

Abstract
Hypoxia plays a role in the deterioration of β-cell function. Hepatocyte nuclear factor 4α (HNF4α) has an important role in pancreatic β-cells, and mutations of the human HNF4A gene cause a type of maturity-onset diabetes of the young (MODY1). However, it remains unclear whether hypoxia affects the expression of HNF4α in β-cells. Here, we report that hypoxia reduces HNF4α protein expression in β-cells. Hypoxia-inducible factor was not involved in the down-regulation of HNF4α under hypoxic conditions. The down-regulation of HNF4α was dependent on the activation of AMP-activated protein kinase (AMPK), and the reduction of HNF4α protein expression by metformin, an AMPK activator, and hypoxia was inhibited by the overexpression of a kinase-dead (KD) form of AMPKα2. In addition, hypoxia decreased the stability of the HNF4α protein, and the down-regulation of HNF4α was sensitive to proteasome inhibitors. Adenovirus-mediated overexpression of KD-AMPKα2 improved insulin secretion in metformin-treated islets, hypoxic islets, and ob/ob mouse islets. These results suggest that down-regulation of HNF4α could be of importance in β-cell dysfunction by hypoxia.
Keywords: AMP-activated kinase (AMPK), β-cell (B-cell), hepatocyte nuclear factor 4 (HNF-4), hypoxia, transcription factor
Introduction
Pancreatic β-cells sense glucose and secrete insulin to maintain normal glucose levels. Because pancreatic β-cells are highly dependent on oxidative phosphorylation for ATP production, they require large amounts of oxygen, especially at increased glucose levels (1, 2). Indeed, we and others demonstrated that pancreatic islets and insulin-producing β-cell lines become hypoxic under high glucose conditions and that the pancreatic islets of diabetic mice are hypoxic (3–5). Furthermore, we recently indicated that moderate hypoxia leads to β-cell dysfunction with a selective down-regulation of genes including Mafa, Pdx1, and Ins1, which play important roles in pancreatic β-cells (6). These results suggest that hypoxia is involved in the deterioration of β-cell function.
Hepatocyte nuclear factor 4α (HNF4α)3 is a transcription factor belonging to the nuclear receptor superfamily and is expressed in the pancreas (including β-cells), liver, kidney, and intestine (7). We found that mutations of the human HNF4A gene cause a particular form of maturity-onset diabetes of the young (MODY), that is, MODY1, which is characterized by autosomal dominant inheritance, early age of onset, and pancreatic β-cell dysfunction (8). In addition, targeted disruption of HNF4α in pancreatic β-cells leads to defective insulin secretion in mice (9, 10). These findings demonstrate the important role of HNF4α in pancreatic β-cells.
In the present study, we investigated the impact of hypoxia on HNF4α expression in MIN6 cells and mouse islets. We demonstrated that hypoxia decreases HNF4α protein expression via proteasome-mediated degradation. The hypoxia-induced down-regulation of HNF4α was regulated by the activation of AMP-activated protein kinase (AMPK). This reduction of HNF4α protein expression was recovered by inactivation of AMPK and re-oxygenation. Our results suggest that down-regulation of HNF4α is a novel mechanism of β-cell dysfunction by hypoxia.
Results
Down-regulation of HNF4α protein expression by hypoxia
MIN6 cells were cultured under moderately hypoxic conditions (3–7% oxygen tension) for 24 h, and HNF4α expression levels were examined by Western blot analysis. Hypoxia significantly decreased HNF4α protein levels, but not β-actin, in a dose-dependent manner (Fig. 1, A and B). Hypoxia did not affect HNF1α/MODY3 (11) and HNF1β/MODY5 (12) protein levels in MIN6 cells (Fig. 1C). Similar to MIN6 cells, decreased HNF4α protein levels by hypoxia were observed in pancreatic islets (Fig. 1D). Hypoxia reduces the expression of Hnf3b/Foxa2 mRNA in MIN6 cells (6). We then examined the expression levels of Hnf4a mRNA. Hypoxia for 12 h had no effect on Hnf4α mRNA expression in MIN6 cells (Fig. 1E). However, a significant down-regulation of HNF4α protein (61.3% of control; p < 0.01) was detected in MIN6 cells following 5% oxygen tension for 12 h (Fig. 1, F and G), suggesting the post-transcriptional regulation of HNF4α expression. In this study, we focused on the hypoxia-induced down-regulation of HNF4α at the protein level.
Figure 1.

Effect of hypoxia on HNF4α expression in β-cells. A, MIN6 cells were exposed to the indicated oxygen tension (% O2) for 24 h, and HNF4α expression was examined by Western blotting. B, relative HNF4α protein levels were calculated (n = 3). C, effect of hypoxia for 24 h on HNF1α and HNF1β expression in MIN6 cells (n = 3). D, isolated mouse islets were incubated at either 5% O2 or 20% O2 for 24 h, and HNF4α protein levels were evaluated by Western blot analysis (n = 3). E, MIN6 cells were cultured at either 5% O2 or 20% O2 for 12 h and 24 h, and Hnf4a mRNA levels were analyzed by qPCR (n = 3). The Hnf4α mRNA level was normalized to that of TBP. F and G, MIN6 cells were cultured at the same conditions as in E, and HNF4α protein levels were examined by Western blot analysis (n = 3). All data are presented as mean ± S.E. (S.E.; error bars). N.S., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Effect of down-regulation of HNF4α expression on β-cells
HNF4α plays an important role in glucose-stimulated insulin secretion by β-cells (8). Suppression of endogenous HNF4α consistently reduced insulin secretion in MIN6 cells (supplemental Fig. S1, A and B). In addition, HNF4α knockdown significantly increased the number of propidium iodide-positive dead cells (supplemental Fig. S1C). We previously reported that hypoxia induces an insulin secretion defect and cell death in MIN6 cells (6). Down-regulation of HNF4α may be involved in this dysfunction by hypoxia. In contrast, cell proliferation rate was unaffected by the down-regulation of HNF4α (supplemental Fig. S1D).
Effect of HIF on HNF4α expression by hypoxia
Hypoxia-inducible factor (HIF) is a transcription factor that regulates the expression of genes mediating adaptive responses to hypoxia. HIF-1α protein is degraded under normoxic conditions, whereas it is stabilized under hypoxic conditions and binds to HIF-1β to act as a transcription factor (13). Hypoxia (5% O2) increased the expression of HIF-1α protein in a time-dependent manner in MIN6 cells (Fig. 2A). We then investigated the role of HIF-1 in the hypoxia-induced down-regulation of HNF4α. Overexpression of a constitutively active form of HIF-1α by retrovirus infection did not affect HNF4α expression (Fig. 2, B and C). Next, we introduced HIF-1α short hairpin RNA (shRNA) into MIN6 cells, and successful HIF-1α knockdown was confirmed under hypoxic conditions (Fig. 2D). Suppression of HIF-1α did not affect the down-regulation of HNF4α at the 5% oxygen condition (Fig. 2, D and E). In addition to HIF-1α, suppression of HIF-2α, HIF-3α, or HIF-1β failed to reverse the down-regulation of HNF4α by hypoxia (supplemental Fig. S2). These findings indicate that HIF is not involved in the down-regulation of HNF4α under hypoxic conditions.
Figure 2.
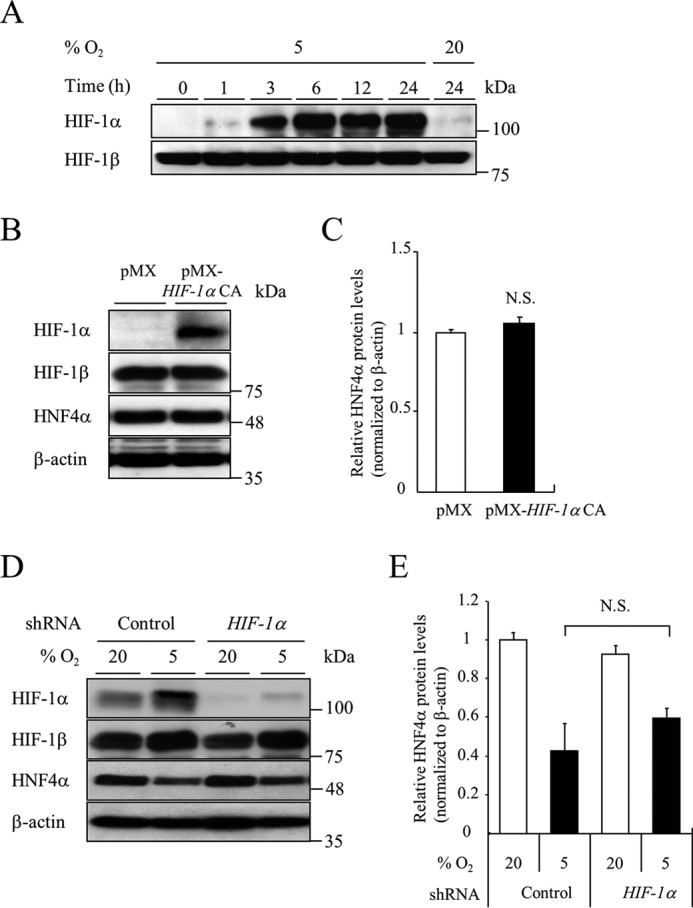
Role of HIF-1α in HNF4α expression by hypoxia. A, MIN6 cells were incubated at either 5% O2 or 20% O2 for the indicated time, and HIF-1α protein levels were detected by Western blotting. B and C, the effect of HIF-1α CA on HNF4α protein levels. MIN6 cells expressing the pMX empty vector or pMX-HIF-1α constitutively active were cultured in normoxia and cell lysates were recovered (n = 3). D and E, MIN6 cells expressing either control shRNA or HIF-1α shRNA were incubated at either 5% O2 or 20% O2 for 24 h, and HNF4α protein levels were examined (n = 3). All data are presented as mean ± S.E. (error bars). N.S., not significant.
Effect of AMPK on HNF4α expression by hypoxia
In addition to the stabilization of HIF-1α, hypoxia is known to activate AMPK (14). Indeed, the phosphorylation of both AMPK and acetyl-CoA carboxylase, a substrate of AMPK, was induced after exposure to hypoxia for 1 h in MIN6 cells (supplemental Fig. S3). Down-regulation of HNF4α was subsequently detected after exposure to hypoxia for 12 h. We then examined whether activation of AMPK could affect HNF4α expression. Both 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR), a cell-permeable activator of AMPK, and metformin decreased the expression of HNF4α in MIN6 cells (Fig. 3A). Similar to MIN6 cells, metformin also decreased HNF4α protein levels in pancreatic islets (Fig. 3, B and C).
Figure 3.

Role of AMPK in HNF4α expression. A, effect of AMPK activators on HNF4α protein levels. MIN6 cells were cultured at the indicated concentration of AICAR or metformin for 24 h, and Western blotting was performed. B and C, isolated mouse islets were treated with 2 mm metformin for 24 h, and HNF4α protein levels were examined. D and E, MIN6 cells expressing the pMX empty vector or pMX-KD-AMPKα2 vector were treated with 2 mm metformin for 20 h, and HNF4α protein levels were examined (n = 3). F and G, an insulin secretion assay was performed (n = 4–5), and insulin concentration was determined by an insulin ELISA. Fold-change in glucose-stimulated insulin secretion (insulin level at 22 mm glucose divided by that at 2.2 mm glucose) is shown (n = 4–5). H, isolated mouse islets expressing either LacZ or KD-AMPKα2 were treated with 2 mm metformin for 20 h, and HNF4α protein levels were examined by Western blot analysis. I and J, islet insulin secretion was examined. Insulin levels are expressed as absolute values or as fold-change (n = 11–12). All data are presented as mean ± S.E. (error bars). N.S., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
The kinase-dead (KD) form of AMPKα2 (Lys-45 was changed to Arg) reportedly functions as a dominant inhibitory protein that eliminates AMPK activity (15). We examined the effect of KD-AMPKα2 overexpression by retrovirus on the metformin-induced down-regulation of HNF4α. Phosphorylation of acetyl-CoA carboxylase was inhibited by KD-AMPKα2 overexpression (Fig. 3D). In addition, HNF4α down-regulation was significantly suppressed by KD-AMPKα2 overexpression (Fig. 3, D and E). We next examined the effect of AMPK activity on insulin secretion in MIN6 cells. Consistent with the decreased expression of HNF4α, metformin reduced insulin secretion at 22 mm glucose (Fig. 3, F and G). In accordance with the increased expression of HNF4α, KD-AMPKα2 improved insulin release by 22 mm glucose in metformin-treated MIN6 cells. Similarly, adenovirus-mediated overexpression of KD-AMPKα2 increased HNF4α protein expression and improved glucose-stimulated insulin secretion in metformin-treated islets (Fig. 3, H–J).
We also investigated the effect of KD-AMPKα2 overexpression on the hypoxia-induced down-regulation of HNF4α. Phosphorylation of AMPK by hypoxia was inhibited by KD-AMPKα2 overexpression (supplemental Fig. S4). Overexpression of KD-AMPKα2 in MIN6 cells suppressed the reduction of HNF4α protein expression by hypoxia, as with metformin (Fig. 4, A and B). Collectively, these findings indicate that hypoxia decreases HNF4α protein levels via the activation of AMPK. Insulin secretion was stimulated 3.4-fold by 22 mm (versus 2.2 mm) glucose in MIN6 cells under 20% O2 tension, whereas hypoxic MIN6 cells exhibited dysregulated insulin secretion (increased insulin secretion at low glucose and blunted insulin secretion at high glucose) (6) (Fig. 4, C and D). However, the diminished glucose-stimulated changes in insulin secretion under 5% O2 tension were significantly increased by overexpression of KD-AMPKα2 (pMX control, 1.9-fold; pMX-KD-AMPKα2, 2.4-fold, p < 0.01) (Fig. 4D). Overexpression of KD-AMPKα2 also increased HNF4α expression and enhanced insulin secretion in response to high glucose in islets under hypoxic conditions (Fig. 4, E–G). These results suggest the functional significance of AMPK- and hypoxia-dependent regulation of HNF4α expression on insulin secretion.
Figure 4.

Role of AMPK in hypoxia-induced HNF4α down-regulation. A and B, MIN6 cells expressing the pMX empty vector or pMX-KD-AMPKα2 vector were cultured at either 5% O2 or 20% O2 for 20 h, and HNF4α protein levels were examined (n = 3). C and D, an insulin secretion assay was performed (n = 6–9), and insulin concentration was determined by an insulin ELISA. Fold-change in glucose-stimulated insulin secretion (insulin level at 22 mm glucose divided by that at 2.2 mm glucose) is shown (n = 6–9). E, mouse isolated islets expressing either LacZ or KD-AMPKα2 were treated with 5% O2 for 20 h, and HNF4α protein levels were examined by Western blot analysis. F and G, islet insulin secretion was examined, and insulin levels are expressed as absolute values or as fold-change (n = 5–10). Data are presented as mean ± S.E. (error bars). N.S., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
HNF4α protein stability under hypoxic conditions
Hypoxia reportedly leads to the degradation of proteins, such as estrogen receptor α and Na,K-ATPase (16, 17). We next evaluated whether HNF4α protein stability is influenced by hypoxia. MIN6 cells were cultured under 20 or 5% oxygen tension in the presence of cycloheximide (CHX), a translational inhibitor. During incubation with CHX, HNF4α protein decayed rapidly in the 5% oxygen condition (Fig. 5A), indicating that hypoxia decreases the stability of HNF4α protein. We also investigated HNF4α stability by metformin treatment. Metformin promoted HNF4α degradation (Fig. 5B). Then, HNF4α protein stability was tested in the presence or absence of the proteasome inhibitor MG132 (Fig. 5C). As shown in Fig. 5, C–F, proteasome inhibition resulted in the stabilization of HNF4α in both hypoxic and metformin-treated conditions (compare lanes 3 and 4 of Fig. 5D). Another proteasome-specific inhibitor, epoxomicin, also stabilized HNF4α protein expression under hypoxia (supplemental Fig. S5). These findings indicate that proteasome-mediated degradation is involved in the hypoxia- and AMPK activation-induced down-regulation of HNF4α.
Figure 5.

Regulation of HNF4α protein stability by hypoxia or metformin. A, MIN6 cells were incubated at 5% O2 or 20% O2 for 18 h, and HNF4α protein stability was examined for another 6 h using CHX (10 μg/ml). Relative HNF4α protein levels are indicated as percentage values (n = 3–4). Half-lives of HNF4α protein are indicated. B, MIN6 cells were incubated with or without metformin (2 mm) for 18 h, and HNF4α protein stability was examined. Relative HNF4α protein levels are indicated as percentage values (n = 3–4). Half-lives of HNF4α protein are shown. C, experimental scheme using the MG132 proteasome inhibitor. D, effect of MG132 (10 μm) on HNF4α expression by hypoxia or metformin treatment. E and F, relative HNF4α protein levels in 5% O2 (hypoxia) (E) or metformin (F) were calculated (n = 3). All data are presented as mean ± S.E. (error bars). N.S., not significant; *, p < 0.05; **, p < 0.01.
AMPK activation leads to decreased levels of proteins by regulating the binding of RNA-binding proteins within the 3′-untranslated region (3′-UTR) of target mRNAs (18). To address the role of the 3′-UTR in HNF4α mRNA, MIN6 cells were infected with a retroviral HNF4α-FLAG expression vector (lacking both the 3′-UTR and 5′-UTR of the HNF4α gene). Both hypoxia and metformin decreased HNF4α protein levels (Fig. 6, A and B), indicating that the 3′-UTR is not involved in down-regulation of HNF4α by hypoxia/AMPK activation in MIN6 cells.
Figure 6.
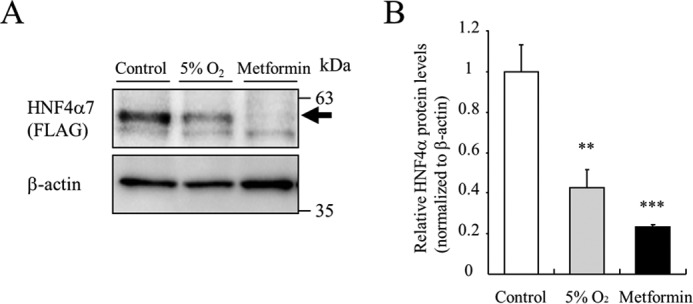
Role of the 3′-UTR in HNF4α protein stability. A, MIN6 cells expressing HNF4α7-FLAG by retrovirus transfection were incubated at either 5% O2 or 2 mm metformin treatment for 24 h, and exogenous HNF4α protein expression was detected using an anti-FLAG antibody. B, relative HNF4α protein levels were calculated (n = 3). All data are presented as mean ± S.E. (error bars). N.S., not significant; **, p < 0.01; ***, p < 0.001.
Phosphorylation of HNF4α by hypoxia
The phosphorylation of proteins triggers protein degradation (19, 20). AMPK activation reportedly leads to the phosphorylation of the liver type of HNF4α at serine 304 (serine 291 of HNF4α7) in vitro (21). Thus, we examined whether this phosphorylation is essential for the hypoxia-induced down-regulation of HNF4α in HNF4α7-FLAG-expressing MIN6 cells. As shown in Fig. 7A, HNF4α7 with a mutation of serine 291 to alanine (S291A) was still down-regulated by hypoxia. Database screening (ppsp.biocuckoo.org/index.php) identified another potential AMPK phosphorylation site (117TRRSSYEDS125, potential phosphorylation serine 121 is underlined) in HNF4α7. However, the replacement of serine 121 with alanine (S121A) also did not restore HNF4α7 instability (Fig. 7A). Because both S121A and S291A mutants were as sensitive to hypoxia-induced down-regulation as wild-type HNF4α7, we investigated whether HNF4α7 was phosphorylated by hypoxia and AMPK activation in MIN6 cells. Phosphorylated proteins show slower migration in Phos-tag SDS-PAGE due to the selective binding of phosphorylated amino acids to the Phos-tag reagent (22). After treatment with metformin or hypoxia, whole cell lysates of MIN6 cells were separated by SDS-PAGE with Phos-tag, and blotted with anti-AMPK and anti-HNF4α antibodies. Treatment with hypoxia or metformin led to AMPK phosphorylation (Fig. 3). Phosphorylated AMPK following hypoxia or metformin treatment was detected as a band with slower migration using an anti-AMPK antibody (Fig. 7B). It has been reported that protein kinase A phosphorylates HNF4α (23). Consistently, a band shift of HNF4α was observed by treatment with forskolin, which activates the protein kinase A pathway via cAMP. In contrast, Western blot analysis with an anti-HNF4α antibody did not show a detectable band shift by treatment with hypoxia or metformin, suggesting that the HNF4α protein is not phosphorylated by metformin and 5% hypoxia in MIN6 cells.
Figure 7.
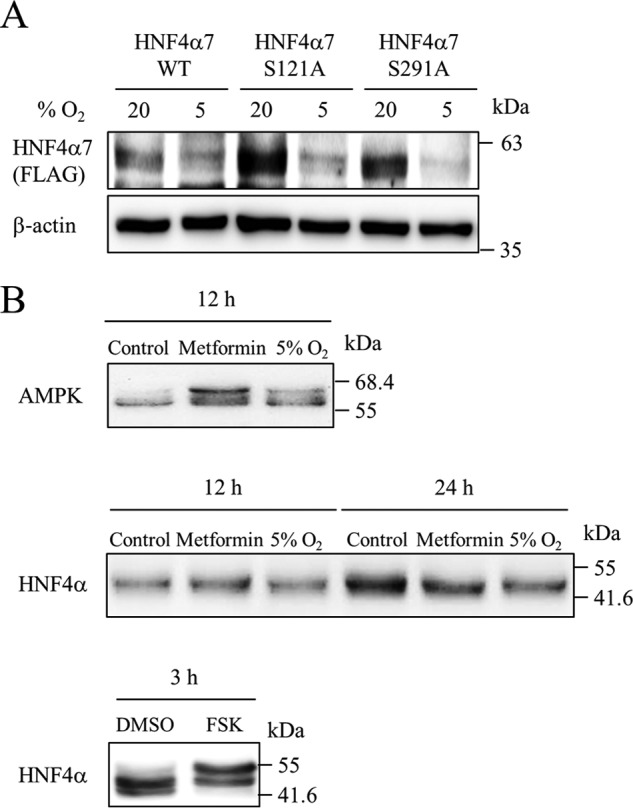
Phosphorylation of HNF4α protein by hypoxia. A, MIN6 cells expressing wild-type (WT) or mutant (S121A or S291A) HNF4α7-FLAG by retrovirus transfection were cultured at either 5% O2 or 20% O2 for 24 h. Both exogenous and endogenous HNF4α proteins were examined using an anti-FLAG antibody and anti-HNF4α antibody, respectively. B, MIN6 cells were treated with metformin (2 mm) or incubated in 5% O2 for 12 and 24 h. Phosphorylation status of both AMPK and HNF4α was evaluated using Phos-tag SDS-PAGE. For a positive control, MIN6 cells were treated with either DMSO or forskolin (FSK, 100 μm) for 3 h.
The effect of ERK5 and E3 ubiquitin ligases on HNF4α expression by hypoxia
A recent study showed that extracellular signal-regulated kinase 5 (ERK5)/mitogen-activated protein kinase 7 (MAPK7) up-regulates proteasome levels upon mTOR (mechanistic target of rapamycin) signaling inhibition (24). Because hypoxia increased the expression of ERK5 protein (Fig. 8A), we examined the effect of ERK5 on HNF4α expression. The hypoxia-induced down-regulation of HNF4α was similar between control and ERK5 knockdown MIN6 cells (Fig. 8, B and C, supplemental Fig. S6, A and B), indicating that ERK5 does not contribute to the down-regulation of HNF4α by hypoxia.
Figure 8.
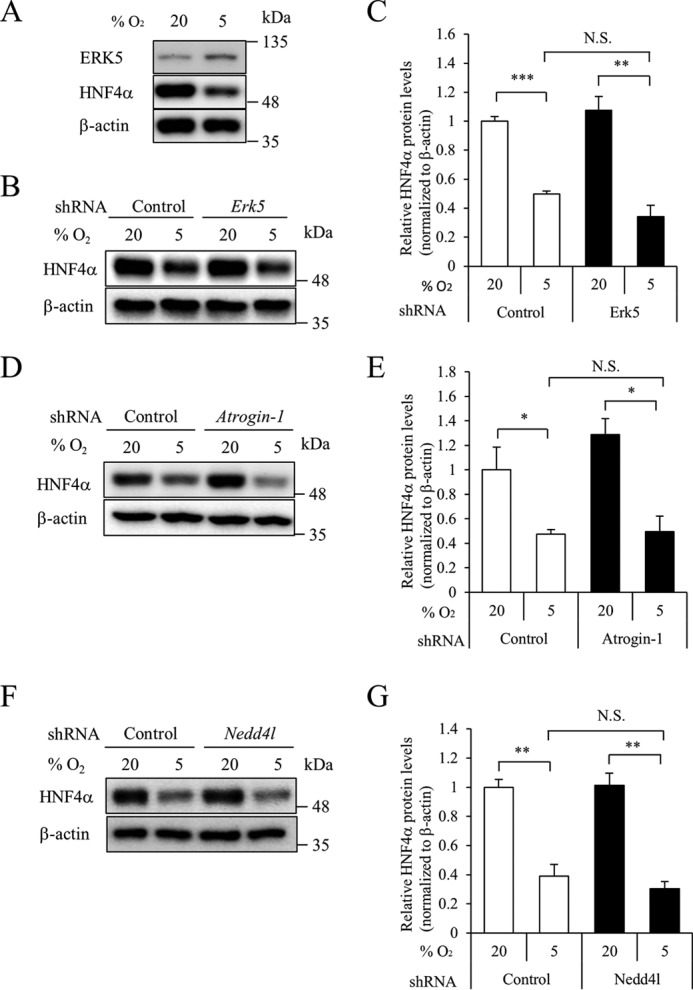
Role of ERK5 and E3 ubiquitin ligases in HNF4α expression by hypoxia. A, MIN6 cells were incubated in either 5 or 20% O2 for 18 h, and expression of ERK5 or HNF4α protein was examined by Western blot analysis. B, MIN6 cells expressing either control shRNA or Erk5 shRNA were incubated in either 5 or 20% O2 for 24 h, and HNF4α protein levels were examined by Western blot analysis. C, relative HNF4α protein levels were calculated (n = 3). D–G, effect of overexpression of atrogin-1 shRNA (D and E) or Nedd4l shRNA (F and G) on HNF4α expression was examined. All data are presented as mean ± S.E. (error bars). N.S., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Activation of AMPK causes proteasomal degradation of proteins via E3 ubiquitin ligases, including atrogin-1, neural precursor cell expressed, developmentally down-regulated gene 4-like (Nedd4l), muscle RING finger-1 (MuRF1), and Smad ubiquitylation regulatory factor-1 (Smurf1) (25–27). Because the expression levels of MuRF1 and Smurf1 mRNA were low in MIN6 cells (data not shown), we examined the effect of atrogin-1 and Nedd4l on HNF4α expression. Suppression of these E3 ubiquitin ligases did not reverse the down-regulation of HNF4α protein under hypoxia (Fig. 8, D–G, supplemental Fig. S6, C and D), suggesting that these ubiquitin ligases are also not involved in the down-regulation of HNF4α.
Restoration of HNF4α protein expression by re-oxygenation or inactivation of AMPK
We next examined the effect of re-oxygenation on HNF4α expression. Expression of HNF4α protein was increased in MIN6 cells that were re-oxygenized for more than 3 h (R3–R12) (Fig. 9A). Re-oxygenation for 12 h (R12) also increased HNF4α expression in pancreatic islets (Fig. 9, B and C). Consistent with our previous finding that pancreatic islets of diabetic ob/ob mice are hypoxic (3), HNF4α protein expression was significantly decreased in ob/ob mouse islets (Fig. 9, D and E). However, expression levels of the HNF4α protein became similar between control and ob/ob islets after incubation at 20% oxygen tension.
Figure 9.

Impact of re-oxygenation on HNF4α expression. A, MIN6 cells were cultured at 5% O2 for 24 h and then cultured at 20% O2 (re-oxygenation) for the indicated time (0.5–12 h). HNF4α expression was examined by Western blot analysis. B and C, isolated mouse islets were cultured at 5% O2 for 20 h and then cultured at 20% O2 for 12 h. HNF4α protein levels were examined (n = 3). D and E, islets were isolated from C57BL/6J or ob/ob mice. HNF4α protein levels were examined immediately after islet isolation and after incubation at 20% O2 for 12 h (n = 4–6). F, islets from ob/ob mice were infected with adenovirus expressing LacZ or KD-AMPKα2 for 2 h, and cultured for 36 h. Expression of AMPKα and HNF4α was examined by Western blotting. G, insulin secretion assay was performed using ob/ob mouse islets expressing either LacZ (n = 13) or KD-AMPKα2 (n = 24). Insulin concentration was determined by an insulin ELISA. H, fold-change in glucose-stimulated insulin secretion (insulin level at 22 mm glucose divided by that at 2.2 mm glucose) is shown. All data are presented as mean ± S.E. (error bars). N.S., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Finally, we investigated the effect of KD-AMPKα2 on insulin secretion in ob/ob islets. Insulin secretion from ob/ob islets was unresponsive to glucose stimulation (28). Consistently, insulin release by high glucose did not differ from that by low glucose in control ob/ob islets. However, overexpression of KD-AMPKα2 increased the expression of HNF4α in ob/ob islets, and insulin secretion by high glucose was significantly improved (Fig. 9, F–H).
Discussion
HNF4α7 and HNF4α8 are major isoforms of HNF4α in pancreatic β-cells, and HNF4α plays important roles in the function of these cells (8, 29, 30). In the present study, we demonstrated that hypoxia leads to the down-regulation of HNF4α protein in β-cells. HNF4α down-regulation was dependent on AMPK activation and was sensitive to proteasome inhibitors, indicating that HNF4α proteasomal degradation is regulated, at least in part, by AMPK activation (Fig. 10). A reduction of hepatic HNF4α (HNF4α1 and HNF4α2 are major hepatic forms) by AICAR treatment has also been reported (31). HNF4α plays an essential role in glucose-stimulated insulin secretion by pancreatic β-cells (8, 9) (supplemental Fig. S1). Metformin treatment attenuated high glucose-stimulated insulin secretion with the decreased expression of HNF4α. Conversely, overexpression of KD-AMPKα2 improved insulin secretion with the increased expression of HNF4α (Fig. 3). Collectively, these results suggest that the regulation of HNF4α expression by AMPK has a functional consequence on insulin secretion. Hypoxia and AMPK activation inhibit multiple steps in the process of insulin secretion (6, 32–36). Our findings uncover a new role for AMPK in impaired insulin secretion by β-cells. Decreased HNF4α expression in β-cells could reduce oxygen consumption by reducing insulin secretion under hypoxic conditions. Thus, the down-regulation of HNF4α may be a fail-safe mechanism against hypoxia. However, prolonged exposure of β-cells to hypoxia, in turn, would have deleterious effects on insulin secretion by reducing HNF4α/MODY1 transcription factor levels and could contribute to the development of type 2 diabetes.
Figure 10.
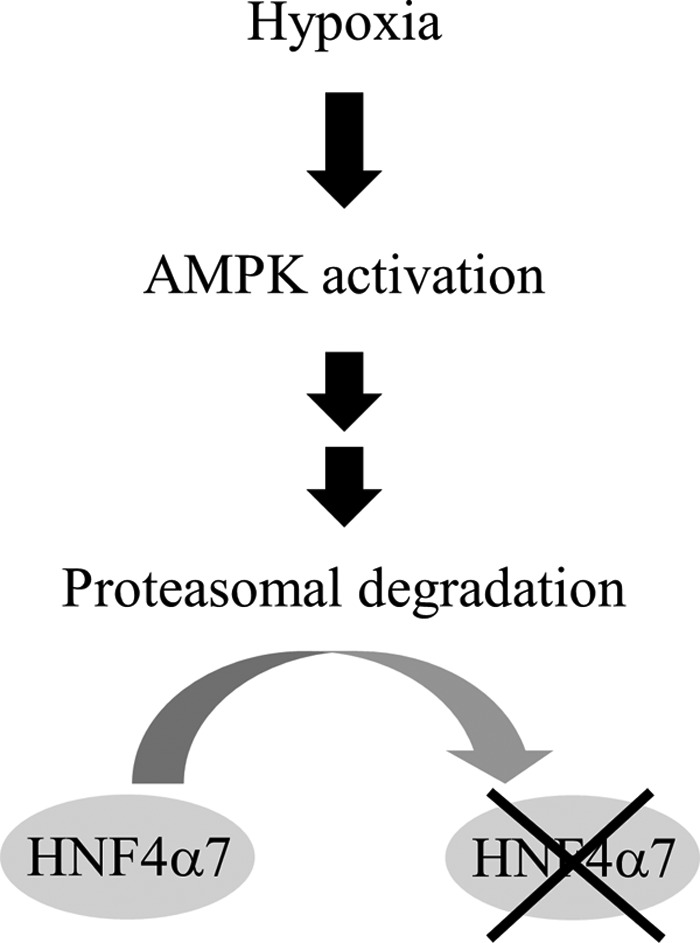
Schematic representation of the hypoxia-induced down-regulation of HNF4α in β-cells.
AMPK activation reportedly induces the phosphorylation of hepatic HNF4α in vitro (21), but the replacement of potential AMPK phosphorylation sites (Ser121 and Ser291) did not affect HNF4α instability in MIN6 cells (Fig. 7). A mobility shift of HNF4α after hypoxia or metformin treatment was not detected using Phos-tag SDS-PAGE. In addition, we could not detect a phosphorylation signal in HNF4α immunoprecipitates that were prepared from hypoxic MIN6 cells using the commercially available anti-phospho-AMPK substrate motif MultiMab antibody (data not shown). Although the possibility that β-cell-type HNF4α is a direct target of AMPK cannot be excluded, these results suggest that HNF4α is not phosphorylated directly by metformin and/or hypoxia in MIN6 cells.
We have demonstrated that hypoxia increases the expression of ERK5, and ERK5 has been shown to up-regulate proteasome levels (24). Moreover, AMPK activation leads to proteasomal degradation of target proteins via the ubiquitin ligases atrogin-1 and Nedd4l (25, 26). However, these molecules do not appear to account for the mechanism of HNF4α degradation by AMPK (Fig. 8). Further studies are necessary to clarify the role of AMPK on HNF4α expression in β-cells.
We found that re-oxygenation restored HNF4α expression in both control and ob/ob islets (Fig. 9). In addition, restoration of HNF4α in ob/ob islets significantly improved insulin secretion. Restoration of HNF4α alone may not be sufficient to normalize completely the impaired secretion of insulin because ob/ob islets exhibit additional abnormalities (37). However, our findings suggest that methods to enhance oxygenation in hypoxic β-cells could be an effective therapeutic approach to remedy insulin secretion defects in type 2 diabetes by restoring HNF4α.
There are limitations in this study. First, the mechanisms by which AMPK activation leads to the down-regulation of HNF4α are unclear. Second, we do not have direct evidence that hypoxia leads to pancreatic β-cell HNF4α protein degradation in vivo. Investigation of HNF4α protein levels of mouse β-cells in a low oxygen environment would be a major undertaking.
In conclusion, we demonstrated that hypoxia decreased HNF4α protein expression via proteasome-mediated protein degradation in β-cells. Furthermore, the hypoxia-induced down-regulation of HNF4α was regulated by the activation of AMPK. Because a small perturbation of HNF4α activity in β-cells results in MODY1 (38), our findings suggest that down-regulation of HNF4α could be of importance in β-cell dysfunction by hypoxia.
Experimental procedures
Reagents
AICAR and 1,1-dimethylbiguanide hydrochloride (metformin) were purchased from Sigma. CHX was obtained from Nacalai Tesque (Kyoto, Japan), MG132 from Calbiochem, and epoxomicin from the Peptide Institute (Osaka, Japan).
Cell culture
The MIN6 β-cell line (39) was a gift from Jun-ichi Miyazaki (Osaka University). MIN6 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum (FBS), 0.1% (v/v) penicillin/streptomycin, and 50 μm β-mercaptoethanol at 37 °C in 5% CO2, 95% air. A multi-gas incubator (APM-300; ASTEC, Fukuoka, Japan) and an INVIVO2400 hypoxia work station (Ruskin Technology, Leeds, UK) were used to render the cells chronically hypoxic (6).
Animals
C57BL/6J and B6.Cg-Lepob/J (ob/ob) mice were purchased from KBT Oriental Co., Ltd. (Saga, Japan). The mice were kept under specific pathogen-free conditions in a 12-h light (7:00–19:00)/12-h dark (19:00–7:00) cycle with free access to water and normal mouse chow (CE-2; CLEA, Tokyo, Japan). Room temperature was maintained at 22 ± 1–2 °C. Handling and killing of the mice by cervical dislocation were in compliance with the animal care guidelines of Kumamoto University. This study was approved by the animal research committee of Kumamoto University and all animal experimental protocols were approved by the Kumamoto University Ethics Review Committee for Animal Experimentation.
Isolation of pancreatic islets
Pancreatic islets were isolated from C57BL/6J or ob/ob mice (20–30 weeks old) as described previously (40). Briefly, after bile duct cannulation and digestion of the pancreas using a mixture of 1.3 mg/ml of collagenase L (Nitta Gelatin, Osaka, Japan), 1.3 mg/ml of hyaluronidase (H3506; Sigma), and 0.1% (v/v) protease inhibitor mixture (Nacalai Tesque), isolated islets were manually picked up and they were maintained in RPMI-1640 medium supplemented with 10% (v/v) FBS, 0.1% (v/v) penicillin/streptomycin, 50 μm β-mercaptoethanol, 10 mm HEPES (Nacalai Tesque), and 1 mm sodium pyruvate (Nacalai Tesque) at 37 °C in 5% CO2, 95% air.
Western blotting
Western blotting was performed as described previously (41). For specific detection of proteins, the following primary antibodies were used: anti-HNF4α (H1415; Perseus Proteomics, Tokyo, Japan), anti-HNF1α (610902; BD Biosciences, San Jose, CA), anti-HNF1β (H85; Santa Cruz Biotechnology, Santa Crux, CA), anti-β-actin (clone AC15; Sigma), anti-GAPDH (Promega, Madison, WI), anti-HIF-1α (NB100–479; Novus Biologicals, Littleton, CO), anti-HIF-1β (#611078; BD Transduction Laboratories, San Jose, CA), anti-phospho-AMPKα (Thr172) (40H9) (number 2535; Cell Signaling, Beverly, MA), anti-AMPK (number 2532; Cell Signaling), anti-phospho-acetyl-CoA carboxylase (Ser79) (number 3661; Cell Signaling), anti-acetyl-CoA carboxylase (number 3662; Cell Signaling), anti-ERK5 (number 3372; Cell Signaling), and anti-DYKDDDDK (FLAG) tag monoclonal (Clone No. 1E6; Wako Pure Chemicals, Osaka, Japan). After incubation with secondary antibodies, the signals were detected by using Chemi-Lumi One L or Chemi-Lumi One Super (Nacalai Tesque).
Quantitative real-time RT-PCR
For quantitative real-time PCR (qPCR), total RNA was extracted from MIN6 cells. qPCR was performed using SYBR Premix Ex TaqII (RR820A; TaKaRa Bio Inc., Shiga, Japan) in an ABI 7300 thermal cycler (Applied Biosystems, Foster City, CA) (42). The mRNA level of each gene was normalized to that of the TATA-binding protein (TBP). The specific primers were as follows: HNF4α: forward, 5′-GCCACAGTTTTCCACCAAGAG-3′, reverse, 5′-AAGGAGGACGTCTGCTTCTGA-3′; HIF-2α: forward, 5′-CCCCGATGAATTCACCCAAA-3′, reverse, 5′-TCTTGGCGTTCTCCCCTGAG-3′; HIF-3α: forward, 5′-CACTTTGTTGAGCAAGGGCC-3′, reverse, 5′-TGCGAGTATGTTGCTCCGTT-3′; atrogin-1: forward, 5′-GTGGCATCGCCCAAAAGAAC-3′, reverse, 5′-CTTGCCGACTCTCTGCACCA-3′; Nedd4l: forward, 5′-TCGGCCAATCATGCAGCTTGC-3′, reverse, 5′-TGTGTTGTGGTTTGGGGGAGTTG-3′; Erk5: forward, 5′-TTGCGCCCCACCTTTTGACTTTG-3′, reverse, 5′-AAGCGGATTTGTTGGCGGATGC-3′; TBP: forward, 5′-CCCCTTGTACCCTTCACCAAT-3′, reverse, 5′-GAAGCTGCGGTACAATTCCAG-3′.
Vector constructs, retrovirus-mediated transduction, and adenovirus-mediated transduction
The pMXs-HIF-1α CA5 and pSUPER.retro-HIF-1α plasmids have been described previously (3, 6). A kinase-dead α2 isoform of K45R AMPK (KD-AMPKα2) was a gift from Morris Birnbaum (Addgene plasmid number 15992, Addgene, Cambridge, MA) (15) and cloned into the pMXs-puro retroviral vector. To introduce an in-frame FLAG tag at the C-terminal of HNF4α7, PCR was performed using pcDNA3-HNF4α7 (42) as a template and the following primers: 5′-CCCCTGCACCCTCACCTGATGCAG-3′ and 5′-GAATTCCTACTTGTCATCGTCGTCCTTGTAGTCGATAACTTCCTGCTTGGTGATGGT-3′ (the underlined nucleotides indicate an EcoRI cloning site, and the bold letters indicate the FLAG sequence). After verification of the sequence, an NdeI/EcoRI fragment of the PCR product and an EcoRI/NdeI fragment of pcDNA3-HNF4α7 were subcloned into the EcoRI site of pcDNA3.1 to generate pcDNA3.1-HNF4α7-FLAG. S121A and S291A mutations were introduced into the pcDNA3.1-HNF4α7-FLAG plasmid using a KOD-Plus Mutagenesis Kit (TOYOBO, Osaka, Japan) according to the manufacturer's protocol. The following primers were used: 5′-TCAGCCTATGAGGACAGCAGCCTG-3′ and 5′-CCTTCGAGTGCTGATCCGGTCCCG-3′ (S121A); 5′-CGTGCCCAGGTGCAGGTGAGCTTG-3′ and 5′-GATCCAGGGAAGATCAAGCGGCTG-3′ (S291A) (the underlined nucleotides indicate the mutated nucleotides for alanine). EcoRI fragments of pcDNA3.1-HNF4α7 (wild-type, S121A, and S291A)-FLAG were cloned into the pMXs-puro retroviral vector. The pSIREN-RetroQ HNF4α shRNA expression vector has been described previously (35). For knockdown of HIF-2α, HIF-3α, atrogin-1, developmentally down-regulated 4-like (Nedd4l), and Erk5, oligonucleotides encoding respective shRNA was cloned into a pSIREN-RetroQ expression vector (Clontech Laboratories, Mountain View, CA). Target sequences were as follows: 5′-GGGACTTACTCAGGTAGAACT-3′ for HIF-2α shRNA, 5′-TTTCCGACAGGTAAGCCATGT-3′ for HIF-3α shRNA, 5′-AAAGTACAGTATCCATGGCGC-3′ for atrogin-1 shRNA, 5′-GGAGACCATTTCAGAGGAAAT-3′ for Nedd4l shRNA, 5′-ATTGTGGCTGAGATTGAGGACTT-3′ for Erk5 shRNA. Retrovirus transfection was performed as described previously (6). Recombinant adenovirus expressing KD-AMPKα2 or LacZ was prepared using Adeno-X Expression System 1 (Clontech Laboratories) per the manufacturer's protocol. Briefly, KD-AMPKα2 was subcloned into Adeno-X viral DNA via the shuttle vector pShuttle2. Then, 10 μg of linearized Adeno-X KD-AMPKα2 vector was transfected to a low passage of HEK293T using JetPRIME transfection reagent (Polyplus-transfection SA, Illkirch, France). Recombinant adenovirus was amplified in HEK293T cells and viral titers were determined by Adeno-X qPCR Titration Kit according to the manufacturer's protocol (Clontech Laboratories).
Insulin secretion assay
Control MIN6 cells or HNF4α knockdown MIN6 cells were seeded in a 24-well plate at 5.0 × 105 cells/well and maintained for 2–3 days. They were preincubated for 1 h in Krebs-Ringer-bicarbonate HEPES (KRBH) buffer (120 mm NaCl, 4.7 mm KCl, 1.2 mm KH2PO4, 2.4 mm CaCl2, 1.2 mm MgCl2, 20 mm NaHCO3, and 10 mm HEPES) containing 2.2 mm glucose and 0.5% (v/v) bovine serum albumin (BSA) at pH 7.4. They were incubated in 2.2 or 22 mm glucose containing KRBH buffer for 1 h, and the culture supernatant was recovered.
Then, mouse pancreatic islets from C57BL/6J mice or ob/ob mice were cultured in RPMI-1640 medium for 2 days, and islets of similar size were randomly picked up, divided into 2 groups (20–30 islets), and separately infected with recombinant adenovirus (LacZ or KD-AMPKα2, 2.0 × 105 infectious units (IFU)) for 2 h. After 24-h incubation in RPMI-1640 medium, these islets were further cultured in 2 mm metformin or 5% O2 for 20 h, and then the insulin secretion assay was performed until 48 h after adenoviral infection. The islets were preincubated for 30 min in KRBH buffer containing 2.2 mm glucose and 0.5% (v/v) BSA. They were incubated in 2.2 or 22 mm glucose containing KRBH buffer for 30 min, and the culture supernatant was recovered to evaluate insulin secretion. Insulin concentration was determined by a mouse insulin ELISA (TMB) kit (AKRIN-011T; Shibayagi Co., Ltd., Gunma, Japan). For MIN6 cells, insulin levels were standardized by whole cell protein content.
Apoptosis assay
Control MIN6 cells or HNF4α knockdown MIN6 cells were incubated for 48 h, and then trypsinized and collected by centrifugation at 6,000 rpm for 10 min. Cells were stained with propidium iodide for 5 min at room temperature in the dark, and stained cells were immediately analyzed using a FACSCalibur flow cytometer (BD Biosciences) and FlowJo software (Tomy Digital Biology, Tokyo, Japan).
Cell proliferation assay
Control or HNF4α knockdown MIN6 cells were seeded in a 96-well plate at 1.5 × 104 cells/well. After 2 days, a cell proliferation assay was performed for 3 consecutive days using cell proliferation reagent WST-1 (Roche Diagnostic, Mannheim, Germany) and the absorbance (450/655) was measured by iMark microplate reader (Bio-Rad).
Protein degradation assay
MIN6 cells were seeded at 2.0 × 105 cells in a 35-mm dish. After MIN6 cells were cultured at 20% O2 or 5% O2 condition for 18 h, they were then treated with 10 μg/ml of CHX for the indicated time (0–6 h). To examine the effects of MG-132 or epoxomicin on HNF4α protein expression, MIN6 cells were cultured at either 20% O2 or 5% O2 for 20 h and then treated with CHX in the presence or absence of MG132 (or epoxomicin) for 4 h. MIN6 cells were also cultured with or without 2 mm metformin for 20 h and then treated with CHX (+)/MG132 (+) or CHX (+)/MG132 (−) for 4 h. HNF4α protein levels were analyzed by Western blotting.
Phos-tag SDS-PAGE
MIN6 cells were treated with 2 mm metformin or cultured under the hypoxic 5% O2 condition for 12 and 24 h. Whole cells were lysed in EDTA (−) RIPA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 20 mg/ml of Na3VO4, 10 mm NaF, 1 mm PMSF, and 1% (v/v) protease inhibitor mixture). Phos-tag-containing polyacrylamide gels were made with Phos-tag acrylamide (40 μm; Wako Pure Chemicals, Osaka, Japan) and MnCl2 (40 μm) according to the manufacturer's recommendation. Twenty micrograms of proteins were subjected to 10% SDS-PAGE. Dr. Western (Oriental Yeast Co., Ltd, Tokyo, Japan) was used for a protein ladder marker. After electrophoresis, the Phos-tag gel was soaked in transfer buffer (25 mm Tris and 192 mm glycine) containing 1 mm EDTA for 10 min to remove the Mn2+ before transferring the proteins to a membrane, and then Western blotting was performed.
Statistical analysis
The significance of differences was assessed with an unpaired t test and a value of p < 0.05 was considered to be statistically significant.
Author contributions
Y. S. and K. Y. designed the study and wrote the paper. Y. S., T. T., C. S., M. F. K., T. Y., and M. I. contributed to the acquisition of data, the statistical data analyses, and drafting of the manuscript. All authors have approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank S. Sato for valuable help with this study. We also thank Prof. J. Miyazaki (Osaka University) for the gift of MIN6 cells, and Prof. T. Kitamura (Tokyo University) for providing the Plat-E cells and retrovirus system.
This work was supported by a Grant-in-Aid for Young Scientists (B) (to Y. S.), a Grant-in-Aid for Scientific Research (B) (to K. Y.), a grant from the Uehara Memorial Foundation (to K. Y.), a grant from the Japan Diabetes Foundation (to K. Y.), and a grant from the Naito Foundation (to K. Y.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S6.
- HNF
- hepatocyte nuclear factor
- AICAR
- 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- AMPK
- adenosine monophosphate-activated protein kinase
- CHX
- cycloheximide
- HIF
- hypoxia inducible factor
- MODY
- maturity-onset diabetes of the young
- Nedd4l
- developmentally down-regulated 4-like
- TBP
- TATA-binding protein
- UTR
- untranslated region
- ERK
- extracellular signal-regulated kinase
- qRT
- quantitative PCR
- KD
- kinase-dead.
References
- 1. Wang W., Upshaw L., Strong D. M., Robertson R. P., and Reems J. (2005) Increased oxygen consumption rates in response to high glucose detected by a novel oxygen biosensor system in non-human primate and human islets. J. Endocrinol. 185, 445–455 [DOI] [PubMed] [Google Scholar]
- 2. Gilbert M., Jung S. R., Reed B. J., and Sweet I. R. (2008) Islet oxygen consumption and insulin secretion tightly coupled to calcium derived from l-type calcium channels but not from the endoplasmic reticulum. J. Biol. Chem. 283, 24334–24342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sato Y., Endo H., Okuyama H., Takeda T., Iwahashi H., Imagawa A., Yamagata K., Shimomura I., and Inoue M. (2011) Cellular hypoxia of pancreatic beta-cells due to high levels of oxygen consumption for insulin secretion in vitro. J. Biol. Chem. 286, 12524–12532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bensellam M., Duvillié B., Rybachuk G., Laybutt D. R., Magnan C., Guiot Y., Pouysségur J., and Jonas J. C. (2012) Glucose-induced O2 consumption activates hypoxia inducible factors 1 and 2 in rat insulin-secreting pancreatic beta-cells. PLOS One 7, e29807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zheng X., Zheng X., Wang X., Ma Z., Gupta Sunkari V., Botusan I., Takeda T., Björklund A., Inoue M., Catrina S. B., Brismar K., Poellinger L., and Pereira T. S. (2012) Acute hypoxia induces apoptosis of pancreatic β-cell by activation of the unfolded protein response and upregulation of CHOP. Cell Death Dis. 3, e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sato Y., Inoue M., Yoshizawa T., and Yamagata K. (2014) Moderate hypoxia induces β-cell dysfunction with HIF-1-independent gene expression changes. PLOS One 9, e114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nammo T., Yamagata K., Tanaka T., Kodama T., Sladek F. M., Fukui K., Katsube F., Sato Y., Miyagawa J., and Shimomura I. (2008) Expression of HNF-4α (MODY1), HNF-1beta (MODY5), and HNF-1α (MODY3) proteins in the developing mouse pancreas. Gene Expr. Patterns 8, 96–106 [DOI] [PubMed] [Google Scholar]
- 8. Yamagata K., Furuta H., Oda N., Kaisaki P. J., Menzel S., Cox N. J., Fajans S. S., Signorini S., Stoffel M., and Bell G. I. (1996) Mutations in the hepatocyte nuclear factor-4α gene in maturity-onset diabetes of the young (MODY1). Nature 384, 458–460 [DOI] [PubMed] [Google Scholar]
- 9. Miura A., Yamagata K., Kakei M., Hatakeyama H., Takahashi N., Fukui K., Nammo T., Yoneda K., Inoue Y., Sladek F. M., Magnuson M. A., Kasai H., Miyagawa J., Gonzalez F. J., and Shimomura I. (2006) Hepatocyte nuclear factor-4α is essential for glucose-stimulated insulin secretion by pancreatic beta-cells. J. Biol. Chem. 281, 5246–5257 [DOI] [PubMed] [Google Scholar]
- 10. Gupta R. K., Vatamaniuk M. Z., Lee C. S., Flaschen R. C., Fulmer J. T., Matschinsky F. M., Duncan S. A., and Kaestner K. H. (2005) The MODY1 gene HNF-4α regulates selected genes involved in insulin secretion. J. Clin. Invest. 115, 1006–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamagata K., Oda N., Kaisaki P. J., Menzel S., Furuta H., Vaxillaire M., Southam L., Cox R. D., Lathrop G. M., Boriraj V. V., Chen X., Cox N. J., Oda Y., Yano H., Le Beau M. M., et al. (1996) Mutations in the hepatocyte nuclear factor-1α gene in maturity-onset diabetes of the young (MODY3). Nature 384, 455–458 [DOI] [PubMed] [Google Scholar]
- 12. Horikawa Y., Iwasaki N., Hara M., Furuta H., Hinokio Y., Cockburn B. N., Lindner T., Yamagata K., Ogata M., Tomonaga O., Kuroki H., Kasahara T., Iwamoto Y., and Bell G. I. (1997) Mutation in hepatocyte nuclear factor-1 β gene (TCF2) associated with MODY. Nat. Genet. 17, 384–385 [DOI] [PubMed] [Google Scholar]
- 13. Semenza G. L. (2009) Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology 24, 97–106 [DOI] [PubMed] [Google Scholar]
- 14. Mihaylova M. M., and Shaw R. J. (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mu J., Brozinick J. T. Jr., Valladares O., Bucan M., and Birnbaum M. J. (2001) A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol. Cell 7, 1085–1094 [DOI] [PubMed] [Google Scholar]
- 16. Stoner M., Saville B., Wormke M., Dean D., Burghardt R., and Safe S. (2002) Hypoxia induces proteasome-dependent degradation of estrogen receptor α in ZR-75 breast cancer cells. Mol. Endocrinol. 16, 2231–2242 [DOI] [PubMed] [Google Scholar]
- 17. Comellas A. P., Dada L. A., Lecuona E., Pesce L. M., Chandel N. S., Quesada N., Budinger G. R., Strous G. J., Ciechanover A., and Sznajder J. I. (2006) Hypoxia-mediated degradation of Na,K-ATPase via mitochondrial reactive oxygen species and the ubiquitin-conjugating system. Circ. Res. 98, 1314–1322 [DOI] [PubMed] [Google Scholar]
- 18. Wang W., Fan J., Yang X., Fürer-Galban S., Lopez de Silanes I, von Kobbe C., Guo J., Georas S. N., Foufelle F., Hardie D. G., Carling D., and Gorospe M. (2002) AMP-activated kinase regulates cytoplasmic HuR. Mol. Cell. Biol. 22, 3425–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown K., Gerstberger S., Carlson L., Franzoso G., and Siebenlist U. (1995) Control of IκBα proteolysis by site-specific, signal-induced phosphorylation. Science 267, 1485–1488 [DOI] [PubMed] [Google Scholar]
- 20. Skowyra D., Craig K. L., Tyers M., Elledge S. J., and Harper J. W. (1997) F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 91, 209–219 [DOI] [PubMed] [Google Scholar]
- 21. Hong Y. H., Varanasi U. S., Yang W., and Leff T. (2003) AMP-activated protein kinase regulates HNF4α transcriptional activity by inhibiting dimer formation and decreasing protein stability. J. Biol. Chem. 278, 27495–27501 [DOI] [PubMed] [Google Scholar]
- 22. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., and Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757 [DOI] [PubMed] [Google Scholar]
- 23. Viollet B., Kahn A., and Raymondjean M. (1997) Protein kinase A-dependent phosphorylation modulates DNA-binding activity of hepatocyte nuclear factor 4. Mol. Cell. Biol. 17, 4208–4219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rousseau A., and Bertolotti A. (2016) An evolutionarily conserved pathway controls proteasome homeostasis. Nature 536, 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baskin K. K., and Taegtmeyer H. (2011) AMP-activated protein kinase regulates E3 ligases in rodent heart. Circ. Res. 109, 1153–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bhalla V., Oyster N. M., Fitch A. C., Wijngaarden M. A., Neumann D., Schlattner U., Pearce D., and Hallows K. R. (2006) AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J. Biol. Chem. 281, 26159–26169 [DOI] [PubMed] [Google Scholar]
- 27. Wei J., Shimazu J., Makinistoglu M. P., Maurizi A., Kajimura D., Zong H., Takarada T., Iezaki T., Lezaki T., Pessin J. E., Hinoi E., and Karsenty G. (2015) Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell 161, 1576–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang C. Y., Parton L. E., Ye C. P., Krauss S., Shen R., Lin C. T., Porco J. A. Jr., and Lowell B. B. (2006) Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced beta cell dysfunction in isolated pancreatic islets. Cell Metab. 3, 417–427 [DOI] [PubMed] [Google Scholar]
- 29. Ihara A., Yamagata K., Nammo T., Miura A., Yuan M., Tanaka T., Sladek F. M., Matsuzawa Y., Miyagawa J., and Shimomura I. (2005) Functional characterization of the HNF4alpha isoform (HNF4α8) expressed in pancreatic beta-cells. Biochem. Biophys. Res. Commun. 329, 984–990 [DOI] [PubMed] [Google Scholar]
- 30. Yamagata K. (2014) Roles of HNF1α and HNF4α in pancreatic β-cells: lessons from a monogenic form of diabetes (MODY). Vitam. Horm. 95, 407–423 [DOI] [PubMed] [Google Scholar]
- 31. Leclerc I., Lenzner C., Gourdon L., Vaulont S., Kahn A., and Viollet B. (2001) Hepatocyte nuclear factor-4α involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes 50, 1515–1521 [DOI] [PubMed] [Google Scholar]
- 32. Zehetner J., Danzer C., Collins S., Eckhardt K., Gerber P. A., Ballschmieter P., Galvanovskis J., Shimomura K., Ashcroft F. M., Thorens B., Rorsman P., and Krek W. (2008) PVHL is a regulator of glucose metabolism and insulin secretion in pancreatic beta cells. Genes Dev. 22, 3135–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cantley J., Selman C., Shukla D., Abramov A. Y., Forstreuter F., Esteban M. A., Claret M., Lingard S. J., Clements M., Harten S. K., Asare-Anane H., Batterham R. L., Herrera P. L., Persaud S. J., Duchen M. R., Maxwell P. H., and Withers D. J. (2009) Deletion of the von Hippel-Lindau gene in pancreatic beta cells impairs glucose homeostasis in mice. J. Clin. Invest. 119, 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. da Silva Xavier G., Leclerc I., Varadi A., Tsuboi T., Moule S. K., and Rutter G. A. (2003) Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. Biochem. J. 371, 761–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsuboi T., da Silva Xavier G., Leclerc I., and Rutter G. A. (2003) 5′-AMP-activated protein kinase controls insulin-containing secretory vesicle dynamics. J. Biol. Chem. 278, 52042–52051 [DOI] [PubMed] [Google Scholar]
- 36. Lim A., Park S. H., Sohn J. W., Jeon J. H., Park J. H., Song D. K., Lee S. H., and Ho W. K. (2009) Glucose deprivation regulates KATP channel trafficking via AMP-activated protein kinase in pancreatic beta-cells. Diabetes 58, 2813–2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang C. Y., Baffy G., Perret P., Krauss S., Peroni O., Grujic D., Hagen T., Vidal-Puig A. J., Boss O., Kim Y. B., Zheng X. X., Wheeler M. B., Shulman G. I., Chan C. B., and Lowell B. B. (2001) Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105, 745–755 [DOI] [PubMed] [Google Scholar]
- 38. Yang Q., Yamagata K., Yamamoto K., Cao Y., Miyagawa J., Fukamizu A., Hanafusa T., and Matsuzawa Y. (2000) R127W-HNF-4α is a loss of function mutation but not a rare polymorphism and causes Type II diabetes in a Japanese family with MODY1. Diabetologia 43, 520–524 [DOI] [PubMed] [Google Scholar]
- 39. Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., and Yamamura K. (1990) Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132 [DOI] [PubMed] [Google Scholar]
- 40. Fukui K., Yang Q., Cao Y., Takahashi N., Hatakeyama H., Wang H., Wada J., Zhang Y., Marselli L., Nammo T., Yoneda K., Onishi M., Higashiyama S., Matsuzawa Y., Gonzalez F. J., et al. (2005) The HNF-1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab. 2, 373–384 [DOI] [PubMed] [Google Scholar]
- 41. Yoshizawa T., Karim M. F., Sato Y., Senokuchi T., Miyata K., Fukuda T., Go C., Tasaki M., Uchimura K., Kadomatsu T., Tian Z., Smolka C., Sawa T., Takeya M., Tomizawa K., et al. (2014) SIRT7 controls hepatic lipid metabolism by regulating the ubiquitin-proteasome pathway. Cell Metab. 19, 712–721 [DOI] [PubMed] [Google Scholar]
- 42. Sato Y., Hatta M., Karim M. F., Sawa T., Wei F. Y., Sato S., Magnuson M. A., Gonzalez F. J., Tomizawa K., Akaike T., Yoshizawa T., and Yamagata K. (2012) Anks4b, a novel target of HNF4α protein, interacts with GRP78 protein and regulates endoplasmic reticulum stress-induced apoptosis in pancreatic β-cells. J. Biol. Chem. 287, 23236–23245 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.