

Abstract
Mitogen-activated protein kinase (MAPK) scaffold proteins, such as IQ motif containing GTPase activating protein 1 (IQGAP1), are promising targets for novel therapies against cancer and other diseases. Such approaches require accurate information about which domains on the scaffold protein bind to the kinases in the MAPK cascade. Results from previous studies have suggested that the WW domain of IQGAP1 binds to the cancer-associated MAPKs ERK1 and ERK2, and that this domain might thus offer a new tool to selectively inhibit MAPK activation in cancer cells. The goal of this work was therefore to critically evaluate which IQGAP1 domains bind to ERK1/2. Here, using quantitative in vitro binding assays, we show that the IQ domain of IQGAP1 is both necessary and sufficient for binding to ERK1 and ERK2, as well as to the MAPK kinases MEK1 and MEK2. Furthermore, we show that the WW domain is not required for ERK-IQGAP1 binding, and contributes little or no binding energy to this interaction, challenging previous models of how WW-based peptides might inhibit tumorigenesis. Finally, we show that the ERK2-IQGAP1 interaction does not require ERK2 phosphorylation or catalytic activity and does not involve known docking recruitment sites on ERK2, and we obtain an estimate of the dissociation constant (Kd) for this interaction of 8 μm. These results prompt a re-evaluation of published findings and a refined model of IQGAP scaffolding.
Keywords: cancer, cell signaling, extracellular-signal-regulated kinase (ERK), mitogen-activated protein kinase (MAPK), protein complex, protein kinase, protein-protein interaction, scaffold protein, IQGAP1
Introduction
The RAS-RAF-MEK-ERK signaling pathway, often referred to as the RAS/MAPK cascade, has been a focus of cancer drug development (1, 2). The success of small-molecule inhibitors of RAF and MEK have validated these efforts; however, the emergence of clinical drug resistance remains a major challenge (3, 4).
Signal propagation through the MAPK cascade is facilitated by scaffold proteins such as KSR, Paxillin, and IQGAP1 (5). Scaffold proteins bind to and assemble multiple elements of signaling/regulatory pathways. They are thought to tether their bound components near each other, thereby increasing the rate at which one activates the other. Furthermore, they operate in distinct subcellular locations and in a spatiotemporally regulated manner (5–7). For these reasons, scaffold proteins provide new therapeutic approaches to cancer and other diseases (8).
IQGAP proteins are evolutionarily conserved in eukaryotes (8–18). They function as scaffold proteins that facilitate the formation of complexes that regulate both cytoskeletal dynamics and intracellular signaling. IQGAP proteins have been highly studied because of their relevance to basic biology and to human disease.
Originally discovered in 1994 (19), IQGAP1 is the founding and best-studied member of a family that includes 3 paralogs in humans (IQGAP1, IQGAP2, and IQGAP3). IQGAP1 overexpression has been implicated in the progression of many cancers (18, 20), and the presence of IQGAP1 has been shown to promote RAS-driven tumorigenesis in mouse models (21). In addition, many bacterial and viral pathogens, including the Ebola and Marburg viruses, have been shown to hijack IQGAP1 during the course of infection (9, 10).
Consistent with their proposed role as scaffold proteins, mammalian IQGAP1 orthologs are over 1600 amino acids long, and contain multiple domains that can mediate protein-protein interactions (Fig. 1A). From amino to carboxyl terminus, these domains include a calponin-homology domain, a region containing several internal repeat sequences that have the capacity to form coiled-coils (IR), a WW domain, an IQ domain (consisting of four closely spaced IQ motifs), a GTPase-activating protein-related domain, and a RasGAP C-terminal domain. Multiple binding partners have been identified for most of these domains (13).
Figure 1.
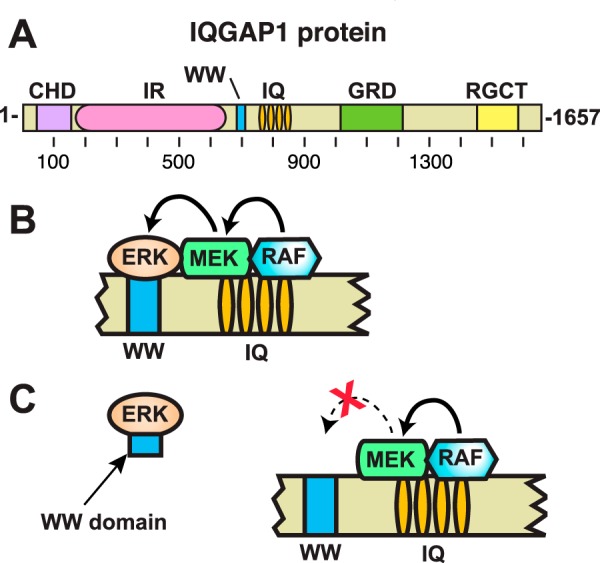
The IQGAP1 scaffold protein. A, schematic depicting full-length human IQGAP1 protein, and the domains it contains. CHD, calponin homology domain; IR, internal repeated sequence/coiled-coil domain; WW, WW domain; IQ, IQ domain; GRD, GTPase-activating protein-related domain; RGCT, RasGAP C-terminal domain. B, schematic interpretation of proposed model of the function of IQGAP1 as a scaffold protein for the MAPK pathway, based on a similar figure in Ref. 8. According to the model, the IQ domain of IQGAP1 binds to RAF and MEK, and the nearby WW domain binds to ERK. These interactions are thought to facilitate RAF phosphorylation of MEK, and MEK phosphorylation of ERK. C, proposed mechanistic model for the anti-tumor efficacy of the isolated IQGAP WW domain studied by Jameson et al. (21). In this model, the WW domain binds to ERK and blocks the ability of ERK to productively interact with IQGAP1 (8, 21). The WW domain fragment studied by Jameson et al. (21) consisted of IQGAP1 residues 680–711, plus N-terminal myc and polyarginine tags.
A scaffolding function for IQGAP1 was first proposed when it was observed to link Ca2+/calmodulin and Cdc42 signaling (22–24). More recent data also suggest that IQGAP can act as a scaffold in the Wnt pathway (25). However, perhaps the best-characterized example of IQGAP1 scaffold function is in its interactions with elements of the RAS/MAPK pathway. As shown in Fig. 1B, both ERK1 and ERK2 (MAP kinases that are activated in numerous human cancers) and MEK1 and MEK2 (MAPK kinases that activate ERK1 and ERK2) have been shown to bind to IQGAP1. Upstream components of the MAPK pathway have also been shown to bind to IQGAP1, including the MEK activator BRAF, as well as multiple receptor tyrosine kinases (9).
For many years, the field has believed that ERK1 and ERK2 bind to the WW domain of IQGAP1 (26, 27). WW domains are compact units that fold into a three-stranded −β-sheet structure (28), and have been shown to bind to Pro-rich sequences such a PPXY and PPPR, or to phospho-Ser/Thr-Pro sequences (29–34). ERK1 and ERK2 are the only proteins purported to interact with the WW domain of IQGAP1 (13). In contrast, the binding of BRAF and MEK1/2 to IQGAP1 requires the presence of the IQ domain (26, 35). The IQ domain of IQGAP1 consists of four tandem IQ motifs (Fig. 1A). IQ motifs are found in many calcium-regulated proteins (36). They consist of a stretch of about 18–25 amino acid residues, and form amphiphilic α helixes that can bind to calmodulin and S100-family proteins, among other ligands.
The assertion that the WW domain of IQGAP1 binds to ERK1 and ERK2 has been widely cited in primary research papers (e.g. see Refs. 21, 26, and 37–50) and reviews (e.g. Refs. 8–18). Indeed, it recently motivated a high-profile translational study in which cancer cells were treated with a cell-permeable version of the WW domain of human IQGAP1 (8, 11, 21). The idea underlying this study was that the WW domain fragment would competitively bind to ERK1/2 and prevent these MAP kinases from interacting with IQGAP1, thus selectively inhibiting MAP kinase activation (Fig. 1C). Indeed, the idea seemed to work, in as much as the WW domain fragment inhibited the proliferation, migration, and tumorigenesis of breast, colorectal, and melanoma tumor cells that contained activating mutations in the RAS/MAPK pathway (8, 11, 21).
Herein we re-examined the binding of ERK1 and ERK2 to IQGAP1. In contrast to previous findings, we show that the WW domain of IQGAP1 is neither necessary nor sufficient for binding to ERK1 and ERK2. Rather, the IQ domain of IQGAP1 is both necessary and sufficient for high-affinity ERK binding. Our results thus prompt a re-evaluation of several highly cited published studies, and suggest a new model for IQGAP scaffolding function.
Results
IQ domain of IQGAP1 is necessary for binding to ERK2
An initial goal of this study was to verify and more precisely delineate the domain(s) of IQGAP1 that were necessary and sufficient for binding to the MAP kinases ERK1 and ERK2. As a first step in this process, we set out to confirm the finding of Roy et al. (27), who first showed that human IQGAP1 binds to ERK2. In this study, Roy et al. (27) used rat ERK in co-sedimentation assays with in vitro translated human IQGAP1. We used a very similar approach; rat ERK2 was fused at its N terminus to Schistosoma japonicum glutathione S-transferase (GST), and the resulting fusion protein (GST-rERK2) was expressed in bacteria and purified by adsorption to glutathione-Sepharose beads. GST-rERK2 (or GST alone as a negative control) was then incubated with full-length human IQGAP1 that had been produced in radiolabeled form by in vitro translation (Fig. 2A). Bead-bound complexes were collected by sedimentation, washed extensively, and analyzed by SDS-PAGE and autoradiography.
Figure 2.

The IQ domain of IQGAP1 is necessary for binding to ERK2; the WW domain is not sufficient. A, rat ERK2, fused to GST, was tested for binding to full-length human IQGAP1, or to truncated derivatives of IQGAP1. Qualitative results of these experiments are shown on the right: +++ indicates strong binding; — indicates minimal binding. B, as shown in A, 35S radiolabeled full-length human IQGAP1 protein and truncated derivatives were prepared by in vitro translation and partially purified by ammonium sulfate precipitation, and portions (10% of the amount added in the binding reactions) were resolved on a 10% SDS-polyacrylamide gel (Input). Samples (∼1 pmol) of the same proteins were incubated with 25 μg of GST or GST-ERK2 bound to glutathione-Sepharose beads, and the resulting bead-bound protein complexes were isolated by sedimentation and resolved by 10% SDS-PAGE on the same gel. The gel was analyzed by staining with GelCode Blue (Thermo Scientific) for visualization of the bound GST fusion protein (a representative example is shown in the lowest panel) and by X-ray film exposure for visualization of the bound radiolabeled protein (upper four panels). C, quantification of the binding of IQGAP1 derivatives to GST or GST-ERK2, normalized to the percent binding of full-length IQGAP1 to GST-ERK2. The results shown are the average of at least 5 independent repetitions of the binding assay shown in A and B, with duplicate points (i.e. technical replicates) in each repetition. S.E. bars are shown (n = 5 to 7). The scatter of the individual normalized data points is also shown for the binding of ERK2 to IQGAP1(1–863). The means for ERK2-IQGAP1 and ERK2-IQGAP1(1–863) binding were significantly different from all other the means shown (p < 0.01), but were not significantly different from each other (p = 0.98, thus the null hypothesis that the population means are the same cannot be rejected with confidence). The minimal binding of ERK2 to IQGAP1(1–719) was not significantly different from that of ERK2 to IQGAP1(1–678) (p = 0.91), nor was it significantly different from the minimal binding of GST alone to IQGAP1(1–719) (p = 0.41).
Human IQGAP1 is a 1632-residue protein, with a calculated molecular mass of 189 kDa. As shown in Fig. 2B (Input lane) IQGAP1 migrated with an apparent molecular mass of 250 kDa on a 10% SDS-PAGE gel. Also as shown in Fig. 2B, full-length IQGAP1 bound efficiently to ERK2. Furthermore, this binding was specific, because only trace precipitation of IQGAP1 occurred when GST was used instead of the GST-rERK2 fusion protein.
To delineate the domain(s) of IQGAP1 involved in binding to ERK2, we utilized a series of C-terminal truncation mutants of IQGAP1 (Fig. 2A). These mutants were constructed using site-directed mutagenesis to introduce translation termination (“stop”) codons after codons 678, 719, or 863. These derivatives all contain the CHD and IR domains, but differ in the presence of the WW and IQ domains. The IQGAP1(1–678) mutant protein lacks both the IQ and WW domains; IQGAP1(1–719) lacks the IQ domain but contains the WW domain; and IQGAP1(1–863) contains both the IQ and WW domains (Fig. 2A).
IQGAP1(1–863) was previously shown to bind rat ERK2 about as well as full-length IQGAP1 did (27). We confirmed this finding (Fig. 2B). Indeed, when we quantified the results of 7 independent binding assays, the binding efficiency of full-length IQGAP1 was not significantly different from that of IQGAP1(1–863) (Fig. 2C).
In stark contrast, both IQGAP1(1–719) and IQGAP1(1–678) exhibited negligible binding to GST-rERK2 (Fig. 2B), and this minimal binding was not significantly different from each other, nor was it significantly different to their binding to GST alone (Fig. 2C). Thus, these results indicate that the IQ domain is necessary for binding to ERK2 (because 1–863 bound whereas 1–719 did not), and also show that the WW domain is not sufficient for binding (because 1–719 did not bind).
Human ERK2 binds to human IQGAP1
As noted above, the original discovery of ERK-IQGAP binding was made using rat ERK2 and human IQGAP1 (27); we used this same cross-species configuration in Fig. 2. To ascertain if the same pattern of interactions seen in Fig. 2 would also be observed using human ERK2, we fused human ERK2 to GST and purified this GST-hERK2 protein from bacteria. As shown in Fig. 3, full-length human IQGAP1 bound equivalently to both rat ERK (rERK2) and human ERK2 (hERK2). Likewise, IQGAP1(1–863) bound equivalently to both rat and human ERK2. Finally, IQGAP1(1–719) and IQGAP1(1–678) bound to neither ERK2 ortholog.
Figure 3.

The IQ domain of IQGAP1 is sufficient for binding to ERK2; the WW domain is not necessary. A, rat or human ERK2, fused to GST, were tested for binding to full-length human IQGAP1, or to fragments of IQGAP1. Qualitative results of these experiments are shown on the right: +++ indicates strong binding; — indicates minimal binding. B, autoradiograms of representative experiments of binding assays are described in A. Each binding assay shown was repeated three separate times (i.e. three independent experiments), with duplicate points (i.e. technical replicates) in each experiment. Other details are as described in the legend to Fig. 2.
We performed 9 independent, quantitative binding assay experiments between human ERK2 and IQGAP1, with technical replicates in each experiment. From these data we were able to obtain an estimate of 7.6 μm for the dissociation constant (Kd) of this interaction (Table 1).
Table 1.
Binding assay data for ERK2-IQGAP1 interaction
| Experimenta | Bindingb | Kdc |
|---|---|---|
| % | μm | |
| A-1 | 37.5 | 3.0 |
| A-2 | 25.1 | 5.3 |
| A-3 | 18.8 | 7.7 |
| A-4 | 16.3 | 9.2 |
| A-5 | 16.7 | 8.9 |
| A-6 | 28.9 | 4.4 |
| A-7 | 11.9 | 13.2 |
| A-8 | 16.4 | 9.1 |
| A-9 | 19.2 | 7.5 |
| Mean | 21.2 | 7.6 |
| Median | 18.8 | 7.7 |
| Standard deviation | 7.9 | 3.1 |
| Standard error | 2.6 | 1.0 |
a Binding reactions (200 μl) contained ∼1 pmol (∼5 nM) 35S-labeled, in vitro translated, full-length IQGAP1 and 25 μg (1.8 μm) of GST-hERK2 fusion protein. Every experiment contained duplicate points (also known as technical replicates); these were averaged to obtain the “% binding” number shown.
b Percent of the input 35S-labeled protein that bound to the GST fusion protein.
c Calculated based on the known input concentrations and percent binding, as described elsewhere (65).
The IQ domain is sufficient for binding to ERK2
To ask if the IQ domain of IQGAP1 is sufficient for binding to ERK2, we made three additional IQGAP1 fragments. As shown in Fig. 3A, IQGAP1(432–863) contains half of the IR domain, the (entire) WW domain, and the IQ domain. IQGAP1(679–863) contains only the WW and IQ domains. Finally, IQGAP1 (720–863) contains just the IQ domain. These mutants were tested for binding both to rat ERK2 (rERK2) and human ERK2 (hERK2).
IQGAP1(432–863) was previously shown to bind to rat ERK2 (27). As shown in Fig. 3B, we confirmed this finding, and extended it to human ERK2. In vitro-translated IQGAP1(432–863) migrated on SDS-PAGE gels as two forms (Fig. 3B): a major, slower migrating form, corresponding to the complete translation product, and a minor, slightly faster migrating form of lower molecular mass. Such faster migrating forms are often seen in cell-free translation reactions, and are typically caused by a low frequency of premature translation termination or internal initiation (51).
Also as shown in Fig. 3B, both IQGAP1(679–863) and IQGAP1(720–863) bound to both rat and human ERK2. Importantly, because IQGAP1(720–863), which contains just the IQ domain, bound to ERK2, we conclude that the IQ domain is sufficient for binding to ERK2.
ERK2 phosphorylation is not required for binding to IQGAP1
ERK2, like most other MAP kinases, is activated by dual phosphorylation at a Thr and Tyr residue in its activation loop. Dual phosphorylation causes remodeling of the activation loop conformation so as to reorganize active site residues, open up substrate specificity determinants, and expose a hydrophobic docking pocket used by some substrates containing FXFP-type docking sites (52–54). Dual phosphorylation of ERK1 and ERK2 is catalyzed by MEK1 and MEK2 during physiological pathway activation. However, ERK2 protein is known to exhibit a low level of autophosphorylation on these residues under various conditions, including bacterial expression (55).
To ascertain whether phosphorylation of ERK2 was necessary for its ability to bind to IQGAP1, we constructed two mutant versions of human ERK2 incapable of undergoing autophosphorylation, and compared their ability to bind IQGAP1 with wild-type ERK2. The first ERK2 mutant, K54A, contains a substitution of a highly conserved catalytic lysine residue; this substitution has been shown to render ERK2 catalytically inactive (56, 57). The second mutant, T185A/Y187F (hereafter designated ERK2-AF), changes the dual phosphorylation sites to non-phosphorylatable residues (54). As shown in Fig. 4, both ERK2-K54A and ERK2-AF bound to full-length IQGAP1 and IQGAP1(1–863) comparably to wild-type ERK2. These results indicate that phosphorylation and activation of ERK2 is not required for its ability to bind to IQGAP1.
Figure 4.

Further characterization of the ERK2-IQGAP1 interaction. A, human ERK2, or mutant derivatives thereof, fused to GST, were tested for binding to full-length human IQGAP1, or to IQGAP1(1–863). The ERK2 alleles tested were the wild-type allele (ERK2), catalytically inactive (K54A mutation, K54A), unphosphorylatable and unactivatable (T185A Y185F mutations, AF), and docking groove mutated (L115A, Q119A, D318A, D321A mutations, ”DGM“). Small circles on the schematics indicate the wild-type residues and the alterations thereof. B, autoradiograms of representative experiments of binding assays described in A. Each binding assay shown was repeated three separate times (i.e. three independent experiments), with duplicate points (i.e. technical replicates) in each experiment. Other details are as described in the legend to Fig. 2.
The docking recruitment site of ERK2 is not involved in IQGAP1 binding
The interaction of MAP kinases with scaffold proteins, other kinases, substrates, and phosphatases often involves the MAPK binding to a short linear motif, a MAPK-docking site, on its binding partner. A well known class of MAPK-docking sites, designated “D-sites,” has the consensus Lys/Arg1–3-X1–6-Φ-X-Φ, where “X” is any residue and “Φ” is a hydrophobic residue. D-sites were first identified in MAPK kinases (58, 59) and certain transcription factors (60, 61), and were subsequently found in numerous other MAPK partners, including scaffold proteins such as yeast Ste5 and mammalian JIP1 and JIP3 (62–64). A second docking motif (consensus Leu-X1–2–Arg/Lys2–5), related to the D-site, is found in MAPK-activated kinases such as RSK1 and MAPKAP2 (65, 66). Both classes of docking sites are known to bind to a charged surface patch and adjacent hydrophobic cleft on MAPKs referred to as the D-recruitment site or “docking groove” (67, 68).
The IQ domain of IQGAP1 contains two stretches that loosely fit the D-site consensus sequence, including 790KQKKAYQDRLAY801 and 822RKRYRDRLQY831. This observation suggested to us the possibility that, as is true of several other MAPK scaffold proteins, ERK2-IQGAP1 binding might be mediated by the docking groove of ERK2. To address this possibility, we constructed a mutant version of human ERK2 that contained 4 amino acid substitutions (L115A, Q119A, D318A, D321A) known to disrupt docking groove-mediated interactions (62, 69–72). As shown in Fig. 4, both full-length IQGAP1 and IQGAP1(1–863) bound comparably to wild-type ERK2 and to the ERK2 docking-groove mutant (DGM).3 Hence, the docking groove of ERK2 does not appear to play a significant role in ERK2-IQGAP1 binding.
Another docking motif found in MAPK binding partners has been named the “DEF motif” (consensus FXFP) (73). The IQ domain of IQGAP1 contains no matches to this consensus. Moreover, the complementary binding site on ERK2, designated the F-recruitment site, is only fully formed upon ERK2 phosphorylation and activation (52, 74), and we showed above that ERK2 phosphorylation and activation are not required for IQGAP1 binding.
To summarize, our results strongly suggest that ERK2-IQGAP1 binding is not mediated by any known MAPK-docking sites on IGQAP1 or recruitment sites on ERK2. Further studies will be required to delineate the region of ERK1/2 that mediates binding to IQGAP1.
The IQ domain is necessary and sufficient for binding to ERK1, MEK1, and MEK2
In prior studies, ERK1 has not been studied as extensively as ERK2 with regard to IQGAP1 binding. ERK1 has been shown to co-immunoprecipitate with full-length IQGAP1 from MCF-7 cells (26), and to increase the binding of MEK1 to IQGAP1 in vitro (26). It is generally assumed that ERK1 binds to the WW domain of IQGAP1, as is purported for ERK2. However, the domain on IQGAP1 to which ERK1 binds has not, to our knowledge, been carefully mapped.
MEK1 and MEK2 have also been shown to bind to IQGAP1 (26). In this case, domain mapping experiments indicated that the IQ domain of IQGAP1 was necessary for MEK binding (26). However, whether or not this domain is sufficient for MEK binding has not been addressed.
To investigate these questions, we expressed and purified full-length human ERK1, MEK1, and MEK2 as GST fusions, and tested them for binding to full-length IQGAP1 and to the panel of IQGAP1 deletion mutants (Fig. 5A).
Figure 5.

The IQ domain of IQGAP1 is sufficient for binding to ERK1, MEK1, and MEK2. A, human ERK1, MEK1, or MEK2, fused to GST, was tested for binding to full-length human IQGAP1, or to fragments of IQGAP1. Qualitative results of these experiments are shown on the right: +++ indicates strong binding; — indicates minimal binding. B, autoradiograms of representative experiments of binding assays described in A. Each binding assay shown was repeated three separate times (i.e. three independent experiments), with duplicate points (i.e. technical replicates) in each experiment. Other details are as described in the legend to Fig. 2.
Like GST-ERK2, GST-ERK1 is efficiently expressed and translated in Escherichia coli, resulting in abundant production of the expected full-length product (Fig. 5B, bottom panels). As shown in Fig. 5B, GST-ERK1 bound efficiently to full-length IQGAP1.
Bacterial production of full-length GST-MEK1 and GST-MEK2 is less efficient (compared with the GST-ERKs), resulting in both the production of the expected full-length product and of a series of lower molecular weight bands (Fig. 5B, lower panels). These bands are presumably attributable to premature transcription and/or translation termination (and possibly also some low level of proteolysis, although the presence/absence of protease inhibitors did not change the pattern appreciably). Despite their less-than-optimal expression, both GST-MEK1 and GST-MEK2 bound to full-length IQGAP1 (Fig. 5B), confirming previous observations (26).
When tested with the IQGAP1 domain-deletion mutants, the three proteins tested (human ERK1, human MEK1, and human MEK2) displayed the same pattern of binding interactions as seen in Fig. 3 for ERK2: they bound to derivatives containing the IQ domain, including the “IQ-domain only” construct 720–863, but did not bind to derivatives lacking the IQ domain (Fig. 5B). This pattern indicates that the IQ domain is necessary and sufficient for the interaction of ERK1, MEK1, and MEK2 with IQGAP1.
The WW domain does not contribute binding energy to the interaction
The results presented above (Figs. 2–5) clearly demonstrate that the IQ domain is necessary and sufficient for high-affinity binding to ERK2, whereas the WW domain is neither necessary nor sufficient. However, these results do not exclude the possibility that the WW domain of IQGAP1 contributes to the binding energy of the ERK-IQGAP interaction.
To further investigate this question, we constructed a mutant version of IQGAP1(1–863) that contained five different substitution mutations, each of which has been shown to be inactivating in other WW domains; this mutant protein is designated “IQGAP1(1–863)wwmut.” The sequence of the core of the WW domain of human IQGAP1 is shown in Fig. 6A, where it is aligned with WW domains from human WWOX1 and human PIN1. A Y33R mutation in WWOX1 was previously shown to abolish its interaction with several ligands (75). At the molecular level, this mutation was interpreted as compromising the “aromatic cradle” structure that is essential for the formation of WW domain-ligand complexes. The first mutation in the IQGAP1(1–863)wwmut is an analogous substitution, Y696R. Jager et al. (76) carried out an extensive substitution analysis of the WW domain of the human PIN1 protein, and identified four positions that partially or completely unfolded the protein when substituted with alanine: Trp11, Tyr24, Asn26, and Pro37. These residues are all highly conserved in the WW domain family, and identical residues are found in homologous positions in human IQGAP1. IQGAP1(1–863)wwmut contains analogous substitutions in each of these four residues: W685A, Y697A, N699A, and P710A.
Figure 6.

The WW domain does not contribute to the ERK-IQGAP1 interaction. A, the top three lines show an alignment of the amino acid sequences of the first WW domain from human WWOX1 (accession number NP_057457, residues shown are 22–47), and the single WW domains in human PIN1 (NP_006212, residues 11–37) and human IQGAP1 (NP_003861, residues 685–710). Residues identical in all three domains are boxed; these include the two tryptophan residues (positions 685 and 707 in IQGAP1) that give the WW domain its name. Residues that were the site of inactivating mutations in other studies (75, 76) are shaded orange. The bottom line shows the sequence of the quintuplely-mutated WW domain in the derivative IQGAP1(1–863)wwmut; residues mutated to alanine are shown in red and underlined. B and C, human ERK1, human ERK2, and rat ERK2, fused to GST, were tested for binding to human IQGAP1(1–863) or IQGAP1(1–863)wwmut. Other details are as described in the legend to Fig. 2. D, quantification of the binding of IQGAP1(1–863) or IQGAP1(1–863)wwmut to GST-hERK2. The results shown are the average of 4 independent repetitions of the binding assay shown in B and C, with duplicate points (i.e. technical replicates) in each repetition. S.E. bars are shown (n = 4). The scatter of the individual data points is also shown. The ERK2-IQGAP1(1–863) and ERK2-IQGAP1(1–863)wwmut interactions were not significantly different from each other (p = 0.57, thus the null hypothesis that the population means are the same cannot be rejected with confidence).
To summarize, the WW domain of IQGAP1(1–863)wwmut contains five different amino acid substitutions, any one of which would be expected to compromise its ability to fold and/or bind ligand. Nevertheless, no obvious difference was seen when this mutant was compared with wild-type IQGAP(1–863) for its ability to bind to rat ERK2, human ERK2, or human ERK1 (Fig. 6, B–D). We conclude that the WW domain contributes little or no binding energy to the ERK-IQGAP1 interaction.
Discussion
IQGAP proteins are highly studied, evolutionarily conserved scaffold proteins that act as integrators for a number of signaling/regulatory pathways, including the RAS/MAPK pathway. Recently, there has been interest in interdicting the IQGAP-MAPK interaction as a therapeutic strategy in cancer (8, 11, 21). Successful efforts in this direction will be contingent upon accurate information regarding which domains of the IQGAPs bind to the various kinases in the MAPK cascade. For example, the precise identification of the JNK-binding site in the JIP1 scaffold protein led to the development of both peptide and small molecule inhibitors of JNK (77–79).
The core of the RAS/MAPK cascade consists of the MAPK kinases MEK1 and MEK2, which phosphorylate and activate the MAPKs ERK1 and ERK2. Here we investigated the binding of human IQGAP1 to these core kinases, and presented five significant findings.
First and most importantly, we showed that, contrary to what the field has longed believed, the IQ domain of IQGAP1 is both necessary and sufficient for high-affinity binding to the ERK1 and ERK2 MAPKs (Figs. 2, 3, and 5). We also showed that the IQ domain of IQGAP1 is necessary and sufficient for binding to the MAPK kinases MEK1 and MEK2 (Fig. 5).
In addition, we quantified the strength of the interaction between ERK2 and IQGAP1, determining that the dissociation constant (Kd) for this interaction is about 8 μm (Table 1). Dissociation constants in the low micromolar range have also been observed for other MAPK-scaffold interactions (62, 77, 80).
Further characterizing the ERK2-IQGAP1 interaction, we showed that it was not dependent on the kinase activity of the ERK2, nor on the activation of ERK2 (Fig. 4). We also demonstrated that it did not involve known MAPK-docking sites in IQGAP1 or docking-recruitment sites on ERK2 (Fig. 4).
Finally, we asked if the WW domain contributed in any significant way to the ERK-IQGAP1 binding interaction. Arguing against this possibility, we found that in constructs lacking the IQ domain, there was only trace binding of WW-containing derivatives to ERK. Furthermore, even this negligible binding was not significantly different from the binding of constructs lacking the WW domain, nor from the binding of either type of construct to GST alone. In other words, in the absence of the IQ domain, there was only minimal background binding in all cases (Figs. 2, 3, and 5).
We also sought to address the question of whether or not the WW domain could help the IQ domain to bind to ERK2 via cooperative interactions. To do this, we compared the binding of an IQGAP1 derivative containing an intact WW domain to an otherwise identical construct containing five WW-domain substitution mutations that have been shown (in other WW domains) to be critical for folding and/or substrate binding. The binding of these two constructs was virtually indistinguishable (Fig. 6). Thus, not only is the WW domain neither necessary nor sufficient for the ERK-IQGAP1 (or MEK-IQGAP1) interaction; we could find no evidence that it contributes to these interactions in any way whatsoever.
In this regard, we note that there is nothing in the primary amino acid sequence of ERK1 and ERK2 that would suggest that they are likely to interact canonically with any WW domain. First, neither ERK protein contains the cognate core motif, PPXY, which is dominant among ligands of WW domains. Moreover, neither ERK protein contains a polyproline stretch of any sort; such stretches, with proper flanking residues, can bind to certain types of WW domains (81). Indeed, ERK1 and ERK2 do not even have 2 prolines in a row anywhere in their sequence.
Comparison with previous results
The assertion that the WW domain of IQGAP1 binds to ERK1 and ERK2 originates from Roy et al. (27). Here we used the same proteins as Roy et al. (27) (rat ERK2 and human IQGAP1), and an extremely similar experimental approach (in vitro binding assays with bacterially expressed ERK proteins and in vitro-translated IQGAP1 derivatives), and reached an opposite conclusion.
Although we confirmed the finding of Roy et al. (27) that ERK2 binds to full-length IQGAP1, as well as to IQGAP1(1–863) and IQGAP1(432–863), we cannot readily explain two of their reported results. First, they reported that a mutant designated “ΔWW” (which is deleted of residues 643–744) did not bind to ERK2. This mutant is missing the entire WW domain (which spans approximately residues 680–710), yet contains essentially all of the IQ domain (which spans approximately residues 744–856). Based on our finding that the IQ domain is necessary and sufficient for ERK binding, we would expect this mutant to bind. However, it is possible that the lack of binding observed by Roy et al. (27) was caused by improper folding of this internally deleted protein.
Roy et al. (27) also reported that a mutant that they designated “ΔIQ” (which is missing residues 699–905) bound to ERK2 as well as full-length IQGAP1 did (27). Remarkably, this derivative also lacks the C-terminal end of the WW domain, including the second of the two defining tryptophan residues. Thus, the WW domain in this mutant is partially deleted and most likely non-functional. Nevertheless, Roy et al. (27) used the positive binding results obtained with this mutant as part of their argument that the WW domain mediates ERK-IQGAP1 binding. Because this derivative lacks the entire IQ domain, we would expect this mutant not to bind. Possibly, this protein also did not fold properly, and did so in a way that made it nonspecifically sticky. Another possibility is that this particular internal deletion caused a conformational change that unmasked or created a second ERK-binding site elsewhere in the protein.
Mechanism of anti-tumor activity of the WW domain
Jameson et al. (21) showed that an IQGAP WW domain fragment (consisting residues 680–711 of human IQGAP1, plus short tags) could inhibit RAS- and RAF-driven tumorigenesis, and could bypass acquired resistance to the RAF inhibitor vemurafenib (21). Our results clearly call into question the mechanistic interpretation that these effects were due to the titration of ERK proteins by the WW domain (that is, our results call into question the model shown in Fig. 1C). An obvious alternative hypothesis is that the anti-tumor activity of the WW domain is attributable to its binding to some other ligand(s). However, no other ligands have been identified for the WW domain of IQGAP1, although we note that MAPKAP2 (a protein kinase regulated by ERK1/2 and p38) was predicted as a WW ligand, as it contains a perfect PPXY motif (82). Given the apparent efficacy of the WW domain of IQGAP1 to inhibit tumor growth and invasiveness, identifying the true ligand(s) of this WW domain should now be prioritized.
Conclusions
Our results suggest a new model of IQGAP scaffolding in which both MEK and ERK bind to the IQ domain, in close proximity for binding sites for RAF (35) and receptor tyrosine kinases (83, 84).
Experimental procedures
Genes
The mammalian genes used in this study were human IQGAP1 (NCBI accession number NM_003870), human ERK1 (MAPK3; NM_002746), human ERK2 (MAPK1, NP_620407), rat Erk2 (NM_053842), human MEK1 (MAPK2K1, NM_002755), and human MEK2 (MAP2K2, NM_030662).
Plasmids for the production of GST fusion proteins
The vector used for generating GST fusion proteins was pGEX-LB, a derivative of pGEX-4T-1 (Amersham Biosciences) (85). In pGEX-LB, an encoded Pro residue is replaced with a Gly-Gly-Gly-Gly-Gly-Ser-Gly coding sequence to promote the independent functioning of the GST and fusion moieties. Plasmid GST-hERK1 encodes a fusion of GST to full-length human ERK1, GST-rERK2 encodes a fusion of GST to full-length rat ERK2 (85). GST-hrERK2 encodes a fusion of GST to a sequence that encodes human ERK2 protein; this was generated for this study from the rat sequence by site-directed mutagenesis (the human and rat protein sequences differ in only 3 positions). The K54A mutant, the T185A/Y187F mutant, and the DGM (D318A/D321A/L115A/Q119A) were generated from this plasmid by site-directed mutagenesis. See Table 2 for primer sequences. The QuikChange and QuikChange Multi kits (Agilent) were used for all site-directed mutagenesis reactions.
Table 2.
Oligonucleotides used in this study
| Name | Sequence (5′ → 3′)a | Use |
|---|---|---|
| LL-hIQG1-P864stop-s | ACAAGACTCTCATCAATGCTGAGGATTAACCTATGGTTGTGGTCC | IQGAP1 (1–863) |
| JB-hIQG1-L720stop-s | ATTTTGTGCAAAATTCTATGCAGTAATCTCGGGAGGAGATCCAGAGTTC | IQGAP1 (1–719) |
| LL-hIQG1-G679stop-s | AGCCAAGAAGAAAAAACTGGCAGTATAAGATAATAACAGCAAG | IQGAP1 (1–678) |
| hIQGAP1(679-X) | GGAGGCGGTGGATCCACCATGGGAGATAATAACAGCAAGTGG | IQGAP1 (679–863) |
| hIQGAP1(720-X) | GGAGGCGGTGGATCCACCATGCTTTCTCGGGAGGAGATCCA | IQGAP1 (720–863) |
| hIQGAP1(X-863) | GCCGCTCGAGTCGACTTAATCCTCAGCATTGATGAGAG | 679 and 720 to 863 |
| LL-hIQGAP1-432-Y | GGTACCCGGGGATCCACCATGGAGCTGGTTACCCTGCAGCG | IQGAP1 (432–863) |
| LL-hIQGAP1-Y-863 | TGCCTGCAGGTCGACTTAATCCTCAGCATTGATGAGAG | IQGAP1 (432–863) |
| JB-W685A-s | TAGGAGATAATAACAGCAAGGCGGTGAAGCACTGGGTAAAAG | IQGAP1 (1–863)wwmut |
| JB-P710A-s | CCAGGAAGGAGGATGGGATGAAGCCCCAAATTTTG | IQGAP1 (1–863)wwmut |
| JB-Y696R-Y697A-N699A-s | GTGAAGCACTGGGTAAAAGGTGGATATTATCGTGCCCACGCTCTGGAGACCCAGGAAGG | IQGAP1 (1–863)wwmut |
| JB-rERK2-L44V-s | GGCATGGTTTGTTCTGCTTATGATAATGTCAACAAAGTTCGA | hERK2 |
| JB-rERK2-ins8AGlong-s | CCACCATGGCGGCGGCGGCGGCGGCGGGCGCGGGCCCGGAGATGGTCCGCGGGCAGGTGT | hERK2 |
| JB-hrERK2-K54A | AACAAAGTTCGAGTTGCTATCGCGAAAATCAGTCCTTTTGAGCAC | hERK2 K54A |
| JB-hrERK2T185A/Y187F | TCATACAGGGTTCTTGGCAGAGTTTGTAGCCACGCGTTGG | hERK2 T185A Y187F |
| JBhrERK2-D318A/D321A-s | CCTGGAGCAGTATTATGCCCCAAGTGCTGAGCCCATTGC | hERK2 DGM |
| JBhrERK2-L115A/Q119A-s | TGGAGACAGATCTTTACAAGGCCTTGAAGACAGCGCACCTCAGCAATGATCATA | hERK2 DGM |
a BamHI and SalI restriction sites used in cloning are underlined. Mutagenized codons are shown in bold. Introduced start and stop codons are also shown in bold.
Plasmids for the production of in vitro translated IQGAP1 and derivatives
A human IQGAP1 cDNA in expression vector pCR-Blunt II-TOPO was obtained from Dharmacon/GE Healthcare; this clone is from the mammalian gene collection, accession number BC139731. In vitro transcription and translation of full-length IQGAP1 was possible directly from the T7 promoter in this plasmid. The IQGAP truncations IQGAP1(1–863), IQGAP1(1–719), and IQGAP1(1–678) were derived from pCR-Blunt II-TOPO-IQGAP1 by introducing stop codons at codons 864, 720, and 679 of the IQGAP1 coding sequence via site-directed mutagenesis. The coding strand primers for these mutagenesis reactions were LL-hIQG1-P864stop-s, JB-hIQG1-L720stop-s, and LL-hIQG1-G679stop-s, respectively (Table 2).
The plasmid encoding IQGAP1(1–863)wwmut was derived from IQGAP1(1–863) by site-directed mutagenesis in two stages. First, the W685A and P710A substitutions were introduced with primers JB-W685A-s and JB-P710A-s. This was done in a single step using QuikChange Multi. Next, this intermediate derivative was used as the template in a mutagenesis reaction using coding strand primer JB-Y696R-Y697A-N699A-s and the corresponding antisense primer. The final product contains the substitutions W685A, Y696R, Y697A, N699A, P710A.
To construct pGEM3Z-IQGAP1(679–863) (used for the in vitro transcription and translation of IQGAP1(679–863)), a polymerase chain reaction (PCR) was performed with primers hIQGAP1(679-X) and hIQGAP1(X-863) (Table 2). The resulting product was then inserted into pGEXLB using recombination-based cloning (Cold Fusion, System Biosciences). Next, the insert was excised from this vector by digestion with restriction enzymes BamHI and SalI, and ligated into plasmid pGEM3Z (Promega), which had been cut with the same enzymes. Plasmid pGEM3Z-IQGAP1(720–863) was constructed using a similar strategy. Plasmid pGEM3Z-IQGAP1(432–863) was also constructed using a similar strategy, except that the PCR product was digested directly with BamHI and SalI prior to insertion into the corresponding sites of pGEM3Z. See Table 2 for primer sequences.
All IQGAP1 derivatives were confirmed by DNA sequencing.
Protein purification
GST fusion proteins were expressed in bacteria, purified by affinity chromatography using glutathione-Sepharose (GE Healthcare), and quantified as described elsewhere (85, 86).
In vitro transcription and translation
Proteins labeled with [35S]methionine were produced by coupled transcription and translation reactions (T7, Promega). Translation products were partially purified by ammonium sulfate precipitation (65), and resuspended in binding buffer (20 mm Tris-HCl, pH 7.5, 125 mm KOAc, 0.5 mm EDTA, 1 mm DTT, 0.1% (v/v) Tween 20, 12.5% (v/v) glycerol) prior to use in binding assays.
Protein binding assays
Protein binding assays were performed as described previously (85, 86). Quantification of binding was performed on a Typhoon TRIO+ Imager using phosphorimaging mode. Percent binding was determined by comparing the input with the amount that was co-sedimented. Each binding assay presented in this paper was repeated at least three separate times (i.e. three independent experiments), with duplicate points (i.e. technical replicates) in each experiment. Technical replicates in a given experiment are averaged together to obtain a single data point. We define “independent experiments” as experiments performed on different days, with fresh batches of GST fusion proteins and in vitro translated proteins.
Statistical analysis
Statistical analysis of binding assay results was performed using Welch's unequal variance t test with two tails (87). This was accomplished in Microsoft Excel using the T.TEST function, setting the “tail” option to 2, and the “type” option to 3.
Author contributions
A. J. B., L. L., and L. B. designed the experiments; A. J. B., L. L., and R. Z. performed the experiments; A. J. B., L. L., R. Z., and L. B. analyzed the results. A. J. B., L. L., and L. B. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
This work was supported in part by National Institutes of Health NIGMS Grant P50 GM76516. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- DGM
- docking-groove mutant.
References
- 1. Dhillon A. S., Hagan S., Rath O., and Kolch W. (2007) MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290 [DOI] [PubMed] [Google Scholar]
- 2. Lawrence M. C., Jivan A., Shao C., Duan L., Goad D., Zaganjor E., Osborne J., McGlynn K., Stippec S., Earnest S., Chen W., and Cobb M. H. (2008) The roles of MAPKs in disease. Cell Res. 18, 436–442 [DOI] [PubMed] [Google Scholar]
- 3. Caunt C. J., Sale M. J., Smith P. D., and Cook S. J. (2015) MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat. Rev. Cancer 15, 577–592 [DOI] [PubMed] [Google Scholar]
- 4. Lito P., Rosen N., and Solit D. B. (2013) Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 19, 1401–1409 [DOI] [PubMed] [Google Scholar]
- 5. Dhanasekaran D. N., Kashef K., Lee C. M., Xu H., and Reddy E. P. (2007) Scaffold proteins of MAP-kinase modules. Oncogene 26, 3185–3202 [DOI] [PubMed] [Google Scholar]
- 6. Bhattacharyya R. P., Reményi A., Yeh B. J., and Lim W. A. (2006) Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 75, 655–680 [DOI] [PubMed] [Google Scholar]
- 7. Langeberg L. K., and Scott J. D. (2015) Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 16, 232–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stuart D. D., and Sellers W. R. (2013) Targeting RAF-MEK-ERK kinase-scaffold interactions in cancer. Nat. Med. 19, 538–540 [DOI] [PubMed] [Google Scholar]
- 9. Hedman A. C., Smith J. M., and Sacks D. B. (2015) The biology of IQGAP proteins: beyond the cytoskeleton. EMBO Rep. 16, 427–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith J. M., Hedman A. C., and Sacks D. B. (2015) IQGAPs choreograph cellular signaling from the membrane to the nucleus. Trends Cell Biol. 25, 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanchez-Laorden B., Viros A., and Marais R. (2013) Mind the IQGAP. Cancer Cell 23, 715–717 [DOI] [PubMed] [Google Scholar]
- 12. Brown M. D., and Sacks D. B. (2006) IQGAP1 in cellular signaling: bridging the GAP. Trends Cell Biol. 16, 242–249 [DOI] [PubMed] [Google Scholar]
- 13. White C. D., Erdemir H. H., and Sacks D. B. (2012) IQGAP1 and its binding proteins control diverse biological functions. Cell Signal. 24, 826–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malarkannan S., Awasthi A., Rajasekaran K., Kumar P., Schuldt K. M., Bartoszek A., Manoharan N., Goldner N. K., Umhoefer C. M., and Thakar M. S. (2012) IQGAP1: a regulator of intracellular spacetime relativity. J. Immunol. 188, 2057–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abel A. M., Schuldt K. M., Rajasekaran K., Hwang D., Riese M. J., Rao S., Thakar M. S., and Malarkannan S. (2015) IQGAP1: insights into the function of a molecular puppeteer. Mol. Immunol. 65, 336–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Osman M. A., Sarkar F. H., and Rodriguez-Boulan E. (2013) A molecular rheostat at the interface of cancer and diabetes. Biochim. Biophys. Acta 1836, 166–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu Y., and Chen Y. C. (2014) Structure and function of IQ-domain GTPase-activating protein 1 and its association with tumor progression (review). Biomed. Rep. 2, 3–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson M., Sharma M., and Henderson B. R. (2009) IQGAP1 regulation and roles in cancer. Cell Signal. 21, 1471–1478 [DOI] [PubMed] [Google Scholar]
- 19. Weissbach L., Settleman J., Kalady M. F., Snijders A. J., Murthy A. E., Yan Y. X., and Bernards A. (1994) Identification of a human rasGAP-related protein containing calmodulin-binding motifs. J. Biol. Chem. 269, 20517–20521 [PubMed] [Google Scholar]
- 20. White C. D., Brown M. D., and Sacks D. B. (2009) IQGAPs in cancer: a family of scaffold proteins underlying tumorigenesis. FEBS Lett. 583, 1817–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jameson K. L., Mazur P. K., Zehnder A. M., Zhang J., Zarnegar B., Sage J., and Khavari P. A. (2013) IQGAP1 scaffold-kinase interaction blockade selectively targets RAS-MAP kinase-driven tumors. Nat. Med. 19, 626–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ho Y. D., Joyal J. L., Li Z., and Sacks D. B. (1999) IQGAP1 integrates Ca2+/calmodulin and Cdc42 signaling. J. Biol. Chem. 274, 464–470 [DOI] [PubMed] [Google Scholar]
- 23. Osman M. A., and Cerione R. A. (1998) Iqg1p, a yeast homologue of the mammalian IQGAPs, mediates cdc42p effects on the actin cytoskeleton. J. Cell Biol. 142, 443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Briggs M. W., and Sacks D. B. (2003) IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep. 4, 571–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carmon K. S., Gong X., Yi J., Thomas A., and Liu Q. (2014) RSPO-LGR4 functions via IQGAP1 to potentiate Wnt signaling. Proc. Natl. Acad. Sci. U.S.A. 111, E1221–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roy M., Li Z., and Sacks D. B. (2005) IQGAP1 is a scaffold for mitogen-activated protein kinase signaling. Mol. Cell. Biol. 25, 7940–7952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roy M., Li Z., and Sacks D. B. (2004) IQGAP1 binds ERK2 and modulates its activity. J. Biol. Chem. 279, 17329–17337 [DOI] [PubMed] [Google Scholar]
- 28. Macias M. J., Wiesner S., and Sudol M. (2002) WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 513, 30–37 [DOI] [PubMed] [Google Scholar]
- 29. Lu P. J., Zhou X. Z., Shen M., and Lu K. P. (1999) Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science 283, 1325–1328 [DOI] [PubMed] [Google Scholar]
- 30. Bedford M. T., Chan D. C., and Leder P. (1997) FBP WW domains and the Abl SH3 domain bind to a specific class of proline-rich ligands. EMBO J. 16, 2376–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen H. I., and Sudol M. (1995) The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc. Natl. Acad. Sci. U.S.A. 92, 7819–7823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hu H., Columbus J., Zhang Y., Wu D., Lian L., Yang S., Goodwin J., Luczak C., Carter M., Chen L., James M., Davis R., Sudol M., Rodwell J., and Herrero J. J. (2004) A map of WW domain family interactions. Proteomics 4, 643–655 [DOI] [PubMed] [Google Scholar]
- 33. Otte L., Wiedemann U., Schlegel B., Pires J. R., Beyermann M., Schmieder P., Krause G., Volkmer-Engert R., Schneider-Mergener J., and Oschkinat H. (2003) WW domain sequence activity relationships identified using ligand recognition propensities of 42 WW domains. Protein Sci. 12, 491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sudol M., Chen H. I., Bougeret C., Einbond A., and Bork P. (1995) Characterization of a novel protein-binding module: the WW domain. FEBS Lett. 369, 67–71 [DOI] [PubMed] [Google Scholar]
- 35. Ren J. G., Li Z., and Sacks D. B. (2007) IQGAP1 modulates activation of B-Raf. Proc. Natl. Acad. Sci. U.S.A. 104, 10465–10469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bähler M., and Rhoads A. (2002) Calmodulin signaling via the IQ motif. FEBS Lett. 513, 107–113 [DOI] [PubMed] [Google Scholar]
- 37. Tekletsadik Y. K., Sonn R., and Osman M. A. (2012) A conserved role of IQGAP1 in regulating TOR complex 1. J. Cell Sci. 125, 2041–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ang B. K., Lim C. Y., Koh S. S., Sivakumar N., Taib S., Lim K. B., Ahmed S., Rajagopal G., and Ong S. H. (2007) ArhGAP9, a novel MAP kinase docking protein, inhibits Erk and p38 activation through WW domain binding. J. Mol. Signal. 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heil A., Nazmi A. R., Koltzscher M., Poeter M., Austermann J., Assard N., Baudier J., Kaibuchi K., and Gerke V. (2011) S100P is a novel interaction partner and regulator of IQGAP1. J. Biol. Chem. 286, 7227–7238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gao C., Frausto S. F., Guedea A. L., Tronson N. C., Jovasevic V., Leaderbrand K., Corcoran K. A., Guzmán Y. F., Swanson G. T., and Radulovic J. (2011) IQGAP1 regulates NR2A signaling, spine density, and cognitive processes. J. Neurosci. 31, 8533–8542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shannon K. B. (2012) IQGAP family members in yeast, Dictyostelium, and mammalian cells. Int. J. Cell Biol. 2012, 894817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim H., White C. D., Li Z., and Sacks D. B. (2011) Salmonella enterica serotype Typhimurium usurps the scaffold protein IQGAP1 to manipulate Rac1 and MAPK signalling. Biochem. J. 440, 309–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gorman J. A., Babich A., Dick C. J., Schoon R. A., Koenig A., Gomez T. S., Burkhardt J. K., and Billadeau D. D. (2012) The cytoskeletal adaptor protein IQGAP1 regulates TCR-mediated signaling and filamentous actin dynamics. J. Immunol. 188, 6135–6144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rigothier C., Auguste P., Welsh G. I., Lepreux S., Deminière C., Mathieson P. W., Saleem M. A., Ripoche J., and Combe C. (2012) IQGAP1 interacts with components of the slit diaphragm complex in podocytes and is involved in podocyte migration and permeability in vitro. PLoS ONE 7, e37695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goto T., Sato A., Shimizu M., Adachi S., Satoh K., Iemura S., Natsume T., and Shibuya H. (2013) IQGAP1 functions as a modulator of dishevelled nuclear localization in Wnt signaling. PLoS ONE 8, e60865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheung K. L., Lee J. H., Shu L., Kim J. H., Sacks D. B., and Kong A. N. (2013) The Ras GTPase-activating-like protein IQGAP1 mediates Nrf2 protein activation via the mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK)-ERK pathway. J. Biol. Chem. 288, 22378–22386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wallrabe H., Cai Y., Sun Y., Periasamy A., Luzes R., Fang X., Kan H. M., Cameron L. C., Schafer D. A., and Bloom G. S. (2013) IQGAP1 interactome analysis by in vitro reconstitution and live cell 3-color FRET microscopy. Cytoskeleton 70, 819–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goto T., Sato A., Adachi S., Iemura S., Natsume T., and Shibuya H. (2013) IQGAP1 protein regulates nuclear localization of β-catenin via importin-β5 protein in Wnt signaling. J. Biol. Chem. 288, 36351–36360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu J., Guidry J. J., and Worthylake D. K. (2014) Conserved sequence repeats of IQGAP1 mediate binding to Ezrin. J. Proteome Res. 13, 1156–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lu R., Herrera B. B., Eshleman H. D., Fu Y., Bloom A., Li Z., Sacks D. B., and Goldberg M. B. (2015) Shigella effector OspB activates mTORC1 in a manner that depends on IQGAP1 and promotes cell proliferation. PLoS Pathog. 11, e1005200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Struhl K. (1991) Reverse biochemistry: methods and applications for synthesizing yeast proteins in vitro. Methods Enzymol. 194, 520–535 [DOI] [PubMed] [Google Scholar]
- 52. Lee T., Hoofnagle A. N., Kabuyama Y., Stroud J., Min X., Goldsmith E. J., Chen L., Resing K. A., and Ahn N. G. (2004) Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol. Cell 14, 43–55 [DOI] [PubMed] [Google Scholar]
- 53. Canagarajah B. J., Khokhlatchev A., Cobb M. H., and Goldsmith E. J. (1997) Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell 90, 859–869 [DOI] [PubMed] [Google Scholar]
- 54. Prowse C. N., Deal M. S., and Lew J. (2001) The complete pathway for catalytic activation of the mitogen-activated protein kinase, ERK2. J. Biol. Chem. 276, 40817–40823 [DOI] [PubMed] [Google Scholar]
- 55. Seger R., Ahn N. G., Boulton T. G., Yancopoulos G. D., Panayotatos N., Radziejewska E., Ericsson L., Bratlien R. L., Cobb M. H., and Krebs E. G. (1991) Microtubule-associated protein 2 kinases, ERK1 and ERK2, undergo autophosphorylation on both tyrosine and threonine residues: implications for their mechanism of activation. Proc. Natl. Acad. Sci. U.S.A. 88, 6142–6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cohen-Armon M., Visochek L., Rozensal D., Kalal A., Geistrikh I., Klein R., Bendetz-Nezer S., Yao Z., and Seger R. (2007) DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol. Cell 25, 297–308 [DOI] [PubMed] [Google Scholar]
- 57. Robinson M. J., Harkins P. C., Zhang J., Baer R., Haycock J. W., Cobb M. H., and Goldsmith E. J. (1996) Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5′-triphosphate. Biochemistry 35, 5641–5646 [DOI] [PubMed] [Google Scholar]
- 58. Bardwell L., and Thorner J. (1996) A conserved motif at the amino termini of MEKs might mediate high-affinity interaction with the cognate MAPKs. Trends Biochem. Sci. 21, 373–374 [PubMed] [Google Scholar]
- 59. Bardwell L., Cook J. G., Chang E. C., Cairns B. R., and Thorner J. (1996) Signaling in the yeast pheromone response pathway: specific and high-affinity interaction of the mitogen-activated protein (MAP) kinases Kss1 and Fus3 with the upstream MAP kinase kinase Ste7. Mol. Cell. Biol. 16, 3637–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kallunki T., Deng T., Hibi M., and Karin M. (1996) c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell 87, 929–939 [DOI] [PubMed] [Google Scholar]
- 61. Yang S. H., Yates P. R., Whitmarsh A. J., Davis R. J., and Sharrocks A. D. (1998) The Elk-1 ETS-domain transcription factor contains a mitogen-activated protein kinase targeting motif. Mol. Cell. Biol. 18, 710–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kusari A. B., Molina D. M., Sabbagh W. Jr, Lau C. S., and Bardwell L. (2004) A conserved protein interaction network involving the yeast MAP kinases Fus3 and Kss1. J. Cell Biol. 164, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kelkar N., Gupta S., Dickens M., and Davis R. J. (2000) Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol. Cell. Biol. 20, 1030–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dickens M., Rogers J. S., Cavanagh J., Raitano A., Xia Z., Halpern J. R., Greenberg M. E., Sawyers C. L., and Davis R. J. (1997) A cytoplasmic inhibitor of the JNK signal transduction pathway. Science 277, 693–696 [DOI] [PubMed] [Google Scholar]
- 65. Bardwell L., and Shah K. (2006) Analysis of mitogen-activated protein kinase activation and interactions with regulators and substrates. Methods 40, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cargnello M., and Roux P. P. (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Roskoski R., Jr. (2012) ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol. Res. 66, 105–143 [DOI] [PubMed] [Google Scholar]
- 68. Peti W., and Page R. (2013) Molecular basis of MAP kinase regulation. Protein Sci. 22, 1698–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fang L., Lu W., Choi H. H., Yeung S. C., Tung J. Y., Hsiao C. D., Fuentes-Mattei E., Menter D., Chen C., Wang L., Wang J., and Lee M. H. (2015) ERK2-dependent phosphorylation of CSN6 is critical in colorectal cancer development. Cancer Cell 28, 183–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chang C. I., Xu B. E., Akella R., Cobb M. H., and Goldsmith E. J. (2002) Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol. Cell 9, 1241–1249 [DOI] [PubMed] [Google Scholar]
- 71. Molina D. M., Grewal S., and Bardwell L. (2005) Characterization of an ERK-binding domain in microphthalmia-associated transcription factor and differential inhibition of ERK2-mediated substrate phosphorylation. J. Biol. Chem. 280, 42051–42060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tanoue T., Adachi M., Moriguchi T., and Nishida E. (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell. Biol. 2, 110–116 [DOI] [PubMed] [Google Scholar]
- 73. Jacobs D., Glossip D., Xing H., Muslin A. J., and Kornfeld K. (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 13, 163–175 [PMC free article] [PubMed] [Google Scholar]
- 74. Piserchio A., Ramakrishan V., Wang H., Kaoud T. S., Arshava B., Dutta K., Dalby K. N., and Ghose R. (2015) Structural and dynamic features of F-recruitment site driven substrate phosphorylation by ERK2. Sci. Rep. 5, 11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Aqeilan R. I., Donati V., Palamarchuk A., Trapasso F., Kaou M., Pekarsky Y., Sudol M., and Croce C. M. (2005) WW domain-containing proteins, WWOX and YAP, compete for interaction with ErbB-4 and modulate its transcriptional function. Cancer Res. 65, 6764–6772 [DOI] [PubMed] [Google Scholar]
- 76. Jäger M., Dendle M., and Kelly J. W. (2009) Sequence determinants of thermodynamic stability in a WW domain: an all-β-sheet protein. Protein Sci. 18, 1806–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Barr R. K., Boehm I., Attwood P. V., Watt P. M., and Bogoyevitch M. A. (2004) The critical features and the mechanism of inhibition of a kinase interaction motif-based peptide inhibitor of JNK. J. Biol. Chem. 279, 36327–36338 [DOI] [PubMed] [Google Scholar]
- 78. Ho D. T., Bardwell A. J., Abdollahi M., and Bardwell L. (2003) A docking site in MKK4 mediates high affinity binding to JNK MAPKs and competes with similar docking sites in JNK substrates. J. Biol. Chem. 278, 32662–32672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stebbins J. L., De S. K., Machleidt T., Becattini B., Vazquez J., Kuntzen C., Chen L. H., Cellitti J. F., Riel-Mehan M., Emdadi A., Solinas G., Karin M., and Pellecchia M. (2008) Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc. Natl. Acad. Sci. U.S.A. 105, 16809–16813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bhattacharyya R. P., Reményi A., Good M. C., Bashor C. J., Falick A. M., and Lim W. A. (2006) The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science 311, 822–826 [DOI] [PubMed] [Google Scholar]
- 81. Chen H. I., Einbond A., Kwak S. J., Linn H., Koepf E., Peterson S., Kelly J. W., and Sudol M. (1997) Characterization of the WW domain of human yes-associated protein and its polyproline-containing ligands. J. Biol. Chem. 272, 17070–17077 [DOI] [PubMed] [Google Scholar]
- 82. Einbond A., and Sudol M. (1996) Towards prediction of cognate complexes between the WW domain and proline-rich ligands. FEBS Lett. 384, 1–8 [DOI] [PubMed] [Google Scholar]
- 83. McNulty D. E., Li Z., White C. D., Sacks D. B., and Annan R. S. (2011) MAPK scaffold IQGAP1 binds the EGF receptor and modulates its activation. J. Biol. Chem. 286, 15010–15021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bañón-Rodríguez I., Gálvez-Santisteban M., Vergarajauregui S., Bosch M., Borreguero-Pascual A., and Martín-Belmonte F. (2014) EGFR controls IQGAP basolateral membrane localization and mitotic spindle orientation during epithelial morphogenesis. EMBO J. 33, 129–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bardwell A. J., Flatauer L. J., Matsukuma K., Thorner J., and Bardwell L. (2001) A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J. Biol. Chem. 276, 10374–10386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gordon E. A., Whisenant T. C., Zeller M., Kaake R. M., Gordon W. M., Krotee P., Patel V., Huang L., Baldi P., and Bardwell L. (2013) Combining docking site and phosphosite predictions to find new substrates: identification of smoothelin-like-2 (SMTNL2) as a c-Jun N-terminal kinase (JNK) substrate. Cell Signal. 25, 2518–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ruxton G. D. (2006) The unequal variance t-test is an underused alternative to Student's t-test and the Mann-Whitney U test. Behav. Ecol. 17, 688–690 [Google Scholar]