed after 6 h of incubation at room temperature. Wells in column 1 contain protein/peptides incubated with RBCs preconditioned with hemin, wells in column 2 contain protein/peptides preincubated with hemin and subsequently added to RBCs, wells in columns 3 and 4 are the same as in columns 1 and 2 after centrifuging at 800 × g, wells in column 5 contain protein/peptides incubated with hemin alone, and wells in column 6 contain protein/peptides incubated with RBCs alone. The proteins/peptides tested by rows were as follows: A, sMF6p/FhHDM-1; B, sFhMF6a; C, sFhMF6c. OVA (D) and LSZ (E) were used as controls. Proteins/peptides were incubated at 40 μm, hemin was incubated at 20 μm, and RBCs were incubated at 0.5% in TBS. Control wells containing RBCs plus hemin and RBCs only were placed in wells 7A and 7B, respectively. B, table depicting the degree of hemolysis of hemin-preconditioned RBCs produced by different combinations of sMF6p/FhHDM-1 protein and hemin concentrations. The values depict the degree of hemolysis, ranging from no hemolysis (−) to intermediate (++ and +++) and complete hemolysis (++++) as determined by macroscopic observation of the turbidity of RBCs (0.5%) preconditioned with 2.5–40 μm hemin and subsequently incubated with different concentrations of the sMF6p/FhHDM-1 protein (2.5–40 μm).
To determine the degree of hemolysis in the above experiments, the microtiter plates were centrifuged at the end of the experiment. The data shown in Fig. 11A, columns 3 and 4, confirmed the above observations but also showed that the pseudoagglutination effect exerted by the peptide sFhMF6a preincubated with hemin (Fig. 11A, well B4) is weak because, unlike LSZ (Fig. 11A, well E4), no differences were observed relative to the control well (Fig. 11A, well B7) after the plate was centrifuged.
When several concentrations of sMF6p/FhHDM-1 were incubated with RBCs preconditioned with different concentrations of hemin, we also observed that any combination of hemin in the range of 5–40 μm and sMF6p/FhHDM-1 in the range of 10–40 μm produced complete hemolysis of RBCs (Fig. 11B, table). Moreover, hemolysis also occurred when RBCs sensitized with 80 μm hemin were washed twice by centrifugation to eliminate free hemin if immediately incubated with the sMF6p/FhHDM-1 protein (10 μm; data not shown). These findings and the fact that sFhMF6a is cationic in nature strongly suggest that this peptide is able to insert itself into and disrupt the cell membrane after interacting electrostatically with hemin.
Bioinformatics tools predict the N-terminal region of the MF6p/HDM family of proteins to have structural similarity to cell-penetrating peptides (CPPs)
CPPs are short peptides (typically of 5–30 residues) capable of crossing the cellular membrane with limited toxicity (25, 26). Structurally, CPPs can be linear, cyclic, cationic (rich in Arg and Lys residues), anionic, hydrophobic, hydrophilic, amphipathic, non-amphipathic, random coiled, α-helical, or β-sheet (26). However, most CPPs are positively charged amphipathic α-helices (25). The use of circular dichroism spectroscopy and bioinformatics tools (6) revealed that the Fasciola MF6p/FhHDM-1 protein adopts a predominant α-helix structure and that its C-terminal region has significant structural homology to antimicrobial peptides (AMPs). As AMPs and CPPs are structurally and functionally related (27, 28) and the cationic N-terminal fragment of MF6p/FhHDM-1 contains a high proportion of Arg and Lys residues (33.4%), we investigated the possibility that this protein may have some of the structural characteristics of CPPs. Interestingly, analysis of these proteins with CPPpred revealed that the cationic N-terminal, but not the C-terminal, fragment of MF6p/FhHDM-1 and related orthologs contains a stretch of 29 residues, each predicted to be a CPP sequence. Among these, those corresponding to C. sinensis and F. hepatica showed the highest probability of actually being CPPs (Fig. 12). In addition, the data obtained with the HeliQuest v2 tool (Fig. 13, A–E) showed that all N-terminal MF6p/HDM sequences are amphiphilic with moderate hydrophobic moments (μH range, 0.338–0.434; Fig. 13, A–E, arrows) and cationic (z values ranging from +4 to +7). Finally, the QUARK bioinformatics tool predicted that all MF6p/HDM proteins are probably composed by two α-helices of different length and connected by a small random coil (see Fig. 13F for 3D representation of MF6p/FhHDM-1), the longer of which includes the FhMF6a sequence.
Figure 12.

Predictive analysis for the presence of CPPs within the N- and C-terminal sequences of the MF6p/HDM family of proteins. The presence of CPP regions was predicted with the CPPpred bioinformatics tool as indicated under “Experimental procedures.” In addition to the N- and C-terminal sequences derived from the trematode MF6p/HDM family of proteins, three well known CPPs were analyzed as controls with the same tool. The score indicates the probability (p range, 0–1) of the peptides being CPPs. Scores of peptides being CPPs with a probability of ≥50% are shaded in green. The positive CPP control sequences derived from the proteins Antennapedia (Penetratin; residues 43–58), human immunodeficiency virus type 1 Tat (residues 48–60), and murine vascular endothelium cadherin (pVEC; residues 615–632), respectively (25).
Figure 13.
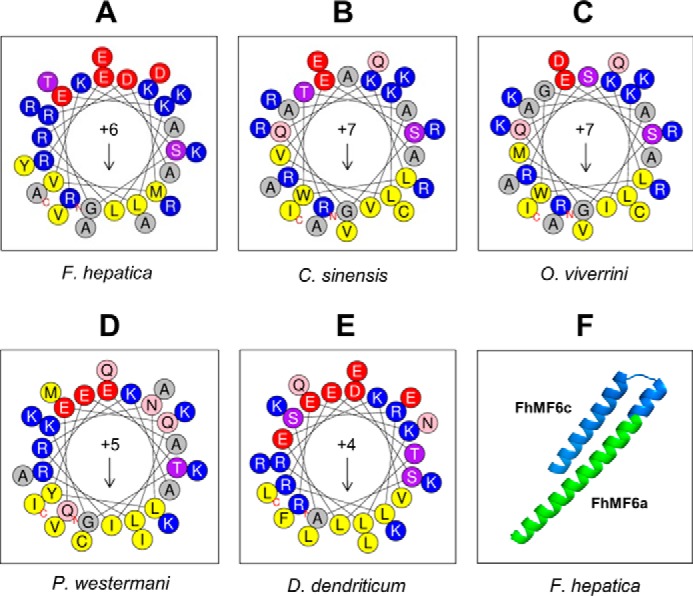
Helical wheel projections of N-terminal sequences derived from the MF6p/HDM family of proteins and predicted 3D structure of the Fasciola MF6p/FhHDM-1 protein. A–E, helical wheel projections corresponding to peptide FhMF6a (33 residues; see Fig. 1) and related orthologs from the trematodes C. sinensis, O. viverrini, P. westermani, and D. dendriticum. The projections were performed with the HeliQuest v2 tool covering the full sequence. The data indicate that all peptides are amphiphilic with moderate hydrophobic moments (arrows; μH range, 0.338–0.434) and have a net positive charge (z values ranging from +4 to +7). F, ab initio calculation of the 3D structure of the full-length mature sMF6p/FhHDM-1 protein with the QUARK bioinformatics tool predicted that it is composed of two α-helices of different length connected by a small random coil. The portion of the sequence corresponding to the peptides FhMF6c and FhMF6a is marked in blue and green, respectively.
Discussion
Since the discovery and molecular characterization of the Fasciola MF6p/FhHDM-1 protein (6, 7), new questions have arisen in relation to the physicochemical properties and biological function(s) of this protein. Given the discordance between the high apparent molecular mass of nMF6p/FhHDM-1 (≥150 kDa) in Fasciola SAs and the molecular mass deduced from its amino acid sequence (7.8 kDa) and Western blot analysis (7), we first tested the role of protein oligomerization and/or the presence of heme in this phenomenon. The findings of the chromatographic analysis with sMF6p/FhHDM-1 (Fig. 3C) show that the protein alone elutes with a higher apparent molecular mass than that of the cathepsin-rich fraction (molecular mass, 25–28 kDa). The displacement of the molecular mass can be explained by the tendency of this protein to form oligomers composed of two to five units as already noted in a study using differential ultracentrifugation with a recombinant form of MF6p/FhHDM-1 (6). However, this does not explain the existence of predominant species of ≥150 kDa in natural Fasciola SAs (Figs. 2 and 3) and suggests the involvement of hemin. Hemin (i.e. the oxidized form of heme) is a negatively charged hydrophobic molecule with a high tendency to form aggregates in aqueous solutions at physiological pH (29, 30). We therefore performed a SEC study under conditions aimed at preventing hemin aggregation (i.e. by adding caffeine to the equilibration buffer) to avoid misinterpretation during assessment of the apparent molecular mass of protein·hemin complexes due to the presence in the sample of hemin aggregates. Under these conditions, we observed that the molecular mass of protein·hemin complexes increased with the ratio of protein to hemin (Fig. 2) and produced a chromatogram resembling that obtained when hemin adsorbed on a Superdex 75 HR column was eluted with sMF6p/FhHDM-1 in the absence of caffeine (see Fig. 3A). Moreover, we also demonstrated that nMF6p/FhHDM-1·hemin complexes present in Fasciola SAs can be disrupted by addition of an apoprotein (apoMYO) with higher affinity for hemin than MF6p/FhHDM-1 (Fig. 9B). Considered together, these data confirm, for the first time, the involvement of hemin molecules in the formation of species of high molecular mass containing the MF6p/FhHDM-1 protein in Fasciola SAs. In addition to MF6p/FhHDM-1, hemin has also been shown to be involved in the oligomerization of other proteins as diverse as human arginyl-tRNA synthetase (31), the heme receptor IsdH-NEAT3 from Staphylococcus aureus (16), and the PrPC cellular normal prion protein (32).
Having demonstrated the capacity of hemin to form high-molecular-mass complexes with MF6p/FhHDM-1, we also investigated the morphology of native protein·hemin aggregates by using cryo-TEM. In the present study, we observed that the native protein·hemin complexes, present in the affinity-purified nMF6p/FhHDM-1 from SAs, tend to self-assemble forming fibril-like and granular nanostructures (Fig. 4), probably reflecting the coexistence in the sample of two or more types or organization states of protein·hemin complexes. The formation of similar fibril-like nanostructures that can be detected by electron microscopy has already been reported for other porphyrin derivatives (33) as well as for synthetic constructs of amphiphilic peptides capable of binding metalloporphyrins (34).
The tendency of hemin and other metalloporphyrins to aggregate in aqueous solutions depends on several physicochemical characteristics, such as ionic strength, pH, and solvent composition (19, 30). The degree of aggregation is highly dependent on the strong π-π interaction caused by the 22 π-electrons of porphyrins (35, 36) that facilitates the formation of two types of aggregates: J-type (side by side) characterized by a spectral UV/visible red shift of B (Soret) and Q bands and H-type (face to face) characterized by a blue shift of B band and red shift of Q bands relative to those of monomers (19, 37). In this study, we observed that the interaction between the sMF6p/FhHDM-1 protein and monodispersed hemin leads to a blue shift of the B band together with a red shift of Q bands in the hemin spectrum (Fig. 6A). These shifts resemble the spectroscopic changes typical of the formation of H-type aggregates of porphyrins (38). Although these protein·hemin complexes cannot be classified as pure H-aggregates because of the presence of MF6p/FhHDM-1 (39), the data obtained by UV/visible spectroscopy together with the findings of the SEC and cryo-TEM analyses suggest that MF6p/FhHDM-1 reorganizes the aggregation state of hemin, perhaps by a stacking mechanism. We also observed that nMF6p/FhHDM-1 maintains dispersal of these pseudo-H-aggregates as a colloid. This is evidenced by the fact that the brownish native protein·heme complexes present in Fasciola SAs tend to sediment in the bottom of tubes when centrifuged; however, these complexes are easily redispersed by weak agitation as occurs with colloidal (nanoparticle) preparations. Similar behavior has also been observed for the intercellular heme carrier HRG-3 from the nematode Caenorhabditis elegans as this protein also tends to precipitate bound to heme as a bright red complex after several hours (40).
Among the high diversity of heme-binding proteins (41), those capable of inducing a blue shift of the Soret band after interaction with hemin are very scarce in nature. Some examples include the iron response regulator protein of the bacterium Bradyrhizobium japonicum (42) and the consensus heme regulatory motif peptide sequence AKRCPVHTM present in several proteins (43). Interestingly, the blue shift observed with iron response regulator and heme regulatory motif depends on the presence of a cysteine, a residue not present in either the Fasciola MF6p/FhHDM-1 protein or in the C-terminal region of any of the parasites under study (see Fig. 1A). Moreover, both the mature Fasciola MF6p/FhHDM-1 protein and the corresponding orthologous sequences also lack histidine and do not share methionine residues, the other typical residues acting as axial ligand of classical heme proteins (41). This emphasizes the unusual characteristics of these proteins and strongly suggests that they are transitory heme ligands according to the classification established by Smith et al. (41). The fact that the presence of apoMYO was sufficient to release heme from sMF6p/FhHDM-1·hemin complexes is consistent with this hypothesis (Fig. 8A) and suggests a possible role for the protein in heme storage and in mediating a transfer mechanism based on affinity gradients. Transfer to proteins with a greater affinity for heme has also been suggested for the IsdH-NEAT3 domain from S. aureus (16), the chlorite dismutase-like protein (LmCld) from Listeria monocytogenes (44), and the novel Cj1386 protein from Campylobacter jejuni (45).
Considering the characteristic spectral changes derived from the interaction of sMF6p/FhHDM-1 with hemin, we aimed to determine which region and residues of these proteins are involved in heme binding. Comparison of the UV/visible absorption spectra obtained by mixing hemin with the full-length sMF6p/FhHDM-1 protein, its N- and C-terminal regions (Fig. 6B), or the truncated sMF6p/HDM C-terminal sequences derived from the trematodes C. sinensis, O. viverrini, and P. westermani (Fig. 6D) revealed that the C-terminal region of the protein is the main area involved in the interaction with hemin. Of the F. hepatica C-terminal truncated peptides sFhMF6c1 and sFhMF6c2, only the latter retained some ability to induce changes in the UV/visible spectrum (Fig. 6C). It therefore appears that some residues present in the 82DRLEKYA88 region are crucial for effective binding. Moreover, after comparing the amino acid sequence variations in the C-terminal region of Fasciola with those of the related trematodes (sPwMF6c from P. westermani, sOvMF6c from O. viverrini, and sCsMF6c from C. sinensis; see Fig. 1B), we concluded that the common residues (65LG, 68KI, Val72, 75IL, 79RLT, Arg83, Glu85, and Tyr87) along the sFhMF6c2 sequence are potentially important for heme binding and/or for maintaining the secondary structure of the peptide.
In addition to being responsible for the blue shift of hemin UV/visible absorption spectrum, the C-terminal region of the Fasciola MF6p/FhHDM-1 protein also causes the ameliorative effect on hemin peroxidase-like activity as reported previously for the whole protein (7). Nevertheless, the activity was much lower than in the parent protein (Fig. 7). The inhibitory effect of the peroxidase-like activity of hemin is probably related to the ability of MF6p/FhHDM-1 protein and its C-terminal region to form hemin pseudo-H-aggregates as observed by UV/visible spectrophotometry (see above). Similarly, heme aggregation has been reported to cause a reduction in the formation of heme-inducing free radicals, probably by a steric hindrance phenomenon that limits access of hydroperoxides to the iron center of heme molecules (46). It is thus possible that the MF6p/FhHDM-1 protein may be able to protect the parasite from the potentially toxic activity of heme by limiting its oxidative activity. This potential antioxidant mechanism has also been demonstrated for other heme-binding proteins, such as the Rhodnius heme-binding protein, the heme-lipoprotein of ticks, and the hemopexin from vertebrates (47).
In accordance with previous findings (6), our data confirm that the entire Fasciola MF6p/FhHDM-1 protein and its C-terminal region are able to bind LPS and, moreover, to compete with PMX for LPS binding. The latter aspect is of major importance because it demonstrates that the binding is specific and because recent reports indicate that proteins and peptides able to interact with the PMX-binding site on LPS are potential candidates for neutralizing LPS toxicity (23). In agreement with this hypothesis, MF6p/FhHDM-1 and its C-terminal region were reported to be able to significantly inhibit the LPS-induced production of TNF by murine macrophages (6, 10). However, unlike in the interaction with hemin, the C-terminal peptides derived from other trematodes, such as C. sinensis, O. viverrini, and P. westermani, were unable to compete with PMX for LPS binding (Fig. 10). Indeed, most of these peptides displayed a significant disaggregating effect, also exerted by the N-terminal cationic fragment of the Fasciola MF6p/FhHDM-1. These results indicate that the ability of the Fasciola MF6p/FhHDM-1 and its C-terminal region to bind and neutralize the LPS activity (referred to as their capacity to compete with PMX for LPS binding) is not a universal property of the MF6p/HDM family of proteins. Moreover, the observation that the peptides sFhMF6a, sOvMF6c, sPwMF6c, and sCsMF6c have significant disaggregating effects on LPS suggests they may enhance the biological activity of LPS. Parallelism between disaggregation and enhancement of the biological activity of LPS has also been suggested for other proteins, such as hemoglobin and serum albumin (48, 49).
The findings of the experiments performed with RBCs preconditioned with hemin (Fig. 11) revealed for the first time that the N- and C-terminal regions of the Fasciola MF6p/FhHDM-1 protein have different biological properties. With this experimental model, we observed that neither the complete Fasciola sMF6p/FhHDM-1 nor its N- and C-terminal fragments promoted relevant hemolytic activity on intact RBCs during the experiment (Fig. 11A). However, the complete protein and its N-terminal region were both able to cause immediate total hemolysis of RBCs preconditioned with hemin, a phenomenon not observed with the C-terminal sFhMF6c peptide or with the control proteins OVA and LSZ. Considering that the peptide corresponding to the heme-binding region of MF6p/FhHDM-1 (sFhMF6c) does not disrupt the hemin-altered membrane of RBCs and that heme-binding serum proteins, such as albumin and hemopexin, are able to draw hemin from the membrane without any damage to RBCs (50), it appears that the sFhMF6a peptide is able to insert itself into the membrane. Moreover, high hemolytic activity was achieved even when hemin was present in the well of the microtiter plate at a 4–8 m excess relative to the sMF6p/FhHDM-1 protein (Fig. 11B). This clearly indicates that the sMF6p/FhHDM-1 protein can interact with the RBC membrane even when saturated by external hemin. In addition, the fact that hemolysis did not occur when the sFhMF6a peptide preincubated with hemin was subsequently added to RBCs suggests that there is also some interaction (probably electrostatic) with hemin. Thus, hemolysis may involve a process whereby sFhMF6a interacts with hemin present in the membrane of RBCs, which would facilitate the penetration of sFhMF6a into the hemin-altered membrane, ultimately causing hemolysis. A similar effect was reported by Carr and Morrison (51), who observed that PMX caused hemolysis of LPS-preconditioned RBCs via ionic association of PMX with LPS bound to the membrane and the subsequent insertion of PMX into the lipid bilayer. Considered together, these results strongly suggest dual biological activity of the MF6p/HDM family of proteins whereby the cationic N-terminal region is able to interact with, and probably cross, the membrane barrier of cells and the neutral C-terminal region acts as a heme carrier/scavenger. Although further experimental evidence is required to confirm this, the possibility that MF6p/FhHDM-1 is able to cross cell membranes is also supported by the fact that all N-terminal sequences derived from the Fasciola MF6p/FhHDM-1 protein and orthologous proteins from related trematodes have a positive charge (+4 to +7), a predicted α-helix secondary structure with amphiphilic nature (Fig. 13), and a predominance of arginine and lysine residues, all of which are characteristics frequently observed in CPPs and AMPs (27, 52). The ability of the Fasciola MF6p/FhHDM-1 protein to interact via electrostatic interactions with the plasma membrane/lipid raft components of murine macrophages has been reported previously (9); however, to our knowledge, the binding properties of the N-terminal fragment have not previously been tested.
In summary, we elucidated the mechanism whereby the F. hepatica heme-binding protein MF6p/FhHDM-1 forms high-molecular-mass complexes and demonstrated that the C- and N-terminal regions have different functions. Specifically, we identified the involvement of the C-terminal region in heme binding and inhibition of the peroxidase-like activity of heme, and we also demonstrated the ability of the N-terminal region to bind to cell membranes. These findings suggest that the MF6p/FhHDM-1 and orthologous proteins may act primarily as heme scavengers, to protect parasite tissues from the deleterious effects of heme, and/or as a source of iron or heme for synthesis of heme proteins.
Experimental procedures
Ethics statement
This study was carried out in strict accordance with the guidelines of the European Directive 2010/63/EU and the Spanish Law (RD 53/2013) on Care and Use of Laboratory Animals. The protocol was approved by the Ethics Committee of the Universidad de Santiago de Compostela and by the Xunta de Galicia (Code 15007AE/12/DIG ENF 06), Spain. The parasite samples used in this study were obtained from local abattoirs.
Native and synthetic proteins/peptides
Fasciola SAs were obtained as reported previously (53). Briefly, live adult flukes were collected from bile ducts of naturally infected cows. The flukes were first washed in sterile saline solution containing antibiotics (penicillin/streptomycin) and glucose (2 g/liter) at 38 °C and then in RPMI 1640 cell culture medium supplemented with 20 mm HEPES, 0.3 g/liter l-glutamine, 2 g/liter sodium bicarbonate, and antibiotics at 38 °C under 5% CO2 in air. The flukes were maintained for 24 h in culture medium (3 ml/fluke) in tissue culture flasks at 38 °C under 5% CO2 in air. The medium containing the SAs was then removed and centrifuged at 10,000 × g for 20 min at 4 °C in the presence of protease inhibitors (SigmaFast Protease Inhibitor Tablets, Sigma-Aldrich). The supernatant was then passed through a 0.45-μm-pore filter disk and stored at −80 °C until required. The protein concentration in the supernatant was determined using the Micro BCA Protein Assay kit (Pierce, Thermo Fisher Scientific).
The synthetic peptides evaluated in the present study (Fig. 1B) were acquired (≥95% pure) from GeneCust Europe (Dudelange, Luxembourg). They include sMF6p/FhHDM-1 from F. hepatica as well as the corresponding N-terminal (sFhMF6a; residues 23–55) and C-terminal (sFhMF6c; residues 56–90) regions and synthetic peptides derived from the C-terminal region (residues 56–89) of orthologous proteins sCsMF6c, sOvMF6c, and sPwMF6c. Two shorter derivatives of the C-terminal fragment of MF6p/FhHDM-1 (sFhMF6c1 and sFhMF6c2; residues 56–81 and 65–88, respectively) and an N-acetylated and C-amidated derivative of sFhMF6c (sFhMF6cm; residues 56–90) were also tested.
Production of mAb MF6
Hybridoma cells secreting mAb MF6 were obtained as described previously (7, 54). The secreting hybridoma cells were grown intraperitoneally in Pristane-primed BALB/c mice. The anti-F. hepatica IgG1/κ antibodies in the ascitic fluid were purified by affinity chromatography on a protein G column (HiTrap Protein G, GE Healthcare) according to the manufacturer's protocol.
Purification of native Fasciola MF6p/FhHDM-1 by affinity chromatography with mAb MF6
The nMF6p/FhHDM-1 protein was isolated from F. hepatica SAs by affinity chromatography with the mAb MF6, which was conjugated on CNBr-activated Sepharose 4B (GE Healthcare) according to the manufacturer's instructions. Once the affinity column was equilibrated with PBS, F. hepatica SAs were applied, and the column was washed with PBS to remove unbound proteins. Bound proteins were eluted with 0.1 m glycine-HCl, pH 2.5, and neutralized with 2 m Tris before being dialyzed against distilled water. The purified proteins were concentrated in a 3K Macrosep Centrifugal Concentrator (Pall Filtron Corp., Port Washington, NY), and the protein content was determined with the Micro BCA Protein Assay kit. Finally, aliquots of the proteins were stored at −80 °C until further analysis.
Size-exclusion chromatography
The SEC experiments were performed using a Superdex 75 HR 10/30 column (Amersham Biosciences) connected to an FPLC system (ÄKTA Basic 10, Amersham Biosciences). Simultaneous monitoring was carried out at 280 and 395 nm or at 395 nm only (for samples and running buffer containing caffeine). The column was calibrated with a mixture of proteins of known molecular mass (Gel Filtration LMW Calibration kit, Amersham Biosciences). Samples subjected to SEC consisted of whole F. hepatica SAs (200 μl/run) diluted in PBS or in TBS-C (Sigma-Aldrich) or sMF6p/FhHDM-1·hemin complexes isolated by two different methods.
Fractionation of samples of hemin preincubated with sMF6p/FhHDM-1
A stock solution of hemin was prepared by dissolving 5 mg of hemin chloride (bovine; Sigma-Aldrich) in 1 ml 0.1 m NaOH. The solution was centrifuged at 12,000 × g for 10 min at room temperature to remove any precipitate. The supernatant was then diluted 1:10 in TBS and centrifuged before the hemin concentration was determined spectroscopically, considering an extinction coefficient of 58.44 mm−1 cm−1 at 385 nm (55). The hemin solution was stored in darkness at 4 °C until use. A stock solution of sMF6p/FhHDM-1 was also prepared by dissolving the protein at 10 mg/ml in distilled water. The solution was stored frozen at −20 °C until use. Samples were prepared for SEC analysis by diluting the stock solutions of hemin and protein in TBS-C and then mixing both dilutions 1:1 to yield a final concentration of 0.06 mm hemin with sMF6p/FhHDM-1 at protein to hemin molar ratios of 0.5:1, 1:1, and 2:1. Samples were incubated at room temperature for at least 30 min and then filtered through a 0.2-μm filter. The column was equilibrated with TBS-C, and 0.2-ml aliquots of the preincubated samples were applied to the Superdex 75 HR column at a flow rate of 0.5 ml/min with continuous monitoring at 395 nm.
Elution of hemin previously adsorbed to the column beads with sMF6p/FhHDM-1
To investigate the ability of sMF6p/FhHDM-1 to associate with hemin to form high-molecular-mass complexes, 0.6 ml of a 0.06 mm solution of hemin was prepared in PBS and filtered through a 0.2-μm filter. Then, a sample of 0.2 ml was injected in the column (previously equilibrated with PBS) and chromatographed under the same conditions as above with simultaneous monitoring at 280 and 395 nm. Hemin was not eluted under these conditions as it was strongly retained at the top of the column. A filtered sample (0.2 ml) of 0.12 mm sMF6p/FhHDM-1 in PBS was applied to the same column and chromatographed as for the hemin alone, and in this case, the adsorbed hemin was eluted together with the protein. Finally, the same amount of protein was loaded onto a clean column equilibrated with PBS, and SEC was conducted as described above.
Cryo-TEM
Plunge freezing
One drop of the immunopurified and dialyzed sample of nMF6p/FhHDM-1 (see above; protein concentration, 1.4 mg/ml) was applied to the carbon surface of a glow-discharged lacey carbon 300-mesh copper grid (Ted Pella Inc., Redding, CA). The sample was cryo-immobilized in a Vitrobot Mark III system (FEI, Eindhoven, Netherlands) by plunge freezing in liquefied ethane. The sample was maintained at 100% humidity, and excess liquid was automatically blotted with filter paper. The vitrified samples were stored in liquid nitrogen until examination in the cryoelectron microscope.
Imaging
A plunge frozen sample was transferred to a Tecnai F20 electron microscope (FEI) in a cryoholder (Gatan, Pleasanton, CA). The sample was examined at 200 kV, at a temperature of between −179 and −170 °C, and under low-dose imaging conditions. Low-dose images were recorded with a 4096 × 4096-pixel charge-coupled device Eagle camera (FEI). The images were acquired at electron doses between 0.8 and 2 e−/Å2 and at 6-nm underfocus values. Images were taken at magnifications of ×25,000–50,000.
UV/visible spectrophotometric analysis
The interactions of hemin with sMF6p/FhHDM-1 and with peptides derived from Fasciola and other related trematodes were determined by measuring the changes in the UV/visible absorption spectra of hemin after incubation with the protein/peptides. As hemin tends to aggregate in aqueous solutions, we first determined the best conditions for dispersing and maintaining the hemin in a monomeric and completely soluble form (29, 32). For this purpose, a stock solution of hemin, prepared as indicated above, was diluted at 12 μm in TBS, TBS-C, or TBS supplemented with 0.1% Tween (TBS-T). The samples were incubated for 30 min at 22 °C before being analyzed by UV/visible spectrophotometry (range, 300–700 nm) at 22 °C in a Multiskan GO UV/visible microplate spectrophotometer (Thermo Fisher Scientific) previously adjusted to zero with the corresponding buffer. TBS-C buffer was used to prepare the samples for the spectrophotometric analysis. In addition to the sMF6p/FhHDM-1 and related peptides, the cationic compounds LSZ (from chicken egg white; Fluka Analytical, Sigma-Aldrich) and PMX (Sigma-Aldrich) and the neutral protein BSA (heat shock fraction; Sigma-Aldrich) were included as controls. Stock solutions of each protein or peptide were prepared in distilled water (5–20 mg/ml) and stored frozen at −20 °C until use. Both hemin and the proteins/peptides were diluted in TBS-C to twice the desired final concentration. A 250-μl aliquot of the hemin solution (24 μm) was then mixed with the same volume of each protein/peptide solution to be tested, or TBS-C only (control), to produce a final concentration of 12 μm hemin and protein to hemin molar ratios ranging from 0.5 to 5. Samples were incubated for 30 min at 22 °C, and the spectra were measured as above.
Inhibition of the peroxidase-like hemin activity
The peroxidase-like activity of hemin alone or hemin complexed with sMF6p/FhHDM-1, sFhMF6a, sFhMF6c, and sFhMF6cm was measured by monitoring the oxidation of TMB by H2O2 at pH 6 (the pH at which maximum catalytic activity of hemin occurs) (56) as described previously (7). Briefly, the peroxidase substrate solution of a commercial two-component TMB system (KPL Inc., Gaithersburg, MD) was diluted 2-fold with TBS containing 0.2% Tween 20 and 0.02% H2O2, and this solution was subsequently mixed with an equal volume of the TMB peroxidase substrate. Hemin (12 μm) was incubated with the peptides/protein at protein to hemin molar ratios of 5:1 or alone (control) in TBS-T for 30 min at room temperature prior to the addition of the substrate. An aliquot (20 μl) of each sample was added to the wells of a 96-well microtiter plate and mixed with 100 μl of the TMB solution. The reaction was measured by monitoring the increase in the absorbance at 650 nm in a UV/visible microplate spectrophotometer at 5-min intervals for 30 min at 22 °C.
Determination of LPS-binding ability by competitive ELISA
A previously reported competitive capture ELISA (23) was used to study the ability of native and synthetic MF6p/FhHDM-1 and derived peptides from Fasciola (sFhMF6a, sFhMF6c, sFhMF6c1, and sFhMF6c2) and other trematodes (sCsMF6c, sOvMF6c and sPwMF6c) to bind and/or to disaggregate LPS. Plates were coated with 100 μl of 10 μg/ml deglycosylated egg white avidin (Fluka Analytical, Sigma-Aldrich) in PBS for 2 h at 37 °C, washed three times with PBS, and blocked with 1% BSA in PBS (200 μl/well) for 1 h at room temperature. The plates were washed three times with PBS and incubated with biotinylated PMX (Hycult Biotech, Uden, Netherlands) diluted 1:50 in PBS containing 0.1% BSA (PBS-BSA) for 1 h at room temperature under shaking at 750 rpm. In parallel, samples of FITC-LPS from E. coli serotypes O111:B4 (1.25 μg/ml) and O55:B5 (2.5 μg/ml) were preincubated with each of the proteins/peptides to be tested or PMX (control for positive inhibition) at 10 and 1 μg/ml in PBS-BSA with 1 mm EDTA for 1 h at room temperature. The plates were then washed five times with PBS, and the preincubated samples were added (100 μl/well) and incubated for 30 min at room temperature with shaking at 750 rpm. The plates were washed five times with PBS-T, and 100 μl/well HRP-conjugated sheep anti-FITC (1:4,000 in PBS-BSA) was added. The plates were then incubated for 30 min at room temperature with shaking at 750 rpm. The plate was washed again and incubated for 20 min at room temperature with 100 μl of substrate/well (SigmaFast OPD, Sigma-Aldrich) before the reaction was stopped with 25 μl of 3 n H2SO4. The absorbance (A) was measured at 492 nm. The percentage of inhibition of LPS binding to captured PMX was calculated as follows: [(Acontrol − Atest/Acontrol)] × 100 where Atest is the average A492 for the wells containing LPS and the inhibitor and Acontrol is the average A492 for LPS alone. The mean A value for wells with all reagents except biotinylated PMX (negative control) was subtracted from all A values obtained.
Hemin transfer assays
Preparation of hemin and apoMYO from horse MYO
For extraction of heme from MYO, 10 mg of MYO from equine skeletal muscle (Sigma-Aldrich) was dissolved in 2 ml of double distilled water in a glass tube and slowly acidulated with 5% HCl under magnetic stirring until a pH of 3 was reached. A 2-ml aliquot of pure methyl ethyl ketone (MEK; Merck, WWR) was then added to the protein sample. The mixture was vortexed for 1 min and centrifuged at 500 × g for 5 min in a centrifuge equipped with a swinging bucket rotor to accelerate formation of the two phases. The upper phase containing MEK and hemin was carefully collected, and the residual hemin still present in the lower phase was re-extracted with another 2 ml of MEK by using the same procedure described above. Finally, the apoMYO present in the lower phase was aspirated with a syringe fitted with an 18-gauge needle. Both the apoMYO (lower aqueous phase) and hemin (upper organic phase) were dried overnight in a centrifugal evaporator (Savant AS160 SpeedVac, Thermo Fisher Scientific).
Hemin transfer from sMF6p/FhHDM-1 to apoMYO
For heme transfer experiments, the previously dried hemin chloride was dissolved at 20 μm in TBS-C in a final volume of 540 μl. A 20-μl aliquot of a stock solution of sMF6p/FhHDM-1 (8 mg/ml in distilled water) was then added to the hemin solution to yield a protein to hemin ratio of ∼2:1. Both spectra (before and after adding the protein) were recorded at between 325 and 650 nm, and the interaction between sMF6p/FhHDM-1 and hemin was confirmed by the blue shift of the Soret band (from 401 to 389 nm). A total volume of 100 μl of a working solution of apoMYO (475 μg/ml in TBS-C) was added in 20-μl aliquots to the 560-μl sample in the spectrophotometer cuvette. After each addition of protein, the sample was gently mixed with the aid of the pipette tip, and the sample was allowed to stand for 5 min at room temperature before the corresponding spectrum was recorded. Another 560-μl sample of hemin with sMF6p/FhHDM-1, prepared as above, was also incubated with increasing amounts of BSA as a control. As BSA has a much lower constant affinity for hemin than MYO, a larger proportion (about t10 times the molarity of apoMYO) of the former was added. Specifically, 100 μl of a working solution of BSA (20 mg/ml in TBS-C) was added in 20-μl aliquots, and the spectra were recorded as above. Although the spectra obtained for both samples were not corrected for dilution, changes in the pH were negligible (pH ∼7.0).
Disruption of nMF6p/FhHDM-1·hemin complexes by apoMYO
An aliquot (200 μl) of whole Fasciola SAs (protein content, 3 mg/ml) was fractionated as above by using a Superdex 75 HR 10/30 column equilibrated with PBS at a flow rate of 0.5 ml/min with continuous monitoring at 282 and 395 nm. Another aliquot of whole SAs (1 ml) was mixed with 3 mg of dried apoMYO, vortexed, and allowed to stand for 5 min at room temperature. The sample was then filtered through a 0.2-μm-pore filter and chromatographed under the same conditions described above. As controls, a sample of apoMYO and an aliquot of native MYO were also chromatographed. The proteins present in each peak obtained after SEC were identified by MALDI-TOF as reported previously (7).
RBC hemolysis assays
Blood was collected from a rabbit (New Zealand strain) into EDTA tubes and centrifuged at 2,000 × g for 10 min at 4 °C. The plasma and the buffy coat were removed, and RBCs were washed thoroughly with TBS and diluted in the same buffer to produce a 2% suspension (4 times the desired final concentration). Stock solutions (×4 concentration) of hemin and the Fasciola protein/peptides sMF6p/FhHDM-1, sFhMF6a, and sFhMF6c and of the proteins OVA (grade V; Sigma-Aldrich) and LSZ (controls) were each prepared in TBS (80 μm hemin and 160 μm each protein/peptide). Two types of incubation with RBCs were then performed: (i) RBCs were first preconditioned with hemin before being incubated with the protein/peptide to be tested and (ii) RBCs were incubated with premixed hemin and protein. In the first assay (i), a volume of the 2% suspension of RBCs was mixed with an equal volume of the 80 μm hemin solution, and 125-μl aliquots of the mixture were placed in the wells of a U-shaped microtiter plate (Sterilin, Thermo Fisher Scientific). The stock solutions of each peptide/protein (160 μm) to be tested were then diluted 2-fold with TBS, and 125 μl of each sample was added to the wells containing the preconditioned RBCs. The solutions were mixed slowly with the aid of the pipette, and the plates were left for 6 h at room temperature. In the second assay (ii), the hemin and each of the protein solutions were mixed 1:1 and incubated for 15 min at room temperature. A 125-μl aliquot of each sample was then added to the wells containing the same volume of a 1% suspension of RBCs and incubated as above. Control wells containing protein plus hemin, RBCs alone, and RBCs plus hemin or protein were also included. The plates were photographed at the end of the incubation period before and after being centrifuged at 800 × g for 10 min. An additional assay with RBCs preconditioned with hemin was carried out as for the first assay (i) to test the effect of using several concentrations of both hemin and sMF6p/FhHDM-1 in the range of 2.5–40 μm.
Bioinformatics sequence analyses
Prediction of the presence of CPP sequences
The presence of CPP regions in the sequences of the MF6p/HDM family of proteins was predicted using the CPPpred tool (57) available online. As CPPs are usually short peptides not exceeding 30 residues (25) and typically cationic (25), we determined which stretches of extension closest to 30 residues (the maximum value accepted by CPPpred) of the N-terminal and C-terminal regions of MF6p/FhHDM-1 (Fig. 1A) yielded the maximum score as CPPs. These regions comprised the Arg27–Arg55 and Met57–Leu84 regions of peptides FhMF6a and FhMF6c from Fasciola MF6p/FhHDM-1, respectively (Fig. 1B). Finally, we selected the peptide sequences of the same extension and position as Fasciola MF6p/FhHDM-1 from other trematode orthologs (7) and analyzed them for the presence of putative CPPs by using the same CPPpred tool.
Predictions of the whole 3D structure of the MF6p/HDM family of proteins and helical wheel models of selected sequences
As 3D structural models of the MF6p/HDM family of proteins are not yet available, we performed ab initio predictions using the QUARK prediction tool (58) available online. The first model obtained with QUARK was used to compare all sequences belonging to the MF6p/HDM family of proteins. Finally, the HeliQuest v2 tool (59) available online was used to construct helical wheel projections corresponding to peptide FhMF6a (33 residues) and related orthologs from the trematodes C. sinensis, O. viverrini, P. westermani, and Dicrocoelium dendriticum (see Fig. 1).
Statistical analysis
An unpaired t test with Welch correction was used to determine the significance of differences between inhibition and control values (without inhibition) in the competitive ELISA. The statistical analysis was conducted using the GraphPad InStat statistical package (GraphPad Software Inc., La Jolla, CA). Differences were considered significant at p < 0.05.
Author contributions
V. M.-S. and F. M. U. designed the study and wrote the paper. V. M.-S. and F. M. U. designed and performed the experiments. V. M.-S., M. M., M. G.-W., M. J. P., T. G., F. R., and F. M. U. analyzed the data. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Lidia Delgado (Unitat de Crio-Microscòpia Electrònica, Centres Científics i Tecnològics) from the Universitat de Barcelona (Spain) for assistance with cryo-TEM analysis. We also thank Rita Giovannetti (University of Camerino, Italy) and Xerardo García Mera (University of Santiago de Compostela, Spain) for helping with data interpretation.
This work was supported in part by Ministerio de Ciencia e Innovación (Spain) Grant AGL2011-30563-C03, Xunta de Galicia (Spain) Grant GPC2014/058, Instituto de Salud Carlos III-Acción Estratégica de Salud Intramural Grant PI14CIII/00076, and the European Fund for Regional Development (FEDER). The authors declare that they have no conflicts of interest with the contents of this article.
- SAs
- secretory antigens
- SEC
- size-exclusion chromatography
- cryo-TEM
- cryogenic transmission electron microscopy
- LSZ
- lysozyme
- PMX
- polymyxin B
- TMB
- 3,3′,5,5′-tetramethylbenzidine
- MYO
- myoglobin
- OVA
- ovalbumin
- CPP
- cell-penetrating peptide
- AMPs
- antimicrobial peptides
- MEK
- methyl ethyl ketone
- HDM
- helminth defense molecule
- Fh
- F. hepatica
- nMF6p
- native MF6p
- sMF6p/FhHDM-1
- synthetic form of MF6p/FhHDM-1
- μH
- mean helical hydrophobic moment
- Cs
- C. sinensis
- Ov
- O. viverrini
- Pw
- P. westermani.
References
- 1. Keiser J., and Utzinger J. (2005) Emerging foodborne trematodiasis. Emerg. Infect. Dis. 11, 1507–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ashrafi K., Bargues M. D., O'Neill S., and Mas-Coma S. (2014) Fascioliasis: a worldwide parasitic disease of importance in travel medicine. Travel Med. Infect. Dis. 12, 636–649 [DOI] [PubMed] [Google Scholar]
- 3. Haçarız O., Sayers G., and Baykal A. T. (2012) A proteomic approach to investigate the distribution and abundance of surface and internal Fasciola hepatica proteins during the chronic stage of natural liver fluke infection in cattle. J. Proteome Res. 11, 3592–3604 [DOI] [PubMed] [Google Scholar]
- 4. Mulvenna J., Sripa B., Brindley P. J., Gorman J., Jones M. K., Colgrave M. L., Jones A., Nawaratna S., Laha T., Suttiprapa S., Smout M. J., and Loukas A. (2010) The secreted and surface proteomes of the adult stage of the carcinogenic human liver fluke Opisthorchis viverrini. Proteomics 10, 1063–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cao X., Fu Z., Zhang M., Han Y., Han Q., Lu K., Li H., Zhu C., Hong Y., and Lin J. (2016) Excretory/secretory proteome of 14-day schistosomula, Schistosoma japonicum. J. Proteomics 130, 221–230 [DOI] [PubMed] [Google Scholar]
- 6. Robinson M. W., Donnelly S., Hutchinson A. T., To J., Taylor N. L., Norton R. S., Perugini M. A., and Dalton J. P. (2011) A family of helminth molecules that modulate innate cell responses via molecular mimicry of host antimicrobial peptides. PLoS Pathog. 7, e1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martínez-Sernández V., Mezo M., González-Warleta M., Perteguer M. J., Muiño L., Guitián E., Gárate T., and Ubeira F. M. (2014) The MF6p/FhHDM-1 major antigen secreted by the trematode parasite Fasciola hepatica is a heme-binding protein. J. Biol. Chem. 289, 1441–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McGonigle S., and Dalton J. P. (1995) Isolation of Fasciola hepatica haemoglobin. Parasitology 111, 209–215 [DOI] [PubMed] [Google Scholar]
- 9. Robinson M. W., Alvarado R., To J., Hutchinson A. T., Dowdell S. N., Lund M., Turnbull L., Whitchurch C. B., O'Brien B. A., Dalton J. P., and Donnelly S. (2012) A helminth cathelicidin-like protein suppresses antigen processing and presentation in macrophages via inhibition of lysosomal vATPase. FASEB J. 26, 4614–4627 [DOI] [PubMed] [Google Scholar]
- 10. Thivierge K., Cotton S., Schaefer D. A., Riggs M. W., To J., Lund M. E., Robinson M. W., Dalton J. P., and Donnelly S. M. (2013) Cathelicidin-like helminth defence molecules (HDMs): absence of cytotoxic, anti-microbial and anti-protozoan activities imply a specific adaptation to immune modulation. PLoS Negl. Trop. Dis. 7, e2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gallagher W. A., and Elliott W. B. (1967) Caffeine derivatives of haematin compounds. Biochem. J. 105, 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pasternack R. F., Gibbs E. J., Hoeflin E., Kosar W. P., Kubera G., Skowronek C. A., Wong N. M., and Muller-Eberhard U. (1983) Hemin binding to serum proteins and the catalysis of interprotein transfer. Biochemistry 22, 1753–1758 [DOI] [PubMed] [Google Scholar]
- 13. Travascio P., Li Y., and Sen D. (1998) DNA-enhanced peroxidase activity of a DNA-aptamer-hemin complex. Chem. Biol. 5, 505–517 [DOI] [PubMed] [Google Scholar]
- 14. Maiti N. C., Mazumdar S., and Periasamy N. (1998) J- and H-aggregates of porphyrin-surfactant complexes: time-resolved fluorescence and other spectroscopic studies. J. Phys. Chem. B 102, 1528–1538 [Google Scholar]
- 15. GE Healthcare Life Science (2000) Size Exclusion Chromatography: Principles and Methods, Publication 18-1022-18, GE Healthcare Life Sciences [Google Scholar]
- 16. Watanabe M., Tanaka Y., Suenaga A., Kuroda M., Yao M., Watanabe N., Arisaka F., Ohta T., Tanaka I., and Tsumoto K. (2008) Structural basis for multimeric heme complexation through a specific protein-heme interaction: the case of the third neat domain of IsdH from Staphylococcus aureus. J. Biol. Chem. 283, 28649–28659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lengyel J., Hnath E., Storms M., and Wohlfarth T. (2014) Towards an integrative structural biology approach: combining cryo-TEM, X-ray crystallography, and NMR. J. Struct. Funct. Genomics 15, 117–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burrows N. D., and Penn R. L. (2013) Cryogenic transmission electron microscopy: aqueous suspensions of nanoscale objects. Microsc. Microanal. 19, 1542–1553 [DOI] [PubMed] [Google Scholar]
- 19. Giovannetti R. (2012) The use of spectrophotometry UV-vis for the study of porphyrins, in Macro to Nano Spectroscopy (Uddin J., ed) pp. 87–108, InTech, Rijeka, Croatia [Google Scholar]
- 20. Rai J., Raghothama S., and Sahal D. (2007) Tyrosine-heme ligation in heme-peptide complex: design based on conserved motif of catalase. J. Pept. Sci. 13, 406–412 [DOI] [PubMed] [Google Scholar]
- 21. Hargrove M. S., Barrick D., and Olson J. S. (1996) The association rate constant for heme binding to globin is independent of protein structure. Biochemistry 35, 11293–11299 [DOI] [PubMed] [Google Scholar]
- 22. Culbertson D. S., and Olson J. S. (2010) Role of heme in the unfolding and assembly of myoglobin. Biochemistry 49, 6052–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martínez-Sernández V., Orbegozo-Medina R. A., Romarís F., Paniagua E., and Ubeira F. M. (2016) Usefulness of ELISA methods for assessing LPS interactions with proteins and peptides. PLoS One 11, e0156530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang F., Wang T., Lai J., Li M., and Zou C. (2006) Vitamin E inhibits hemolysis induced by hemin as a membrane stabilizer. Biochem. Pharmacol. 71, 799–805 [DOI] [PubMed] [Google Scholar]
- 25. Bechara C., and Sagan S. (2013) Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 587, 1693–1702 [DOI] [PubMed] [Google Scholar]
- 26. Milletti F. (2012) Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov. Today 17, 850–860 [DOI] [PubMed] [Google Scholar]
- 27. Henriques S. T., Melo M. N., and Castanho M. A. (2006) Cell-penetrating peptides and antimicrobial peptides: how different are they? Biochem. J. 399, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu X., Cao R., Wang S., Jia J., and Fei H. (2016) Amphipathicity determines different cytotoxic mechanisms of lysine- or arginine-rich cationic hydrophobic peptides in cancer cells. J. Med. Chem. 59, 5238–5247 [DOI] [PubMed] [Google Scholar]
- 29. Kawamura-Konishi Y., and Suzuki H. (1985) Binding reaction of hemin to globin. J. Biochem. 98, 1181–1190 [DOI] [PubMed] [Google Scholar]
- 30. Srinivas V., and Rao C. M. (1990) Time profile of hemin aggregation: an analysis. Biochem. Int. 21, 849–855 [PubMed] [Google Scholar]
- 31. Yang F., Xia X., Lei H. Y., and Wang E. D. (2010) Hemin binds to human cytoplasmic arginyl-tRNA synthetase and inhibits its catalytic activity. J. Biol. Chem. 285, 39437–39446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee K. S., Raymond L. D., Schoen B., Raymond G. J., Kett L., Moore R. A., Johnson L. M., Taubner L., Speare J. O., Onwubiko H. A., Baron G. S., Caughey W. S., and Caughey B. (2007) Hemin interactions and alterations of the subcellular localization of prion protein. J. Biol. Chem. 282, 36525–36533 [DOI] [PubMed] [Google Scholar]
- 33. Fuhrhop J.-H., Bindig U., and Siggel U. (1993) Micellar rods and vesicular tubules made of 14‴,16‴-diaminoporphyrins. J. Am. Chem. Soc. 115, 11036–11037 [Google Scholar]
- 34. Fry H. C., Garcia J. M., Medina M. J., Ricoy U. M., Gosztola D. J., Nikiforov M. P., Palmer L. C., and Stupp S. I. (2012) Self-assembly of highly ordered peptide amphiphile metalloporphyrin arrays. J. Am. Chem. Soc. 134, 14646–14649 [DOI] [PubMed] [Google Scholar]
- 35. Hunter C. A., and Sanders J. K. M. (1990) The nature of π-π interactions. J. Am. Chem. Soc. 112, 5525–5534 [Google Scholar]
- 36. Giovannetti R., Alibabaei L., and Petetta L. (2010) Aggregation behaviour of a tetracarboxylic porphyrin in aqueous solution. J. Photochem. Photobiol. A 211, 108–114 [Google Scholar]
- 37. Verma S., and Ghosh H. N. (2012) Exciton energy and charge transfer in porphyrin aggregate/semiconductor (TiO2) composites. J. Phys. Chem. Lett. 3, 1877–1884 [DOI] [PubMed] [Google Scholar]
- 38. Gregg B. A., Fox M. A., and Bard A. J. (1989) Effects of order on the photophysical properties of the liquid crystal zinc octakis(β-octoxyethyl) porphyrin. J. Phys. Chem. 93, 4227–4234 [Google Scholar]
- 39. Berlepsch H. V., and Böttcher C. (2015) H-aggregates of an indocyanine Cy5 dye: transition from strong to weak molecular coupling. J. Phys. Chem. B 119, 11900–11909 [DOI] [PubMed] [Google Scholar]
- 40. Chen C., Samuel T. K., Sinclair J., Dailey H. A., and Hamza I. (2011) An intercellular heme-trafficking protein delivers maternal heme to the embryo during development in. C. elegans. Cell 145, 720–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smith L. J., Kahraman A., and Thornton J. M. (2010) Heme proteins-diversity in structural characteristics, function, and folding. Proteins 78, 2349–2368 [DOI] [PubMed] [Google Scholar]
- 42. Qi Z., Hamza I., and O'Brian M. R. (1999) Heme is an effector molecule for iron-dependent degradation of the bacterial iron response regulator (Irr) protein. Proc. Natl. Acad. Sci. U.S.A. 96, 13056–13061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang L., and Guarente L. (1995) Heme binds to a short sequence that serves a regulatory function in diverse proteins. EMBO J. 14, 313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hofbauer S., Hagmüller A., Schaffner I., Mlynek G., Krutzler M., Stadlmayr G., Pirker K. F., Obinger C., Daims H., Djinović-Carugo K., and Furtmüller P. G. (2015) Structure and heme-binding properties of HemQ (chlorite dismutase-like protein) from Listeria monocytogenes. Arch. Biochem. Biophys. 574, 36–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Flint A., and Stintzi A. (2015) Cj1386, an atypical hemin-binding protein, mediates hemin trafficking to KatA in Campylobacter jejuni. J. Bacteriol. 197, 1002–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oliveira M. F., Timm B. L., Machado E. A., Miranda K., Attias M., Silva J. R., Dansa-Petretski M., de Oliveira M. A., de Souza W., Pinhal N. M., Sousa J. J., Vugman N. V., and Oliveira P. L. (2002) On the pro-oxidant effects of haemozoin. FEBS Lett. 512, 139–144 [DOI] [PubMed] [Google Scholar]
- 47. Graça-Souza A. V., Maya-Monteiro C., Paiva-Silva G. O., Braz G. R., Paes M. C., Sorgine M. H., Oliveira M. F., and Oliveira P. L. (2006) Adaptations against heme toxicity in blood-feeding arthropods. Insect Biochem. Mol. Biol. 36, 322–335 [DOI] [PubMed] [Google Scholar]
- 48. Gioannini T. L., Zhang D., Teghanemt A., and Weiss J. P. (2002) An essential role for albumin in the interaction of endotoxin with lipopolysaccharide-binding protein and sCD14 and resultant cell activation. J. Biol. Chem. 277, 47818–47825 [DOI] [PubMed] [Google Scholar]
- 49. Kaca W., Roth R. I., and Levin J. (1994) Hemoglobin, a newly recognized lipopolysaccharide (LPS)-binding protein that enhances LPS biological activity. J. Biol. Chem. 269, 25078–25084 [PubMed] [Google Scholar]
- 50. Solar I., Muller-Eberhard U., Shviro Y., and Shaklai N. (1991) Long-term intercalation of residual hemin in erythrocyte membranes distorts the cell. Biochim. Biophys. Acta 1062, 51–58 [DOI] [PubMed] [Google Scholar]
- 51. Carr C. Jr., and Morrison D. C. (1985) Mechanism of polymyxin B-mediated lysis of lipopolysaccharide-treated erythrocytes. Infect. Immun. 49, 84–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schmidt N., Mishra A., Lai G. H., and Wong G. C. (2010) Arginine-rich cell-penetrating peptides. FEBS Lett. 584, 1806–1813 [DOI] [PubMed] [Google Scholar]
- 53. Mezo M., González-Warleta M., and Ubeira F. M. (2003) Optimized serodiagnosis of sheep fascioliasis by Fast-D protein liquid chromatography fractionation of Fasciola hepatica excretory-secretory antigens. J. Parasitol. 89, 843–849 [DOI] [PubMed] [Google Scholar]
- 54. Mezo M., González-Warleta M., Carro C., and Ubeira F. M. (2004) An ultrasensitive capture ELISA for detection of Fasciola hepatica coproantigens in sheep and cattle using a new monoclonal antibody (MM3). J. Parasitol. 90, 845–852 [DOI] [PubMed] [Google Scholar]
- 55. Choi C. Y., Cerda J. F., Chu H. A., Babcock G. T., and Marletta M. A. (1999) Spectroscopic characterization of the heme-binding sites in Plasmodium falciparum histidine-rich protein 2. Biochemistry 38, 16916–16924 [DOI] [PubMed] [Google Scholar]
- 56. Li B., Du Y., Li T., and Dong S. (2009) Investigation of 3,3′,5,5′-tetramethylbenzidine as colorimetric substrate for a peroxidatic DNAzyme. Anal. Chim. Acta 651, 234–240 [DOI] [PubMed] [Google Scholar]
- 57. Holton T. A., Pollastri G., Shields D. C., and Mooney C. (2013) CPPpred: prediction of cell penetrating peptides. Bioinformatics 29, 3094–3096 [DOI] [PubMed] [Google Scholar]
- 58. Xu D., and Zhang Y. (2012) Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80, 1715–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gautier R., Douguet D., Antonny B., and Drin G. (2008) HELIQUEST: a web server to screen sequences with specific α-helical properties. Bioinformatics 24, 2101–2102 [DOI] [PubMed] [Google Scholar]